Modifying graphite oxide nanostructures in various media by high-energy irradiation
Lei Chen,
Zhiwei Xu*,
Jialu Li,
Baoming Zhou,
Mingjing Shan,
Yinglin Li,
Liangsen Liu,
Baodong Li and
Jiarong Niu
Key Laboratory of Advanced Braided Composites, Ministry of Education, School of Textiles, Tianjin Polytechnic University, Tianjin 300160, People's Republic of China. E-mail: xuzhiwei@tjpu.edu.cn; Fax: +86 022 83955231; Tel: +86 022 83955231
First published on 13th November 2013
Abstract
The alterations of GO nanostructures after γ-ray irradiation in water, air and styrene with an absorbed dose of 200 kGy are systematically investigated. The interlayer structures of the ultimate products are confirmed to be remarkably different from each other due to the distinct changes of functional groups on single-sheets in various media. After irradiation in water, oxygen groups in graphite oxide are shown to be obviously decreased owing to the generation of reductive radicals by the decomposition of water molecules, which is reflected in the decrease of graphite oxide interlayer spacing. The interlayer distance of graphite oxide irradiated in air is found to be significantly increased, which may be attributed to the increase of the hydroxyl groups and the topological defects. However, the graphite oxide seems to be mainly exfoliated and functionalized by the intercalation of the monomers and the grafting of polystyrene chains when irradiated in styrene. It is expected that γ-ray irradiation in different media should be a promising strategy for manipulating nanostructures and properties of graphite oxide for improving its applicability in fields of composites, catalysts and sensors.
1. Introduction
Since it was first synthesized in 1855 by Brodie, graphite oxide (GO) has attracted the recurring interest of the chemical community.1,2 It is synthesized by strong acid/base treatments of graphite resulting in the incorporation of oxygen in many forms. Although the exact composition is difficult to determine, it is clear that the previously contiguous aromatic lattice of graphene is interrupted by epoxides, alcohols, ketone carbonyls, and carboxylic groups.3 Numerous studies were done throughout the decades to modify its structures and properties for stretching its applications in fields of optoelectronics, biodevices, drug delivery systems, and composites.4 Regardless of the preparation method,4 GO composition (group types and concentrations) and structure changed obviously with the addition of oxidizing agent before achieving some threshold oxidation,5 which is considered as a strategy to modify the structure of GO.6 For further applications, exfoliation of GO in aqueous solutions7 or high temperature8,9 to produce monolayer graphene sheets is also widely used for its high specific area and fascinating electrical properties. In addition, the intercalation of polymer chains or metallics could improve structures and properties for nanocomposites, surfactants and catalytic agents.10–16 However, in the above modification methods, the numbers and the types of oxygen groups are hard to be monitored and the modification uniformity cannot be guaranteed within the process of adding chemical agents.17 Many impurities are also introduced in the interlayer structures for some special applications, which results in that the universality for the using in various fields is very limited. Furthermore, in traditional methods mentioned above, high cost and low efficiency are serious problems that we should pay lots of attentions. Accordingly, manipulating the functional groups and the structures of GO variably with a versatile, low-cost and high-efficiency strategy is concluded to be very important for improving its ultimate applicability in various fields.High energy radiation, such as γ-ray irradiation, has been used for many years as a competitive method to modify structures of carbon systems for its low-cost, large-scale, high-energy and ultra-uniformity.11,18–20 It is naturally produced on earth by decay of high energy states in atomic nuclei with high frequency. In our previous work, the interlayer structure of multi-walled carbon nanotubes (MWCNTs) was found to be manipulated when irradiated in different media.11 Functionalization of MWCNTs with styrene was successfully achieved under γ-rays.21 The graphene oxide sheets was also decorated with poly(vinyl acetate) successfully by a γ-ray irradiation-induced graft polymerization.22 In addition, Zhang has demonstrated that functionalization and exhaustive reduction of graphene oxide could be implemented by γ-ray irradiation in the presence of different media.22,23 According to these works, it is reasonable to expect that the γ-ray irradiation should be a promising method for manipulating the functional groups and interlayer structures of GO in different media.
