A general and green procedure for the synthesis of organochalcogenides by CuFe2O4 nanoparticle catalysed coupling of organoboronic acids and dichalcogenides in PEG-400†
Debasish
Kundu
,
Nirmalya
Mukherjee
and
Brindaban C.
Ranu
*
Department of Organic Chemistry, Indian Association for the Cultivation of Science, Jadavpur, Kolkata, 700 032, India. E-mail: ocbcr@iacs.res.in; Fax: +913324732805; Tel: +913324734971
First published on 30th October 2012
Abstract
A general and efficient procedure has been developed for the synthesis of organochalcogenides (selenides and tellurides) by a simple reaction of organoboronic acids and dichalcogenides catalysed by CuFe2O4 nanoparticles in PEG-400 without any ligand. This protocol offers the scope for access to a wide spectrum of chalcogenides including diaryl, aryl–heteroaryl, aryl–styrenyl, aryl–alkenyl, aryl–allyl, aryl–alkyl and aryl–alkynyl versions. The catalyst is magnetically separable and recyclable eight times without any loss of appreciable catalytic activity. The products are obtained in high purities after evaporation of solvent followed by filtration column chromatography.
1. Introduction
The organochalcogenides (selenides and tellurides) have received renewed interest as these compounds play an important role in organic synthesis as useful intermediates1 and in pharmaceutical industries for their potential biological activities such as antiviral, antihypertensive, antioxidant, antimicrobial and anticancer properties.2 They have important applications as materials too.3 Thus several methods have been developed for their synthesis, primarily through transition metal catalysed aryl–chalcogen bond formation.4 Various metals including palladium,5 nickel,6 and copper7 have been employed to catalyze the aryl–chalcogen bond forming reaction by treatment of aryl halides with thiol and selenol/PhSeNa under basic conditions. However, because of the instability and toxicity of these reagents, chalcogenide synthesis by the reaction of stable and easily available diaryl dichalcogenides and aryl halides is widely employed.8 The organoboronic acids have received much attention as a reagent of choice because of their easy accessibility, stability, nontoxicity and compatibility with a variety of functional groups.9 Thus, a few metal-catalysed aryl carbon–chalcogen bond forming reactions using aryl boronic acids have been reported.10 These include iron,10a CuI,10b CuO nanoparticle,10c and InBr310d catalysed couplings of aryl boronic acids with diselenides and ditellurides. Notably, these reactions addressed primarily the synthesis of aryl–aryl selenides and tellurides. The coupling with alkyl boronic acids was not successful10a,10d except for one case in the presence of ligand,10e and coupling with heteroaryl, allyl, alkenyl and alkynyl boronic acids was not addressed. Moreover, a high temperature and long reaction period were employed. Thus a green and convenient protocol for the synthesis of these compounds with general applicability and high efficiency is of much importance.We recently reported a procedure for the synthesis of diaryl chalcogenides by a reaction of aryl diazonium fluoroborate and diaryl dichalcogenides in the presence of Zn in dimethyl carbonate.11 However, this procedure is limited for access to diaryl chalcogenides only. Thus to have a better and more general method we report here a CuFe2O4 nanoparticle12 catalysed coupling of boronic acid with ditellurides and diselenides in PEG-400 (polyethylene glycol-400) in the presence of DMSO as an additive (Scheme 1).
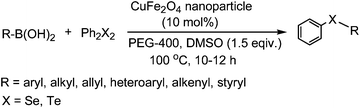 | ||
| Scheme 1 Coupling of boronic acids with diaryl diselenides and ditellurides. | ||
2. Results and discussion
To standardise the reaction conditions a series of experiments were performed under varying reaction parameters such as solvent, temperature, time and additive for a representative reaction of 4-methoxyphenyl boronic acid and diphenyl ditelluride in the presence of CuFe2O4 nanoparticles. Among a number of solvents such as DMSO, DMF, NMP, CH3CN, toluene, EtOH and dioxane (Table 1, entries 1–8) studied in the first phase, DMSO was found to provide the best yields in a relatively short period of 10 h. However, we sought to have a greener reaction medium than DMSO and we found that PEG-400 (polyethylene glycol) in combination with a small amount of DMSO as an additive furnished a high yield of product in 10 h (Table 1, entry 11). The use of DMSO as an additive in glycerol required 30 h for completion of a similar reaction.10b The reaction in PEG-400 in the absence of DMSO gave only 32% yield. The amount of DMSO was optimised to 1.5 equivalents with respect to the boronic acid for the best results. The reaction did not initiate at all in the absence of CuFe2O4 catalyst. The amount of catalyst was optimised to 10 mol%. The PEG-400 was also found to work better than PEG-600 (Table 1, entry 15). Thus, in a typical experimental procedure, a mixture of an organoboronic acid and diphenyl ditelluride in PEG-400 was heated at 100 °C in the presence of CuFe2O4 as the catalyst and DMSO as an additive for the required period of time (TLC). Extraction of crude product by ethyl acetate followed by evaporation of solvent and column chromatography provided the pure product. This procedure was also found to work efficiently for the reactions of boronic acid and diphenyl diselenide, boronic esters and diphenyl ditelluride/diselenide, trifluoroborates and diphenyl ditelluride/diselenide. The CuFe2O4 nanoparticles were commercially available (Aldrich) and were of 20 nm size.|
|
||||
|---|---|---|---|---|
| Entry | Solvent | T/°C | Time/h | Yield (%)a |
| a Yields of isolated pure products with 0.1 mmol catalyst loading (1H and 13C). b 1.5 mmol DMSO was used. c No catalyst was used. d 0.05 mmol catalyst was used. e 1.0 mmol of DMSO was used. | ||||
| 1 | DMSO | 100 | 10 | 95 |
| 2 | DMSO | 80 | 20 | — |
| 3 | DMF | 100 | 20 | 30 |
| 4 | NMP | 100 | 20 | 38 |
| 5 | CH3CN | 100 | 20 | 52 |
| 6 | Toluene | 100 | 20 | 36 |
| 7 | EtOH | 100 | 20 | 42 |
| 8 | Dioxane | 100 | 20 | — |
| 9 | DMC | 100 | 20 | 28 |
| 10 | PEG-400 | 100 | 10 | 32 |
| 11 | PEG-400 | 100 | 10 | 96 |
| 12 | PEG-400 | 100 | 10 | —c |
| 13 | PEG-400 | 100 | 10 | 78b,d |
| 14 | PEG-400 | 100 | 10 | 80e |
| 15 | PEG-600 | 100 | 10 | 84b |
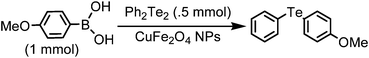
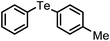
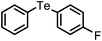
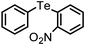
A wide range of diversely substituted organoboronic acids underwent reactions with diphenyl ditelluride by this procedure to produce the corresponding organotellurides. The results are summarized in Table 2. Several electron-withdrawing groups such as COMe, COOEt, NO2, CHO, CN, NHCOMe and electron-donating functionalities including OMe, OCF3, OH on the phenyl ring of the boronic acids are compatible in this reaction and the yields and reaction time remained uniform irrespective of the nature of the substituents. The heteroaryl boronic acids (Table 2, entries 12 and 18) which were not addressed in telluride coupling earlier,10 also underwent facile reaction to provide the corresponding products. The reaction of alkenyl and alkynyl boronic acids with diphenyl ditellurides provided equally high yields of products (Table 2, entries 14, 15, 16 and 17). A hindered 2,4,6-trimethylphenyl boronic acid also underwent coupling with no difficulty (Table 2, entry 7). The 4,4′-biphenyl diboronic acid provided the corresponding bis-telluride (Table 2, entry 13) which might be of potential interest as this class of compounds shows antioxidant properties.13 The styrenyl and phenyl acetylene tellurides (Table 2, entries 15–17) obtained easily by this procedure may be of interest in the pharmaceutical industry.14,15
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Product | Yield (%)a | Time/h | Ref. |
| a Isolated yields of pure products (1H and 13C). | |||||
| 1 | (4-OMe-C6H4)– |
|
96 | 10 | 10a |
| 2 | (4-COMe-C6H4)– |
|
92 | 12 | 11 |
| 3 | (4-COOEt-C6H4)– |
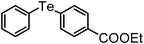
|
90 | 10 | 11 |
| 4 | (3-NHCOMe-C6H4)– |
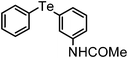
|
92 | 10 | — |
| 5 | (2-CHO-C6H4)– |
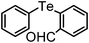
|
86 | 10 | 16a |
| 6 | (2-naphthyl)– |
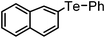
|
94 | 10 | 16b |
| 7 | (2,4,6-trimethyl-C6H2)– |
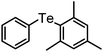
|
93 | 12 | 16c |
| 8 | (2-NO2-C6H4)– |
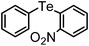
|
79 | 12 | 11 |
| 9 | (2-OH-C6H4)– |
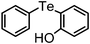
|
73 | 12 | 11 |
| 10 | (2-SMe-C6H4)– |
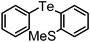
|
90 | 10 | — |
| 11 | (4-CN-C6H4)– |
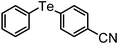
|
89 | 11 | 11 |
| 12 | (3-pyridinyl)– |
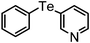
|
87 | 10 | 16d |
| 13 | (4,4′-biphenyl)– |
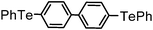
|
85 | 12 | 16e |
| 14 | (n-pentenyl)–(E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z = 30:70) :Z = 30:70) |
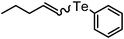
|
82 (E:Z = 30:70) |
12 | 16f |
| 15 | (styryl)– |
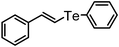
|
81 | 12 | 10e |
| 16 | (4-methylstyryl)– |
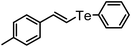
|
78 | 12 | 16g |
| 17 | (phenyl acetenyl)– |

|
82 | 12 | 16h |
| 18 | (3-thiophenyl)– |
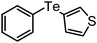
|
85 | 10 | — |
| 19 | (4-Me-C6H4)– |
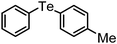
|
90 | 10 | 11 |
| 20 | (4-OCF3-C6H4)– |
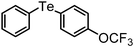
|
92 | 10 | 11 |
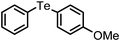
The substituted phenyl boronic acids, when subjected to reactions with diphenyl diselenides by this procedure, produced the corresponding organoselenides. The results are reported in Table 3. A wide range of electron withdrawing and electron donating substituents on the phenyl ring of boronic acids are well tolerated in this reaction too. As in telluride synthesis, a series of diaryl, aryl-heteroaryl, aryl-alkenyl, aryl-alkynyl unsymmetric selenides were obtained efficiently by this procedure. The reactions of alkyl boronic acids which were not successful in absence of ligand by other procedures10a,10b were achieved efficiently by our protocol (Table 3, entries 13 and 18). The reaction of dialkyl diselenide and phenyl boronic acid also proceeded without any difficulty by this protocol (Table 3, entry 19). Significantly, both electron withdrawing and electron donating group substituted diphenyl diselenides participated in this reaction (Table 3, entries 6, 11 and 18).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Product | Yield (%)a | Time/h | Ref. |
| a Isolated yields of pure products (1H and 13C). b Di-(4-methoxyphenyl)-diselenide was used. c Di-(4-chlorophenyl)-diselenide was used. d Di-(4-methylphenyl)-diselenide was used. e Dimethyl diselenide was used. | |||||
| 1 | (C6H5)– |
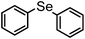
|
92 | 12 | 11 |
| 2 | (2-OH-C6H4)– |
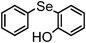
|
79 | 12 | 16i |
| 3 | (2-CHO-C6H4)– |
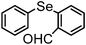
|
90 | 12 | 10a |
| 4 | (3,5-dimethyl-C6H3)– |
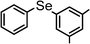
|
86 | 12 | 8c |
| 5 | (1-naphthyl)– |
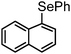
|
92 | 10 | 11 |
| 6b | (C6H5)– |
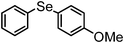
|
95 | 10 | 11 |
| 7 | (3-pyridinyl)– |
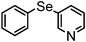
|
83 | 10 | 16d |
| 8 | (3-quinoline)– |
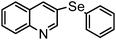
|
87 | 12 | 16j |
| 9 | (2-thiophenyl)– |
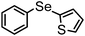
|
89 | 10 | 8c |
| 10 | (2-CN-C6H4)– |
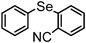
|
87 | 12 | 8c |
| 11c | (4-Cl-C6H4)– |
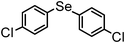
|
83 | 12 | 16k |
| 12d | (allyl)– |
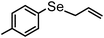
|
78 | 12 | 8c |
| 13 | (n-butyl)– |
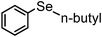
|
83 | 11 | 8c |
| 14 | (n-pentenyl)–(E:Z = 30:70) |
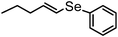
|
87 (E:Z = 30:70) |
12 | 16l |
| 15 | (4-methyl styryl)– |
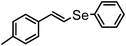
|
80 | 12 | 16g |
| 16 | (phenyl acetenyl)– |
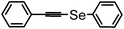
|
83 | 12 | 16h |
| 17b | (phenyl acetynyl)– |
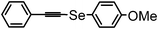
|
78 | 12 | 16h |
| 18b | (methyl)– |
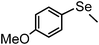
|
87 | 12 | 16m |
| 19e | (C6H5)– |
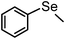
|
87 | 12 | 10e |
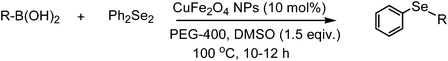
To expand the scope of this coupling, boronic esters and trifluoroborates were also employed as alternatives to boronic acid in these tellurylation and selenylation reactions. Several substituted boronic acid pinacol esters and aryl trifluoroborates underwent reactions with diphenyl ditellurides and diselenides by the same procedure to produce the corresponding products. The coupling of boronic esters is summarized in Table 4 and that of trifluoroborates is presented in Table 5.
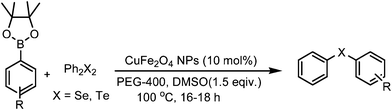
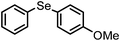
In general, the reactions are very clean and high yielding. All the reactions are complete within 10–12 h except for those of boronic acid pinacol esters (16–18 h). The products are obtained in high purity just by filtration chromatography. The nature and position of the substituents did not have any effect on the reaction rates and yields. Functionalization of products with a wide range of groups such as OMe, COOEt, COMe, CHO, CN, NO2, OCF3, SMe, NHCOMe, Cl, F has been successfully achieved by this procedure. Thus this procedure offers scope for further manipulation of products. The CuFe2O4 nanoparticle catalyst is recyclable up to eight runs without any considerable loss of reactivity (Fig. 1). However, after each cycle the CuFe2O4 nanoparticles which were initially of 20 nm size grew slightly bigger because of their inherent agglomeration tendency. After the 8th cycle, the particles attained 78 nm sizes and their catalytic activity was reduced substantially (Fig. 2).
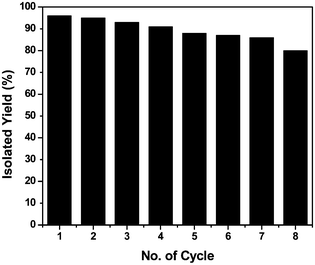 | ||
| Fig. 1 Recyclability of the catalyst. | ||
 | ||
| Fig. 2 TEM images of CuFe2O4 NPs. | ||
A recent report10f described C–Se bond formation by reaction of PhSeBr and organoboronic acids catalysed by CuFe2O4. However, our work deals with the reaction of commercially available and robust diphenyldiselenides/tellurides with boronic acids involving both selenide/telluride moieties. Thus it is essentially different and more practical as it uses diaryl dichalcogenides directly. Notably, PhSeBr was obtained from PhSeSePh.
To have an understanding of the active catalytic centre on CuFe2O4, when the reaction of diphenyl ditelluride and 4-methoxyphenyl boronic acid was performed using Fe3O4 nanoparticles the progress of the reaction was found to be only 25% in 12 h time. Thus, it is more likely that copper is the active catalytic centre as observed in similar reactions,10b,10c,10e although iron may have some role in enhancing the catalytic activity of Cu.
A possible reaction pathway is outlined in Scheme 2. Initially, in cycle I the CuFe2O4 nanoparticle undergoes oxidative addition to diphenyl ditelluride/diselenide to form an intermediate A which then undergoes transmetallation with phenyl boronic acid to provide the intermediate B. In a subsequent step this intermediate leads to the product, with telluride/selenide regenerating the catalyst via reductive elimination. On the other hand in cycle II the boronic acid interacts with CuFe2O4 to form the intermediate C which then reacts with [PhTe]−, another half of Ph2Te2 leading to B which finally produces the product. Thus both telluride/selenide moieties of (PhTe)2/(PhSe)2 are consumed in the reaction, making it atom-efficient. The DMSO present in the reaction mixture is reduced to dimethyl sulfide under the reaction conditions and it is assumed that dimethyl sulfide stabilizes the boronic acid formed in the transmetallation step and thus the presence of DMSO accelerates the process.10d
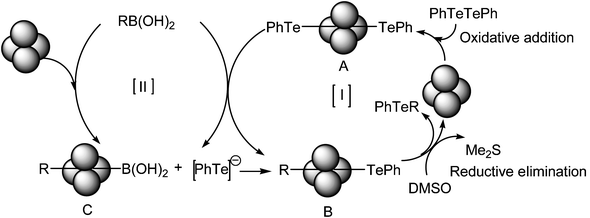 | ||
| Scheme 2 Possible mechanism | ||
3. Experimental
IR spectra were recorded on a Shimadzu 8300 FTIR spectrometer. 1H-NMR and 13C-NMR spectra were run on Bruker DPX-300 and DPX-500 instruments. HRMS were acquired on a microtek Qtof Micro YA263 spectrometer. The size of the nanoparticles (catalyst) was determined by HR-TEM experiment. All commercial reagents were distilled before use. CuFe2O4 nanoparticle, DMSO (dimethylsulfoxide), PEG-400 (polyethylene glycol- 400), boronic acids, boronic esters and trifluoroborates were purchased from Aldrich.Representative experimental procedure for the reaction of 4-methoxyphenyl boronic acid and diphenyl ditelluride (Table 2, entry 1)
A mixture of 4-methoxyphenyl boronic acid (152 mg, 1 mmol), diphenyl ditelluride (200 mg, 0.5 mmol), CuFe2O4 nanoparticles (24 mg, 0.1 mmol) and DMSO (120 mg, 1.5 mmol) in PEG-400 (3 mL) was heated at 100 °C for 10 h (TLC). After being cooled to room temperature the reaction mixture was extracted with ethyl acetate and the extract was evaporated to give the crude product which was purified by column chromatography to provide the product as a white solid (299 mg, 96%), mp 60–62 °C, IR (KBr) 3062, 1880, 1585, 1488 cm−1; 1H NMR (500 MHz, CDCl3) δ 3.76 (s, 3H), 6.78 (d, J = 8.5 Hz, 2H), 7.13–7.19 (m, 3H), 7.55 (d, J = 7.5 Hz, 2H), 7.71 (d, J = 8.5 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 55.2, 103.3, 115.6 (2C), 116.0, 127.4, 129.4 (2C), 136.5 (2C), 141.3 (2C), 160.1. These data are in good agreement with the reported values11 for (4-methoxyphenyl)(phenyl)tellane.This procedure was followed for all the reactions in Tables 2, 3, 4 and 5. A few of these products are known compounds (see the references in Tables 2, 3, 4 and 5) and were easily identified by comparison of their spectroscopic data with those previously reported. The unknown compounds were fully characterized by their IR, 1H NMR, 13C NMR and HRMS spectra, and C, H, N-analyses. These data are given below in order of their entries in Table 2.
N-(3-(Phenyltellanyl)phenyl)acetamide (Table 2, entry 4)
Yellow viscous liquid; IR (neat) 3296, 3257, 3064, 2991, 2850, 1668, 1581 cm−1; 1H NMR (300 MHz, CDCl3) δ 2.08 (s, 3H), 7.08–7.28 (m, 4H), 7.37 (d, J = 7.34 Hz, 1H), 7.52 (d, J = 7.90 Hz, 1H), 7.68 (d, J = 7.01 Hz, 2H), 7.77 (s, 1H), 8.13 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 24.4, 114.5, 115.1, 119.7, 128.0, 128.9, 129.6 (2C), 129.9, 133.5, 138.2 (2C), 138.8, 169.1; HRMS calcd. for C14H13OTe [M + Na]+: 363.9957; Found: 363.9926.Methyl(2-(phenyltellanyl)phenyl)sulfane (Table 2, entry 10)
Yellow liquid; IR (neat) 3049, 2916,1562, 1433 cm−1; 1H NMR (500 MHz, CDCl3) δ 2.42 (s, 3H), 6.84 (t, J = 8.0 Hz, 1H), 6.96 (d, J = 7.5 Hz, 1H), 7.07 (t, J = 7.5 Hz, 1H), 7.12–7.24 (m, 3H), 7.31 (t, J = 7.5 Hz, 2H), 7.79 (d, J = 8.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 19.3, 115.0, 124.5, 127.8, 128.8, 129.9 (2C), 130.1, 134.3 (2C), 140.5, 140.7; anal calc. for C13H12STe: C 47.62, H 3.69; found: C 47.45, H 3.72%.3-Phenyltellanyl-thiophene (Table 2, entry 18)
Yellow liquid; IR (neat) 3064, 2989, 1573, 1473 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.19–7.31 (m, 5H), 7.59–7.62 (m, 3H); 13C NMR (125 MHz, CDCl3) 104.0, 115.4, 127.3, 127.9, 129.5 (2C), 134.6, 136.6 (2C), 136.9; anal calc. for C10H8STe: C 41.73, H 2.80; found: C 41.66, H 2.92%.Procedure for recyclability of catalyst
After completion of the reaction the magnetic bar covered with the used catalyst was collected by a magnetic rod and the bar was successively washed with ethanol and acetone before being poured into another reaction pot for the next cycle of reaction.4. Conclusion
In conclusion, we have developed an efficient procedure for the synthesis of organotellurides and selenides by the reaction of boronic acid/boronic ester/trifluoroborate with diphenyl ditelluride/diselenide catalysed by CuFe2O4 nanoparticles in PEG-400 in the presence of a small amount of DMSO as an additive. The simplicity in operation, general applicability to various types of boronic acids including heteroaryl, alkynyl, alkenyl and particularly alkyl boronic acids which were reported to be inactive,10a,10d and wide scope of the functionalization make this protocol more attractive than the existing ones. In addition, recyclability of the catalyst for eight runs, use of PEG-400 as the reaction medium and high yields of products in a relatively short period make this procedure greener and more cost effective. We believe, this will provide a practical solution to the synthesis of difficult to access organotellurides and selenides.Acknowledgements
We are pleased to acknowledge the financial support from DST, New Delhi under J. C. Bose Fellowship grant (SR/S2/JCB-11/2008) to B. C. Ranu. DK thanks CSIR, New Delhi for his fellowship. We acknowledge support of Nanoscience Project Unit at IACS, funded by DST, New Delhi.References
- (a) C. Paulmier, in Selenium Reagents and Intermediates in Organic Synthesis, Organic Chemistry Series 4; J. E. Baldwin, Ed.; Pergamon Press Ltd.: Oxford, 1986 Search PubMed; (b) N. Petragnani, Tellurium in Organic Synthesis, A. R. Katritzky, O. Meth-Cohn and C. W. Rees, ed.; Academic Press: San Diego, 1994 Search PubMed.
- (a) G. Mugesh, W. W. duMont and H. Sies, Chem. Rev., 2001, 101, 2125 CrossRef CAS; (b) C. W. Nogueira, G. Zeni and J. B. T. Rocha, Chem. Rev., 2004, 104, 6255 CrossRef CAS.
- (a) Y. Okamoto, in The Chemistry Of Organic Selenium and Tellurium Compounds, ed. S. Patai and Z. Rappoport, Wiley, Chichester, U. K., 1986, 1, 10 Search PubMed; (b) J. Hellberg, T. Remonen, M. Johansson, O. Inganas, M. Theander, L. Engman and P. Eriksson, Synth. Met., 1997, 84, 251 CrossRef CAS; (c) T. Ando, T. S. Kwon, A. Kitagawa, T. Tanemura, S. Kondo, H. Kunisada and Y. Yuki, Macromol. Chem. Phys., 1996, 197, 2803 CrossRef CAS.
- (a) T. Kondo and T. Mitsudo, Chem. Rev., 2000, 100, 3205 CrossRef CAS; (b) S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400 CrossRef CAS.
- (a) Y. Nishiyama, K. Tokunaga and N. Sonoda, Org. Lett., 1999, 1, 1725 CrossRef CAS; (b) T. Itoh and T. Mase, Org. Lett., 2004, 6, 4587 CrossRef CAS; (c) D. Barranano and J. F. Hartwig, J. Am. Chem. Soc., 1995, 117, 2937 CrossRef.
- (a) K. Takagi, Chem. Lett., 1987, 2221 CrossRef CAS; (b) H. J. Cristau, B. Chaband, A. Chene and H. Christol, Synthesis, 1981, 892 CrossRef CAS.
- (a) R. K. Gujadhur and D. Venkataraman, Tetrahedron Lett., 2003, 44, 81 CrossRef CAS; (b) H. Suzuki, H. Abe and A. Osuka, Chem. Lett., 1981, 151 CrossRef CAS; (c) F. Y. Kwong and S. L. Buchwald, Org. Lett., 2002, 4, 3517 CrossRef CAS.
- (a) D. Sing, E. E. Alberto, O. E. D. Rodrigues and A. L. Braga, Green Chem., 2009, 11, 1521 RSC; (b) N. Taniguchi, J. Org. Chem., 2004, 69, 6904 CrossRef CAS; (c) S. Bhadra, A. Saha and B. C. Ranu, J. Org. Chem., 2010, 75, 4864 CrossRef CAS and references cited therein; (d) H. Wang, L. Jiang, T. Chen and Y. Li, Eur. J. Org. Chem., 2010, 2324 CrossRef CAS.
- N. Miyayura, Bull. Chem. Soc. Jpn., 2008, 81, 1535 CrossRef.
- (a) M. Wang, K. Ren and L. Wang, Adv. Synth. Catal., 2009, 351, 1586 CrossRef CAS; (b) V. G. Ricordi, C. S. Freitas, G. Perin, E. J. Lenardao, R. G. Jacob, L. Savegnago and D. Alves, Green Chem., 2012, 14, 1030 RSC; (c) D. Alves, C. G. Santos, M. W. Paixao, L. C. Soares, D. de Souza, O. E. D. Rodrigues and A. L. Braga, Tetrahedron Lett., 2009, 50, 6635 CrossRef CAS; (d) K. Ren, M. Wang and L. Wang, Org. Biomol. Chem., 2009, 7, 4858 RSC; (e) N. Taniguchi, J. Org. Chem., 2007, 72, 1241 CrossRef CAS; (f) K. H. V. Reddy, B. Satish, K. Ramesh, K. Karnakar and Y. V. D. Nageswar, Chem. Lett., 2012, 41, 585 CrossRef CAS.
- D. Kundu, S. Ahammed and B. C. Ranu, Green Chem., 2012, 14, 2024 RSC.
- (a) K. Swapna, S. N. Murthy, T. M. Jyoti and Y. V. D. Nageswar, Org. Biomol. Chem., 2011, 9, 5989 RSC; (b) N. Panda, A. K. Jena and S. Mahapatra, Appl. Catal., A, 2012, 433, 258 CrossRef; (c) R. Zhang, J. Liu, S. Wang, I. Niu, P. Xia and W. Sun, ChemCatChem, 2011, 3, 146 CrossRef CAS.
- (a) H. Xu, Y. Wang, Z. Wang, J. Liu, S. Mario and D. Wim, Chin. Sci. Bull., 2006, 51, 2315 CrossRef CAS; (b) U. Jkilgore, J. A. Karly, M. Pink, X. Gao and D. J. Mindola, Angew. Chem., Int. Ed., 2009, 48, 2394 CrossRef.
- (a) Y. Mitsuhiro and F. Hataaka, Chem. Pharm. Bull., 2004, 52, 248 CrossRef; (b) J. V. Comasseto, W. L. Ling, N. Pelnagnani and H. A. Stefani, Synthesis, 1997, 373 CrossRef.
- (a) E. A. Okoronkwo, R. A. Rosario, D. Alves, L. Sawegnago, W. C. Noguerina and G. Zeni, Tetrahedron Lett., 2008, 49, 3252 CrossRef; (b) E. A. Okoronkwo, B. Godoi, F. R. Schumacher, S. J. Santosneto, C. Luchese, M. Pnigol, W. C. Nogueina and G. Zeni, Tetrahedron Lett., 2009, 50, 909 CrossRef.
- (a) J. L. Piette, M. C. Pardon, R. Weber, M. Baiwir and G. Llabres, Bull. Soc. Chim. Belg., 1986, 95, 247 CrossRef CAS; (b) H. Rheinboldt and G. Vicentini, Chem. Ber., 1956, 89, 624 CrossRef CAS; (c) T. Arnauld, D. H. R. Barton and J.-F. Norman, J. Org. Chem., 1999, 64, 3722 CrossRef CAS; (d) S. Kumar and L. Engman, J. Org. Chem., 2006, 71, 5400 CrossRef CAS; (e) C. Degrand, Tetrahedron, 1990, 46, 5237 CrossRef CAS; (f) C. C. Silveira, A. L. Braga and R. C. Guadagnin, Tetrahedron Lett., 2003, 44, 5703 CrossRef CAS; (g) A. L. Braga, T. Barcellos, M. W. Paixao, A. M. Deobald, M. Godoi, H. A. Stefani, R. Cella and A. Sharma, Organometallics, 2008, 27, 4011 CrossRef; (h) A. Sharma, R. Schwab, A. L. Braga, T. Barcellos and M. W. Paixao, Tetrahedron Lett., 2008, 49, 5172 CrossRef CAS; (i) J. Oddershede, L. Henriksen and S. Larsen, Org. Biomol. Chem., 2003, 1, 1053 RSC; (j) D. H. Barton, X. Lusinchi and P. Milliet, Tetrahedron, 1985, 41, 4727 CrossRef CAS; (k) Y. Li, C. Nie, H. Wang, X. Li, F. Verpoort and C. Duan, Eur. J. Org. Chem., 2011,(36), 7331 CrossRef CAS; (l) C. C. Silveira, G. Perin and A. L. Braga, J. Chem. Res., Synopses, 1994, 12, 492 Search PubMed; (m) L. M. Bouchet, A. B. Penenory and J. E. Angueello, Tetrahedron Lett., 2011, 52, 969 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: 1H and 13C NMR spectra of all products. See DOI: 10.1039/c2ra22415a |
| This journal is © The Royal Society of Chemistry 2013 |