Reversible swelling transitions in stimuli-responsive layer-by-layer films containing block copolymer micelles†
Julia
Gensel
a,
Inna
Dewald
a,
Johann
Erath
a,
Eva
Betthausen
b,
Axel. H. E.
Müller
b and
Andreas
Fery
*a
aDepartment of Physical Chemistry II, University of Bayreuth, Universitätsstr. 30, 95440 Bayreuth, Germany. E-mail: andreas.fery@uni-bayreuth.de
bDepartment of Macromolecular Chemistry II, University of Bayreuth, Universitätsstr. 30, 95440 Bayreuth, Germany
First published on 6th September 2012
Abstract
We present a new nanoporous multilayer system with a reversible pH-triggered swelling transition. Using the layer-by-layer approach, pH-responsive block copolymer micelles with a hydrophobic core, a weak polyanion shell and a strong polycation corona formed from an ABC triblock terpolymer are included within multilayer films. The approach of complexing the strong polycationic corona with a strong polyanion leads to the creation of novel double-end-tethered polyelectrolyte brush structures confined between the hydrophobic micellar cores and the interpolyelectrolyte complexes. The swelling degree, morphology as well as the mechanical properties of the coatings are reversibly tunable by the solution pH due to the ionization-induced swelling of the pH-sensitive polyelectrolyte-brush-like shell of the incorporated micelles resulting in large-scale volumetric changes of the film. Moreover, controlling the internal film architecture by the number of deposition steps allows tuning the properties of the porous multilayers such as the density of incorporated micelles, the porosity, and the equilibrium swelling degree to more than 1200%.
Introduction
The design of “smart” coatings, which can reversibly switch their physico-chemical characteristics in reaction to external stimuli is a very attractive research field regarding its diverse applications, e.g. in drug delivery and microfluidic systems, cell tissue engineering, as well as sensing, or actuation (see reviews 1–10). The adsorption of stimuli-responsive soft matter systems on a solid support is an effective and simple way to fabricate switchable surfaces. Coatings which respond to changes in pH or ionic strength can be obtained by chemical grafting of polymer or polyelectrolyte brushes onto planar11–14 or curved15 surfaces. Alternatively, thin films of polymer networks can be used as smart coatings. In most of these cases hydrogel-like films can uptake large amounts of water, whereby their swelling degree (the ratio of swollen to dry volume), which can be controlled by external physical or chemical signals, determines their mechanical and optical properties, permeability, adhesion etc. Such a modulation of the mechanical properties of coatings can be used for example to control cell adhesion.16Another elegant approach to design smart surfaces is the adsorption of block copolymer micelles. Several research groups have studied the stimuli-responsive behavior of an adsorbed monolayer of micelles.17–21 Recently, we reported on the immobilization of micelles of a linear ABC triblock terpolymer consisting of polybutadiene (B), poly(methacrylic acid) (MAA), and quaternized poly(2-(dimethylamino)ethyl methacrylate) (Dq), BMAADq on silica.22 We have found that by controlling the solution pH at the solid–liquid interface of the adsorbed micelles, it is possible to reversibly switch the micellar morphology and charge density of the corona. This pH-responsive behavior can be controlled by the ionization degree of the weak polyelectrolyte middle block (MAA) and is completely reversible on a short time scale. More recently, we have successfully used these switchable coatings as active surfaces for bio-applications, in particular for controlled self-regulated bacteria release.23 However, the long-term treatment at pH < pKa,apparent of MAA causes irreversible changes in the morphology of the immobilized BMAADq micelles.22 In general, the instability of a monolayer of stimuli-responsive micelles is a challenging aspect17,19,22,24,25 which is critical for many applications. One way to improve the resilience of such structures is the chemical cross-linking of the hydrophobic cores.22 Sukhishvili and co-workers reported another facile method for stabilization against environmental influences for otherwise instable surface-attached micelles via self-assembly of the micelles with a polyelectrolyte layer using physical cross-linking.24 They observed irreversible morphological changes and desorption of a significant fraction of temperature-responsive micelles of poly(N-vinylpyrrolidone)-b-poly(N-isopropylacryl-amide) into the solution, while the same surface-adsorbed micelles covered with a top layer of poly(methacrylic acid) maintained their original structural integrity.
Besides the improved stability, this strategy allows coverage of any type of substrate and fine-tuning the thickness and nanostructure of the stimuli-responsive film via layer-by-layer (LbL) assembly of oppositely charged polymers.26–28 In particular, Tan et al. reported that temperature-sensitive core–shell micelles of ABA triblock copolymers can be incorporated within the LbL films while retaining their stimuli-responsive properties.29,30 The obtained films are generally very thin (10–100 nm) enabling a rapid response to environmental changes as compared to bulk materials.
The incorporation of charged nanoparticles31,32 as well as diblock copolymer micelles into multilayers with integrated stimuli-responsive properties has been previously reported for one-type-micellar33–37 or micelle–micelle-systems.38–40 These types of systems are particularly suitable for the fabrication of hydrogel-like films35 for drug delivery,24,34,36 coatings with tunable optical properties,33,38 or antibacterial coatings.41 Furthermore, LbL assembly of micelles offers the advantage of fabricating porous thin films38 without any of the additional post-treatment steps usually required for their preparation.42 A special class of stimuli-responsive porous hydrogel-like thin films combines fast response and precise control over the pore closing/opening by an external signal.7,8
Here, we study a new LbL system, consisting of ABC block terpolymer micelles with a hydrophobic polybutadiene (B) core, a pH-sensitive poly(methacrylic acid) (MAA) shell, and a cationic corona of quaternized poly(2-(dimethylamino)ethyl methacrylate) (Dq) (BMAADq) and linear strong polyanion poly(sodium 4-styrene sulfonate) (PSS). Due to the pH-sensitive character of the incorporated micelles, the system allows the ionization degree of the weak polyelectrolyte block (MAA) to be varied by changing the pH. One remarkable feature of the system is that the pH-sensitive block is not a component of the multilayer complexes, but is covalently bound to the micellar core on the one side and to the multilayer-forming corona on the other side. This approach leads to the formation of a double-end-tethered weak polyelectrolyte brush-like shell. Hence, we investigated the effect of solution pH on the swelling behavior as well as on the mechanical properties of the hydrogel film. In addition, the influence of the swelling on the film porosity was studied.
Materials and methods
Materials
The triblock terpolymer consisting of polybutadiene (B), poly(methacrylic acid) (MAA), and quaternized poly(2-(dimethylamino)ethyl methacrylate) (Dq), B800MAA200Dq285 (subscripts denoting the degrees of polymerization of the respective blocks) with an Mn of ∼110![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and a PDI of 1.10 was synthesized via sequential living anionic polymerization followed by polymer-analogous modifications as described elsewhere.43 In particular, the poly(2-(dimethylamino)ethyl methacrylate) block was exhaustively quaternized with dimethyl sulfate. After quaternization in a dioxane–water mixture (1:1, v/v), the BMAADq terpolymer was dialyzed against pH 10 buffer solution to obtain a micellar stock solution with a concentration of 0.5 g L−1. From this stock solution changes in pH were performed by dialyzing against the corresponding buffer solutions (pH 4, VWR, AVS Titrinorm).43 The obtained micelles have a spherical shape as confirmed by cryogenic TEM and DLS experiments.43 The hydrodynamic radius Rh of the micelles in pH 4 buffer was determined by DLS to 107 nm. According to the core radius obtained via cryo-TEM, the aggregation number, Nagg, of the BMAADq micelles can be calculated with eqn (1):
000 g mol−1 and a PDI of 1.10 was synthesized via sequential living anionic polymerization followed by polymer-analogous modifications as described elsewhere.43 In particular, the poly(2-(dimethylamino)ethyl methacrylate) block was exhaustively quaternized with dimethyl sulfate. After quaternization in a dioxane–water mixture (1:1, v/v), the BMAADq terpolymer was dialyzed against pH 10 buffer solution to obtain a micellar stock solution with a concentration of 0.5 g L−1. From this stock solution changes in pH were performed by dialyzing against the corresponding buffer solutions (pH 4, VWR, AVS Titrinorm).43 The obtained micelles have a spherical shape as confirmed by cryogenic TEM and DLS experiments.43 The hydrodynamic radius Rh of the micelles in pH 4 buffer was determined by DLS to 107 nm. According to the core radius obtained via cryo-TEM, the aggregation number, Nagg, of the BMAADq micelles can be calculated with eqn (1):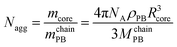 | (1) |
000 g mol−1) was purchased from Sigma-Aldrich. HCl and NaOH solution (0.1 mol L−1, Grüssing) were used to adjust the pH of water.
Multilayer deposition
The LbL films were assembled on silicon wafers (CrysTec) from 0.45 g L−1 polymer solutions via dip coating. The substrates were cleaned using the RCA technique44 (sonication in a 1:1 mixture of water and 2-propanol for 15 min, followed by heating at 70 °C in a 5:1:1 mixture of water, 25% ammonia solution, and 30% hydrogen peroxide solution for 10 min). The freshly cleaned substrates were then dipped into a solution of BMAADq micelles in pH 4 buffer solution (VWR, AVS Titrinorm, ionic strength ∼ 0.05 M) for 15 min before rinsing with water. Next, the substrates were dipped into an aqueous solution of PSS (adjusted to pH 4 with 0.1 M HCl) for 15 min. The films were dried in a stream of nitrogen before characterization.
Methods
Ellipsometry measurements in air were performed with a Sentech SE 850 spectroscopic ellipsometer at a constant incidence angle of 70°. A home-built liquid cell45 was used for in situ ellipsometry in water of a different pH at a constant incidence angle of 65°. Measurements were performed after a minimum equilibration time of 20 min.Atomic force microscopy (AFM) images were taken with a commercial AFM (Dimension™ 3100 equipped with a NanoScope® V controller, both from Bruker AXS Inc., USA) operating in TappingMode™ using Si3N4 cantilevers from Olympus with a typical spring constant of ∼42 N m−1 and a typical resonance frequency of 300 kHz (OMCL-AC160TS).
Scanning in fluid was performed with a direct driven TappingMode™ probe holder (DTFML-DD-HE, Bruker, AXS Inc., USA) using cantilevers with a spring constant of ∼0.06 N m−1 and a resonance frequency of 12–24 kHz (Bruker, SNL-10).
Scanning electron microscopy (SEM) measurements were obtained on a Gemini Leo 1550 instrument operating at 3 keV. Samples were sputtered with a 1.3 nm thin platinum layer.
The colloidal probe atomic force microscopy (CP AFM) measurements were performed on an Asylum MFP 3D AFM (Mannheim, Germany) in a water droplet at the corresponding pH. Glass particles (Polysciences, Germany) were used as force sensors. The CPs, made of SiO2, were glued with an epoxy resin (UHU schnellfest, Germany) to pre-calibrated cantilevers (force constant ∼0.1 N m−1, NSC 12, tipless, noAl, Micromash, Estonia) using a micromanipulator (MP-285, Shutter Instrument, USA) and an inverted optical microscope (Axiovert 200, Zeiss, Germany). Force constants of the cantilevers were determined by the thermal noise method introduced by Hutter and Bechhoefer.46 For all the presented data, a cantilever with a force constant of 0.125 N m−1 and a CP with a radius of R = 23 μm was used. The optical lever sensitivity was always detected prior to recording the data by reference measurements on a hard glass substrate.
Results and discussion
Layer-by-layer assembly
To develop pH-responsive layer-by-layer hydrogel coatings, we used a linear ABC triblock terpolymer consisting of polybutadiene (B), poly(methacrylic acid) (MAA), and quaternized poly(2-(dimethylamino)ethyl methacrylate) (Dq), BMAADq. The molecular structure is given in Fig. 1a. Details regarding the synthesis and characterization of the polymer can be found elsewhere.43 In aqueous solution, BMAADq self-assembles into core–shell–corona micelles with a hydrophobic B core, a pH-sensitive MAA shell and a strong cationic Dq corona.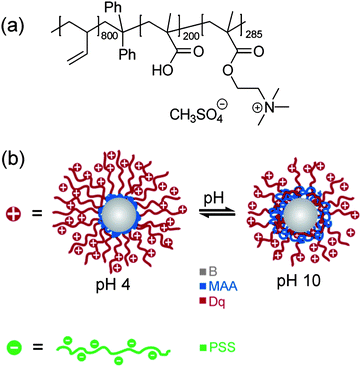 | ||
| Fig. 1 Chemical structure of the BMAADq triblock terpolymer (a), schematic representation of the solution structure of positively charged BMAADq micelles at pH 4 and pH 10, and negatively charged PSS (b). | ||
At low pH (pH < pKa,app of MAA ≈ 5.5 (ref. 47)), the pH-sensitive MAA block is uncharged and does not form an IPEC, but rather phase separates from the corona as shown in Fig. 1b. At high pH, this block is negatively charged through the deprotonation of the carboxylic acid groups leading to intramicellar interpolyelectrolyte complex (im-IPEC) formation with the cationic corona of Dq. Hence, the composition of the micellar shell as well as the charge density of the corona can be controlled by the solution pH (Fig. 1b).
Due to the cationic character of the micelles, we chose anionic poly(sodium 4-styrene sulfonate) (PSS) as the counterpart for the layer-by-layer assembly. PSS is known to form stable complexes/multilayers with strongly charged polycations (like Dq) because of its permanently high charge density.48
We prepared LbL films at pH 4. At this pH, the MAA shell is protonated and does not form an im-IPEC with the Dq corona. Therefore, all of the coronal chains are expected to form stable complexes with PSS. Consequently, the MAA block should form an uncharged shell around the B core at this pH. Fig. 2a displays the ellipsometric dry film thickness plotted versus the number of triblock terpolymer/polyanion deposition steps, x. The film growth follows a square root dependency (Fig. 2b). This behavior indicates a diffusion-limited assembly process, which may originate from the diffusion of the micelles into the pores of the film: further deposited micelles do not adsorb on top of the film, but diffuse through the pores to fill them and thus increase the micelle density in the multilayers.
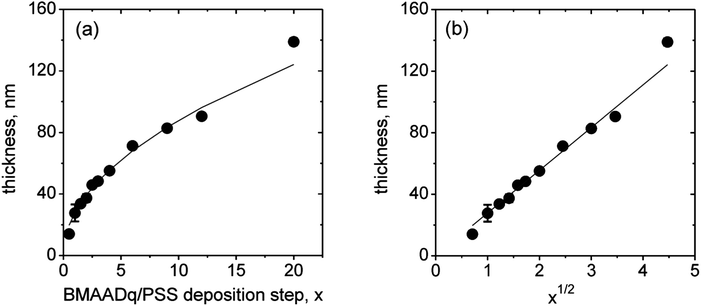 | ||
| Fig. 2 Ellipsometric dry thickness vs. the number of BMAADq/PSS deposition steps, x (a) and vs. the square root of x (b). The lines are a square root fit and a linear fit to the data, respectively. | ||
To obtain detailed structural information about the LbL multilayers, atomic force microscopy (AFM) and scanning electron microscopy (SEM) measurements were performed on dried samples. From the AFM height image of one bilayer of BMAADq/PSS (denoted as (BMAADq/PSS)1) (Fig. 3a), the average diameter and the height of the spherical-cap-like structures (based on the average of at least 30 features) were estimated to be (125 ± 8) nm and (49 ± 5) nm, respectively. The dimensions obtained by AFM correspond to the topography image including the core, the shell, and the IPEC between the Dq corona and the extrinsic homopolymer PSS. In contrast, the SEM image of one bilayer in the dry state (Fig. 3c) shows the hydrophobic cores (bright) surrounded by a dark shell and the Dq/PSS IPEC indicating the retained core–shell–IPEC-structure upon incorporation of the micelles into multilayers. The diameter of the hydrophobic core is (71 ± 8) nm (based on the average of at least 30 micelles), which is comparable with the diameter of the cores in pH 4 buffer solution (∼64 nm).43 In our previous work, we have shown that the adsorption of BMAADq micelles onto a silica surface follows the random sequential adsorption (RSA) model with a maximum surface coverage of 0.54.22 Therefore, micelles can only be randomly dispersed on the substrate for the first layer leading to a porous structure of the resulting LbL films (Fig. 3). The adsorption of the same micelles on the substrates covered with a layer of negatively charged PSS resulted in similar adsorption kinetics as well as the saturation value of ∼0.5 (Fig. S1, ESI†). A further example is the random adsorption of the micelles studied on ultrasonically formed mesoporous aluminum.23 These examples confirm that the system studied here can be used to cover different substrates in a controlled and reproducible manner. Note that since this hit-stick RSA adsorption behavior is associated with the immobilization of micelles, rearrangements inside the film should not be possible. This is consistent with the work of Kabanov and co-workers: the high affinity of PSS to polycations imposes kinetic restrictions on the exchange reactions of PSS/Dq IPECs.50 Therefore, to the best of our knowledge, the non-linear buildup and the reduction of porosity do not occur as a result of the rearrangement of micelles during the deposition.
 | ||
| Fig. 3 1.5 μm × 1.5 μm AFM height images (a and b) (color equates to z = 0–100 nm) and SEM images (c–f) of (BMAADq/PSS)x porous films. x is the number of build-up steps. Arrows indicate the formation of hydrophobic bridges. | ||
The assumption of micelles filling the void space in the film instead of attaching on the top (resulting in a non-linear buildup behavior (Fig. 2)) is supported by a detailed study of the nanostructure (Fig. 3). The AFM height and SEM images in Fig. 3 demonstrate a significant increase in the micelle density in the film and therefore a decreased porosity with increasing number of deposition steps.
The porosity (P) of the films was evaluated from refractive index measurements applying the mixing rule to the Lorentz–Lorenz equation51
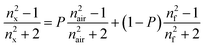 | (2) |
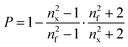 | (3) |
Fig. 4 shows the refractive indices nx of porous films measured by ellipsometry and the resulting porosity values obtained from (3), assuming that the refractive index of the dense film nf = 1.51 (estimated by ellipsometry of drop-coated PSS (n = 1.51) and BMAADq (n = 1.51) films).
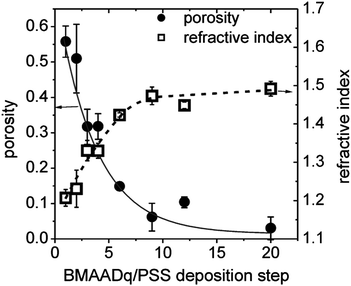 | ||
| Fig. 4 Porosity (●) and refractive index (□) vs. the number of BMAADq/PSS deposition steps. The lines are guides to the eye. | ||
The porosity of the multilayers can be easily tuned between ∼50% and 0% by the number of deposition steps. Such interconnected micellar network formation can be explained by the generation of hydrophobic bridges during the assembly process as indicated by the arrows in the SEM-images (Fig. 3c–e). Hydrophobic bridges were also found in aqueous solutions of the same micelles43 as well as those of other block terpolymer micelles with a low glass transition temperature of the core-forming block.52,53 Still, mainly spherical particles are found in the multilayers indicating that the core–shell structure remains intact upon integration into multilayers.
Stimulus response
On the basis of the retained core–shell structure of the LbL-incorporated micelles and due to the pH-responsive character of the polyelectrolyte brush-like MAA shell, we investigated the swelling behavior of these films by in situ ellipsometry. To confirm data obtained from ellipsometry, we additionally measured the height difference at the edge of a scratch by AFM in a liquid cell. Exemplarily, results for a three-bilayer-film (BMAADq/PSS)3 at different pH between pH 4 and 12 are shown in Fig. 5a. Films with differing number of bilayers follow the same trend with varying pH.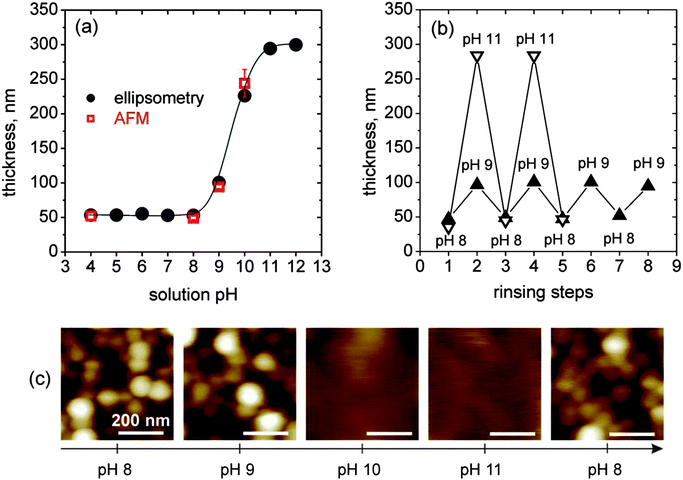 | ||
Fig. 5
In situ measurements of the ellipsometric thickness (●) and liquid cell AFM height difference at a scratch ( ) of (BMAADq/PSS)3vs. the solution pH (a), in situ ellipsometry measurements of reversible pH-triggered swelling and contraction of (BMAADq/PSS)3 film (b) and corresponding 500 nm × 500 nm AFM height images (color equates to z = 0–100 nm) in water at different pH values (c). ) of (BMAADq/PSS)3vs. the solution pH (a), in situ ellipsometry measurements of reversible pH-triggered swelling and contraction of (BMAADq/PSS)3 film (b) and corresponding 500 nm × 500 nm AFM height images (color equates to z = 0–100 nm) in water at different pH values (c). | ||
Swelling of the film is observed when the pH is increased to alkaline values with a transition at pH ∼ 9.5. The increase in thickness corresponds to the deprotonation of the carboxylic groups of the MAA shell and can be tuned by the degree of dissociation α. In acidic solutions at α ∼ 0, the film is in a contracted state. With increasing pH, α increases leading to a higher charge density and therefore increased repulsive interactions between the COO− groups. This results in a higher osmotic pressure of trapped counter ions, a stretching of the MAA chains and a swelling of the film.
The transition region at pH ∼ 9.5 (corresponding to the pH at which α ∼ 0.5) is significantly higher than the apparent pKa value of PMAA reported in the literature (pKa,app ∼ 5.5 (ref. 47)) indicating that when confined into the micellar multilayer film, MAA becomes a weaker polyacid in comparison to its behavior in dilute solution. The shift in the apparent pKa values of weak polyelectrolytes upon incorporation into multilayered films is also known from other work.54–58 In contrast to our results, in the latter cases, the apparent pKa values of incorporated weak polyacids and polybases were shifted by ∼1 to 4 pH units to the acidic or alkaline values, respectively. The above cited results indicate that the polyacid becomes a stronger acid if it is the component of a polyelectrolyte multilayer film.
However, we observed an opposite effect, which originates from the fact that the MAA block is not a component of the multilayer complexes. In our system, the MAA block, which is covalently bound to the core-forming B block on the one side and to the Dq block (forming stable IPECs with PSS) on the other side, can be described as a spherical polyelectrolyte brush (around the micellar core), confined between electrostatically assembled layers. In fact, the shift to higher pH values agrees with experiments on polyelectrolyte brushes. The swelling transition of grafted PMAA brush layers with a high grafting density was found to be shifted to pH 9 as a result of the Coulombic repulsion of neighboring charges.59 Currie et al. reported similar pKa shifts of poly(acrylic acid) brushes that became more pronounced with increasing grafting density.60 In potentiometric titrations of multi-arm star-shaped poly(acrylic acid) we also observed an increase in the pKa,app with increasing arm number.61 These findings are additionally supported by theoretical predictions.62
The pH switch is fully reversible after several pH 8/pH 9 and pH 8/pH 11 cycling steps (Fig. 5b). The MAA domains respond to pH switching by changing between high and low ionized states resulting in the swelling and shrinking of the multilayers.
Although a single layer of micelles studied here shows irreversible morphological changes at pH 4 or lower,22 micelles that are covered with a layer of PSS are very robust and stable after long-term treatment (in the order of several hours) at pH values between pH 4 and pH 12. This is due to the formation of a stable IPEC of PSS and the cationic corona of the micelles. A similar method for the stabilization against environmental influences for otherwise instable surface-attached micelles was reported by Sukhishvili and Zhu.24 They observed that surface-adsorbed micelles covered with a top layer of poly(methacrylic acid) remained stable and maintained their original structural integrity, while the same uncovered micelles showed irreversible morphological changes and desorption into the solution.
The corresponding AFM images in water at different pH values are shown in Fig. 5c. At slightly alkaline pH (pH 8), the film maintains its porous micellar morphology with an average pore diameter of ∼70 nm ranging from 30 to 160 nm. At higher pH (pH 9), the micellar diameter increases while the pore diameter decreases to a mean value of ∼50 nm. Here, the pores range in diameter from 20 to 120 nm. At even higher pH (pH 10 and pH 11), the volume filling factor V = Vmicelle/Vtotal, film is changed significantly upon swelling. After decreasing the pH to 8, the film regained its original porous structure indicating the reversible morphology changes triggered by pH. On the basis of the ellipsometric and AFM studies, we propose a schematic illustration of the reversible pH-triggered swelling and contraction of BMAADq/PSS multilayers (Fig. 6).
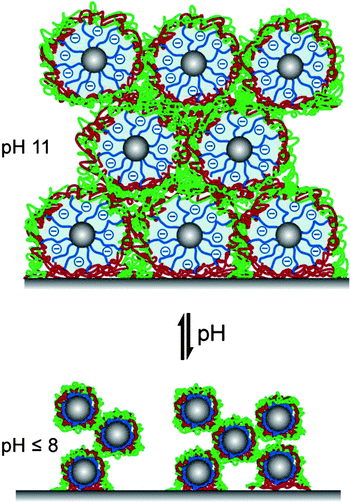 | ||
| Fig. 6 Proposed schematic illustration of reversible pH-triggered swelling and contraction of BMAADq/PSS multilayers. | ||
As IPECs have an important and characteristic feature of competition (polyion exchange) and replacement (polyion substitution) reactions, the ionized MAA brush at pH 11 might compete with PSS in participating complexation with quaternized amine groups of the coronal chains. However, the rate and position of the equilibrium of these reactions is strongly dependent on the nature of the polyelectrolyte pair within a polyelectrolyte complex or multilayer (e.g. reviews 49, 50). A comparable polyion interchange reaction in polyelectrolyte multilayers (PSS or PMAA and a polycation containing quaternized amine groups) was studied by Jomaa and Schlenoff.63 They demonstrated that PSS irreversibly replaced all PMAA chains that were already part of the multilayer film at alkaline pH, when PMAA groups are completely ionized. Exposure of the PSS-containing multilayers to an alkaline solution of PMAA yielded no incorporation of PMAA as this would result in the formation of the energetically less favorable complex. This is as well in line with earlier findings of Kabanov and coworkers that in mixtures of polyanions, a polycation preferentially binds with sulfonate-containing polyanions.49,50,64 PSS is one of the strongest competitors for associating with polycations, whereas PMAA is known to form weakly bound complexes.49,63,65 Dubas and Schlenoff reported the difference in free energy of complexation of a quaternized amine group with an acrylic acid group and a sulfonate group to be 14.9 kJ mol−1.65 As a consequence of this strong binding of sulfonate-containing polyanions with polycations, the interpolyelectrolyte chain exchange is inhibited. Thus, for our system, the positively charged Dq corona will preferentially associate with PSS rather than PMAA, even at pH 11.
Mechanical characterization
As the BMAADq/PSS multilayers swell upon increasing the pH of the solution, we expect that the mechanical properties change with the degree of swelling. To study these changes we used the colloidal probe technique introduced by Butt66 and Ducker.67 Data were recorded by force-mapping measurements with an AFM. The detected force–distance curves were transformed into force–indentation curves of the coatings by subtracting the effect of the cantilever deflection.Fig. 7a shows the recorded force–indentation data for different swelling states of a (BMAADq/PSS)3 multilayer. As upper force threshold we defined 20 nN which corresponds to deformations of around 80% of the film thickness. Measurements on the same spot of the sample show that the film is not plastically deformed (see Fig. S2 in the ESI†).
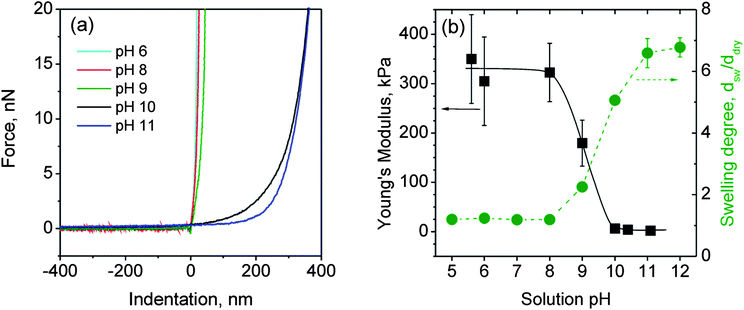 | ||
Fig. 7 Force–indentation data for different swelling states (a) and Young's modulus (■) and swelling degree ( ) (determined by ellipsometric measurements) as a function of the solution pH for a (BMAADq/PSS)3 film (b). ) (determined by ellipsometric measurements) as a function of the solution pH for a (BMAADq/PSS)3 film (b). | ||
Since the adhesion in liquid is low and the films are not plastically deformed during the measurement, we used the linear elasticity theory to evaluate the mechanical properties of the system. The indentation of a sphere into a linear elastic infinite half space can be described by the Hertz model68
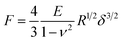 | (4) |
To avoid substrate effects, only data below 30% of indentation were used for the Hertz analysis. However, the values should be treated only as a rough estimation because the Hertz model is not exactly suited for inhomogeneous systems as in our case. Still, our data is well represented by the Hertz model as shown by the comparison of force curves plotted in log–log scale together with the power function F = δ3/2, confirming the Hertzian power law (Fig. S3, ESI†).
The Young's modulus was estimated using eqn (4). Fig. 7b summarizes the obtained data of the Young's modulus and the swelling degree as a function of the solution pH. Each value for the Young's modulus contains at least 300 data points.
The swelling degree was estimated from the ellipsometric measurements as dsw/ddry, where dsw and ddry are the film thicknesses in the swollen and dry state, respectively. As can be clearly seen, the Young's modulus is inversely proportional to the degree of swelling. The modulus decreases with increasing pH (6 to 11) by at least 2 orders of magnitude from around 100 to 1 kPa. Surface force spectroscopy studies performed by Tsukruk and co-workers have also shown a softening of pH-sensitive LbL capsules by 2 orders of magnitude (in the range of 0.1–1 MPa in the contracted and 10 kPa in the swollen state) within a narrow pH range.69–71 Their results are comparable to our system: for the quenched state (pH ≤ 8), the modulus is nearly constant at around 300 kPa, which is typical for partially swollen hydrogel films16 or LbL capsules.69 The transition between the contracted and swollen states occurs at pH ∼ 9 corresponding to the estimated apparent pKa value of the MAA brush. At pH ≥ 10 (highly swollen state), the modulus converges to the low kPa range. This low kPa range of the Young's modulus is characteristic for highly swollen hydrogel materials.69–72
Effect of film thickness on swelling behavior
Due to significant differences in the density of the incorporated micelles and the porosity of the (BMAADq/PSS)x layers (Fig. 3), we studied the effect of film microstructure on the macroscopic swelling behavior. The swelling degree (the ratio of swollen to dry volume) of surface-attached films equals the linear degree of swelling73 and can be calculated using ellipsometric thickness measurements. The equilibrium swelling degrees of the hydrogel-like films as a function of the dry thickness are summarized in Fig. 8a.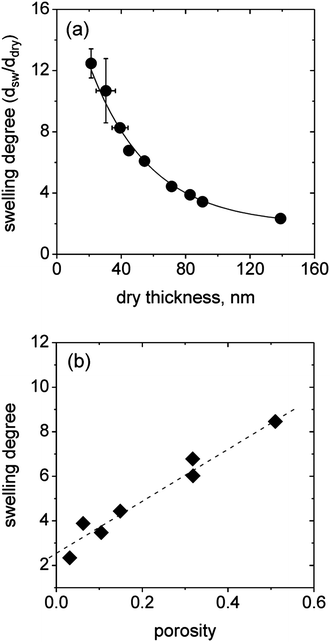 | ||
| Fig. 8 Swelling degree vs. dry thickness determined by ellipsometric measurements (a) and swelling degree vs. porosity showing a linear dependency (b). | ||
Ellipsometric measurements show that the swelling degree is strongly dependent on the LbL film thickness and therefore on the porosity of the multilayers, which can be tuned by the number of LbL deposition steps. The maximum swelling degree of ∼12 is obtained for the thinnest films made by one deposition step. With increasing thickness (decreased porosity), the swelling degree drops to a value of 2 for the thickest film studied.
Taking the differences in the microstructure into account, the observed trend of decreased swelling degree with increasing dry thickness is reasonable. The equilibrium swelling degree is a balance between two opposing forces: on the one hand the electrostatic self-repulsion and osmotic pressure of the trapped counter ions, which favor swelling, and on the other hand the elastic free energy, which opposes swelling. Thus, with increasing dry thickness the decreasing void fraction confines the particle volume change to one dimension perpendicular to the substrate. Consequently, steric effects may lead to a dominating contribution of the stretching entropy resulting in decreased swelling.
Additionally, the solvent gradient induced by the accumulation of solvent at the outmost layers may impact the swelling behavior. A similar trend of decreased swelling degree with increasing thickness was found for block copolymer films74 and for polyelectrolyte multilayers in saturated solvent vapor75 and solvents.76
Recalculation of the data to the water content in a swollen film (1 − ddry/dsw) results in a linear decrease with increasing dry thickness for all films with more than one bilayer (Fig. S4†). The highly swollen films exhibit a maximum water content of ∼90%. Interestingly, the plot of the swelling degree versus the porosity of the films yields a linear dependence (Fig. 8b), reconfirming the dominating elastic energy contribution with decreasing porosity. The non-zero intercept means that a close-packed film with a porosity of 0 would still swell by a factor of 2 due to the contribution of the PSS/Dq IPEC and the swelling of the MAA brush. Note that in the case of zero porosity, only 1D swelling perpendicular to the substrate is possible.
Conclusions
Using the layer-by-layer technique, ABC block terpolymer micelles with a hydrophobic B core, a pH-sensitive MAA shell and a charged Dq corona were included within multilayer films with tailored porous nanostructure and integrated pH-responsive properties. The approach of ionic cross-linking of the cationic corona with a strong polyanion leads to the creation of a novel polyelectrolyte brush-like structure, which is double-end-tethered between the hydrophobic cores and the flexible IPECs of the Dq corona and PSS. The purpose of the latter is to prevent the dissolution of incorporated micelles providing the stability of the multilayers on the one hand. On the other hand, IPECs are penetrable for water and electrolytes enabling high pH-induced volumetric changes. The medium surrounding the (BMAADq/PSS)x multilayers strongly influences the ionization degree of the double-end-tethered MAA brush and therefore its degree of swelling and mechanical properties.The film swelling degree and morphology, as well as the mechanical properties of the coatings, are reversibly tunable by the solution pH. Furthermore, the swelling behavior and the water content in the hydrogel-like films can be tuned by the porosity of the multilayers, which can be adjusted by the number of micelles/PSS deposition steps to more than 1200% swelling degree and 90% water content in the swollen films. With increasing thickness (decreased porosity), the swelling degree drops since the contribution of the elastic energy dominates the osmotic pressure.
Surface-attached pH-responsive hydrogel-like LbL films can be assembled from weak polyelectrolytes.77 Although these films are highly swellable, they are often unstable with respect to pH variations.78 Other LbL-derived stimuli-responsive hydrogel-like systems, whose structure is stabilized by covalent cross-links often show considerably lower swelling degrees.79 In our case, the stabilization results from the formation of IPECs, which are covalently bound to the pH-sensitive component. Thus, no stabilization by covalent bonding is needed to trap the PMAA chains and significantly higher swelling degrees are observed.
High swelling due to the separation of the binding and responsive components within the LbL films is also known for multilayers with other incorporated core–shell ABA triblock copolymer micelles with temperature-sensitive cores.29,30 In this particular case, Tan et al. reported on the temperature-driven swelling behavior of LbL films with swelling degrees ranging from 4 to 10.
Our results demonstrate that significant (2–12 fold), reversible, and controllable (by pH and bilayer number) swelling can be realized via the incorporation of core–shell–corona micelles into the LbL films. Here we take advantage of the separation of the functional components and the retained internal structure consisting of hydrophobic core, a pH-sensitive double-end-tethered polyelectrolyte brush shell and a binding corona.
Acknowledgements
This research was supported by SFB840, COST D43, and COST Action CM1101. The authors thank S. Sukhishvili (University of New Jersey) and F. Boulmedais (University of Strasbourg) for fruitful discussions. They are also thankful to C. Kunert (University of Bayreuth) for conducting the SEM experiments.Notes and references
- Y. Liu, L. Mu, B. H. Liu and J. L. Kong, Chem.–Eur. J., 2005, 11, 2622–2631 CrossRef CAS.
- K. Glinel, C. Dejugnat, M. Prevot, B. Schöler, M. Schönhoff and R. V. Klitzing, Colloids Surf., A, 2007, 303, 3–13 CrossRef CAS.
- J. F. Mano, Adv. Eng. Mater., 2008, 10, 515–527 CrossRef CAS.
- V. Kozlovskaya, E. Kharlampieva, I. Erel and S. A. Sukhishvili, Soft Matter, 2009, 5, 4077–4087 RSC.
- P. M. Mendes, Chem. Soc. Rev., 2008, 37, 2512–2529 RSC.
- I. Tokarev and S. Minko, Soft Matter, 2009, 5, 511–524 RSC.
- I. Tokarev and S. Minko, Adv. Mater., 2010, 22, 3446–3462 CrossRef CAS.
- I. Tokarev and S. Minko, Adv. Mater., 2009, 21, 241–247 CrossRef CAS.
- I. Tokarev, M. Motornov and S. Minko, J. Mater. Chem., 2009, 19, 6932–6948 RSC.
- M. A. Cohen Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef.
- E. B. Zhulina, O. V. Borisov and T. M. Birshtein, J. Phys. II, 1992, 2, 63–74 CrossRef CAS.
- S. Minko, Polym. Rev., 2006, 46, 397–420 Search PubMed.
- A. Synytska, M. Stamm, S. Diez and L. Ionov, Langmuir, 2007, 23, 5205–5209 CrossRef CAS.
- P. Uhlmann, H. Merlitz, J. U. Sommer and M. Stamm, Macromol. Rapid Commun., 2009, 30, 732–740 CrossRef CAS.
- M. Ballauff, Prog. Polym. Sci., 2007, 32, 1135–1151 CrossRef CAS.
- S. Schmidt, M. Zeiser, T. Hellweg, C. Duschl, A. Fery and H. Möhwald, Adv. Funct. Mater., 2010, 20, 3235–3243 CrossRef CAS.
- G. B. Webber, E. J. Wanless, V. Bütün, S. P. Armes and S. Biggs, Nano Lett., 2002, 2, 1307–1313 CrossRef CAS.
- G. B. Webber, E. J. Wanless, S. P. Armes, Y. Q. Tang, Y. T. Li and S. Biggs, Adv. Mater., 2004, 16, 1794–1798 CrossRef CAS.
- G. B. Webber, E. J. Wanless, S. P. Armes and S. Biggs, Faraday Discuss., 2005, 128, 193–209 RSC.
- K. Sakai, E. G. Smith, G. B. Webber, M. Baker, E. J. Wanless, V. Bütün, S. P. Armes and S. Biggs, Langmuir, 2006, 22, 8435–8442 CrossRef CAS.
- K. Sakai, E. G. Smith, G. B. Webber, M. Baker, E. J. Wanless, V. Bütün, S. P. Armes and S. Biggs, J. Colloid Interface Sci., 2007, 314, 381–388 CrossRef CAS.
- J. Gensel, E. Betthausen, C. Hasenöhrl, K. Trenkenschuh, M. Hund, F. Boulmedais, P. Schaaf, A. H. E. Müller and A. Fery, Soft Matter, 2011, 7, 11144–11153 RSC.
- J. Gensel, T. Borke, N. Pazos Pérez, A. Fery, D. V. Andreeva, E. Betthausen, A. H. E. Müller, H. Möhwald and E. V. Skorb, Adv. Mater., 2012, 24, 985–989 CrossRef CAS.
- Z. C. Zhu and S. A. Sukhishvili, ACS Nano, 2009, 3, 3595–3605 CrossRef CAS.
- B. Mahltig, P. Müller-Buschbaum, M. Wolkenhauer, O. Wunnicke, S. Wiegand, J.-F. Gohy, R. Jérôme and M. Stamm, J. Colloid Interface Sci., 2001, 242, 36–43 Search PubMed.
- G. Decher and J. D. Hong, Ber. Bunsen-Ges. Phys. Chem., 1991, 95, 1430–1434 CAS.
- G. Decher and J. Schmitt, Prog. Colloid Polym. Sci., 1992, 89, 160–164 CAS.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- W. S. Tan, R. E. Cohen, M. F. Rubner and S. A. Sukhishvili, Macromolecules, 2010, 43, 1950–1957 CrossRef CAS.
- W. S. Tan, Z. C. Zhu, S. A. Sukhishvili, M. F. Rubner and R. E. Cohen, Macromolecules, 2011, 44, 7767–7774 CrossRef CAS.
- N. A. Kotov, I. Dekany and J. H. Fendler, J. Phys. Chem., 1995, 99, 13065–13069 CrossRef CAS.
- F. G. Aliev, M. A. Correa-Duarte, A. Mamedov, J. W. Ostrander, M. Giersig, L. M. Liz-Marzan and N. A. Kotov, Adv. Mater., 1999, 11, 1006–1010 CrossRef CAS.
- N. Ma, Y. P. Wang, Z. Q. Wang and X. Zhang, Langmuir, 2006, 22, 3906–3909 CrossRef CAS.
- B. S. Kim, S. W. Park and P. T. Hammond, ACS Nano, 2008, 2, 386–392 CrossRef CAS.
- K. Emoto, M. Iijima, Y. Nagasaki and K. Kataoka, J. Am. Chem. Soc., 2000, 122, 2653–2654 CrossRef CAS.
- T. Addison, O. J. Cayre, S. Biggs, S. P. Armes and D. York, Langmuir, 2008, 24, 13328–13333 CrossRef CAS.
- Z. C. Zhu and S. A. Sukhishvili, J. Mater. Chem., 2012, 22, 7667–7671 RSC.
- J. H. Cho, J. K. Hong, K. Char and F. Caruso, J. Am. Chem. Soc., 2006, 128, 9935–9942 CrossRef CAS.
- S. Biggs, K. Sakai, T. Addison, A. Schmid, S. P. Armes, M. Vamvakaki, V. Bütün and G. Webber, Adv. Mater., 2007, 19, 247–250 CrossRef CAS.
- T. Addison, O. J. Cayre, S. Biggs, S. P. Armes and D. York, Langmuir, 2010, 26, 6281–6286 CrossRef CAS.
- P. M. Nguyen, N. S. Zacharia, E. Verploegen and P. T. Hammond, Chem. Mater., 2007, 19, 5524–5530 CrossRef CAS.
- D. A. Bernards and T. A. Desai, Soft Matter, 2010, 6, 1621–1631 RSC.
- E. Betthausen, M. Drechsler, M. Förtsch, F. H. Schacher and A. H. E. Müller, Soft Matter, 2011, 7, 8880–8891 RSC.
- W. Kern and D. A. Puotinen, RCA Rev., 1970, 31, 187–206 Search PubMed.
- H. Elbs and G. Krausch, Polymer, 2004, 45, 7935–7942 CrossRef CAS.
- J. L. Hutter and J. Bechhoefer, Rev. Sci. Instrum., 1993, 64, 1868–1873 CrossRef CAS.
- H. Dautzenberg, W. Jaeger, J. Kötz, B. Philipp, C. Seidel and D. Stscherbina, Polyelectrolytes, Carl Hanser Verlag, München, 1994 Search PubMed.
- N. G. Hoogeveen, M. A. C. Stuart, G. J. Fleer and M. R. Bohmer, Langmuir, 1996, 12, 3675–3681 CrossRef CAS.
- S. A. Sukhishvili, E. Kharlampieva and V. Izumrudov, Macromolecules, 2006, 39, 8873–8881 CrossRef.
- V. A. Kabanov, Russ. Chem. Rev., 2005, 74, 3–20 Search PubMed.
- M. Born and E. Wolf, Principles of Optics, Cambridge University Press, Cambridge, 1999 Search PubMed.
- A. Walther and A. H. E. Müller, Chem. Commun., 2009, 1127–1129 RSC.
- C. V. Synatschke, F. H. Schacher, M. Förtsch, M. Drechsler and A. H. E. Müller, Soft Matter, 2011, 7, 1714–1725 RSC.
- J. D. Mendelsohn, C. J. Barrett, V. V. Chan, A. J. Pal, A. M. Mayes and M. F. Rubner, Langmuir, 2000, 16, 5017–5023 CrossRef CAS.
- T. Mauser, C. Dejugnat and G. B. Sukhorukov, Macromol. Rapid Commun., 2004, 25, 1781–1785 CrossRef CAS.
- H. H. Rmaile and J. B. Schlenoff, Langmuir, 2002, 18, 8263–8265 CrossRef CAS.
- S. S. Shiratori and M. F. Rubner, Macromolecules, 2000, 33, 4213–4219 CrossRef CAS.
- Z. J. Sui and J. B. Schlenoff, Langmuir, 2004, 20, 6026–6031 CrossRef CAS.
- A. J. Parnell, S. J. Martin, C. C. Dang, M. Geoghegan, R. A. L. Jones, C. J. Crook, J. R. Howse and A. J. Ryan, Polymer, 2009, 50, 1005–1014 CrossRef CAS.
- E. P. K. Currie, A. B. Sieval, G. J. Fleer and M. A. Cohen Stuart, Langmuir, 2000, 16, 8324–8333 CrossRef CAS.
- F. A. Plamper, H. Becker, M. Lanzendörfer, M. Patel, A. Wittemann, M. Ballauff and A. H. E. Müller, Macromol. Chem. Phys., 2005, 206, 1813–1825 CrossRef CAS.
- E. B. Zhulina, T. M. Birshtein and O. V. Borisov, Macromolecules, 1995, 28, 1491–1499 CrossRef CAS.
- H. W. Jomaa and J. B. Schlenoff, Langmuir, 2005, 21, 8081–8084 CrossRef CAS.
- V. A. Kabanov, A. B. Zezin, V. A. Izumrudov, T. K. Bronich and K. N. Bakeev, Makromol. Chem., 1985, 13, 137–155 CrossRef CAS.
- S. T. Dubas and J. B. Schlenoff, Langmuir, 2001, 17, 7725–7727 CrossRef CAS.
- H. J. Butt, Biophys. J., 1991, 60, 1438–1444 CrossRef CAS.
- W. A. Ducker, T. J. Senden and R. M. Pashley, Nature, 1991, 353, 239–241 CrossRef CAS.
- H. Hertz, J. Reine Angew. Math., 1881, 92, 156–171 Search PubMed.
- M. O. Lisunova, I. Drachuk, O. A. Shchepelina, K. D. Anderson and V. V. Tsukruk, Langmuir, 2011, 27, 11157–11165 CrossRef CAS.
- O. Shchepelina, M. O. Lisunova, I. Drachuk and V. V. Tsukruk, Chem. Mater., 2012, 24, 1245–1254 CrossRef CAS.
- I. Drachuk, O. Shchepelina, M. Lisunova, S. Harbaugh, N. Kelley-Loughnane, M. Stone and V. V. Tsukruk, ACS Nano, 2012, 6, 4266–4278 Search PubMed.
- C. Picart, B. Senger, K. Sengupta, F. Dubreuil and A. Fery, Colloids Surf., A, 2007, 303, 30–36 CrossRef CAS.
- R. Toomey, D. Freidank and J. Rühe, Macromolecules, 2004, 37, 882–887 CrossRef CAS.
- J. Gensel, C. Liedel, H. G. Schoberth and L. Tsarkova, Soft Matter, 2009, 5, 2534–2537 RSC.
- J. E. Wong, F. Rehfeldt, P. Hanni, M. Tanaka and R. V. Klitzing, Macromolecules, 2004, 37, 7285–7289 CrossRef CAS.
- S. E. Burke and C. J. Barrett, Biomacromolecules, 2005, 6, 1419–1428 CrossRef CAS.
- J. Hiller and M. F. Rubner, Macromolecules, 2003, 36, 4078–4083 CrossRef CAS.
- S. T. Dubas and J. B. Schlenoff, Macromolecules, 2001, 34, 3736–3740 CrossRef CAS.
- E. Kharlampieva, I. Erel-Unal and S. A. Sukhishvili, Langmuir, 2007, 23, 175–181 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: details on the adsorption kinetics of BMAADq micelles onto different substrates, colloidal probe measurements confirming the validity of the Hertz model, and information about water content of (BMAADq/PSS)x films in a swollen state. See DOI: 10.1039/c2sc20836a |
| This journal is © The Royal Society of Chemistry 2013 |