In this work, water, air and styrene are chosen as three media to assist the irradiation of GO. The differences of functional groups and structures between the pristine GO and irradiated GO (such as GO-WATER, GO-AIR and GO-STYRENE) are investigated in detail. To assure the effect mechanisms of irradiation media on GO, the pristine and irradiated samples are characterized by means of Fourier transform infrared (FT-IR) spectra, X-ray photoelectron spectroscopy (XPS), Raman spectra, X-ray diffraction (XRD), UV-visible (UV-Vis) absorption spectra and thermogravimetric analysis (TGA). Atomic force microscopy (AFM), transmission electron microscope (TEM) and high resolution TEM (HR-TEM) are used to observe the morphologies of products. The potential applications of these irradiated GO are also illustrated eventually.
2. Experimental section
2.1. Preparation of GO
Pristine GO is prepared according to the method in 6. A 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of concentrated H2SO4/H3PO4 (720:80 mL) is added to a mixture of graphite flakes (6.0 g, carbon content >99.99%) and KMnO4 (18.0 g), producing a slight exotherm to 35–40 °C. The solution is then heated to 50 °C, stirred for 12 h and then poured onto ice (800 mL) with 30% H2O2 (6 mL). For workup, the mixture is filtered over a PTFE membrane with a 0.22 μm pore size. The remaining solid is then washed in succession with 2000 mL deionized water. For each wash, the mixture is centrifuged at a speed of 8000 rpm. The solid obtained in the centrifuge tube is then vacuum-dried overnight at room temperature to get GO.
:1 mixture of concentrated H2SO4/H3PO4 (720:80 mL) is added to a mixture of graphite flakes (6.0 g, carbon content >99.99%) and KMnO4 (18.0 g), producing a slight exotherm to 35–40 °C. The solution is then heated to 50 °C, stirred for 12 h and then poured onto ice (800 mL) with 30% H2O2 (6 mL). For workup, the mixture is filtered over a PTFE membrane with a 0.22 μm pore size. The remaining solid is then washed in succession with 2000 mL deionized water. For each wash, the mixture is centrifuged at a speed of 8000 rpm. The solid obtained in the centrifuge tube is then vacuum-dried overnight at room temperature to get GO.
2.2. Preparation of irradiated GO
The 100 mg GO are firstly dissolved in water (100 mL) and styrene/toluene (50:50 mL) by weak sonication (5 min). These two as-prepared mixtures and 100 mg GO are then exposed to 60Co γ-ray source in an absorbed dose 200 kGy in air at room temperature. The air humidity is 55%. After irradiation, the suspension of GO irradiated in water is centrifugated and the precipitations were then also dried in a vacuum oven at 50 °C for 24 h to get GO-WATER. GO-AIR is taken out directly without any treatment. GO-STYRENE is purified repeatedly by cycling-membrane filtration/tetrahydrofuran(THF)-redispersion until no white emulsion precipitated when the filtrate is added to water. The precipitate is then dried in a vacuum oven at 50 °C for 24 h.
2.3. Characterization
GO and irradiated GO are characterized by X-ray diffraction (XRD, 1.54059 Å Cu Kα 1 as wavelength), Raman (RENISHAW in Via Raman Microscope, recorded using 514 nm laser excitation with a power of 5 mW), XPS (Thermo ESCALAB 250), FT-IR spectra (Bruker VECTOR-22 IR spectrometer) and TGA (NETZSCH STA 409 PC/PG), respectively. In addition, GO, GO-WATER and GO-AIR are sonicated in the deionized water for 2 h to get the nanosheets. For GO-STYRENE, the sheets are just got by dissolving GO-STYRENE in THF with weak sonication of 5 min. The morphologies of them are analyzed by atomic force microscope (AFM, CSPM5500) and high resolution transmission electron microscope (HR-TEM, Tecnai G2 F20). The samples for AFM analyses are precisely prepared by depositing the sheets on cleaved mica surfaces. The UV-visible absorption spectra of the dispersions are measured on a HITACHI UV-3310 spectrophotometer within the wavelength region 220–700 nm.3. Results and discussion
3.1. Compositions of irradiated GO
XPS is employed to determine the surface chemical composition of GO and irradiated GO. Curve fitting of C1s spectra is performed using a Gaussian–Lorentzian peak shape after performing a linear background correction and the results and detailed information are shown in Fig. 1 and Table 1. The C1s of GO consists of six different chemically shifted components and can be deconvoluted into: sp2 C–C, sp3 C–C in aromatic rings, C–OH, C–O–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and –COOH. These assignments agree with previous work.24–26
O and –COOH. These assignments agree with previous work.24–26
 | ||
| Fig. 1 The C1s peaks in the XPS spectra of (a) GO, (b) GO-WATER, (c) GO-AIR and (d) GO-STYRENE. | ||
| C–C | Oxygen groups | ||||||
|---|---|---|---|---|---|---|---|
| sp2 | sp3 | C–H | C–OH | C–O–C | CO |
–COOH | |
| (284.0–284.4) | (284.5–284.9) | (285.0–285.3) | (286.2–286.7) | (286.8–287.3) | (287.5–288) | (288.5–289) | |
| GO | 9.34 | 27.97 | — | 23.27 | 32.72 | 3.05 | 3.65 |
| GO-WATER | 9.42 | 37.75 | — | 16.64 | 26.02 | 4.90 | 5.26 |
| GO-AIR | 9.57 | 29.78 | — | 36.43 | 18.03 | 4.48 | 1.71 |
| GO-STYRENE | 18.85 | 18.77 | 41.94 | 20.44 | |||
After irradiation in water (Fig. 1b and Table 1), the percents of sp3 C–C is significantly increased and the content of oxygen groups is decreased. According to the radiation chemistry of water,27 γ-ray can decompose the water molecules to H˙, e−aq. and ˙OH, which makes an reductive agent in water to transform the functional groups to C–C bonding.23 In the spectra of GO-AIR, an increased component for C–OH and a significantly decreased component for C–O–C can be observed. To our knowledge, GO is a kind of hygroscopic material and some water molecules may not be eliminated exhaustively under the GO drying conditions.28
When irradiated in air (Fig. 1c), the C–O–C groups on the basal plane are seemed to be unstable and the rings are easy to be opened under the γ-rays in air. The bonding between ring-opening carbon-bear free radicals and decomposed ˙OH would make the dramatic increase of C–OH (Fig. 1c and Table 1). For GO-STYRENE, the percent of C–H is found to increase to 41.94% (Fig. 1d and Table 1), speculating the covalent bonding of polystyrene (PS) chains on the sheets under the γ-rays in styrene.29 The plentiful oxygen-containing groups on GO may provide active sites for chemical grafting of polymer chains on GO surface under γ-rays,30–32 which is reflected by the significant decrease of oxygen group content.
Fig. 2 shows the XPS survey spectra of GO and irradiated GO. A trend of decrease in O/C is exhibited after irradiation in all media. For GO-WATER (Fig. 2b), the oxygen groups are not eliminated exhaustively as that mentioned in ref. 23, which should be attributed to the fact that the surrounding environment is not deoxygenated by nitrogen bubbling.23 In addition, the O/C in GO-AIR (Fig. 2c) is seemed to be a little bigger than that of GO-WATER. This weaker reduction effect in GO-AIR should be explained by the small quantity of reduction media and lots of air around GO. With regard to GO-STYRENE, owing to the bonding of PS chains, Fig. 2d exhibits a notable decrease of the O/C.
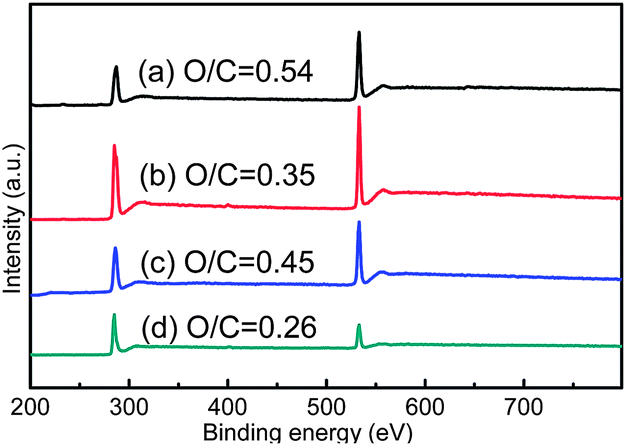 | ||
| Fig. 2 XPS full scan spectra of (a) GO, (b) GO-WATER, (c) GO-AIR and (d) GO-STYRENE. | ||
The FT-IR spectra of GO (Fig. 3) indicates the presence of CO (vCO at 1728 cm−1), C–O(vC–O at 1218 cm−1), alkoxy (valkoxy at 1060 cm−1) and C–OH(vC–OH at 3380 cm−1), and demonstrates that the GO has abundant oxygen containing groups.33 After irradiation in water, the intensity of all FT-IR peaks correlated to the oxygen containing groups decreases dramatically (Fig. 3b), which confirms the decrease of oxygen groups. Considering about GO-AIR (Fig. 3c), the spectrum exhibits characteristic vibration bands at 3000–2800 cm−1 and 696 cm−1, which is attributed to the stretching vibration of C–H, indicating the attachment of alkyl groups on the nanosheets under γ-ray irradiation. Furthermore, the characteristic peak at 1060 cm−1, which is corresponded to alkoxy, is significantly increased by γ-rays. This is in good agreement with XPS results and should be caused by the bonding of carbon-bare free radicals with H˙ and ˙OH.23 The spectrum of GO-STYRENE exhibits characteristic vibration bands for PS (absorbance peaks at 3026, 1597, 1491, 750, and 696 cm−1 correspond to the phenyl group, the peaks at 2920 and 2850 cm−1 represent the emergence of methylene and methenyl groups, and the peaks at 1448 and 1404 cm−1 could be assigned to saturated and unsaturated C–H bonds) (Fig. 3d), which confirms the successful functionalization.21,34
 | ||
| Fig. 3 FT-IR spectra of (a) GO, (b) GO-WATER, (c) GO-AIR and (d) GO-STYRENE. | ||
TGA curves in the temperature range from 25 to 700 °C are shown in Fig. 4. The weight of GO starts to lose at around 90 °C, corresponds to the liberation of absorbed water steam in pristine samples (Fig. 4a). In the temperature range of 100 °C and 250 °C, the major weight loss of GO occurs due to the exhaustive evaporation of water molecules and release of CO, CO2 from the decomposition of most labile functional groups (such as epoxides).22,28 The mass loss of GO-WATER shows a similar trend as that of GO at first (Fig. 4b). However, owing to the decomposition of increased carbon radicals, the curve shows a sharp loss after 180 °C.35 The thermal stability of GO-AIR is found to be obviously improved when the temperature is lower than 180 °C due to fact that the thermally labile oxygen functional groups (such as epoxides) and water molecules are prematurely decomposed by γ-rays in air (see Fig. 4c). Nevertheless, GO-AIR has a bigger weight loss compared with that of GO from 25 °C and 700 °C. This may be caused by the dramatic increase of –OH groups, which will take more carbon atoms during the heating process than another oxygen groups. Two clearly separated weight loss stages in the range of 120–220 °C and 320–460 °C for GO-STYRENE can be observed in Fig. 4d, corresponding to GO and grafted PS in GO-STYRENE.21 The grafting ratio is evaluated to be ∼53% as calculated in our previous work,36 which is a little bigger than that in ref. 22 due to the higher irradiation dose and more oxygen groups on GO sheets.
 | ||
| Fig. 4 TGA curves (in N2) of (a) GO, (b) GO-WATER, (c) GO-AIR and (d) GO-STYRENE. | ||
The UV-vis spectra of GO and irradiated GO are recorded for an equal concentration of each material (Fig. 5). The degree of remaining conjugation can be determined by the λmax of each UV-vis spectrum in the 227–231 nm for GO.37,38 Irradiated GO shows similar spectra with those of GO, indicating that the irradiation do not change the structure of GO basal plane significantly. However, the λmax of GO-WATER and GO-AIR (Fig. 5b and c) is seemed to be a little bigger than that of GO, suggesting that the more ordered structure of GO-WATER and GO-AIR due to greater retention of carbon rings in the basal planes after irradiation,6 which is in good agreement with the results of XPS. In Fig. 5d, a new absorbance peak emerged at ∼260 nm, indicating the decreased conjugation in GO sheet by the grafting PS chains.39 The dispersion extent of irradiated GO can be determined by their UV-vis absorption spectra.40 Through Bee–Lambert law (eqn (1)), we could establish a relationship between the exact concentrations of suspended irradiated GO and their absorbance values.
| A = εlc | (1) |
 | ||
| Fig. 5 UV-vis spectra of (a) GO, (b) GO-WATER, (c) GO-AIR and (d) GO-STYRENE. | ||
3.2. Interlayer structures of irradiated GO
Fig. 6 shows the XRD patterns of GO and irradiated GO with an absorbed dose of 200 kGy. The interlayer distance obtained from the (001) peak is 0.93 nm (2θ = 9.56°) in pristine GO. After irradiation in water, the distance is obviously contracted due to the decrease of oxygen groups as shown in Fig. 6b.41 However, the interlayer distance is significantly expanded to 2.11 nm (2θ = 4.22°) for GO-AIR (Fig. 6c). This may be due to the significant increase of –OH groups between the interlayers as that indicated in XPS results. Furthermore, the increased topological defects and the improvement of steric hindrance in interlayers may also result in this expansion.42 With regard to GO-STYRENE, GO is shown to be exfoliated when irradiated in styrene (Fig. 6d). The van der Waals forces seem to be insufficient to maintain GO stacking under the high-energy γ-rays. The (002) peak is almost going to be disappeared in GO-STYRENE, presumably because of introduction of styrene between the GO layers, grafting of PS chains on the sheets and then exfoliation of GO induced by the γ-ray irradiation.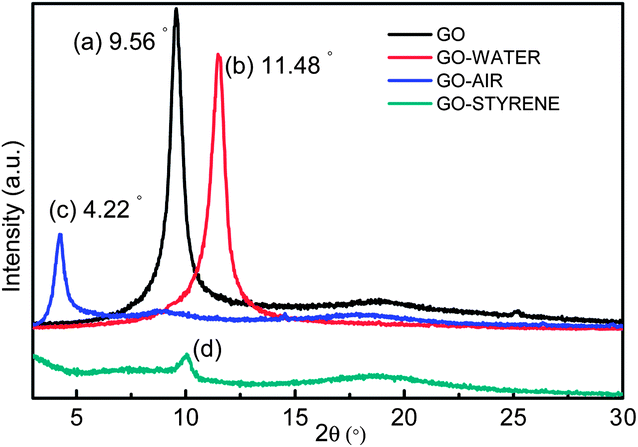 | ||
| Fig. 6 XRD patterns of (a) GO, (b) GO-WATER, (c) GO-AIR and (d) GO-STYRENE. | ||
Evidences for the alteration of GO interlayer structures after irradiation in different media were shown by the TEM and HR-TEM images in Fig. 7. To our knowledge, the interlayer distance of the conventional GO should be ∼0.9 nm. After irradiation in air, the interlayer distance is found to be increased to ∼2.0 nm (see the insert in Fig. 7b), which is in good agreement with XRD results. For GO-WATER, the oxygen groups are proved to be partially reduced, which result in the decrease of GO-WATER d-spacing (Fig. 7d). Thus, the sheets are found to be hardly separated with each other so that many sheets with multilayers are exhibited (Fig. 7c). In addition, GO-STYRENE can be dissolved in THF without powerful sonication owing to the intercalation of styrene, grafting of PS chains on the nanosheets and ultimate exfoliation of GO. Moreover, some dark features are observed on the GO-STYRENE sheets in Fig. 7e due to the pisolitic aggregates formed on surface of GO sheet by encircling with PS chains and single-layer functionalized graphene sheets can be detected (Fig. 7f). The exact mechanisms for alterations of single-layers and the differences between the ultimate irradiated GO structures are illustrated in the schematic drawing in Fig. 8.
 | ||
| Fig. 7 TEM images of (a) sonicated GO, (c) sonicated GO-WATER and (e) dissolved GO-STYRENE and HR-TEM image of (b) GO-AIR, (d) GO-WATER and (f) GO-STYRENE. | ||
 | ||
| Fig. 8 Schematic drawing of the effects of various media on the eventual nanostructure of irradiated GO. The alterations in each single layer are stressed in dashed box to state the exact mechanisms. | ||
AFM images of sonicated GO, GO-WATER and GO-AIR and dissolved GO-STYRENE are presented in Fig. 9. The thicknesses of pristine GO single-layer sheet we prepared are mostly within a range of 1.2–1.5 nm (Fig. 9a and b), which is a little higher than that reported.43,44 Some sheets are shown as multi-layers from Fig. 9a because of the overlapping or aggregation in sonicated GO samples. After irradiation in water, the thickness of monolayer sheet seems to be decreased, indicating the decrease of oxygen groups by γ-rays. However, Fig. 9b and d show that the heights of GO-STYRENE monolayer sheets are about 1.9 nm to 2.4 nm. It is noteworthy that the height value may be higher than that of the pristine GO monolayers due to the PS chains grafted on nanosheet surfaces or trapped underneath the drop-dried SP-graphene flake ‘coffee rings’.45 In addition, after sonicating GO-AIR, the monolayer percent is significantly increased and the nanosheets are found to be dispersed uniformly owing to the dramatic increase in the d-spacing and alteration of the oxygen groups. Furthermore, some vacancies are found to be formed after irradiation in all of these media, making the boundaries and the surface of the sheets a little more ‘bumpy’ than the pristine ones due to the cutting effect of γ-rays (see Fig. 9b–d).46,47
 | ||
| Fig. 9 AFM tapping mode and height images of (a) GO, (b) GO-WATER and (c) GO-AIR sheets that sonicated in water for 2 h and (d) sheets from the weakly sonicated GO-STYRENE in THF for 5 min. | ||
The structure of GO can also be monitored by the variation of relative intensities of G band at 1596 cm−1 corresponding to the first-order scattering of the E2g mode, and D band at 1357 cm−1, arising from a breathing mode of k-point phonons of A1g symmetry in the Raman spectra.48 From Fig. 10, the intensity ratio of D band to G band (ID/IG) of irradiated GO don't show an obvious change compared with that of GO, which may be attributed to the combined effect of the change of oxygen groups and the restoration or destruction of carbon systems under irradiation. With the reduction progressing (Fig. 10b), the intensity ratio of D band to G band (ID/IG) is increased, implying the forming of new domains of conjugated carbon atoms (bonded in sp2 hybridization) accompanying the removal of oxygen-containing groups.33 The alteration of epoxide groups to hydroxyl should be the main mechanism for the decrease of ID/IG in GO-AIR (Fig. 10c). The ID/IG of GO-STYRENE is shown to be increased from 0.93 to 0.95, suggesting a decrease in the average size of sp2 carbon domains, which is induced by the increased number of smaller graphitic domains formed during the irradiation process (Fig. 10d).49 Moreover, this weak increase may also suggest the bonding of PS chains on graphene nanosheets and support the cutting effects (from the AFM results) under irradiation.
 | ||
| Fig. 10 Raman spectra of (a) GO, (b) GO-WATER, (c) GO-AIR and (d) GO-STYRENE. | ||
4. Conclusions
In summary, irradiated GO show different compositions and nanostructures from each other due to the intrinsic properties of the media. The interlayer distance of GO-AIR is found to be dramatically increased due to significant increase of –OH group. However, GO-WATER d-spacing is confirmed to be diminished because of the decrease of oxygen groups under the irradiation-induced reductive agent in water. With regard to GO-STYRENE, intercalation of styrene and grafting of PS chains result in a self-exfoliation of the functionalized GO, which are confirmed by the improvement of GO-STYRENE monolayer thickness. Accordingly, γ-ray irradiation should be an effective strategy for decreasing the GO d-spacing for GO/hydrophilic composites, increasing the GO d-spacing for catalytic agents and exfoliating and functionalizing GO for nanocomposites and sensors.Acknowledgements
The authors acknowledge support from the National Natural Science Foundation of China (11175130), Natural Science Foundation of Tianjin, China (10JCYBJC02300) and China Postdoctoral Science Foundation (2012M520578).Notes and references
- B. C. Brodie, Philos. Trans. R. Soc. London, 1859, 149, 249–259 CrossRef.
- A. Dimiev, D. V. Kosynkin, L. B. Alemany, P. Chaguine and J. M. Tour, J. Am. Chem. Soc., 2012, 134, 2815–2822 CrossRef CAS PubMed.
- S. Stankovich, R. D. Piner, X. Q. Chen, N. Q. Wu, S. T. Nguyen and R. S. Ruoff, J. Mater. Chem., 2006, 16, 155–158 RSC.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- C. Shi, L. Chen, Z. Xu, Y. Jiao, Y. Li, C. Wang, M. Shan, Z. Wang and Q. Guo, Physica E, 2012, 44, 1420–1424 CrossRef CAS PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- T. Zhou, F. Chen, K. Liu, H. Deng, Q. Zhang, J. Feng and Q. Fu, Nanotechnology, 2011, 22, 045704 CrossRef PubMed.
- H. B. Zhang, J. W. Wang, Q. Yan, W. G. Zheng, C. Chen and Z. Z. Yu, J. Mater. Chem., 2011, 21, 5392–5397 RSC.
- M. J. McAllister, J.-L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car, R. K. Prud'homme and I. A. Aksay, Chem. Mater., 2007, 19, 4396–4404 CrossRef CAS.
- J. Barkauskas, I. Stankeviciene, J. Daksevic and A. Padarauskas, Carbon, 2011, 49, 5373–5381 CrossRef CAS PubMed.
- Z. Xu, L. Chen, L. Liu, X. Wu and L. Chen, Carbon, 2011, 49, 350–351 CrossRef CAS PubMed.
- P. G. Liu, K. C. Gong, P. Xiao and M. Xiao, J. Mater. Chem., 2000, 10, 933–935 RSC.
- Y. Matsuo, K. Tahara and Y. Sugie, Carbon, 1996, 34, 672–674 CrossRef CAS.
- T. Cassagneau, F. Guerin and J. H. Fendler, Langmuir, 2000, 16, 7318–7324 CrossRef CAS.
- T. Kyotani, H. Moriyama and A. Tomita, Carbon, 1997, 35, 1185–1187 CrossRef CAS.
- G. C. Wang, Z. Y. Yang, X. W. Li and C. Z. Li, Carbon, 2005, 43, 2564–2570 CrossRef CAS PubMed.
- L. Zhang, X. Li, Y. Huang, Y. Ma, X. Wan and Y. Chen, Carbon, 2010, 48, 2367–2371 CrossRef CAS PubMed.
- Z. W. Xu, Y. D. Huang, C. H. Zhang, L. Liu, Y. H. Zhang and L. Wang, Compos. Sci. Technol., 2007, 67, 3261–3270 CrossRef CAS PubMed.
- S. A. Vitusevich, V. A. Sydoruk, M. V. Petrychuk, B. A. Danilchenko, N. Klein, A. Offenhausser, A. Ural and G. Bosman, J. Appl. Phys., 2010, 107, 063701 CrossRef.
- Z. Xu, L. Chen, B. Zhou, Y. Li, B. Li, J. Niu, M. Shan, Q. Guo, Z. Wang and X. Qian, RSC Adv., 2013, 3, 10579–10597 RSC.
- H. X. Xu, X. B. Wang, Y. F. Zhang and S. Y. Liu, Chem. Mater., 2006, 18, 2929–2934 CrossRef CAS.
- B. W. Zhang, Y. J. Zhang, C. Peng, M. Yu, L. F. Li, B. Deng, P. F. Hu, C. H. Fan, J. Y. Li and Q. Huang, Nanoscale, 2012, 4, 1742–1748 RSC.
- B. W. Zhang, L. F. Li, Z. Q. Wang, S. Y. Xie, Y. J. Zhang, Y. Shen, M. Yu, B. Deng, Q. Huang, C. H. Fan and J. Y. Li, J. Mater. Chem., 2012, 22, 7775–7781 RSC.
- C. C. Teng, C. C. M. Ma, C. H. Lu, S. Y. Yang, S. H. Lee, M. C. Hsiao, M. Y. Yen, K. C. Chiou and T. M. Lee, Carbon, 2011, 49, 5107–5116 CrossRef CAS PubMed.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS PubMed.
- S. Park, K. S. Lee, G. Bozoklu, W. Cai, S. T. Nguyen and R. S. Ruoff, ACS Nano, 2008, 2, 572–578 CrossRef CAS PubMed.
- A. R. Anderson and E. J. Hart, J. Phys. Chem., 1962, 66, 70–75 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS PubMed.
- Y. L. Liu and W. H. Chen, Macromolecules, 2007, 40, 8881–8886 CrossRef CAS.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- H. Bai, C. Li and G. Q. Shi, Adv. Mater., 2011, 23, 1089–1115 CrossRef CAS PubMed.
- Y. W. Cao, J. C. Feng and P. Y. Wu, Carbon, 2010, 48, 1683–1685 CrossRef CAS PubMed.
- X. J. Zhou, J. L. Zhang, H. X. Wu, H. J. Yang, J. Y. Zhang and S. W. Guo, J. Phys. Chem. C, 2011, 115, 11957–11961 CAS.
- M. Steenackers, A. M. Gigler, N. Zhang, F. Deubel, M. Seifert, L. H. Hess, C. H. Y. X. Lim, K. P. Loh, J. A. Garrido, R. Jordan, M. Stutzmann and I. D. Sharp, J. Am. Chem. Soc., 2011, 133, 10490–10498 CrossRef CAS PubMed.
- H. F. Yang, F. H. Li, C. S. Shan, D. X. Han, Q. X. Zhang, L. Niu and A. Ivaska, J. Mater. Chem., 2009, 19, 4632–4638 RSC.
- L. Chen, Z. Xu, J. Li, Y. Li, M. Shan, C. Wang, Z. Wang, Q. Guo, L. Liu, G. Chen and X. Qian, J. Mater. Chem., 2012, 22, 13460–13463 RSC.
- H. B. Yu, X. Y. Mo, J. Peng, M. L. Zhai, J. Q. Lia, G. S. Wei, X. H. Zhang and J. L. Qiao, Radiat. Phys. Chem., 2008, 77, 656–662 CrossRef CAS PubMed.
- X. F. Gao, J. Jang and S. Nagase, J. Phys. Chem. C, 2010, 114, 832–842 CAS.
- W. N. Zhang, W. He and X. L. Jing, J. Phys. Chem. B, 2010, 114, 10368–10373 CrossRef CAS PubMed.
- Y. Z. Pan, H. Q. Bao and L. Li, ACS Appl. Mater. Interfaces, 2011, 3, 4819–4830 CAS.
- H. J. Shin, K. K. Kim, A. Benayad, S. M. Yoon, H. K. Park, I. S. Jung, M. H. Jin, H. K. Jeong, J. M. Kim, J. Y. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987–1992 CrossRef CAS.
- O. C. Compton and S. T. Nguyen, Small, 2010, 6, 711–723 CrossRef CAS PubMed.
- J. Inhwa, M. Vaupel, M. Pelton, R. Piner, D. A. Dikin, S. Stankovich, A. Jinho and R. S. Ruoff, J. Phys. Chem. C, 2008, 112, 8499–8506 Search PubMed.
- Z. H. Ni, H. M. Wang, J. Kasim, H. M. Fan, T. Yu, Y. H. Wu, Y. P. Feng and Z. X. Shen, Nano Lett., 2007, 7, 2758–2763 CrossRef CAS PubMed.
- Z. Jin, T. P. McNicholas, C. J. Shih, Q. H. Wang, G. L. C. Paulus, A. Hilmer, S. Shimizu and M. S. Strano, Chem. Mater., 2011, 23, 3362–3370 CrossRef CAS.
- J. Y. Li and Y. F. Zhang, Appl. Surf. Sci., 2006, 252, 2944–2948 CrossRef CAS PubMed.
- J. Peng, X. X. Qu, G. S. Wei, J. Q. Li and J. L. Qiao, Carbon, 2004, 42, 2741–2744 CrossRef CAS PubMed.
- S. Reich and C. Thomsen, Philos. Trans. R. Soc., A, 2004, 362, 2271–2288 CrossRef CAS PubMed.
- M. S. Dresselhaus, A. Jorio, M. Hofmann, G. Dresselhaus and R. Saito, Nano Lett., 2010, 10, 751–758 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |