Acceleration of the Eschenmoser coupling reaction by sonication: efficient synthesis of enaminones†
Naga Durgarao
Koduri
a,
Bethany
Hileman
a,
Justin D.
Cox
a,
Halee
Scott
a,
Phuong
Hoang
a,
Alexa
Robbins
a,
Kyle
Bowers
a,
Lemma
Tsebaot
a,
Kun
Miao
a,
Maria
Castaneda
a,
Michael
Coffin
a,
Guan
Wei
a,
Tim D. W.
Claridge
b,
Kenneth P.
Roberts
a and
Syed Raziullah
Hussaini
*a
aDepartment of Chemistry and Biochemistry, The University of Tulsa, Keplinger Hall, 800 South Tucker Drive, Tulsa, Oklahoma 74104, United States. E-mail: syed-hussaini@utulsa.edu; Fax: +1-918-631-3404; Tel: +1- 918-631-3520
bDepartment of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, United Kingdom
First published on 14th November 2012
Abstract
Enaminones are commonly prepared by the Eschenmoser coupling reaction. The duration of the reaction is often long. Here, we describe how sonication can accelerate this reaction. The reaction conditions provide an efficient method for the coupling of primary, secondary and tertiary thioamides with α-bromocarbonyl compounds.
Introduction
Enaminones are enamines of a 1,3-diketone, β-ketoester or similar 1,3-difunctional reagents.1 Enaminones are important building blocks in organic synthesis. They are used in the synthesis of natural products2–4 and utilized in the development of pharmaceuticals,5,6 peptidomimetics,7,8 ligands for catalysis9,10 and chiral auxiliaries.11 Additionally, enaminones form part of biologically active compounds. These include compounds with anticonvulsive, anti-inflammatory, antitumor and antibiotic activity.1,5 Enaminones can act either as electrophiles or nucleophiles and have been used widely in organic synthesis. A number of approaches are available for the synthesis of these compounds1,2,12,13 and the development of new and efficient methods remains an active area of research.3,14,15The Eschenmoser coupling reaction has been extensively employed in the construction of enaminones.1,2,4,13 The reaction couples a thioamide and an α-halocarbonyl compound (Scheme 1). The reaction generally requires a base, can be carried out with or without the need of a thiophile, and the reaction is known to work well with a range of different substrates.4
 | ||
| Scheme 1 The Eschenmoser coupling reaction. | ||
The Eschenmoser coupling reaction often requires a long reaction time16–18 and primary thioamides are not acceptable substrates as they tend to convert into nitriles under the reaction conditions.4 The Eschenmoser coupling reaction conducted under heterogeneous conditions4,16,19 could benefit from sonication as sonication reduces the particle size and provides better mass and heat transfer compared to conventional heating.20 In this paper, the effects of sonication on such heterogeneous Eschenmoser coupling reactions, and how it addresses the common issue of long reaction times and allows for the use of primary thioamides, are discussed.
Results and discussion
Usually, carbonate and bicarbonate bases have been used when the Eschenmoser coupling reaction is conducted under heterogeneous conditions. Both have been used in the coupling of 1,3-dicarbonyl compounds with thioamides.4,16,19 Since the aim was to couple both mono- and di-carbonyl compounds with the thioamides, the stronger base Na2CO3 was selected for the present study. The pKa of sodium carbonate (∼10) is close to the pKa of amine bases (∼10) that have been used successfully for the coupling of monocarbonyl halides with thioamides under non-heterogeneous conditions.4Synthesis of starting materials
The synthesis of thioamides 1 (Scheme 1) was carried out using literature conditions.14,21 The synthesis of α-bromocarbonyl compounds 2 however, gave some unexpected challenges (Fig. 1). Attempts to prepare 2b by reacting ethyl acetoacetate with NaH and Br2,22 TsOH and NBS,23 or NH4OAc and NBS24 did not give a pure product. Efforts to purify the product by flash chromatography also failed as 2b decomposed on the column. Finally, the use of bromodimethylsulfonium bromide (BDMS)25 provided 2b in pure form. We did not succeed in isolating 2c under solvent free conditions.26 The bromide 2c was obtained in pure form by reacting ethyl benzoylacetate with the in situ generated bromo ion (Br+)27 and isolating the product by column chromatography. Synthesis of 2d by the reaction of NBS on Meldrum's acid provided low yields (5%) of pure product.28 A better yield of 2d (up to 95%) was obtained by reacting Meldrum's acid with Br2 and NaOH.29 However, the purity of the product varied from experiment to experiment. All of the other bromides were obtained from commercial sources. | ||
| Fig. 1 α-Bromocarbonyl compounds used in this study. | ||
Synthesis of enaminones with the help of an ultrasonic bath
Ultrasonic baths are usually available in organic laboratories. They are the most economical and commercially available sources of ultrasonic irradiation for the chemical laboratory.20 As such, they have the greatest potential to be used in organic synthesis. This type of equipment was initially used for the Eschenmoser coupling reaction. Both the primary and the secondary thioamides provided the enaminones after only 2.5 h of sonication (Table 1, entries 1–4). In comparison, a low product conversion was observed when the reaction was refluxed for 2.5 h through conventional heating (entry 4).
| Entry | Thioamide | Product | Yield (%) | Time allowed |
|---|---|---|---|---|
| a Reaction was carried out in the absence of base. b Refluxed in a pressure vessel immersed in a 47 °C oil bath. c Product was obtained as an inseparable mixture of diastereomers. d Probe sonication was used. e Reaction was cooled by a (−8 °C) condenser. | ||||
| 1 |

|

|
||
| 3a R = OEt (90) | 2.5 h | |||
| 3b R = CH3 (60) | 2.5 h | |||
| 1a | ||||
| 2 |
|

|
||
| 3c R = OEt (98) | 2.5 h | |||
| (27)a | 2.5 h | |||
| 3d R = CH3 (84) | 2.5 h | |||
| 1b | ||||
| 3 |

|

|
||
| 3e R = OEt (80) | 2.5 h | |||
| 3f R = CH3 (47) | 2.5 h | |||
| 1c | ||||
| 4 |

|

|
||
| 3g R = OEt (73) | 2.5 h | |||
| (24)b | 2.5 h | |||
3h R = Ph (74 E/Z 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)c :1)c |
2.5 h | |||
| 1d | ||||
| 5 |

|

|
||
| 3i R = OEt (74) | 20 h | |||
| (97)d | 8.5 h | |||
| 3j R = Ph (99)d | 3.0 h | |||
| 1e | ||||
| 6 |
|

|
||
| 3k (85) | 46 h | |||
| (89)d | 7.0 h | |||
| (67 conversion by NMR)d,e | 7.0 h | |||
| 1f | ||||
| 7 |

|
|
||
| 3l R = OEt (94) | 5.0 days | |||
| (87)d | 11.5 h | |||
|
3m R = Ph (100 dr 2.4:1)c,d |
6.0 h | |||
| 1g | ||||
The mechanism of the Eschenmoser coupling reaction consists of two main steps (Scheme 2). The first step is the S-alkylation of thioamides with an electrophile. In the second step the resulting thioether gets deprotonated at the α-proton which is captured by the iminium 6 or the imine 7 to form the episulfide 8. The episulfide collapses, with or without the help of a thiophile, to give enaminones 3. When monoesters are used, tertiary thioamides tend to give higher yields of product with shorter reaction times. This rate and yield enhancement is explained as a result of a greater acidity of the side-chain α-proton. The greater acidity is due to the presence of the positive charge in the thioiminium salts 4. In the case of secondary thioamides, the iminium nitrogen does not retain its charge. Under reaction conditions, it loses its proton and is converted to an imine 5.4,30
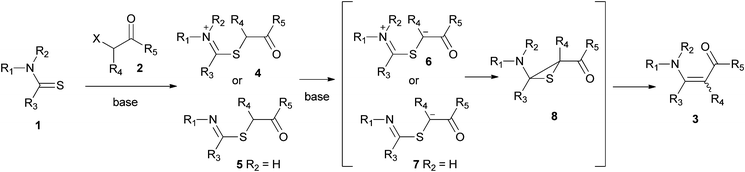 | ||
| Scheme 2 Accepted mechanism of the Eschenmoser coupling reaction. | ||
In contrast to the above reports, in this study the coupling reaction of tertiary thioamides proved to be more challenging than the coupling of secondary thioamides and required longer reaction times (compare entries 1–4 with entries 5–7). This could be attributed to the higher acidity of α-bromodicarbonyl compounds compared to α-monocarbonyl compounds. Greater acidity could make the conversion of 1→4 or 1→5 the rate determining step, resulting in the reactions of tertiary thioamides being slower as greater steric hindrance would pose greater energy requirements in the alkylation step (1→4, Scheme 2).31 Nonetheless, in all of the reactions of tertiary thioamides, good yields were obtained and the reaction was suitable for acyclic (entry 1), cyclic (entries 2–6) and acyclically positioned thiocarbonyl groups (entry 7). The α-bromoketoesters are generally considered unsuitable coupling partners for providing enaminones with primary thioamides. They instead give thiozoles.32–34 Nevertheless, a primary thioamide did give the coupled product with α-bromodicarbonyls (entry 1), and both the α-bromodiester and α-bromoketoesters were also viable coupling partners for secondary and tertiary thioamides (entries 2–7).
The reaction of α-bromoketoesters with secondary thioamides (entries 2–4) provided predominantly E diastereomers. The E stereochemistry in such compounds provides better hydrogen bonding between the N–H and the ketone oxygen and lowers the energy of the product. The coupling of 1a and 2b (R4 = OEt, R5 = CH3) (entry 1) gave a single diastereomer 3b. The stereochemistry of 3b was tentatively assigned as E on the basis of the H-bonding argument. The stereochemistry in the secondary enaminones was assigned by comparing the chemical shifts of the N–H protons, and by comparing the signals of the carbonyl ester groups in the IR of relevant compounds. The coupling of 1e with 2c (entry 5) provided just the E diastereomer 3j. Such selectivity has been observed before.4,35,36 The reaction of 1g with 2c was less selective and produced an inseparable mixture of diastereomers 3m in a 2.4:1 ratio.
Synthesis of enaminones with the help of probe sonication
Ultrasonic probes are less common in organic laboratories than ultrasonic baths, even though they are not very expensive.37 Furthermore, they are considered standard equipment for biochemistry and analytical chemistry laboratories. The ultrasonic probe provides much more acoustic energy than an ultrasonic bath. With an ultrasonic bath, the amount of energy that reaches the reaction is around 1–5 W cm−2, while an ultrasonic probe can provide several hundred W cm−2 to the reaction.20 We hypothesized that the reaction time for the coupling of tertiary thioamides could be reduced by providing more energy to the reaction. Thus, the use of an ultrasonic probe for the coupling of tertiary thioamides was explored. Table 1 (entries 5–7) shows that probe sonication reduces reaction times from days to hours.Reasons for the acceleration in reaction rates
The ultrasonic energy accelerates reactions primarily as a result of cavitation bubbles. These bubbles can act as micro-reactors which generate temperatures of several thousand degrees and pressures greater than one thousand atmospheres.20 In heterogeneous liquid–solid ionic reactions, cavitation bubbles can form at or near a solid surface. When such a bubble collapses, a liquid jet is formed which is targeted at the solid surface. The liquid jet disrupts interfacial boundary layers, causing an increase in the amount of mass and heat transfer to the surface. The collapse of bubbles can also generate shock waves. These shock waves can cause pressure and temperature increases and other mechanical effects.38 Additionally, cavitation has the ability to reduce the size of particles, which can also improve the mass transfer to the surface.20 So, which of these factors are more important than others in the sonication promoted Eschenmoser coupling reaction? We designed a few experiments to answer this question.Since α-halodicarbonyl species can exist in the enol form, they have the potential to react in the absence of a base to form enaminones.4 Hence, it was important to ascertain the impact of the use of a base in this transformation. When 1b was sonicated in a water bath with 2a in the absence of a base, only 27% of 3c was isolated after 2.5 h (Table 1 entry 2). The rest of the material remained in the thioether form indicating that without the base, sonication alone cannot complete this transformation in 2.5 h and the use of the base accelerates the conversion of 5 into enaminones (Scheme 2).
Thioamide 1f was reacted next with 2a using probe sonication (Table 1, entry 6). A short condenser (−8 °C) was attached to cool the vapor. After 7 h of sonication, 67% of 1f was converted to the enaminones 3k. Similar results were obtained when the reaction was cooled from outside by immersing the reaction vessel in a room temperature water bath. In contrast, without a condenser, 100% conversion of 1f and 89% isolated yield was obtained for 3k (Table 1, entry 6). This suggests that high temperatures and pressures are important factors and that mechanical effects are not solely responsible for the rate enhancement.20
Particle distribution analysis (Fig. 2, see also the ESI†) was performed on commercial, water bath sonicated and probe sonicated Na2CO3. It showed that the probe sonicated base had the smallest mean volume diameter (MV) of particles (646 nm) and the narrowest range of particle size distribution (428–1060 nm). The cleaning bath sonicated base had finer particles and a smaller range of particle sizes (761–1450 nm) than the commercial base (1232–5640 nm).
 | ||
| Fig. 2 Particle distribution analysis of sodium carbonate. | ||
The bath sonicated base was used in the reaction of 1b with 2a. After stirring for 2.5 h at room temperature all of 1b was converted to 3c, suggesting that reduction in particle size was the only factor responsible for the rate increase (compare with Table 1, entry 2). However, when probe sonicated Na2CO3 (sonicated for 8.5 h) was used to couple 1e and 2a, less than 50% conversion of 1e into product 3i was observed even after 8.5 h of stirring at room temperature (compare Table 1, entry 5). These results indicate that, for tertiary thioamides, a reduced particle size of the base is not the sole reason for the fast reaction. It seems that both a reduction in the size of particles and an increased temperature are important factors in the rate enhancement of this reaction.
Limitations and side reactions
When 2d was reacted with 1b, only a 24% yield of the enaminone 3n was obtained (Scheme 3). It was observed that bromide 2d reacts rapidly with 1b without the need for sonication. Immediate heating of the reaction vessel and effervescence were observed upon the addition of 2d to the CH2Cl2 solution of 1b and Na2CO3. TLC analysis suggested the complete disappearance of 1b within the amount of time it took to check the TLC (<5 min.). The fast reactivity of 2d is expected as it is sterically less hindered and more acidic than the open chain bromoesters and ketones (for comparison: the pKa of Meldrum's acid is 7.339 while the pKa of ethyl acetoacetate and diethyl malonate are 14.240 and 16.439 respectively). These attributes should enable 2d to alkylate easily 1→5 and deprotonate and cyclize efficiently 5→8 (Scheme 2). The low yield is possibly due to the thermolysis of the Meldrum's acid moiety of the enaminones 3n. It is known that such enaminones can decompose readily.41 A more controlled reaction and much better yield were possible with Et3N. | ||
| Scheme 3 Coupling conditions for the cyclic diester 2d. | ||
The coupling reaction of 1c and 2c provided the single diastereomer of the enaminone 3o as a minor product (Scheme 4).42 On the basis of 1D and 2D NMR spectroscopy and mass spectrometry, structure 9 was assigned to the major product. The formation of 9 can be explained as a result of the breaking of the episulfide bond and proton transfer. Production of 9 in this reaction provides strong evidence for the presence of an episulfide intermediate. Such intermediates 8 (Scheme 4) have never been isolated, but have been proposed in the mechanism of the Eschenmoser reaction.
 | ||
| Scheme 4 Formation of the unexpected thiol. | ||
The reaction of monocarbonyl halides was not successful under standard reaction conditions. When 1d was reacted with 2e the reaction did not proceed beyond the thioether stage 10 after 2.5 h of bath sonication (Scheme 5). However, on probe sonication, in the presence of the thiophile PPh3 and xylene as a solvent, the reaction provided the enaminone 3p as a single diastereomer after 11 h.43 In contrast, under these conditions, coupling of less acidic 1b with 2f remained unsuccessful. Even after 20 h of probe sonication, only thioether 11 could be recovered.
 | ||
| Scheme 5 Issues with the sulphide contraction of monocarbonyl thioethers. | ||
Conclusions
It has been shown that sonication can accelerate the Eschenmoser coupling reaction. The method can be applied to the efficient coupling of primary, secondary and tertiary thioamides with α-bromodiesters and α-bromoketones. In the case of secondary thioamides, the rate enhancement is mainly due to the reduction in the size of Na2CO3 particles. For tertiary thioamides, other factors, such as heat and mechanical effects that are produced due to the rapid collapse of bubbles in sonication, are also important in the coupling process. In the future, the methodology will be expanded to include the coupling of α-halomonoesters and thioamides. The reaction will also be used in the synthesis of biologically important enaminones and natural products.Experimental
General
1H and 13C NMR spectra were obtained using a Varian 400/54 (400 MHZ) spectrometer. The residual solvent signals were used as references (CDCl3: δH = 7.26 ppm, δC = 77.16 ppm). Infrared spectra were recorded on an Avatar 360 FT-IR. High resolution mass spectra and liquid chromatography were performed on a Thermo Scientific Exactive LC-MS instrument. The instrument was operated in a positive ion electrospray mode by Dr K. P. Roberts and J. Holland at the Department of Chemistry and Biochemistry, The University of Tulsa. Chromatographic separation was performed on this instrument with Thermo Scientific Hypersil Gold HPLC Column (50 × 2.1 mm I.D.; particle size 1.9 μm) coupled to a UV detector. Thin layer chromatography (TLC) was performed on 250 μm glass or aluminium-backed silica gel plates. Visualization was accomplished using short wavelength UV light (254 nm), an iodine chamber, or basic aqueous KMnO4 solution. Flash column chromatography was performed using Sorbent silica gel 60 Å (40–63 μm). Petroleum ether refers to those fractions that distil at 30–60 °C. Anhydrous Na2CO3 was purchased from Fisher (LOT AD-9195-27). Unless specified, all chemical reagents and solvents were obtained from commercial sources. Aldrich's Zerostat® 3 gun was used to neutralize static electricity during the Na2CO3 weighing and transfer. Sonication was conducted either by an ultrasonic bath (VWR, model no. 97043-968) or probe sonicator (Sonics, Vibra cell, model no. VC X 130) equipped with a standard probe (tip dia. 6mm, length 113 mm). The pulse settings were 7 s on and then 1 s off with a 100% amplitude.General procedure A: synthesis of enaminones using an ultrasonic bath
A solution of 1 (157 μmol) in 0.40 mL CH2Cl2 was added to a test tube containing Na2CO3 (3.2 eq). Bromide 2 (2 eq) was dissolved in 0.40 mL CH2Cl2 and added to the reaction vessel. Each vial, which originally contained 1 and 2, was washed twice with 0.10 mL CH2Cl2 and the contents were transferred to the reaction mixture. The test tube was capped and sonicated for the specified period of time (Table 1).General procedure B: synthesis of enaminones using probe sonication
A solution of 1 (157 μmol) in 0.40 mL CH2Cl2 was added to a test tube containing Na2CO3 (3.2 eq). Bromide 2 (2 eq) was dissolved in 0.40 mL CH2Cl2 and added to the reaction vessel. Each vial, which originally contained 1 and 2, was washed twice with 0.10 mL CH2Cl2 and the contents were transferred to the reaction mixture. The sonication probe was inserted into the reaction tube and the tube was covered with paraffin. The reaction was sonicated for the specified period of time (Table 1). During sonication, the solvent level was monitored. If the level reduced to ∼0.40 mL, additional CH2Cl2 (0.80 mL) was added to make up the difference in lost solvent.Diethyl 2-(1-amino-3-phenylpropylidene)malonate (3a)14
The reaction was performed according to the general procedure A. It was sonicated for 2.5 h to convert 1a14 (20.0 mg, 121 μmol) into crude 3a. Column chromatography (CH2Cl2 then 10% ethyl acetate in CH2Cl2) provided pure 3a (31.6 mg, 108 μmol, 90%) as yellow oil. Rf 0.23 (CH2Cl2). IR (neat) νmax/cm−1: 3416, 3314, 3028, 2981, 2936, 2904, 1664, 1614, 1526, 1497, 1454, 1366, 1253, 1072, 798; 1H NMR (400 MHz, CDCl3) δ: 8.78 (s, broad, 1H), 7.31–7.26 (m, 2H), 7.23–7.19 (m, 3H), 4.90 (s, broad, 1H), 4.23 (q, J = 7.2 Hz, 2H), 4.17 (q, J = 7.2 Hz, 2H), 2.93 (t, J = 7.6 Hz, 2H), 2.69 (t, J = 7.6 Hz, 2H), 1.30 (t, J = 7.2 Hz, 3H), 1.27 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 168.9, 168.5, 165.8, 140.5, 128.8, 128.5, 126.6, 93.4, 60.6, 59.8, 37.1, 34.8, 14.4; HRMS (ESI+): Calcd. for C16H21NO4 (M + H+): 292.1543, found 292.1544; m/z (ESI+): 292 (M + H+, 100%), 246 (38%); HPLC, Rt = 8.44 min.(E)-Ethyl 2-acetyl-3-amino-5-phenylpent-2-enoate (3b)
The reaction was performed according to the general procedure A. It was sonicated for 2.5 h to convert 1a (20.0 mg, 121 μmol) into crude 3b. Column chromatography (30% ethyl acetate in CH2Cl2) provided pure 3b (19.0 mg, 72.5 μmol, 60%) as a single diastereomer. Rf 0.36 (dichloromethane:EtOAc 7:3). IR (neat) νmax/cm−1: 3370, 2921, 2844, 1710, 1645, 1551, 1320, 1260, 1093, 913; 1H NMR (400 MHz, CDCl3) δ: 7.32–7.27 (m, 2H), 7.25–7.20 (m, 3H), 4.31 (q, J = 7.2 Hz, 2H), 3.28 (t, J = 7.8 Hz, 2H), 3.10 (t, J = 7.8 Hz, 2H), 2.71 (s, 3H), 1.35 (t, J = 7.2 Hz, 3H), 1.25 (s, broad, 2H); 13C NMR (100 MHz, CDCl3) δ: 128.8, 35.7, 35.0, 29.8, 14.4; HRMS (ESI+): Calculated for C15H19NO3, (M + H+): 262.1437 found 262.1438; m/z (ESI+) 262 (M + H+); HPLC, Rt = 9.29 min.
Diethyl 2-(pyrrolidin-2-ylidene)malonate (3c)14,42
The reaction was performed according to the general procedure A. It was sonicated for 2.5 h to convert 1b14 (12.0 mg, 114 μmol) into crude 3c. Column chromatography (CH2Cl2 then ethyl acetate) provided pure 3c (25.4 mg, 112μmol, 98%). Rf 0.37 (hexanes:EtOAc 3:2). IR (neat) νmax/cm−1: 3317, 2980, 1685, 1643, 1577, 1437, 1366, 1334, 1247, 1084, 1038, 797; 1H NMR (400 MHz, CDCl3) δ: 9.52 (s, broad 1H), 4.18 (q, J = 7.2 Hz, 2H), 4.17 (q, J = 7.2 Hz, 2H), 3.58 (t, J = 7.2 Hz, 2H), 3.09 (t, J = 8.0 Hz, 2H), 2.02 (tt, J = 7.2, 8.0 Hz, 2H), 1.27 (t, J = 7.2 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 173.4, 170.1, 167.9, 87.4, 59.8, 59.7, 47.5, 34.3, 21.8, 14.5; HRMS (ESI+): Calcd. for C11H17NO4 (M + H+): 228.1230, found 228.1220; m/z (ESI+): 228 (M + H+, 100%), 182 (79%); HPLC, Rt = 6.59 min.
(E)-Ethyl 3-oxo-2-(pyrrolidin-2-ylidene)butanoate (3d)14,35
The reaction was performed according to the general procedure A. It was sonicated for 2.5 h to convert 1b14 (20.0 mg, 198 μmol) into crude 3d. Column chromatography (40% ethyl acetate in petroleum ether) provided pure 3d (32.9 mg, 167μmol, 84%). Rf 0.31 (petroleum ether:EtOAc 3:2). IR (neat) νmax/cm−1: 3316, 2981, 1743, 1691, 1652, 1570, 1367, 1247, 1017, 1035, 798; 1H NMR (400 MHz, CDCl3) δ: 11.59 (s, br, 1H), 4.21 (q, J = 7.2 Hz, 2H), 3.63 (t, J = 7.6 Hz, 2H), 3.15 (t, J = 8.0 Hz, 2H), 2.41 (s, 3H), 2.02 (t, J = 7.6, 8.0 Hz, 2H), 1.31 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 197.8, 174.7, 169.0, 99.0, 59.6, 48.0, 35.2, 30.9, 21.2, 14.6; HRMS (ESI+): Calcd. for C10H15NO3 (M + H+): 198.1125, found 198.1115; m/z (ESI+): 198 (M + H+, 100%), 152 (17%); HPLC, Rt = 6.02 min.
Diethyl 2-(piperidine-2-ylidene)malonate (3e)14,19
The reaction was performed according to the general procedure A. It was sonicated for 2.5 h to convert 1c14 (30.0 mg, 173 μmol) into crude 3e. Column chromatography (17%, then 20% ethyl acetate in hexanes) provided pure 3e (33.7 mg, 140 μmol, 80%). Rf 0.26 (hexanes:EtOAc 4:1). IR (neat) νmax/cm−1: 3248, 2979, 1697, 1643, 1600, 1480, 1281, 1225, 1071; 1H NMR (400 MHz, CDCl3) δ: 10.08 (s, broad 1H), 4.18–4.11 (m, 4H), 3.35–3.32 (m, 2H), 2.64 (t, J = 6.4 Hz, 2H), 1.78–1.68 (m, 4H), 1.30–1.22 (m, 6H); 13C NMR (100 MHz, CDCl3) δ: 169.5, 168.9, 165.7, 89.8, 60.2, 59.3, 41.6, 27.2, 21.8, 19.4, 14.5, 14.4; HRMS (ESI+): Calcd. for C12H19NO4 (M + H+): 242.1386, found 242.1377; m/z (ESI+): 242 (M + H+) ; HPLC, Rt = 9.29 min.
(E)-Ethyl 3-oxo-2-(piperidine-2-ylidene)butanoate (3f)42
The reaction was performed according to the general procedure A. It was sonicated for 2.5 h to convert 1c14 (20.0 mg, 174 μmol) into crude 3f. Column chromatography (40% ethyl acetate in petroleum ether) provided pure 3f (17.4 mg, 82.2 μmol, 47%) as a single diastereomer. Rf 0.31 (petroleum ether:EtOAc 3:2). IR (neat) νmax/cm−1: 3398, 2928, 2855, 1692, 1599, 1463, 1448, 1358, 1332, 1307, 1278, 1192, 1108, 1070, 1054, 1025; 1H NMR (400 MHz, CDCl3) δ: 12.77 (s, br, 1H), 4.19 (q, J = 7.2 Hz, 2H), 3.40–3.38 (m, 2H), 2.70 (t, J = 6.2 Hz, 2H), 2.25 (s, 3H), 1.81–1.67 (m, 4H), 1.31 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 195.4, 170.1, 168.7, 101.8, 60.1, 41.7, 27.8, 21.3, 19.2, 14.4; HRMS (ESI+): Calcd. for C11H17NO3 (M + H+): 212.1281, found 212.1270; m/z (ESI+): 212 (M + H+, 100%), 166 (16%); HPLC, Rt = 6.41min.
(S)-5-(Di(ethoxycarbonyl)methylidene)-pyrrolidine-2-carboxylic acid ethyl ester (3g)44
The reaction was performed according to the general procedure A. It was sonicated for 2.5 h to convert 1d44 (30.0 mg, 173 μmol) into crude 3g. Column chromatography (40% ethyl acetate in petroleum ether) provided pure 3g (37.5 mg, 125 μmol, 73%). Rf 0.59 (hexanes:EtOAc 3:2). IR (neat) νmax/cm−1: 3316, 2981, 1743, 1691, 1652, 1570, 1441, 1417, 1367, 1247, 1077, 1036; 1H NMR (400 MHz, CDCl3) δ: 9.62 (s, br 1H), 4.45 (dd, J = 6.0, 8.8 Hz, 1H), 4.23–4.14 (m, 6H), 3.21–3.06 (m, 2H), 2.34 (dddd, J = 7.2, 8.8, 8.8, 13.2 Hz, 1H), 2.16 (dddd, J = 6.0, 6.0, 9.2, 13.2 Hz, 1H), 1.31–1.22 (m, 9H); 13C NMR (100 MHz, CDCl3) δ: 172.1, 171.2, 169.5, 167.5, 89.0, 60.9, 60.8, 59.9, 59.8, 33.2, 26.0, 14.4, 14.2; HRMS (ESI+): Calcd. for C14H21NO6 (M + H+): 300.1442, found 300.1417; m/z (ESI+): 300 (M + H+, 100%), 254 (42%), 225 (60%); HPLC, Rt = 6.99 min.
(S)-Ethyl 5-(1-ethoxy-1,3-dioxo-3-phenylpropan-2-ylidene)pyrrolidine-2-carboxylate (3h)
The reaction was performed according to the general procedure A. It was sonicated for 2.5 h to convert 1d44 (30.0 mg, 173 μmol) into crude 3h. Column chromatography (columned packed in petroleum ether and run in 20%, then 30% ethyl acetate in petroleum ether) provided pure 3h (42.5 mg, 128μmol, 74%) as an inseparable mixture of diasteromers in a ratio of 1:2 (Z:E). Rf 0.56 (petroleum ether:EtOAc 3:2). IR (neat) νmax/cm−1: 3314, 2981, 1742, 1685, 1593, 1573, 1537, 1230; 1H NMR (400 MHz, CDCl3) δ: 10.98 (s, br, 1H, major), 9.48 (s, br, 1H, minor), 7.61 (d, J = 6.8 Hz, 1H), 7.46–7.31 (m, 4H), 4.55–4.43 (m, 1H), 4.23 (d, J = 7.2 Hz, 2H), 3.89–3.79 (m, 2H), 3.34–3.14 (m, 2H), 2.46–2.36 (m, 1H), 2.28–2.15 (m, 1H), 1.29 (t, J = 7.2 Hz, 3H), 0.73–0.70 (m, 3H); 13C NMR (100 MHz, CDCl3) δ: (major) 195.3, 173.4, 170.8, 169.1, 143.3, 129.8, 127.8, 126.9, 98.9, 62.0, 61.6, 59.6, 33.2, 25.5, 14.2, 13.5; (minor) 195.3, 172.2, 171.1, 169.7, 142.6, 130.9, 128.1, 127.8, 96.8, 62.0, 61.0, 59.4, 32.5, 26.2, 14.2, 13.5; HRMS (ESI+): Calcd. for C18H21NO5 (M + H+): 332.1465, found 332.1492; m/z (ESI+) 332 (M + H+, 100%), 207 (56%), 172 (24%); HPLC, Rt = 7.87 min.
Diethyl 2-(methylpyrrolidin-2-ylidene)malonate (3i)14,36
:EtOAc 3:2). IR (neat) νmax/cm−1: 2965, 2926, 2852, 1680, 1574, 1450, 1366, 1286, 1223, 1189, 1069, 915; 1H NMR (400 MHz, CDCl3) δ: 4.14 (q, J = 7.2 Hz, 4H), 3.48 (t, J = 7.2 Hz, 2H), 3.13 (t, J = 7.6 Hz, 2H), 2.82 (s, 3H), 1.94 (tt, J = 7.2, 7.6 Hz, 2H), 1.24 (t, J = 7.2 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 168.3, 166.7, 89.2, 59.9, 57.2, 36.8, 35.1, 20.6, 14.4; HRMS (ESI+): Calcd. for C12H19NO4 (M + H+): 242.1386, found 242.1375; m/z (ESI+) 242 (M + H+, 43%), 196 (100%); HPLC, Rt = 6.33 min.
(E)-Ethyl 2-(methylpyrrolidin-2-ylidene)-3-oxo-3-phenylpropanoate (3j)36
The reaction was performed according to the general procedure B. It was sonicated for 3 h to convert 1e14 (20.0 mg, 174 μmol) into crude 3j. Column chromatography (40% ethyl acetate in petroleum ether) provided pure 3j (47.1 mg, 172 μmol, 99%) as a single diastereomer. Rf 0.16 (petroleum ether:EtOAc 3:2). IR (neat) νmax/cm−1: 2925, 2854, 1682, 1615, 1598, 1558, 1447, 1408, 1364, 1316, 1285, 1233, 1173, 1123, 1094, 1060, 910, 880, 827; 1H NMR (400 MHz, CDCl3) δ: 7.73–7.69 (m, 2H), 7.43–7.39 (m, 1H), 7.37–7.33 (m, 2H), 3.82 (q, J = 7.2 Hz, 2H), 3.65 (t, J = 7.2 Hz, 2H), 3.25 (t, J = 7.6 Hz, 2H), 2.74 (s, 3H), 2.05 (tt, J = 7.2, 7.6 Hz, 2H), 0.70 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 194.0, 170.7, 169.3, 142.6, 130.8, 128.2, 127.9, 96.9, 59.3, 57.7, 38.9, 35.6, 20.7, 13.6; HRMS (ESI+): Calcd. for C16H19NO3 (M+H)+: 274.1437, found 274.1425; m/z (ESI+) 274 (M + H+, 100%), 228 (83%), 105 (17%); HPLC, Rt = 6.85 min.
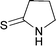
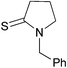
Diethyl 2-(benzylpyrrolidin-2-ylidene)malonate 3k
:EtOAc 3:2). IR (neat) νmax/cm−1: 2979, 1684, 1570, 1454, 1364, 1234, 1190, 1147, 1091, 1048, 955; 1H NMR (400 MHz, CDCl3) δ: 7.34–7.25 (m, 3H), 7.19–7.17 (m, 2H), 4.40 (s, 2H), 4.06 (q, J = 7.2 Hz, 4H), 3.31 (t, J = 7.2 Hz, 2H), 3.23 (t, J = 7.6 Hz, 2H), 1.92 (tt, J = 7.2, 7.6 Hz, 2H), 1.17 (t, J = 7.2 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 168.5, 165.0, 135.8, 128.8, 127.8, 127.7, 90.6, 60.1, 54.3, 52.6, 35.4, 20.8, 14.3; HRMS (ESI+): Calcd. for C18H23NO4 (M + H+): 318.1700, found 318.1674; m/z (ESI+) 318 (M + H+, 100%), 272 (90%); HPLC, Rt = 7.85 min.
Diethyl 2-(phenyl(piperidine-1-yl)methylene)malonate (3l)14,45
:EtOAc 3:2). IR (neat) νmax/cm−1: 2925, 2854, 1690, 1524, 1446, 1365, 1291, 1253, 1222, 1155, 1113, 1078, 1027, 858, 765; 1H NMR (400 MHz, CDCl3) δ: 7.45–7.42 (m, 3H), 7.39–7.35 (m, 2H), 3.98 (q, J = 7.2 Hz, 4H), 3.14–3.11 (m, br, 4H), 1.72–1.63 (m, br, 6H), 1.10 (t, J = 7.2 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 169.0, 167.9, 138.1, 130.5, 129.8, 128.5, 98.4, 60.0, 52.7, 27.0, 23.9, 14.2; HRMS (ESI+): Calcd. for C19H25NO4 (M + H+): 332.1856, found 332.1836; m/z (ESI+) 332 (M + H+, 53%), 286 (100%); HPLC, Rt = 8.68 min.
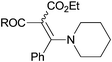
Ethyl 2-benzoyl-3-phenyl(piperidine-1-yl)acrylate (3m)
The reaction was performed according to the general procedure B. It was sonicated for 6 h to convert 1g14 (20.0 mg, 97.4 μmol) into crude 3m. Column chromatography (40% petroleum ether in CH2Cl2) provided pure 3m (35.4 mg, 97.4 μmol, 100%) as an inseparable mixture of diastereomers (2.4:1). Rf 0.14 (petroleum ether:EtOAc 9:1). IR (neat) νmax/cm−1: 2935, 2855, 1670, 1668, 1507, 1446, 1364, 1273, 1243, 1220, 1208, 1168, 1156, 1110, 1059, 1026, 1000, 904, 772, 761; 1H NMR (400 MHz, CDCl3) δ: (Major) 7.77 (d, J = 7.6 Hz, 2H), 7.37–7.28 (m, 8H), 4.20 (q, J = 7.2 Hz, 2H), 3.27–3.26 (m, 4H), 1.70–1.68 (m, 6H), 1.24 (t, J = 7.2 Hz, 3H); (Minor) 8.07 (d, J = 7.2 Hz, 2H), 7.61–7.40 (m, 8H), 3.73 (q, J = 7.2 Hz, 2H), 3.20 (m, br, 4H), 1.59 (m, br, 6H), 0.69 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: (Major) 193.5, 170.2, 166.6, 137.8, 137.3, 132.8, 130.3, 129.5, 128.5, 128.3, 127.3, 81.0, 60.3, 53.2, 27.1, 27.0, 14.8; (Minor) 191.8, 171.6, 169.2, 139.5, 136.9, 133.3, 130.3, 129.9, 128.7, 128.3, 127.6, 85.1, 60.1, 52.5, 29.8, 26.9, 13.7; HRMS (ESI+): Calcd. for C23H25NO3 (M + H+): 364.1907, found 364.1892; m/z (ESI+) 364 (M + H+); HPLC, Rt = 10.38 min.
Acknowledgements
Financial support was provided by The University of Tulsa new faculty Lab-startup funds. B.H. and H.S. gratefully acknowledge both the TURC and the CSURP scholarship funds from The University of Tulsa. The authors thank J. Holland for her help with the spectroscopic measurements. This material is based upon work supported by the National Science Foundation under Grant No. (CHE-1048784).References
- G. Negri, C. Kascheres and A. J. Kascheres, J. Heterocycl. Chem., 2004, 41, 461–491 CrossRef CAS
.
- M. C. Elliott, J. L. Wood and S. V. Wordingham, Trends in Heterocyclic Chemistry, 2005, 10, 73–95 CAS
- B. Pettersson, V. Hasimbegovic and J. Bergman, J. Org. Chem., 2011, 76, 1554–1561 CrossRef CAS
-
K. Shiosaki, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, Oxford, vol. 2, 1991, ch. 3.7, pp. 865–892 Search PubMed
- I. O. Edafiogho, S. B. Kombian, K. V. V. Ananthalakshmi, N. N. Salama, N. D. Eddington, T. L. Wilson, M. S. Alexander, P. L. Jackson, C. D. Hanson and K. R. Scott, J. Pharm. Sci., 2007, 96, 2509–2531 CrossRef CAS
- C. M. Park and D. J. Jeon, Org. Biomol. Chem., 2012, 10, 2613–2620 CAS
- J. Svete, ARKIVOC, 2006, vii, 35–56 Search PubMed
- A. B. Smith III, R. Akaishi, D. R. Jones, T. P. Keenan, M. C. Guzman, R. C. Holcomb, P. A. Sprengeler, J. L. Wood, R. Hirschmann and M. K. Holloway, Biopolymers, 1995, 37, 29–53 CrossRef
- U. Beckmann, E. Eichberger, M. Lindner, M. Bongartz and P. C. Kunz, Eur. J. Org. Chem., 2008, 4139–4147 CrossRef CAS
- C. Cheng, G. Sun, J. Wan and C. Sun, Synlett., 2009, 2663–2668 Search PubMed
- N. G. Ramesh, F. J. A. D. Bakkeren, D. d. Groot, U. Passamonti, A. J. H. Klunder and B. Zwanenburg, Tetrahedron, 2001, 57, 9877–9887 CrossRef CAS
- A.-Z. A. Elassar and A. A. El-Khair, Tetrahedron, 2003, 59, 8463–8480 CrossRef CAS
- H. M. C. Ferraz and E. R. S. Goncalo, Quim. Nova, 2007, 30, 957–964 CrossRef CAS
- N. D. Koduri, H. Scott, B. Hileman, J. D. Cox, M. Coffin, L. Glicksberg and S. R. Hussaini, Org. Lett., 2012, 14, 440–443 CrossRef CAS
- M. K. Samantaray, C. Dash, M. M. Shaikh, K. Pang, R. J. Butcher and P. Gosh, Inorg. Chem., 2011, 50, 1840–1848 CrossRef CAS
- M. D. Bachi, R. Breiman and H. Meshulam, J. Org. Chem., 1983, 48, 1439–1444 CrossRef CAS
- J. A. Campbell and H. Rapoport, J. Org. Chem., 1996, 61, 6313–6325 CrossRef CAS
- S. Singh, J. M. Köhler, A. Schober and G. A. Groß, Beilstein J. Org. Chem., 2011, 7, 1164–1172 CrossRef CAS
- J. P. Michael, C. B. de Koning, C. W. v. de Westhuyzen and M. A. Fernandes, J. Chem. Soc., Perkin Trans. 1, 2001, 2055–2062 RSC
- T. J. Mason, Chem. Soc. Rev., 1997, 26, 443–451 RSC
- S. R. Hussaini and M. G. Moloney, Tetrahedron Lett., 2004, 45, 1125–1127 CrossRef CAS
- J. F. Okonya, R. V. Hoffman and M. C. Johnson, J. Org. Chem., 2002, 67, 1102–1108 CrossRef CAS
- J. C. Lee, Y. H. Bae and S.-K. Chang, Bull. Korean Chem. Soc., 2003, 24, 407–408 CrossRef CAS
- K. Tanemura, T. Suzuki, Y. Nishida, K. Satsumabayashi and T. Horaguchi, Chem. Commun., 2004, 470–471 RSC
- A. T. Khan, M. A. Ali, P. Goswami and L. H. Choudhury, J. Org. Chem., 2006, 71, 8961–8963 CrossRef CAS
- I. Pravst, M. Zupan and S. Stavber, Green Chem., 2006, 8, 1001–1005 RSC
- M. Kirihara, S. Ogawa, T. Noguchi, K. Okubo, Y. Monma, I. Shimizu, R. Shimosaki, A. Hatano and Y. Hirai, Synlett, 2006, 2287–2289 CrossRef CAS
- O. V. Kayukova, Y. S. Kayukov, A. N. Nikolaev and O. E. Nasakin, Russ. J. Org. Chem., 2004, 40, 1382–1383 CrossRef CAS
- V. A. Nikolaev, V. V. Shevchenko, M. S. Platz and N. N. Khimich, Russ. J. Org. Chem., 2006, 42, 815–827 CrossRef CAS
- R. E. Ireland and F. R. Brown Jr., J. Org. Chem., 1980, 45, 1868–1880 CrossRef CAS
- K. Shiosaki, G. Fels and H. Rapoport, J. Org. Chem., 1981, 46, 3230–3234 CrossRef CAS
- E. J. Freyne, J. F. Lacrampe, F. Deroose, G. M. Boeckx, M. Willems, W. Exbrechts, E. Coesemans, J. J. Willems, J. M. Fortin, Y. Ligney, L. L. Dillen, W. F. Cools, J. Goossens, D. Corens, A. D. Groot and J. P. V. Wauwe, J. Med. Chem., 2005, 48, 2167–2175 CrossRef CAS
-
E. J. Freyne, F. D. Deroose, J. F. Lacrampe, W. C. Embrechts and J. M. Fortin, WO Patent, 00/37451, 2000 Search PubMed
-
J. F. Lacrampe, E. J. Freyne, F. D. Deroose and E. Coesemans, WO Patent, 01/10866 A1, 2001 Search PubMed
- G. Dannhardt and A. Bauer, Die Pharmazie, 1996, 51, 805–810 CAS
- S. R. Hussaini and G. B. Hammond, ARKIVOC, 2008, xiii, 129–136 Search PubMed
- Current Price of Probe Sonicator (Sonics, Vibra cell, model no. VCX 130), $3,080.
- G. Cravotto and P. Cintas, Chem. Soc. Rev., 2006, 35, 180–196 RSC
- E. M. Arnett, S. G. Maroldo, S. L. Schilling and J. A. Harrelson, J. Am. Chem. Soc., 1984, 106, 6759–6767 CrossRef CAS
- F. G. Bordwell and H. E. Fried, J. Org. Chem., 1981, 46, 4327–4331 CrossRef CAS
- J. P. Cèlèrier, G. Lhommet and P. Maitte, Tetrahedron Lett., 1981, 22, 963–964 CrossRef
- P. Brunerie, J.-P. Cèlèrier, H. Petit and G. Lohmmet, J. Heterocycl. Chem., 1986, 1183–1187 CrossRef CAS
- M. C. Elliott and S. V. Wordingham, Synthesis, 2006, 1162–1170 CrossRef CAS
- S. R. Hussaini and M. G. Moloney, Org. Biomol. Chem., 2006, 4, 2600–2615 CAS
- E. Z. Schottland and Z. Rappoport, J. Org. Chem., 1996, 61, 8536–8543 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures for benzylpyrrolidin-2-one, 1f, 2c, 3n, 3o, 9, 10, 3p and 11, preparation of bath sonicated and probe sonicated Na2CO3 and related experiments, particle distribution graphs and 1H and 13C NMR spectra of all the relevant compounds. See DOI: 10.1039/c2ra22033d |
| This journal is © The Royal Society of Chemistry 2013 |