Semi-interpenetrating polymer networks prepared from in situ cationic polymerization of bio-based tung oil with biodegradable polycaprolactone
Samy A. Madboulyac,
Kunwei Liua,
Ying Xiaa and
Michael R. Kessler*b
aDepartment of Materials Science and Engineering, Iowa State University, Ames, IA 50011, USA
bSchool of Mechanical and Materials Engineering, Washington State University, Pullman, WA, USA. E-mail: MichaelR.Kessler@wsu.edu
cDepartment of Chemistry, Faculty of Science, Cairo University, Orman-Giza, Egypt
First published on 3rd January 2014
Abstract
In situ cationic polymerization of bio-based tung oil in the presence of poly(ε-caprolactone), a crystallizable, biodegradable, and biocompatible polymer, was performed to produce novel semi-interpenetrating polymer networks (IPNs). The macromolecular structure and properties of these IPNs were investigated as a function of composition using small amplitude oscillatory shear flow rheology, FT-IR spectroscopy, dynamic mechanical analysis (DMA), differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), and scanning electron microscopy (SEM). This versatile and low-cost strategy successfully produced bio-polymer blends with various degrees of miscibility, morphology, and crystallization behavior. The carbon–carbon double bonds in tung oil were consumed quickly after adding the cationic initiator to form a three-dimensional (3D) crosslinked network in all measured samples as confirmed by FT-IR. A complete miscible structure with a single glass transition temperature and one-phase morphology was observed for a tung oil/PCL 90/10 blend. On the other hand, a two-phase structure exhibiting a nanoscale morphology of the dispersed minor phase as small as 100 nm was observed for blends with 20 and 30 wt% PCL. For a 50 wt% PCL blend, an interconnected, co-continuous microstructure of the two phases was also detected. DMA and DSC measurements confirmed the miscibility (or partial miscibility) of the blends by following the changes in the glass transitions of phases as a function of the composition. The value of the elastic modulus (E′) in the glassy state as obtained from the DMA measurements was strongly dependent on the composition, reaching a maximum at 20 wt% PCL.
Introduction
Increases in environmental awareness, together with shortages and high costs of finite petroleum resources, have led to an increased emphasis on the use of alternative, sustainable, environmentally-friendly products from biorenewable resources (i.e. biomass).1–7 Exploring these biorenewable resources with green products suitable for generating new classes of engineering materials has received considerable attention in recent years.8–15 Natural vegetable oils are considered to be one of the cheapest and most abundant biorenewable resources worldwide.16–22 They are available in large quantities and have been widely used to synthesize bio-based polymers that offer a variety of advantages, such as low toxicity and inherent biodegradability.23–26 Saturated and unsaturated fatty acids are the main components of the triglyceride vegetable oils that are the platform chemicals for polymer synthesis.Tung oil can be obtained in large quantities from the seeds of the tung tree. This oil has approximately 84% α-elaeostearic acid (cis-9, trans-11, trans-13-octadecatrienoic acid) triglyceride. Because of its high number of conjugated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds that provide for rapid polymerization and excellent drying at room temperature,27 tung oil can be found in many industrial applications, such as paints, varnishes, and related materials. Tung oil can be polymerized to create bio-based thermoset polymers via thermal, free radical, or cationic polymerizations.28–30 Dimerization of elaeostearic acid or formation of dienoic acids caused by exposing tung oil to high temperatures (200–300 °C) creates weakly rubbery materials.31 Brittle, bio-based tung oil thermoset polymers with dark brown or black color can be obtained by cationic polymerization of tung oil using boron trifluoride diethyl etherate (BFE) as initiator. The mechanical properties of tung oil thermosets were improved greatly by thermal copolymerization of tung oil with styrene and divinylbenzene via free radical polymerization.16 The copolymerization process was carried out at 85–160 °C with variations in stoichiometry, oxygen uptake, peroxides, and metallic catalysts.16 A variety of material properties ranging from rubbery to tough and rigid plastics were prepared using copolymerization processes. However, although the mechanical properties of tung oil thermosets were modified to a great extent by the copolymerization process, both styrene and divinylbenzene are nondegradable petroleum-based products.
C bonds that provide for rapid polymerization and excellent drying at room temperature,27 tung oil can be found in many industrial applications, such as paints, varnishes, and related materials. Tung oil can be polymerized to create bio-based thermoset polymers via thermal, free radical, or cationic polymerizations.28–30 Dimerization of elaeostearic acid or formation of dienoic acids caused by exposing tung oil to high temperatures (200–300 °C) creates weakly rubbery materials.31 Brittle, bio-based tung oil thermoset polymers with dark brown or black color can be obtained by cationic polymerization of tung oil using boron trifluoride diethyl etherate (BFE) as initiator. The mechanical properties of tung oil thermosets were improved greatly by thermal copolymerization of tung oil with styrene and divinylbenzene via free radical polymerization.16 The copolymerization process was carried out at 85–160 °C with variations in stoichiometry, oxygen uptake, peroxides, and metallic catalysts.16 A variety of material properties ranging from rubbery to tough and rigid plastics were prepared using copolymerization processes. However, although the mechanical properties of tung oil thermosets were modified to a great extent by the copolymerization process, both styrene and divinylbenzene are nondegradable petroleum-based products.
Poly(ε-caprolactone) (PCL) represents an important class of semicrystalline, biodegradable, and biocompatible polyester with melting point and glass transition temperature of approximately 60 and −60 °C, respectively.32 The degradation process of PCL is initiated by the hydrolysis of its ester linkages in physiological media such as the human body. The biocompatibility and non-toxicity of PCL and its degraded fragments make PCL an excellent candidate for a wide range of biomedical applications, such as sutures, adhesion barriers, scaffolds for tissue engineering, and long-term implants.33,34 In addition, PCL was also used in controlled drug delivery (drugs encapsulated within PCL beads). Furthermore, PCL has been approved by the Food and Drug Administration (FDA) for a variety of applications, particularly in biomedical and regenerative medicine. PCL is also miscible with various polymers and exhibits low temperature adhesiveness.35 Numerous studies have been carried out to modify PCL by blending or copolymerizing it with other polymers.36–40
Mixing two or more polymer components of different physical, biological, and mechanical properties is an excellent and convenient way to produce new materials with diverse, tailored properties compared to those of the pure polymer components or the traditional way of polymer synthesis. Mixtures of immiscible polymers typically exhibit phase-separated morphologies with coarse, irregular, and unstable domain sizes. Furthermore, the interfaces between the domains of immiscible polymers are sharp and weak, leading to poor properties that limit the applications of these blends. However, compatible polymer blends have relatively good mechanical properties and finer phase morphologies compared to those of immiscible blends. Issues related to the phase separation and domain size precluded such polymer blends from many applications due to poor physical properties. Incorporation of small amounts of block, graft, or star copolymers as compatibilizing agents is a well-established approach to enhance the compatibility of immiscible polymer mixtures. During mechanical mixing, the compatibilizing agents migrate to the interface, reduce the surface tension and thus increase thermodynamic stability of such immiscible polymer blends.41–43 Unfortunately, most of these copolymers are expensive, synthesized from non-renewable resources, not available commercially, or cannot be handled or prepared in large quantities, making their industrial use unfeasible. Our main goal here is to incorporate PCL as a crystallizable, biodegradable, and biocompatible polymer component during the cationic polymerization of tung oil. This relatively new strategy for an in situ polymerization and compatibilization process allows for improved control of the phase domain interface, consequently yielding small domain sizes and enhancing the properties of the resulting semi-interpenetrating polymer network.
In the following sections, the morphology of the tung-oil/PCL blends will be investigated as a function of composition using scanning electron microscopy (SEM). Dynamic mechanical analysis (DMA) will be used to investigate the partial miscibility and the molecular dynamics of the blends. The effect of tung oil on the crystallization behavior of PCL will be studied using differential scanning calorimetry (DSC). In addition, small amplitude oscillatory shear flow experiments will be used to evaluate the kinetics of cationic polymerization of bio-based tung oil and its blends with PCL.
Experimental section
Materials
Tung oil and chloroform used in this study were obtained from Sigma-Aldrich and used as received. Tung oil consists primarily of glycerides of α-elaeostearic fatty acid (cis-9, trans-11, trans-13-octadecatrienoic acid). It has a yellow color and a specific gravity of 0.937 at 25 °C. The cationic initiator, boron trifluoride diethyl etherate (BFE), was obtained from Aldrich and used as received. PCL was provided by Union Carbide Corporation (PCL-767). The molecular weight and polydispersity of the PCL were 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 g mol−1 and 2.61, respectively.
400 g mol−1 and 2.61, respectively.
Sample preparation
Tung oil, PCL, and the cationic polymerization initiator BFE were dissolved in chloroform to yield 25 wt% solutions. The concentration of BFE was kept constant at 2 wt% relative to the concentration of tung oil in the blends. Depending on the concentration of tung oil in the blends, the homogenous solutions of PCL, tung oil, and BFE formed transparent, yellow gels after a certain time (i.e., the gelation time was inversely proportional to the concentration of tung oil). The obtained gels were left to dry for about 3 days under vacuum at room temperature. After an additional 2 days in vacuum at 60 °C, evaporation of the solvent to a constant weight was completed. The evaporated chloroform was recycled with a cold trap and reused in the blend preparation process. Two reasons led to the use of chloroform in this reaction: first and foremost the very rapid cationic polymerization process of tung oil. We found that a black material with very poor mechanical properties can be obtained immediately by adding a very small amount of BFE (less than 0.1 wt%) directly to tung oil. The kinetics of this cationic polymerization reaction of tung oil decreased dramatically by adding chloroform as will be explained in the discussion section. In addition, chloroform is necessary to dissolve PCL, tung oil, and BFE to produce homogenous solutions and consequently miscible semi-interpenetrating polymer networks or compatible blends.Characterization of the blends
C bonds before and after the cationic polymerization of tung oil in the blends. A tung oil/PCL 70/30 blend with no cationic initiator was dissolved in chloroform and placed on a KBr salt plate. After evaporating the chloroform, the FT-IR spectrum of the unreacted blend was recorded on a Bruker IFS-66V spectrometer (Billerica, MA). A completely reacted tung oil/PCL 70/30 blend was crushed into powder in liquid nitrogen, and the powder/KBr mixture was compressed to a plate, which was characterized with the same spectrometer.DMA measurements
DMA measurements were carried out using a Q800 dynamic mechanical analyzer from TA Instruments in the three point bending mode. Rectangular samples with approximately 0.9 mm thickness, 7 mm width, and 12 mm length were heated from −90 to 150 °C at a heating rate of 2 °C min−1. A frequency of 1 Hz and displacement amplitude of 5 μm were used in all DMA measurement. The miscibility of tung oil/PCL blends were evaluated from the temperature and composition dependence of the glass relaxation processes of each mixture.DSC and TGA measurements
DSC measurements were conducted using a TA Instruments Q2000. All DSC measurements were carried out in an atmosphere of dry nitrogen. The samples were rapidly heated to 110 °C and annealed for 5 min to erase thermal history. The samples were cooled to −90 °C at a cooling rate of 10 °C min−1 and then heated up to 100 °C at a rate of 10 °C min−1. In the second heating run, the glass transition temperature (Tg) was recorded at the half height of the corresponding heat capacity jump, and the melting temperature (Tm) of all samples was evaluated using the maximum of the endothermic melting peak. The TGA (thermogravimetric analysis) measurements for tung oil/PCL blends were carried out using a Q50 thermogravimetric analyzer (TA Instruments, New Castle, DE). About 5 mg of the blends of different concentrations was heated from 50 to 600 °C under nitrogen atmosphere at 20 °C min−1.Morphology of the blends
Morphological inspection of the polymer blends was performed by scanning electron microscopy (SEM). The materials were fractured in liquid nitrogen, fixed on special SEM holders, and then sputtered with gold. The prepared samples were investigated using a field-emission scanning electron microscope (FE-SEM, FEI Quanta 250) operating at 10 kV under high vacuum.Results and discussion
FT-IR spectra analysis
Chemically, tung oil is a triglyceride of mainly elaeostearic acid, a conjugated triene. During the cationic polymerization process of tung oil, the number of CC bonds decreases substantially with the formation of a three-dimensional crosslinked network structure (Scheme 1). Since PCL was blended with tung oil before adding the initiator, unreacted PCL chains were randomly penetrated into the three-dimensional tung oil network in the final blends, creating a semi-penetrating polymer network. Fig. 1 demonstrates the FT-IR spectra of unreacted tung oil/PCL and polymerized tung oil/PCL blends. Before adding initiator, unreacted tung oil shows a peak at 3013 cm−1, resulting from stretching of C–H bonds. This peak disappears when tung oil is polymerized, indicating most carbon–carbon double bonds in tung oil were consumed through the cationic polymerization process. We presented here just one example for the FT-IR of tung oil/PCL 30/70 blend to prove that the PCL has almost no influence on the cationic polymerization of tung oil. Identical FT-IR spectra have been observed for other compositions.
 | ||
| Scheme 1 Cationic polymerization of tung oil. | ||
 | ||
| Fig. 1 FT-IR spectra of tung oil/PCL 30/70 blend before (A) and after (B) gelation. | ||
Rheology of cationic polymerization of tung oil and its blends
The time evolution of the viscoelastic properties during the cationic polymerization of tung oil and PCL in chloroform were investigated at 20 °C as a function of composition and shear frequency. Fig. 2 depicts the gelation time dependence of G′ and G′′ at different angular frequencies for 15 wt% tung oil in chloroform at 20 °C. Clearly, the values of G′ at different frequencies are much lower than that of G′′ during the early stage of the gelation process. A dramatic increase in both G′ and G′′ was observed with increasing time due to the formation of a crosslinked network structure of tung oil polymer. The increase in G′ and G′′ plateaued at about 70 min (a typical criterion for the formation of a crosslinked structure).44 It is also clear that G′ and G′′ coincided at tgel = 5 min and G′ increases with gelation time more rapidly than G′′ (i.e. G′ is almost two orders of magnitude higher than G′′ at approximately 70 min) which is a criterion for the formation of three-dimensional thermoset polymer. In addition, the value of tgel determined from the crossover of G′ and G′′ is angular frequency independent. The stress induced in the system by the branching and the formation of a crosslinked polymer is primarily elastic in origin; this is why G′ is much more sensitive to the gelation process than G′′ (i.e., G′ increases more rapidly than G′′). Elastic in origin means that the obtained gels have predominant elastic behavior. On the other hand, η* behaves differently with increasing angular frequency as clearly seen in Fig. 3. During the early stages of the gelation process, η* is angular frequency independent. At longer gelation time, η* increases abruptly and becomes strongly angular frequency dependent as the gelation process evolves into the formation of a three-dimension thermoset polymer. | ||
| Fig. 2 Variation of G′ (solid symbols) and G′′ (open symbols) for 15 wt% tung oil in chloroform as a function of gelation time at 20 °C for different constant angular frequencies. The arrow shows the value of tgel. | ||
 | ||
| Fig. 3 Time dependence of dynamic complex viscosity, η*, for 15 wt% tung oil in chloroform at 20 °C for different constant angular frequencies. | ||
The value of tgel obtained from the crossover point of G′ and G′′ was also found to be strongly dependent on the concentration of tung oil in chloroform. Fig. 4a shows the gelation time dependence of G′ and G′′ at 1 rad s−1 and 20 °C for 13 and 15 wt% tung oil in chloroform. The value of tgel is approximately 5 and 43 min for 15 and 13 wt% tung oil, respectively, at a constant concentration of 2 wt % BFE. It is also clear that the values of G′ and G′′ reached plateau much faster for 15 wt% than for 13 wt%. In addition, the magnitudes of G′ and G′′ are much higher for 15 wt% than for 13 wt%. Fig. 4b shows the rheological tgel of tung oil as a function of concentration at 1 rad s−1 and 20 °C. The gelation time decreases exponentially with increasing concentration of tung oil in chloroform.
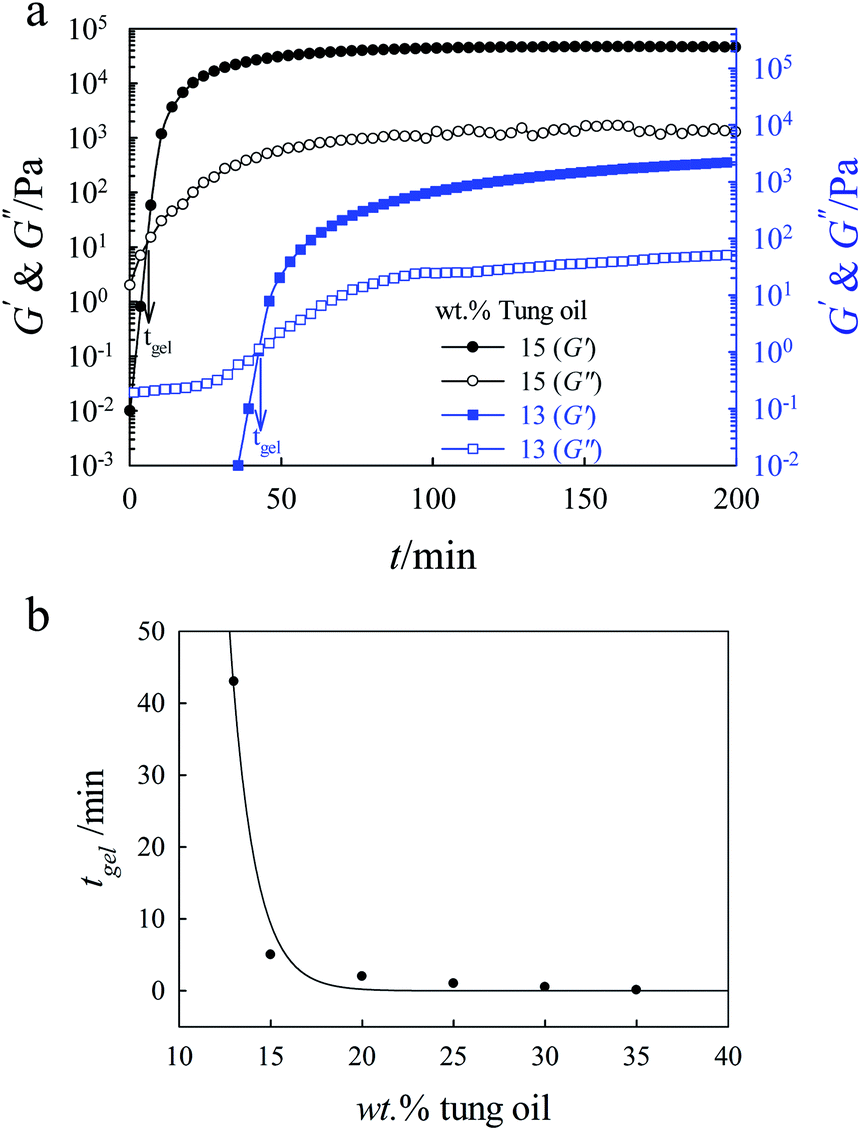 | ||
| Fig. 4 (a) Gelation time dependence of G′ and G′′ for 13 and 15 wt% tung oil in chloroform at 20 °C and 1 rad s−1. The arrows show the values of tgel for each concentration. (b) Composition dependence of gel point for tung oil in chloroform while keeping the concentration of BFE (cationic initiator) constant at 2 wt% with respect to tung oil. | ||
Similar rheological behavior was observed for the tung oil/PCL blends. Fig. 5a shows the gelation time dependence of G′ and G′′ at different angular frequencies and 20 °C for tung oil/PCL 70/30 blend (25 wt% solid content in chloroform). Similar to the rheological behavior of tung oil, the tgel can be evaluated accurately from the crossover of G′ and G′′ and the value of tgel is angular frequency independent. It is also apparent that, addition of PCL leads to a slow gelation process and a decrease in the values of both G′ and G′′.
 | ||
| Fig. 5 (a) Variation of G′ and G′′ for tung oil/PCL 70/30 blend (25 wt% solid content in chloroform) as a function of gelation time at 20 °C for different constant angular frequencies. The arrow shows the value of tgel. (b) Schematic diagram for tung oil/PCL network structure in three-dimension. | ||
Fig. 5b shows a schematic diagram of the tung oil/PCL network structure. This schematic diagram combines the interconnected three-dimension network of tung oil thermoset (green color) and the entangled PCL thermoplastic chains (blue color). This schematic diagram demonstrates how the thermoplastic PCL incorporated into the crosslinked structure of tung oil. The number of net points or crosslinking density increases with increasing concentration of tung oil.
Miscibility of the blends
As already mentioned in the experimental section, the obtained gels were left to dry for about 3 days at room temperature and 2 days in vacuum at 60 °C to a constant weight for complete drying of the sample. Fig. 6 shows the dynamic storage modulus E′ and the loss tangent tanδ as a function of temperature for different blend compositions. For pure tung oil thermoset, a sharp glass relaxation processes (α-relaxation) at approximately 33 °C was observed. The α-relaxation process of pure PCL was detected at approx. −38 °C; it was very broad with low intensity compared to that of the pure tung oil thermoset. In general, α-relaxation processes are attributed to the cooperative reorientation of the polymeric chains and are related to the Tg of the respective material. In tung oil/PCL blends, the α-relaxation process of the tung oil thermoset shifted systematically to the low temperature range with a considerably decrease in its intensity with increasing concentration of PCL up to 30 wt%. This observation suggests that PCL and tung oil are miscible, or partially miscible, in this composition range. For tung oil/PCL 50/50 blends, the shift in the α-relaxation process of the tung oil-rich phase to lower temperature is only 5 °C compared to 20 °C for the tung oil/PCL 70/30 blends, indicating that the miscibility/partial miscibility of PCL and tung oil decreases with increasing concentration of PCL above 30 wt%. The inset-plot of tanδ versus T showed very weak relaxation peaks in the vicinity of the relaxation peaks of pure PCL, indicating that PCL and tung oil are partially miscible but not completely miscible. On the other hand, the broadness of these peaks and their low intensity make it difficult to come to an accurate conclusion regarding the miscibility of the two polymer components. Therefore, the miscibility of these blends will be further investigated using DSC and SEM in the next sections. The value of E′ in the glassy state at −90 °C was also found to be strongly dependent on composition, i.e. E′ increased with increasing concentration of PCL and reached a maximum at 20 wt% PCL. For blends with higher concentrations of PCL, E′ decreased and attained a minimum for tung oil/PCL 50/50 blends (see Figs. 6 and 7). Based on this experimental fact it is apparent that PCL has a significant influence on the mechanical properties of tung oil thermosets. The reinforcement effect caused by adding PCL to tung oil up to 30 wt% may be related to a particular morphology or compatibility at this critical composition level.
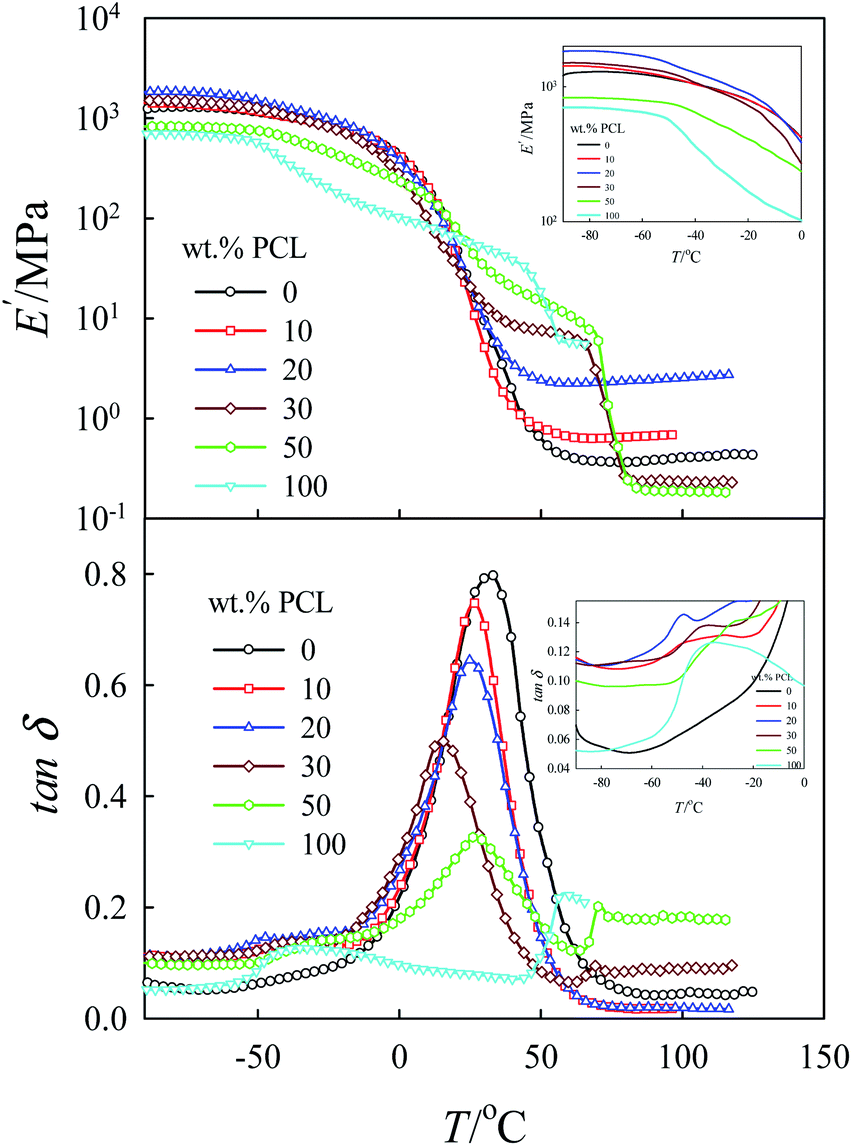 | ||
| Fig. 6 Dynamic storage modulus, E′, and tanδ as a function of temperature for tung oil/PCL blends of different compositions at a frequency of 1 Hz and a heating rate of 2 °C min−1. The inset-plots focus on the data in the low temperature range. | ||
 | ||
| Fig. 7 Composition dependence of E′ at −90 °C for tung oil/PCL blends. | ||
The poor mechanical properties of tung oil/PCL 50/50 blends may be related to low compatibility or macrophase separated structures for this composition. At elevated temperatures (for example, at 100 °C), the melting behavior of PLC causes a dramatic decrease in E′ for 100, 50, and 30 wt% PCL. As seen in Fig. 6, the value of E′ for each composition shows a plateau at 100 °C. This plateau is related to the crosslinked structure of tung oil and its value is a function of the crosslinking density. The plateau reaches a maximum value at 20 wt% PCL and decreases significantly once the concentration of PCL is higher than 30 wt%, similar to the composition dependence of E′ at −90 °C (Fig. 6).
The DSC thermographs for tung oil/PCL blends of different compositions are shown in Fig. 8. The inset plot in Fig. 8 shows the DSC thermograph for a pure tung oil thermoset. The two pure components (PCL and tung oil thermoset) and tung oil/PCL 90/10 blends displayed a single Tg for each sample at −63, 15, and −10 °C, respectively. All other blends showed two Tgs for PCL-rich and tung oil-rich phases at low and high temperatures, respectively. This experimental fact suggests that the in situ cationic polymerization of tung oil in the presence of PCL produced miscible blends up to 10 wt% PCL and partially miscible blends with higher concentrations of PCL.
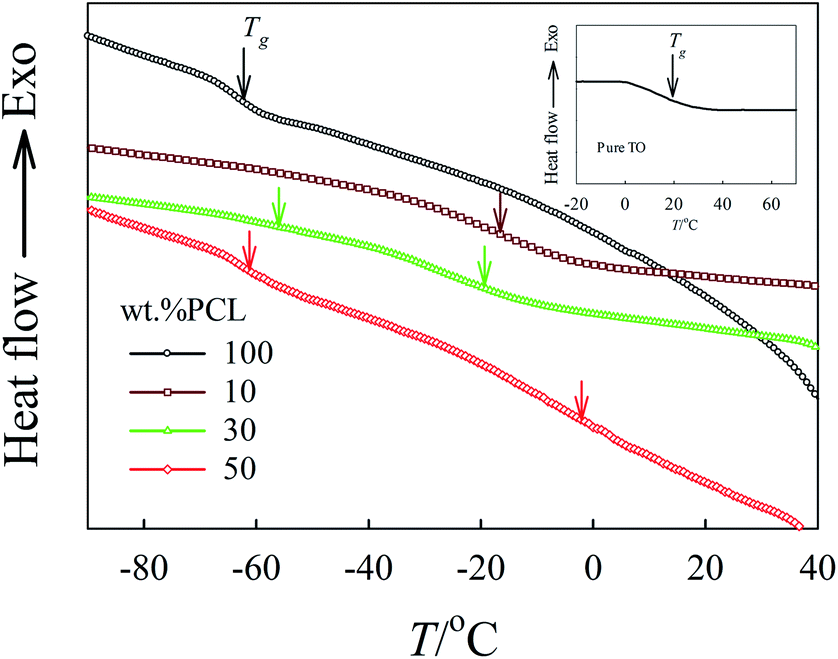 | ||
| Fig. 8 DSC thermograms for tung oil/PCL blends of different compositions. The measurements were carried out at a heating rate of 10 °C min−1. The arrows denote the values of Tg for each composition. The inset-plot shows the DSC thermogram for a pure tung oil thermoset. | ||
Fig. 9 shows the composition dependence of Tg obtained from the DSC measurements (Fig. 8). One-phase regimes (single Tg) can be observed at a concentrations lower than 20 wt% PCL, while two-phase regimes (two Tgs) can be seen at concentrations ranging ≥20 wt% PCL. It is also clear that the difference between the two Tgs of the PCL-rich and the tung oil-rich phases (i.e., ΔTg) increases with increasing concentration of PCL, indicating that the partial miscibility of the two polymer components decreases with increasing concentration of PCL. A deeper understanding of the phase behavior and miscibility of these systems can be obtained from their internal structure or morphology.
 | ||
| Fig. 9 Composition dependence of Tg for PCL-rich and tung oil-rich phases. ΔTg represents the difference in the Tgs between the two phases. | ||
SEM of tung oil/PCL blends was investigated to understand the morphology of the in situ polymerized tung oil and its miscibility with PCL. SEM micrographs of tung oil/PCL blends of different compositions are shown in Fig. 10. Tung oil/PCL 90/10 blends exhibit no morphological structure because of their complete miscibility, which is in good agreement with the DSC measurement (only one Tg was observed for this blend at −10 °C). Nanoscale morphology with an average particle size of the dispersed minor phase as small as 100 nm was observed for 20, 30, and 40 wt% PCL blends, as seen in Fig. 10. The white dispersed particles and the dark matrix represent the PCL and tung oil phases, respectively. For tung oil/PCL 50/50 blend, an interconnected, co-continuous microstructure of the two phases was observed. The two rich phases are highly interconnected. Based on the DMA, DSC, and SEM observation, one can identify three regimes for the phase behavior and morphology of tung oil/PCL blends as a function of blend composition. The first regime with up to 10 wt% PCL represents a complete miscible structure (i.e., single Tg with no morphological structure). In the second regime, partially miscible blends with nanoscale morphologies (as small as 100 nm) were observed in the composition range of 20 ≤ wt% < 50. Partially miscible blends with highly interconnected, co-continuous structures of PCL-rich and tung-oil-rich phases were observed in the third regime at 50 wt% PCL. It must be mentioned here that the cationic polymerization of tung oil in the presence of PCL in concentrations higher than 50 wt% is very slow and the obtained blends exhibit macrophase separation with very poor mechanical properties. Based on the preceding discussion, it is clear that blends with different phase behaviors, including miscible and partially miscible with nanostructured or interconnected co-continuous morphologies with unique combination of properties, can be easily obtained by simply changing the blend composition. Similar results cannot be achieved with classical blends of other pre-made polymers.45 In contrast to classical polymer blends, tung oil thermosets provide remarkable stability to the nanoscale and interconnected, co-continuous morphologies, even at elevated temperatures. The unique morphology and its stability of this polymer blend suggests that the simple strategy discussed in this article can be used to prepare new, functional polymeric systems with a number of beneficial applications, such as drug delivery systems, tissue engineering, and implantation.
 | ||
| Fig. 10 SEM photographs for tung oil/PCL blends of different concentrations. | ||
Fig. 11 demonstrates the TGA measurements for pure tung oil thermoset, PCL and tung oil/PCL 50/50 blend. The TGA measurements were carried out for all blends and we presented the behavior of only three samples in Fig. 11 just for clarity. Fig. 11 shows the percent mass loss as a function of temperature at 20 °C min−1 heating rate under a nitrogen atmosphere. As can be seen in the figure, the PCL undergoes thermal degradation beginning at approximately 325 °C, almost 40 °C lower than pure tung oil, while the tung oil/PCL 50/50 blend begins to degrade around 340 °C. In addition, only one degradation process was observed for all measured blends; its onset thermal degradation temperature is concentration dependent indicating that the blends are highly compatible or miscible and behaves as a single homogenous material as already discussed above.
 | ||
| Fig. 11 TGA measurements for tung oil/PCL blends of different concentrations at 20 °C min−1 heating rate under a nitrogen atmosphere. | ||
Crystallization behavior
The crystallization and melting behavior of crystallizable polymers are often influenced by the presence of other components. It is well established that structural parameters, such as lamellar thickness, crystal interphase, and spherulitic growth rates are substantially modified by additional components.46–48 Moreover, from a thermodynamic point of view, a decrease in the melting point typically occurs as a result of specific interaction between the two components of the blend and because of changes in the free energy required for the formation of crystals. The miscibility of the two blend components is also confirmed by the decrease in Tm and melting enthalpy (ΔHm) or in the degree of crystallinity (DOC) of the crystalline component. The DSC thermographs of tung oil/PCL blends for different concentrations are shown in Fig. 12. The curves (second heating run) displayed endothermic peaks that were slightly shifted to lower temperatures in the composition range of 10 < PCL wt% < 50 because of the compatibility and partial miscibility of the blends. For 10 wt% PCL blends no endothermic peak was observed because of the complete miscibility of this composition. The interconnected, co-continuous morphology of tung oil/PCL 50/50 blend enhances the crystallization behavior of PCL and consequently the endothermic peak slightly shifted to higher temperatures. It is also clear that all blends displayed bimodal melting peaks that may result from melt-recrystallization mechanisms. During heating the sample in DSC, the less perfect crystals melted at lower temperatures and then the samples reorganized into more perfect crystals that melt at higher temperatures. The degree of relative crystallinity and crystallization kinetics can be significantly influenced by the degree of miscibility in amorphous/semicrystalline binary polymer blends. | ||
| Fig. 12 Development of melting temperature for tung oil/PCL blends of different compositions. The DSC measurements were carried out at a heating rate of 10 °C min−1 (second heating run). The inset-plot shows the composition dependence of the degree of crystallinity percentage (DOC%). | ||
The DOC was calculated from the melting endotherm of each sample with respect to the PCL composition in the blend normalized by the heat of fusion of 100% crystalline PCL (136 J g−1). The inset-plot in Fig. 12 shows the DOC for tung oil/PCL as a function of blend composition. The DOC increases with increasing concentration of PCL in the blend, reaching a maximum value for 50 wt% PCL, and then the DOC slightly decreasing until it reaches the value for pure PCL. This behavior is related to the different degrees of miscibility and the morphologies that are obtained with various blend compositions as mentioned earlier and shown in Fig. 10.
Conclusion
Incorporation of PCL as a crystallizable, biodegradable, and biocompatible polymer component to bio-based tung oil during its cationic polymerization was found to have a substantial influence on the miscibility, morphology, and crystallization behavior of the mixed blends. This new strategy of in situ polymerization and compatibilization produced miscible blends for up to 10 wt% PCL and partially miscible blends of tung oil and PCL for higher concentrations of PCL. It also allowed for improved control of the phase domain interface, consequently yielding small domain sizes with enhanced benefits. Nanoscale morphologies with average particle sizes of the dispersed minor phase as small as 100 nm were observed for 20, 30, and 40 wt% PCL blends, while interconnected, co-continuous microstructures of the two rich phases was clearly seen for tung oil/PCL 50/50 blends. In addition, a complete miscible morphology was observed for blends with up to 10 wt% PCL content. In contrast to classical polymer blends, the tung oil thermosets provide a remarkable stability to the nanoscaled, interconnected, co-continuous morphologies even at elevated temperatures. The value of E′ calculated from DMA measurements in the glassy state was also found to be strongly composition dependent, i.e., E′ increased with increasing concentration of PCL and reached a maximum/minimum at 20 and 50 wt% PCL, respectively. This behavior was caused by the different degrees of miscibility of the blends. Miscibility and partial miscibility of these novel blends were confirmed using DSC and DMA measurements and following the changes in the Tgs and the α-relaxation processes of the two rich phases, respectively. With the appropriate selection of starting materials and a prescribed in situ cationic polymerization of tung oil in the presence of PCL, this simple, versatile, low cost strategy for preparing nanoscaled and interconnected, co-continuous morphologies with desired properties should be widely applicable.References
- D. T. Vu, A. K. Kolah, N. S. Asthana, L. Peereboom, C. T. Lira and D. J. Miller, Fluid Phase Equilib., 2005, 236, 125–135 CrossRef CAS PubMed.
- F. Bornke and I. Broer, Curr. Opin. Plant Biol., 2010, 13, 354–362 CrossRef PubMed.
- M. Dash, F. Chiellini, R. M. Ottenbrite and E. Chiellini, Prog. Polym. Sci., 2011, 36, 981–1014 CrossRef CAS PubMed.
- H. Jia and P. Wang, J. Biotechnol., 2007, 132, 314–317 CrossRef CAS PubMed.
- S. Bhuyan, L. S. Holden, S. Sundararajan, D. Andjelkovic and R. Larock, Wear, 2007, 263, 965–973 CrossRef CAS PubMed.
- G. A. R. Martinez, A. J. R. Lasprilla, B. H. Lunelli, A. L. Jardini and R. Maciel, J. Biotechnol., 2010, 150, S97 CrossRef PubMed.
- M. Kunioka, F. Ninomiya and M. Funabashi, Polym. Degrad. Stab., 2006, 91, 1919–1928 CrossRef CAS PubMed.
- N. Hameed, Q. P. Guo, F. H. Tay and S. G. Kazarian, Carbohydr. Polym., 2011, 86, 94–104 CrossRef CAS PubMed.
- U. Konwar, N. Karak and M. Mandal, Prog. Org. Coat., 2010, 68, 265–273 CrossRef CAS PubMed.
- G. Das, N. K. Bordoloi, S. K. Rai, A. K. Mukherjee and N. Karak, J. Hazard. Mater., 2012, 209, 434–442 CrossRef PubMed.
- G. Booth, D. E. Delatte and S. F. Thames, Ind. Crops Prod., 2007, 25, 257–265 CrossRef CAS PubMed.
- M. Black and J. W. Rawlins, Eur. Polym. J., 2009, 45, 1433–1441 CrossRef CAS PubMed.
- V. Sharma and P. P. Kundu, Prog. Polym. Sci., 2008, 33, 1199–1215 CrossRef CAS PubMed.
- M. Sacristan, J. C. Ronda, M. Galia and V. Cadiz, Polymer, 2010, 51, 6099–6106 CrossRef CAS PubMed.
- M. I. Aranguren, J. F. Gonzalez and M. A. Mosiewicki, Polym. Test., 2012, 31, 7–15 CrossRef CAS PubMed.
- Y. Xia and R. C. Larock, Polymer, 2010, 51, 2508–2514 CrossRef CAS PubMed.
- P. P. Kundu and R. C. Larock, Biomacromolecules, 2005, 6, 797–806 CrossRef CAS PubMed.
- F. Li, D. W. Marks, R. C. Larock and J. U. Otaigbe, Polymer, 2000, 41, 7925–7939 CrossRef CAS.
- D. D. Andjelkovic, M. Valverde, P. Henna, F. K. Li and R. C. Larock, Polymer, 2005, 46, 9674–9685 CrossRef CAS PubMed.
- P. H. Henna, D. D. Andjelkovic, P. P. Kundu and R. C. Larock, J. Appl. Polym. Sci., 2007, 104, 979–985 CrossRef CAS.
- M. Valverde, D. Andjelkovic, P. P. Kundu and R. C. Larock, J. Appl. Polym. Sci., 2008, 107, 423–430 CrossRef CAS.
- D. D. Andjelkovic and R. C. Larock, Biomacromolecules, 2006, 7, 927–936 CrossRef CAS PubMed.
- F. K. Li, J. Hasjim and R. C. Larock, J. Appl. Polym. Sci., 2003, 90, 1830–1838 CrossRef CAS.
- H. Miyagawa, M. Misra, L. T. Drzal and A. K. Mohanty, Polym. Eng. Sci., 2005, 45, 487–495 CAS.
- S. J. Park, F. L. Jin and J. R. Lee, Macromol. Rapid Commun., 2004, 25, 724–727 CrossRef CAS.
- B. Cakmakli, B. Hazer, I. O. Tekin, S. Kizgut, M. Koksal and Y. Menceloglu, Macromol. Biosci., 2004, 4, 649–655 CrossRef CAS PubMed.
- R. G. Kinabrew, Tung oil in Mississippi; the competitive position of the industry, Bureau of Business Research, University of Mississippi, MS, 1952 Search PubMed.
- F. K. Li and R. C. Larock, J. Appl. Polym. Sci., 2000, 78, 1044–1056 CrossRef CAS.
- F. K. Li and R. C. Larock, Biomacromolecules, 2003, 4, 1018–1025 CrossRef CAS PubMed.
- D. P. Pfister, J. R. Baker, P. H. Henna, Y. Lu and R. C. Larock, J. Appl. Polym. Sci., 2008, 108, 3618–3625 CrossRef CAS.
- C. CJ, Chem. Soc. Jpn.,Ind. Chem. Sect., 1950, 53, 281 Search PubMed.
- Y. Tokiwa, B. P. Calabia, C. U. Ugwu and S. Aiba, Int. J. Mol. Sci., 2009, 10, 3722–3742 CrossRef CAS PubMed.
- G. H. Kim, Biomed. Mater., 2008 Search PubMed, 3, 025010, 1–8.
- J. Han, T. X. Chen, C. J. Branford-White and L. M. Zhu, Int. J. Pharm., 2009, 382, 215–221 CrossRef CAS PubMed.
- M. E. Broz, D. L. VanderHart and N. R. Washburn, Biomaterials, 2003, 24, 4181–4190 CrossRef CAS.
- Z. G. Wang and B. Z. Jiang, Macromolecules, 1997, 30, 6223–6229 CrossRef CAS.
- P. Svoboda, L. Keyzlarova, P. Saha, F. Rybnikar, T. Chiba and T. Inoue, Polymer, 1999, 40, 1459–1463 CrossRef CAS.
- Y. Aoki and O. Arendt, J. Appl. Polym. Sci., 2001, 82, 2037–2041 CrossRef CAS.
- N. Higashida, J. Kressler and T. Inoue, Polymer, 1995, 36, 2761–2764 CrossRef CAS.
- P. Svoboda, J. Kressler and T. Inoue, J. Macromol. Sci., Part B: Phys., 1996, B35, 505–525 CrossRef CAS.
- K. R. Shull and E. J. Kramer, Macromolecules, 1990, 23, 4769–4779 CrossRef CAS.
- G. H. Fredrickson and F. S. Bates, J. Polym. Sci., Part B: Polym. Phys., 1997, 35, 2775–2786 CrossRef CAS.
- C. W. Macosko, P. Guegan, A. K. Khandpur, A. Nakayama, P. Marechal and T. Inoue, Macromolecules, 1996, 29, 5590–5598 CrossRef CAS.
- S. A. Madbouly, Y. Xia and M. Kessler, Macromolecules, 2012, 45, 7729–7739 CrossRef CAS.
- B. D. Favis and J. P. Chalifoux, Polymer, 1988, 29, 1761–1767 CrossRef CAS.
- G. C. Alfonso and T. P. Russell, Macromolecules, 1986, 19, 1143–1152 CrossRef CAS.
- M. T. C. Brana, J. I. I. Sainz, B. Terselius and U. W. Gedde, Polymer, 1989, 30, 410–415 CrossRef.
- G. Defieuw, G. Groeninckx and H. Reynaers, Polymer, 1989, 30, 2158–2163 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |