
Unraveling the mode of binding of the anticancer drug topotecan with dsDNA†
Abstract
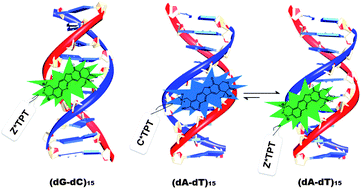
The binding interactions between antitumor drugs and DNA are of burgeoning interest due to increasing demand in medicinal science. In the present work, we have tried to examine the mode of binding of topotecan (TPT) with DNA. TPT, an eminent anti-cancer drug from the Camptothecin family, is found to interact with DNA Topoisomerase-I and inhibits the DNA replication process. Steady state, time resolved fluorescence, circular dichroism and thermal melting studies have been utilized to explore the mode of binding of TPT with synthetic polynucleotides ((dA-dT)15, (dG-dC)15) and natural DNA (CT-DNA). The mode of binding of TPT with the DNA double helix has been substantiated to be principally groove binding. It is found that even though the ground state cationic form (C) of the drug binds to dsDNA irrespective of DNA sequences, the emission mainly appears from Z*, and it is attributed to the intermolecular excited state proton transfer (ESPT) reaction between the drug and surrounding water molecules. However, in the case of (dA-dT)15, the emission profile indicates the existence of a small population of excited state cationic form (C*) of the drug in the minor groove of DNA. The different photophysical behavior of TPT in the case of (dA-dT)15 compared to others is attributed to the narrower and deeper minor groove of (dA-dT)15 than that of the others. The exact molecular picture of the binding interaction between the drug and DNAs has been explored from molecular modeling studies.
 Please wait while we load your content...
Please wait while we load your content...

