Efficient ruthenium-catalysed oxidative cleavage of methyl oleate with hydrogen peroxide as oxidant
Arno
Behr
*,
Nils
Tenhumberg
and
Andreas
Wintzer
Technische Universität Dortmund, Lehrstuhl für Technische Chemie A, Emil-Figge Str. 66, Dortmund, 44227, Germany. E-mail: behr@bci.tu-dortmund.de; Fax: +49-231-755-2311
First published on 16th November 2012
Abstract
The oxidative cleavage of alkenes leads to the formation of carboxylic acids. One of the few technical processes using this reaction is the production of azelaic acid via the ozonolysis of oleic acid. Because of the need for stoichiometric amounts of the expensive oxidant ozone, together with safety hazards, there is still a requirement for a catalytic process using a cheap and environmentally friendly oxidant. In the present work, the oxidative cleavage of methyl oleate by hydrogen peroxide was catalysed by an easily available ruthenium precursor with dipicolinic acid as ligand. The systematic optimisation of the reaction led to the formation of azelaic acid monomethyl ester in high yields amounting to 86%. The investigation of the reaction pathway showed that the reaction proceeds via a tandem reaction of epoxidation and hydrolysis of the epoxide and oxidative cleavage of the vic-diol.
Introduction
The oxidative cleavage of alkenes is one of the paramount reactions of organic chemistry, and is widely investigated. Depending on the reaction conditions, aldehydes or ketones, as well as carboxylic acids, can be formed.The common method for the direct oxidative cleavage of alkenes is ozonolysis.1 Besides ozone, direct cleavage of alkenes can also be performed with catalytic amounts of transition metals such as ruthenium,2–18 osmium,19–24 palladium,25 manganese,26 tungsten,13,27–37 molybdenum15,34,38–41 and rhenium13,42–44 in combination with numerous oxidants for example NaOCl,3,4 NaIO4,3,5–7,20 peracetic acid,3,13 hydrogen peroxide3,13,15–18,24,27–39,42–44 or oxygen.14,23,25,26,45–51
One of the rare industrial applications of the oxidative cleavage of alkenes is the production of azelaic acid from oleic acid via ozonolysis (Fig. 1).52 Pelargonic acid is formed as a by-product in stoichiometric amounts. The total amount of azelaic acid produced is several 1000 tons/year.53 Azelaic acid is industrially used in the manufacture of polyamides, laminates, adhesives, plasticisers and hydraulic fluids.52 Pelargonic acid is also used in the manufacture of lubricants and plasticisers.54
 | ||
| Fig. 1 Ozonolysis of oleic acid.52 | ||
Although ozonolysis has been established as an industrial process for a long time,55,56 the oxidative cleavage of oleic acid, as well as the ozonolysis itself, is still the subject of research and development. Because of the need for stoichiometric amounts of the expensive oxidant ozone, and the safety hazards associated with the process, ozone is not a large-scale oxidant in the chemical industry. Hence, there is a need for a selective, economical and energy-efficient, as well as safe oxidation process for the production of azelaic acid with the use of an environmentally friendly and cheap oxidant.
The transition-metal catalysed oxidative cleavage of oleic acid is often valued as a promising alternative process for the industrial production of azelaic acid. Molecular oxygen seems to be the obviously ideal oxidant, since it is the most important and cheapest oxidant in the chemical industry. However, oxidations with molecular oxygen are sometimes difficult to control and are less selective in comparison to other oxidants, especially in the direct oxidation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds.57,58 Similarly, the aerobic catalytic cleavage of oleic acid or methyl oleate is only rarely described in the literature. Dapurkar et al.59 used microporous and mesoporous catalysts containing chromium whereas Köckritz et al.23 utilised an osmium catalyst in combination with an aldehyde as cooxidant.
C bonds.57,58 Similarly, the aerobic catalytic cleavage of oleic acid or methyl oleate is only rarely described in the literature. Dapurkar et al.59 used microporous and mesoporous catalysts containing chromium whereas Köckritz et al.23 utilised an osmium catalyst in combination with an aldehyde as cooxidant.
According to the limited selectivity of the oxidation of CC bonds with molecular oxygen, different two step processes were also examined for the oxidative cleavage of unsaturated fatty acids with both oxygen and H2O2 as oxidants. The first step is usually the dihydroxylation of the unsaturated fatty acid to the vic-diol with H2WO4/H2O2 or formic acid/H2O2, followed by the aerobic cleavage of the vic-diol with H2WO4/Co(acac)3/N-hydroxyphthalimide60,61 or Co/Mn/Br62 as catalysts.
Apart from molecular oxygen, H2O2 is the cheapest, and also an environmentally friendly oxidant since only water is formed as a by-product. Therefore, it is also a suitable oxidant for the catalytic cleavage of oleic acid. For instance, Re2O7,13 H2WO4,13,37 a tungsten heteropoly acid27 or molybdenum compounds39 were used as a catalyst with H2O2 as oxidant. The cleavage of oleic acid with a two-metal system containing MoO3 and RuCl3 under phase transfer conditions is described by Service et al.15 It is assumed that MoO3 catalysed the dihydroxylation of oleic acid, whereas RuCl3 catalysed the cleavage of the formed vic-diol. Recently, a review about the synthesis of azelaic acid was published by Köckritz and Martin.53 However, up to the present time, no alternative to industrially employed ozonolysis of oleic acid has been developed.
The ruthenium catalysed cleavage of alkenes is usually performed with catalytic amounts of high-valent RuO4 and NaOCl,3,4 NaIO43,6,7 or peracetic acid3,13 as stoichiometric oxidants. In contrast, the ruthenium catalysed cleavage of alkenes with H2O2 as an oxidant is only rarely described in the literature, because H2O2 is unable to oxidise low-valent ruthenium compounds into the catalytic active species RuO4. Only the cleavage of alkenes to carboxylic acids with [(Me3tacn) (CF3CO2)2Ru(III)(OH2)]CF3CO2 (Me3tacn = 1,4,7-trimethyl-1,4,7-cyclononane)16 and the cleavage of primary alkenes to aldehydes with [cis-Ru(II)(dmp)2(H2O)2][PF6]2 (dmp = 2,9-dimethylphenanthroline),17 are described. To the best of our knowledge, with the exception of cleavage of fatty acids with the two-metal system MoO3/RuCl3, the ruthenium catalysed cleavage of fatty acids by H2O2 has not been described.15
In the present study, we report the selective oxidative cleavage of methyl oleate to azelaic acid monomethyl ester by H2O2, with a simple ruthenium salt and with a special emphasis on the reaction mechanism. The reaction conditions were systematically optimised with respect to precursor, ligand, temperature, Ru/ligand ratio, solvent, reaction time and pH-value.
Results and discussion
Since we were interested in the research and development of a new catalyst for the oxidative cleavage of methyl oleate 1 by H2O2, we decided to develop the desired catalyst by performing the reaction with the formation of the epoxide 2 and the vic-diol 3 as intermediate (see Fig. 2). This reaction pathway is also known as hydrolytic pathway (see Fig. 5, pathway A).27 With respect to the reaction mechanism, we assumed that we needed a catalyst which can be used for the epoxidation and/or dihydroxylation of olefins, as well as for the oxidative cleavage of vic-diols. Thus we were also interested in these individual reactions.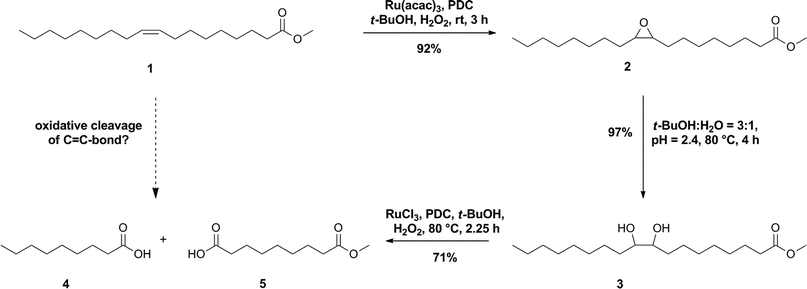 | ||
| Fig. 2 Suggested reaction pathway for the oxidative cleavage of methyl oleate by Ru/PDC with optimized reaction conditions. | ||
In our investigations concerning the oxidation of methyl oleate 1 with H2O2, we found that the catalyst system Ru(acac)3/dipicolinic acid (pyridine-2,6-dicarboxylic acid) (Ru(acac)3/PDC) is an efficient catalyst for the epoxidation of methyl oleate 1.63 Under our optimised reaction conditions for the oxidation of methyl oleate 1, the epoxide 2 was obtained in quantitative yield with acetonitrile (CH3CN) as solvent and at 92% with tert-butyl alcohol (t-BuOH).
We also found that epoxide 2 can be easily converted into the diol 3 in nearly quantitative yields in an aqueous t-BuOH solution, with H2SO4 as catalyst. As shown in Table 1, with a pH-value from 2.0 to 3.1, vic-diol 3 could be obtained with high yields and high selectivity. In contrast, the epoxide 2 seemed to be stable under basic conditions because the hydrolysis does not occur in significant amounts at pH ≥ 4.0.
Ruthenium, especially Ru(PPh3)3Cl2, has been observed to be an effective catalyst for the cleavage of vic-diols by H2O2, as well as for the cleavage of vic-diol 3 into carboxylic acids 4 and 5.64 Our investigation shows that RuCl3 as well as the catalyst system RuCl3/PDC, which we already used for the epoxidation of methyl oleate 1, could also cleave the vic-diol 3 into carboxylic acids 4 and 5 (Table 2). As by-products, the aldehydes nonanal 6 and 9-oxononanoic acid methyl ester 7 as well as the acyloins 8 and the 9,10-diketostearic acid methyl ester 9, are also formed (structures are shown in Fig. 5).
| Yield of oxidation products [%] | |||||||
|---|---|---|---|---|---|---|---|
| Catalyst | Conversion 3 [%] | Y 4 | Y 5 | Y 6 | Y 7 | Y 8 | Y 9 |
| a Reaction conditions: 2 mmol 3, 0.004 mmol Ru (0.2 mol%), 0.012 mmol ligand, 10 ml t-BuOH, 80 °C, 120 min: slow addition of 16 mmol 35% H2O2, total reaction time: 135 min. | |||||||
| Ru(PPh3)3Cl2 | 100 | 63 | 74 | 0 | 0 | 1 | 12 |
| RuCl3/PPh3 | 100 | 64 | 75 | 0 | 0 | 0 | 12 |
| RuCl3/PDC | 98 | 58 | 71 | 6 | 3 | 4 | 21 |
| RuCl3 | 100 | 66 | 79 | 0 | 0 | 0 | 10 |
Our investigation of these three individual reactions showed that the epoxidation of 1, and the oxidative cleavage of the vic-diol 3, can be catalysed by the same catalyst, and also that the hydrolysis of epoxide 2 could be performed under similar reaction conditions. We therefore assumed that Ru(acac)3/PDC would also be an effective catalyst for oxidative cleavage by performing the epoxidation, the hydrolysis and oxidative cleavage of vic-diol 3 consecutively in situ as an auto-tandem reaction. The suggested reaction pathway for the oxidative cleavage of 1 with Ru(acac)3/PDC, and the optimised reaction conditions of the three individual reactions, are shown in Fig. 2.
As a starting point for the reaction conditions for the oxidative cleavage of methyl oleate 1, we modified the reaction conditions of the epoxidation. A metal–ligand ratio of Ru(acac)3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PDC = 1:10 and a mixture of t-BuOH:H2O = 3:1 as solvent was chosen, because the water is necessary for the hydrolysis of the epoxide 2. Due to the fact that H+ is already formed by dissolving the Ru(acac)3, and PDC in the solvent (pH-value ≈ 2.9), no additional H2SO4 was added. The H2O2 (3.3 eq. or 8 eq.) was slowly added to the reaction mixture over a period of 12 h by a syringe pump to avoid unproductive decomposition of the H2O2. A total reaction time of 16 h was chosen. At first, the reaction was performed at different temperatures (Table 3).
:PDC = 1:10 and a mixture of t-BuOH:H2O = 3:1 as solvent was chosen, because the water is necessary for the hydrolysis of the epoxide 2. Due to the fact that H+ is already formed by dissolving the Ru(acac)3, and PDC in the solvent (pH-value ≈ 2.9), no additional H2SO4 was added. The H2O2 (3.3 eq. or 8 eq.) was slowly added to the reaction mixture over a period of 12 h by a syringe pump to avoid unproductive decomposition of the H2O2. A total reaction time of 16 h was chosen. At first, the reaction was performed at different temperatures (Table 3).
| Yield of oxidation products [%] | |||||
|---|---|---|---|---|---|
| T/°C | Conversion 1 [%] | Y 2 | Y 3 | Y 4 | Y 5 |
|
a Reaction conditions: 5 mmol 1, 0.05 mmol Ru(acac)3, 0.5 mmol dipicolinic acid, 20 ml solvent mixture (t-BuOH:H2O = 3:1), pH = 2.9, 12 h: slow addition of 16 mmol 35% H2O2.
b 40 mmol 35% H2O2 in 4 ml t-BuOH, total reaction time: 16 h.
|
|||||
| rt | 94 | 74 | 5 | 0 | 0 |
| 40 | 100 | 65 | 18 | 0 | 0 |
| 60 | 99 | 20 | 24 | 25 | 20 |
| 80 | 99 | 5 | 1 | 50 | 51 |
| 80b | 99 | 2 | 2 | 66 | 65 |
In agreement with the suggested reaction pathway, the conversion of methyl oleate 1 under the chosen reaction conditions led to the formation of the carboxylic acids 4 and 5. At 80 °C, the carboxylic acids 4 and 5 were formed as the major products with yields of 51% each. With eight equivalents of H2O2 (40 mmol), the carboxylic acids 4 and 5 were observed with a yield of 66%. In contrast, no carboxylic acids were formed at room temperature or 40 °C, since the hydrolysis of the epoxide 2, as well as the oxidative cleavage of vic-diol 3, is preferred at higher temperatures. As expected at lower temperature, epoxide 2 is formed as a major compound with yields of up to 74%, whereas vic-diol 3 yields only small amounts, because the hydrolysis of epoxide 2 proceeds slowly under these reaction conditions.
Thus, we were able to present Ru(acac)3/PDC as a new catalyst system for the oxidative cleavage of methyl oleate 1 by analysing the reaction mechanism and investigation of the three single reactions of the hydrolytic reaction pathway (e.g. epoxidation, hydrolysis, cleavage of vic-diol). After establishing the oxidative cleavage with Ru(acac)3/PDC, the reaction conditions were optimised.
Different ligands instead of PDC were tested, but with respect to the fact that the epoxidation of methyl oleate 1 only works quite well with PDC or 2,6-pyridinedimethanol,63 we only obtained the carboxylic acids 4 and 5 with these two ligands (Table 4). Therefore, DPC or 2,6-pyridinedimethanol as ligands as well as Ru(acac)3 or RuCl3 as ruthenium sources are essential for this reaction.
| Yield of oxidation products [%] | |||||
|---|---|---|---|---|---|
| Ligand | Conversion 1 [%] | Y 2 | Y 3 | Y 4 | Y 5 |
|
a Reaction conditions: 5 mmol 1, 0.05 mmol Ru(acac)3, 0.5 mmol ligand, 20 ml solvent mixture (t-BuOH:H2O = 3:1), 80 °C, 12 h: slow addition of 40 mmol 35% H2O2 in 4 ml t-BuOH, total reaction time: 16 h.
|
|||||
| PDC | 99 | 2 | 2 | 66 | 65 |
| 2,6-Pyridine-dimethanol | 99 | 4 | 4 | 44 | 34 |
| N-Methylimino-diacetic acid | 24 | 7 | 2 | 0 | 0 |
| Picolinic acid N-oxide | 24 | 10 | 0 | 0 | 0 |
Further investigation shows that different organic solvents can be used for the oxidative cleavage of methyl oleate 1 (Table 5). Best results are obtained with aqueous mixtures of t-BuOH and CH3CN, which are also the preferred solvents for ruthenium catalysed epoxidation of methyl oleate 1,63 whereas with 1,4-dioxane smaller amounts of the carboxylic acids 4 and 5 are obtained. In aqueous dimethylformamide (DMF), no carboxylic acids are observed, because the hydrolysis of epoxide 2 does not occur in this solvent mixture. Accordingly, epoxide 2 is formed as a major product.
| Solvent | Conversion 1 [%] | Yield of oxidation products [%] | |||
|---|---|---|---|---|---|
| Y 2 | Y 3 | Y 4 | Y 5 | ||
|
a Reaction conditions: 5 mmol 1, 0.05 mmol Ru(acac)3, 0.5 mmol dipicolinic acid, 20 ml solvent mixture (3:1), 80 °C, 12 h: slow addition of 40 mmol 35% H2O2 in 4 ml t-BuOH, or respective organic solvent, total reaction time: 16 h.
|
|||||
|
t-BuOH:H2O |
99 | 2 | 2 | 66 | 65 |
| CH3CN:H2O |
99 | 0 | 4 | 57 | 65 |
| 1,4-Dioxane:H2O |
98 | 5 | 4 | 48 | 50 |
| DMF:H2O |
88 | 42 | 5 | 0 | 0 |
Next, the effect of different concentrations of H2O in the solvent mixture was investigated, with the knowledge that H2O is mandatory for the hydrolysis of epoxide 2, but inhibits the epoxidation of methyl oleate. Table 6 shows the important influence of the water content in the solvent mixture towards the selectivity of the desired acids 4 and 5. The highest yields of the carboxylic acids 4 and 5 are obtained with a t-BuOH:H2O ratio of between 4:1 and 2:1.
| Ratio | Yield of oxidation products [%] | ||||
|---|---|---|---|---|---|
|
t-BuOH:H2O |
Conversion 1 [%] | Y 2 | Y 3 | Y 4 | Y 5 |
| a Reaction conditions: 5 mmol 1, 0.05 mmol Ru(acac)3, 0.5 mmol dipicolinic acid, 20 solvent mixture, 80 °C, 12 h: slow addition of 40 mmol 35% H2O2 in 4 ml t-BuOH, total reaction time: 16 h. | |||||
| pure t-BuOH | 98 | 32 | 10 | 14 | 10 |
| 19:1 |
98 | 13 | 6 | 35 | 36 |
| 4:1 |
99 | 3 | 3 | 64 | 64 |
| 3:1 |
99 | 2 | 2 | 66 | 65 |
| 2:1 |
99 | 1 | 2 | 63 | 64 |
| 1:1 |
99 | 6 | 2 | 54 | 57 |
In contrast with the small amount of water (t-BuOH:H2O = 19:1), only 36% of carboxylic acids are formed. In pure t-BuOH, epoxide 2 is formed as a major compound with an amount of 32%, because there is probably not enough water for the quantitative hydrolysis of epoxide 2. Accordingly, carboxylic acid 4 and 5 are only formed in minor amounts. The optimal reaction mixture t-BuOH:H2O = 3:1 presents a compromise between an effective epoxidation which is preferred by a small amount of water, and the effective hydrolysis of the epoxide 2 to diol 3 which is benefited by higher amounts of water.
Subsequently the Ru:PDC ratio (Table 7) and the catalyst concentration were optimised. Good yields of carboxylic acids 4 and 5 are obtained with an excess of the ligand (M:L < 1:10). The highest yields of carboxylic acids 4 and 5 are observed with a Ru:PDC ratio of 1:20, which conforms to the optimised reaction condition for the epoxidation of methyl oleate 1. With small amounts of PDC (M:L = 1:2), epoxide 2 was formed as the main product, whereas carboxylic acids 4 and 5 are only obtained in small amounts. We believe that an increasing concentration of PDC leads to lower pH-values, which will have a positive effect on the hydrolysis of epoxide 2. The effect of the pH-value on the reaction is described later.
| Yield of oxidation products [%] | |||||
|---|---|---|---|---|---|
| Ru:PDC | Conversion 1 [%] | Y 2 | Y 3 | Y 4 | Y 5 |
|
a Reaction conditions: 5 mmol 1, 0.05 mmol Ru(acac)3, dipicolinic acid, 20 ml solvent mixture (t-BuOH:H2O = 3:1), 80 °C, 12 h: slow addition of 40 mmol 35% H2O2 in 4 ml t-BuOH, total reaction time: 16 h.
|
|||||
| 1:2 |
97 | 37 | 4 | 11 | 12 |
| 1:5 |
98 | 11 | 5 | 46 | 49 |
| 1:10 |
99 | 2 | 2 | 66 | 65 |
| 1:20 |
100 | 1 | 3 | 70 | 73 |
| 1:30 |
100 | 1 | 3 | 67 | 68 |
Variation of the catalyst concentration shows that the reaction could produce good yields with low catalyst concentrations from 0.50 mol% to 1.00 mol% of Ru(acac)3.
Further exploration showed that the yield of carboxylic acids 4 and 5 could be clearly increased by increasing the time t for adding the H2O2 as well as the total reaction time tt (Table 8). High yields of carboxylic acids 4 and 5 up to 86% were achieved by adding the H2O2 slowly over about 20 h. In contrast with a low total reaction time of 6 h, only 40% of 4 and 5 together with broad amounts of epoxide 2 and vic-diol 3 as by-products, were formed. The results indicated that the slow addition of the oxidant by a syringe pump leads to a better efficiency, and avoids the nonproductive decomposition of H2O2. Also, a higher selectivity towards the desired products, the carboxylic acids 4 and 5, is achieved.
| Added H2O2 | tt | Yield of oxidation products [%] | ||||
|---|---|---|---|---|---|---|
| t [h] | [h] | Conversion 1 [%] | Y 2 | Y 3 | Y 4 | Y 5 |
|
a Reaction conditions: 5 mmol 1, 0.05 Ru(acac)3, 1.0 mmol dipicolinic acid, 20 ml solvent mixture (t-BuOH:H2O = 3:1), slow addition of 40 mmol 35% H2O2 in 4 ml t-BuOH, 80 °C.
|
||||||
| 3 | 6 | 98 | 16 | 9 | 37 | 40 |
| 6 | 10 | 99 | 6 | 4 | 55 | 58 |
| 12 | 16 | 100 | 1 | 3 | 70 | 73 |
| 16 | 20 | 99 | 1 | 3 | 77 | 78 |
| 20 | 24 | 99 | 0 | 4 | 81 | 86 |
An interesting and important parameter in the oxidative cleavage of methyl oleate 1 is the pH-value (Fig. 3). As already described in Table 1, the hydrolysis of the epoxide 2 to the vic-diol 3 only occurs in high yields at pH-values < 4.0. This explains why also at pH-values < 4.0 high amounts of carboxylic acids 4 and 5 are formed, whereas with pH-values ≥ 4.0 epoxide 2 is formed as the main product.
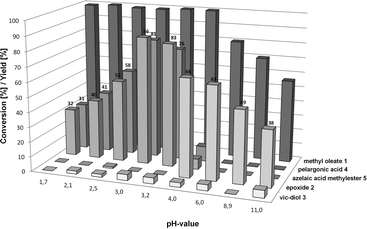 | ||
| Fig. 3 Influence of the pH-value on the ruthenium-catalysed oxidation of methyl oleate. Reaction conditions: 5 mmol 1, 0.05 Ru(acac)3, 1.0 mmol dipicolinic acid, 20 ml solvent mixture (t-BuOH:H2O = 3:1), 80 °C, H2SO4 or NaOH, 20 h: slow addition of 40 mmol 35% H2O2 in 4 ml t-BuOH, total reaction time: 24 h. | ||
 | ||
| Fig. 4 Oxidation of methyl oleate: Conversion-time-plot. Reaction conditions: 15 mmol 1, 0.15 Ru(acac)3, 3.0 mmol dipicolinic acid, 60 ml solvent mixture (t-BuOH:H2O = 3:1), pH = 2.92, 80 °C, 20 h: slow addition of 120 mmol H2O2 (35% aq), in 12 ml t-BuOH, total reaction time: 24 h. | ||
Thus, preceding the reaction with a pH ≥ 4.0, the reaction stops at the epoxide stage because the hydrolysis of epoxide 2 does not take place in a significant amount and therefore only small amounts of carboxylic acids or vic-diol 3 were formed. The results showed clearly that the pH-value can affect the product composition between the carboxylic acids 4 and 5 and the epoxide 2. The highest yields are obtained at a pH-value of 3.0, which is achieved by preparing the reaction solution without adding any acid or base. Similar high yields were only achieved at pH = 3.2. When the pH-value was decreased from 3.0 to 1.7, the yield of the carboxylic acids 4 and 5 also decreased from 86% to 32%. In more acidic reaction mediums, more side reactions occur, leading to the formation of undesired by-products and to a lower selectivity towards the carboxylic acids 4 and 5.
With more than eight equivalents of H2O2, no higher yields of carboxylic acids 4 and 5 were achieved. The reaction could also precede with similar yields of carboxylic acids 4 and 5 using 60% H2O2 instead of 35% H2O2.
To extend the scope of this methodology, we tested other unsaturated compounds such as oleic acid, high oleic sunflower oil (HOSO), as well as the assumed intermediates, epoxide 2 and vic-diol 3 under our optimised reaction conditions (Table 9). All compounds could be converted to azelaic monomethyl ester 5 or azelaic acid, in good yields. In some cases, nonanal 6 and 9-oxononanoic acid methyl ester 7 were formed as by-products in small amounts. The oxidative cleavage of the ester 1 and HOSO led to comparable amounts of cleavage products, whereas with oleic acid smaller amounts of azelaic acid were formed. The existence of the free carboxylic acid obviously led to more side reactions and to the formation of higher molecular by-products.
| Yield of oxidation products [%] | ||||||
|---|---|---|---|---|---|---|
| Educt | Conversion [%] | Y 4 | Y 5 | Y 6 | Y 7 | |
|
a Reaction conditions: 5 mmol educt, 0.05 mmol Ru(acac)3, 0.5 mmol dipicolinic acid, 20 ml solvent mixture (t-BuOH:H2O = 3:1), 80 °C, 20 h: slow addition of 40 mmol 35% H2O2 in 4 ml t-BuOH, total reaction time: 24 h.
b Cleavage products of high oleic sunflower oil (HOSO) and oleic acid were analysed as methyl esters (see experimental section).
|
||||||
| Methyl oleate 1 | 99 | 81 | 86 | 0 | 0 | |
| Epoxide 2 | 98 | 82 | 90 | 4 | 5 | |
| vic-Diol 3 | 100 | 77 | 75 | 0 | 0 | |
| HOSO | 100 | 82b | 78b | 4 | 4 | |
| Oleic acid | 100 | 59b | 66b | 4 | 5 | |
| 9-Decanoic acid methyl ester | 100 | — | 53 | 0 | 0 | |
Epoxide 2 and vic-diol 3 could also convert to carboxylic acids 4 and 5 in high yields, proving that they are intermediates in the oxidative cleavage of 1. 9-Decanoic acid methyl ester could also be cleaved to azelaic monomethyl ester 5 and formic acid as by-product.
This demonstrated that not only unsaturated fatty acids with internal double bonds, but also unsaturated compounds with terminal double bonds, can be cleaved to carboxylic acids.
Reaction mechanism
The reaction pathway of the oxidative cleavage of methyl oleate 1 was investigated with the help of a conversion-time-plot. Fig. 4 clearly shows that the reaction pathway proceeds mainly via the presumed hydrolytic pathway, with the formation of epoxide 2 as well as the vic-diol 3 as intermediates (Fig. 2). After a reaction time of 3 h, a conversion of 94% of 1 was already observed. A maximum yield of 28% of epoxide 2 was reached after 2 h, and after 3 h vic-diol 3 was formed with a maximum yield of 48%, indicating that the hydrolysis of epoxide 2 works fairly fast. After 3 h, the formation of the carboxylic acids 4 and 5 begins, and a maximum yield was achieved after 12 h. In addition to the already mentioned products, the aldehydes 6 and 7 and the acyloins 8a and 8b were also observed to be formed in minor amounts. These products were obviously formed as intermediates in the cleavage process. The acyloins 8 are formed by oxidation of the vic-diol 3. Because acyloin 8 is only formed in minor amounts, we are not able to estimate if it is an important intermediate at the cleavage of ester 1 or not. Therefore, the reaction mechanism for the cleavage of the vic-diol is hard to interpret.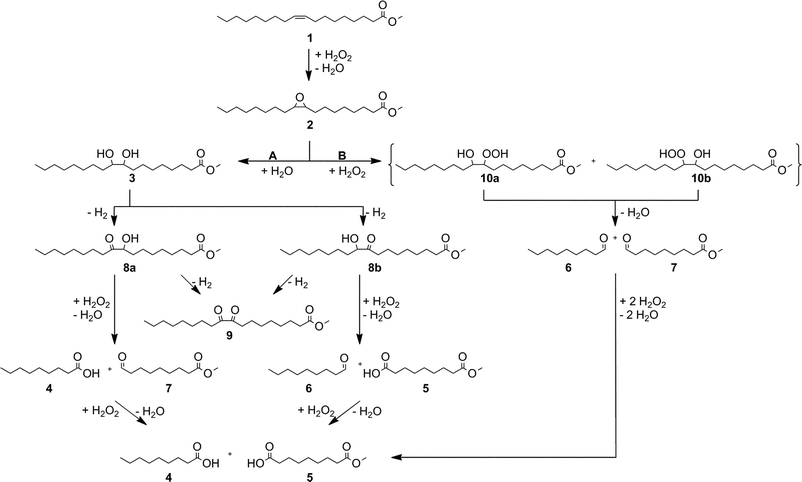 | ||
| Fig. 5 Oxidation of oleic acid methyl ester: Reaction mechanism. | ||
A supposable possibility is the cleavage of the acyloin 8 into a carboxylic acid and an aldehyde which can also be further oxidised to a carboxylic acid by hydrogen peroxide (Fig. 5, A). The mechanism of the cleavage of acyloins into an aldehyde and a carboxylic acid is described by Venturello65 and Ogata66.
The formation of the aldehydes 6 and 7 is already observed after a short reaction time of 2 h, and thus in advance of carboxylic acids 4 and 5, as well as acyloins 8, which are not formed until 3 h. Therefore, the hydrolytic pathway seems not to be the only pathway for the formation of the aldehydes 6 and 7. We assume that these small amounts of aldehydes 6 and 7 were formed via formation of the hydroxyperoxystearic acid methyl esters 10a and 10b as intermediate, which are formed by nucleophilic attack of H2O2 on the epoxide 2. Thermal decomposition of the reactive peroxy-compounds 10 leads to the formation of the aldehydes 6 and 7, which can be further oxidised to the carboxylic acid 4 and the monoester 5. This pathway is known as perhydrolytic pathway (Fig. 5, B).27 Unfortunately, we were not able to observe the formation of the unstable hydroxyperoxystearic acid methyl esters 10a and 10b.
In general the conversion-time plot definitely showed that the hydrolytic pathway is the preferred route for the oxidative cleavage of methyl oleate 1 with Ru(acac)3/PDC, since both the epoxide 2 and vic-diol 3 are formed as intermediates in moderate yields during the reaction. However it is probable that the hydrolytic and the perhydrolytic pathway are running in parallel. There is no indication neither for the cleavage nor for the cis-dihydroxylation of methyl oleate 1 by RuO4, RuO42− or any cis-dioxoruthenium species.
The presented catalyst system Ru(acac)3/PDC presents an alternative method and a different reaction mechanism for the ruthenium-catalysed oxidative cleavage of alkenes in comparison to the RuO4-chemistry and enables also the use of H2O2 as oxidant.
Conclusions
In this work, the oxidative cleavage of methyl oleate 1 by hydrogen peroxide was investigated in detail. After discussing the reaction mechanism of the transition-metal catalysed cleavage of alkenes by hydrogen peroxide, a new ruthenium catalyst for the oxidative cleavage of alkenes was developed. The active catalyst is formed in situ from an easily available ruthenium precursor, and a commercially available and cheap ligand, dipicolinic acid. Systematic investigation of the reaction parameters showed that the oxidative cleavage of methyl oleate 1 was significantly influenced by the ligand, the ratio of Ru-precursor to ligand, the temperature, the solvent and the reaction time. The product distribution between the azelaic acid monomethyl ester and the epoxidised methyl oleate could be affected by changing the pH-value. Under optimised reaction conditions, azelaic acid monomethyl ester was obtained at a high yield of 86%. Investigations of the reaction pathway showed that the reaction proceeds via a tandem reaction with the corresponding epoxide and the vic-diol of the unsaturated compound as intermediates. Besides fatty esters, also fatty acids, fats and terminal olefins were cleaved to carboxylic acids, demonstrating the general application of this process.In summary, a simple procedure for the oxidative cleavage of methyl oleate 1 to azelaic acid monomethyl ester, under mild reaction conditions using hydrogen peroxide as an oxidant, has been developed. The major advances of this method are simplicity, easy preparation of the catalyst and the use of aqueous hydrogen peroxide as an environmentally friendly oxidant.
Experimental section
Methyl oleate (97%+) was prepared by transesterification of high oleic sunflower oil (Emery oleochemicals) with methanol, using catalytic amounts of sulphuric acid. 9,10-Epoxystearic acid methyl ester 2 was prepared by epoxidation of methyl oleate 1 with Ru(acac)3/dipicolinic acid.63 All other chemicals were purchased from commercial suppliers at the highest purity available. They were used as received without further purification.The product samples were analysed by gas chromatography on a Hewlett-Packard 6890 instrument with a HP-5 capillary column (30 m × 0.25 mm × 0.25 μm) using a flame ionization detector. Pentadecane was used as internal standard. Gas chromatography-mass spectroscopy (GC-MS) chromatograms were recorded using a Hewlett-Packard 5973 instrument (HP-5 column 30 m × 0.25 mm × 0.25 μm) with an ionization energy of 70 eV.
1H- and 13C-NMR spectra were recorded on a Bruker model DPX500 spectrometer at room temperature. 1H- and 13C-NMR chemical shifts were reported on the δ-scale (ppm) relative to Me4Si as an external standard.
General procedure for the hydrolysis of 9,10-epoxystearic acid methyl ester 2
In a typical experiment (0.78 g, 2.5 mmol) 9,10-epoxystearic acid methyl ester 2 was dissolved in t-BuOH (5.85 g, 7.5 ml) and H2O (2.50 g, 2.5 ml). The pH-value was adjusted to 2.4 by adding H2SO4 (2 M), and the mixture was stirred magnetically at 80 °C for 4 h. Conversion and yield analyses were performed by GC.General procedure for the oxidative cleavage of 9,10-dihydroxystearic acid methyl ester 3
The oxidative cleavage of 9,10-dihydroxystearic acid methyl ester 3 was performed according to Herrmann et al.64 9,10-Dihydroxystearic acid methyl ester 3 (661 mg, 2.00 mmol), Ru(acac)3 (1.05 mg, 0.004 mmol, 0.2 mol%) and dipicolinic acid (2.01 mg, 0.012 mmol, 0.6 mol%) were dissolved in t-BuOH (7.8 g, 10.0 ml). 35% aqueous H2O2 (1.55 g, 16 mmol) was added slowly to the reaction solution by a syringe pump (ISMATEC, REGLO Digital) with a flow rate of 11.5 μl min−1 over a period of 120 min. The reaction mixture was stirred magnetically at 80 °C for 135 min. Conversion and yield analyses were performed by GC.General procedure for the oxidative cleavage of methyl oleate 1
In a typical experiment, methyl oleate 1 (1.48 g, 5.00 mmol), Ru(acac)3 (19.9 mg, 0.05 mmol, 1 mol%) and dipicolinic acid (167.1 mg, 1.00 mmol, 20 mol%) were dissolved in t-BuOH (11.7 g, 15.0 ml) and H2O (5.0 g, 5.0 ml). If required, the pH-value was adjusted by adding H2SO4 (2 M or 5 M) or NaOH (2 M or 5 M). 35% Aqueous H2O2 (3.88 g, 40 mmol) was dissolved in t-BuOH (3.1 g, 4.0 ml) and added slowly to the reaction solution by a syringe pump (ISMATEC, REGLO Digital) with a flow rate of 7.03 μl min−1 over a period of 20 h. The reaction mixture was stirred magnetically at 80 °C for 24 h. Conversion and yield analyses were performed by GC.Purification and characterization of the products
9,10-Dihydroxystearic acid methyl ester 3, nonanoic acid 4, azelaic acid methyl ester 5, nonanal 6 and 9-oxononanoic methyl ester 7 were purchased from commercial suppliers. 9,10-Diketostearic acid methyl ester 9 was identified by comparison of the retention time in the gas chromatogram with a synthesised sample. The sample was synthesised from methyl oleate 1 by oxidation with potassium permanganate and identified by 1H-NMR and MS.67Characterization of the ketohydroxystearic acid methyl esters 8a/8b
9-Hydroxy-10-ketostearic acid methyl ester 8a and 10-hydroxy-9-ketostearic acid methyl ester 8b were isolated as a mixture of both isomers by column chromatography (silica gel, cycloxhexane:ethyl acetate = 4:1) from the reaction solution of the oxidative cleavage of methyl oleate 1 and identified by 1H- and 13C-NMR and MS.
1H-NMR (400 MHz, CDCl3): δ = 4.14 (m, 1H, –CH(OH)), 3.60 (s, 3H, –OCH3), 2.43 (m, 2H, –CH2CO–), 2.28 (t, 2H, CH2CO2CH3), 1.41–1.61 (m, 6H, –CH2CH2CO2CH3, CH2CH2CO–, –CH2CH(OH)–), 1.26–1.29 (m, 18 H), 0.86 (s, 3H, –CH3);
13C-NMR (400 MHz, CDCl3): δ = 212.47, 212.42, 174.21, 174.17, 76.35, 76.28, 51.4, 37.8, 37.7, 33.99, 33.96, 33.72, 33.66, 31.79, 31.75, 29.42, 29.38, 29.26, 29.18, 29.05, 29.03, 28.96, 28.85, 24.82, 24.78, 24.73, 23.60, 23.48, 22.59, 14.06;
MS: m/z = 279 (2%), 187 (28), 159 (7), 158 (33), 156 (11), 155 (100), 141(5), 129 (7), 115 (22), 109 (17), 87 (48), 83 (18), 74 (30), 71 (12), 69 (24), 67 (17), 59 (13), 57 (30), 55 (53)
The GC-samples for the oxidative cleavage of oleic acid were prepared by transesterification with MeOH/BF3. The preparation of the GC-samples for the oxidative cleavage of high oleic sunflower oil (HOSO) was carried out by saponification with KOH and transesterification with MeOH/BF3.
Acknowledgements
This work was financially supported by the German Federal Ministry of Food, Agriculture and Consumer Protection (represented by the Fachagentur Nachwachsende Rohstoffe) and Emery Oleochemicals. The authors thank Dr Alfred Westfechtel for helpful discussions.References
- F. E. Kühn, R. W. Fischer, W. A. Herrmann and T. Weskampin Transition Metals for Organic Synthesis, ed. M. Beller and C. Bolm, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2 edn, 2004, vol. 2, ch. 2.12, pp. 427-436 Search PubMed.
- L. M. Berkowitz and P. N. Rylander, J. Am. Chem. Soc., 1958, 80, 6682–6684 CrossRef CAS.
- S. Warwel and M. Rüsch gen. Klaas, Lipid Technology, 1997, 10–14 CAS.
- B. Zaidman, A. Kisilev, Y. Sasson and N. Garti, J. Am. Oil Chem. Soc., 1988, 65, 611–615 CrossRef CAS.
- K. R. Rao, V. A. Kumar, V. P. Reddy, R. Sridhar and B. Srinivas, Synlett, 2009, 739–742 Search PubMed.
- K. B. Sharpless, V. S. Martin, T. Katsuki and P. H. J. Carlsen, J. Org. Chem., 1981, 46, 3936–3938 CrossRef.
- C. M. Che, C. M. Ho and W. Y. Yu, Angew. Chem., Int. Ed., 2004, 43, 3303–3307 CrossRef.
- T. Kojima, Y. Hirai, Y. Mizutani, Y. Shiota, K. Yoshizawa and S. Fukuzumi, Angew. Chem., Int. Ed., 2008, 47, 5772–5776 CrossRef.
- W. P. Griffith and E. Kwong, Synth. Commun., 2003, 33, 2945–2951 CrossRef CAS.
- W. P. Griffith, A. G. Shoair and M. Suriaatmaja, Synth. Commun., 2000, 30, 3091–3095 CrossRef CAS.
- A. G. F. Shoair and R. H. Mohamed, Synth. Commun., 2006, 36, 59–64 CrossRef CAS.
- D. Yang and C. Zhang, J. Org. Chem., 2001, 66, 4814–4818 CrossRef CAS.
- M. Rüsch gen. Klaas, P. Bavaj and S. Warwel, Fett. Wiss. Technol., 1995, 97, 359–367 Search PubMed.
- K. Kaneda, S. Haruna, T. Imanaka and K. Kawamoto, J. Chem. Soc., Chem. Commun., 1990, 1467–1468 RSC.
- A. Johnstone, P. J. Middleton, W. R. Sanderson, M. Service and P. R. Harrison, Stud. Surf. Sci. Catal., 1994, 82, 609–614 CrossRef CAS.
- C. M. Che, W. P. Yip and W. Y. Yu, Chem.–Asian J., 2006, 1, 453–458 CrossRef CAS.
- R. Neumann, V. Kogan and M. M. Quintal, Org. Lett., 2005, 7, 5039–5042 CrossRef.
- G. Barak and Y. Sasson, J. Chem. Soc., Chem. Commun., 1987, 1266–1267 RSC.
- T. A. Foglia, P. A. Barr, A. J. Malloy and M. J. Costanzo, J. Am. Oil Chem. Soc., 1977, 54, A870–A872 CrossRef.
- Z. D. Jin, W. S. Yu, Y. Mei, Y. Kang and Z. M. Hua, Org. Lett., 2004, 6, 3217–3219 CrossRef.
- B. Borhan, D. C. Whitehead and B. R. Travis, Tetrahedron Lett., 2006, 47, 3797–3800 CrossRef.
- B. Borhan, B. R. Travis and R. S. Narayan, J. Am. Chem. Soc., 2002, 124, 3824–3825 CrossRef.
- A. Köckritz, M. Blumenstein and A. Martin, Eur. J. Lipid Sci. Technol., 2010, 112, 58–63 CrossRef.
- B. Borhan, S. R. Hart, D. C. Whitehead and B. R. Travis, Org. Biomol. Chem., 2011, 9, 4741–4744 Search PubMed.
- H. F. Jiang and A. Wang, J. Org. Chem., 2010, 75, 2321–2326 CrossRef.
- C. C. Guo, Y. F. Li, X. H. Yan and Q. Liu, J. Porphyrins Phthalocyanines, 2006, 10, 942–947 CrossRef.
- E. Antonelli, R. D'Aloisio, M. Gambaro, T. Fiorani and C. Venturello, J. Org. Chem., 1998, 63, 7190–7206 CrossRef CAS.
- T. Oguchi, T. Ura, Y. Ishii and M. Ogawa, Chem. Lett., 1989, 18, 857–860 CrossRef.
- Y. Q. Deng, Z. F. Ma, K. Wang and J. Chen, Green Chem., 1999, 1, 275–276 RSC.
- J. F. Deng, X. H. Xu, H. Y. Chen and A. R. Jiang, Tetrahedron, 1992, 48, 3503–3514 CrossRef CAS.
- Y. Ishii, K. Yamawaki, T. Ura, H. Yamada, T. Yoshida and M. Ogawa, J. Org. Chem., 1988, 53, 3587–3593 CrossRef CAS.
- Z. P. Pai, A. G. Tolstikov, P. V. Berdnikova, G. N. Kustova, T. B. Khlebnikova, N. V. Selivanova, A. B. Shangina and V. G. Kostrovskii, Russ. Chem. Bull., 2005, 54, 1847–1854 CrossRef CAS.
- M. Schwegler, M. Floor and H. Van Bekkum, Tetrahedron Lett., 1988, 29, 823–826 CrossRef CAS.
- R. A. W. Johnstone, C. D. Brooks, L. C. Huang and M. McCarron, Chem. Comm., 1999, 37–38 Search PubMed.
- B. Ondruschka, J. Freitag and M. Nuchter, Green Chem., 2003, 5, 291–295 RSC.
- R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977–1986 RSC.
- JP Pat., 2003342255, 2003 Search PubMed.
- H. Furukawa, T. Nakamura, H. Inagaki, E. Nishikawa, C. Imai and M. Misono, Chem. Lett., 1988, 17, 877–880 CrossRef.
- S. E. Turnwald, M. A. Lorier, L. J. Wright and M. R. Mucalo, J. Mater. Sci. Lett., 1998, 17, 1305–1307 CrossRef CAS.
- P. Chaumette, H. Mimoun, L. Saussine, J. Fischer and A. Mitschler, J. Organomet. Chem., 1983, 250, 291–310 CrossRef CAS.
- R. D. Rapp and I. J. Borowitz, J. Chem. Soc., Chem. Comm., 1969, 1202–1203 RSC.
- W. A. Herrmann, T. Weskamp, J. P. Zoller and R. W. Fischer, J. Mol. Catal. A: Chem., 2000, 153, 49–52 CrossRef CAS.
- WO Pat., 98/47852, 1998 Search PubMed.
- US Pat., US 5321158, 1994 Search PubMed.
- J. E. Lyons and J. O. Turner, Tetrahedron Lett., 1972, 13, 2903–2906 CrossRef.
- T. Yamada, T. Takai, O. Rhode and T. Mukaiyama, Chem. Lett., 1991, 1–4 CrossRef CAS.
- G. Ragagnin and P. Knochel, Synlett, 2004, 951–954 CAS.
- R. Hunter, P. Turner and S. Rimmer, Synth. Commun., 2000, 30, 4461–4466 CrossRef CAS.
- A. K. Mandal and J. Iqbal, Tetrahedron, 1997, 53, 7641–7648 CrossRef CAS.
- J. Estrada, I. Fernandez, J. R. Pedro, X. Ottenwaelder, R. Ruiz and Y. Journaux, Tetrahedron Lett., 1997, 38, 2377–2380 CrossRef CAS.
- I. Fernandez, J. R. Pedro and R. de la Salud, Tetrahedron, 1996, 52, 12031–12038 CrossRef CAS.
- B. Cornils, P. Lappe, in Ullmann's Encyclopedia of Industrial Chemistry, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 7th edn, 2005 Search PubMed.
- A. Köckritz and A. Martin, Eur. J. Lipid Sci. Technol., 2011, 113, 83–91 CrossRef.
- W. Riemenschneider, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 7th edn, 2005 Search PubMed.
- D. J. Anneken, S. Both, R. Christoph, G. Fieg, U. Steinbrecher, A. Westfechtel, in Ullmann's Encyclopedia of Industrial Chemistry, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 7th edn, 2006 Search PubMed.
- N. G. Janakiefski, NLGI Spokesman, 1997, 61, 14–24 CAS.
- G. Franz, R. A. Sheldon, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 7th edn, 2005 Search PubMed.
- G. Sienel, R. Rieth, K. T. Rowbottom, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 7th edn, 2005 Search PubMed.
- S. E. Dapurkar, H. Kawanami, T. Yokoyama and Y. Ikushima, Top. Catal., 2009, 52, 707–713 CrossRef CAS.
- E. Santacesaria, M. Ambrosio, A. Sorrentino, R. Tesser and M. Di Serio, Catal. Today, 2003, 79, 59–65 CrossRef.
- E. Santacesaria, A. Sorrentino, F. Rainone, M. Di Serio and F. Speranza, Ind. Eng. Chem. Res., 2000, 39, 2766–2771 CrossRef CAS.
- K. Fujitani, H. Manami, M. Nakazawa, T. Oida and T. Kawase, J. Oleo Sci., 2009, 58, 629–637 CrossRef CAS.
- A. Behr, N. Tenhumberg and A. Wintzer, Eur. J. Lipid Sci. Technol., 2012 DOI:10.1002/ejlt.201200036.
- DE Pat., 19724736C1, 1997 Search PubMed.
- C. Venturello and M. Ricci, J. Org. Chem., 1986, 51, 1599–1602 CrossRef CAS.
- Y. Ogata, Y. Sawaki and M. Shiroyama, J. Org. Chem., 1977, 42, 4061–4066 CrossRef CAS.
- G. Knothe, Chem. Phys. Lipids, 2002, 115, 85–91 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |