Synthesis of the C1–C15 fragment of elaiolide†
Yadav
Jhillu Singh
*,
Gangadhara
Rao Yarrapothu
and
Rajender
Vemula
Indian Institute Chemical Technology – Natural Product Chemistry Division, Hyderabad, Andhra Pradesh, 500007, India. E-mail: yadavpub@iict.res.in; Fax: +91 40 27160387; Tel: +91 40 27193128
First published on 14th November 2012
Abstract
The synthesis of the C1–C15 fragment of elaiolide was achieved by successfully utilizing the desymmetrization strategy, zirconium catalyzed C–C bond formation and double stereodifferentiating aldol reactions.
Elaiophylin (1), a glycosidic polyketide, was first isolated from the cultures of Streptomyces melanosporus by Arcamone et al.1a and by Arai1b from a related microorganism. Elaiophylin is a 16-membered macrolide which displays a wide range of bioactivities such as antimicrobial, cell cycle inhibition, apoptosis induction, immunosuppressive, anthelmintic, inhibition of K+-dependent adenosine triphosphatases, and plant growth inhibition.2–4 The C2-symmetric structure of 1 including its absolute configuration was ascertained by chemical degradation,5 spectroscopic methods6 and X-ray crystallographic analysis.7 An impressive biological activity profile, structural intricacy, and the presence of acid and base sensitive hemiketal units enticed the synthetic community to attempt its total synthesis. Following the report on the synthesis of the tetramethyl derivative of elaiolide by Seebach and co-workers8a and the first total synthesis of elaiophylin in 1986 by Kinoshita and co-workers,8b notable synthetic studies have been carried out by several groups.8c–m
As part of our ongoing programme to synthesize natural products comprised of polyketide units by a desymmetrization strategy,9 we aspired to demonstrate its versatility by synthesizing the C1–C15 unit 3 of elaiolide (1a), which is an aglycon of elaiophylin (1) (Fig. 1).
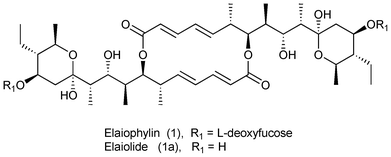 | ||
| Fig. 1 Structure of elaiophylin and elaiolide. | ||
Our retrosynthetic analysis of elaiolide 1a, as illustrated in Scheme 1, suggests a monomeric unit 2 that further led to the protecting group translocated tautomeric aldol adduct 3, which was anticipated to be derived from keto ester 4 and aldehyde 6, whose origins can be traced back to lactone 510 and D-mannitol, respectively, with the former being synthesized earlier by us following the desymmetrization strategy developed in our laboratory.
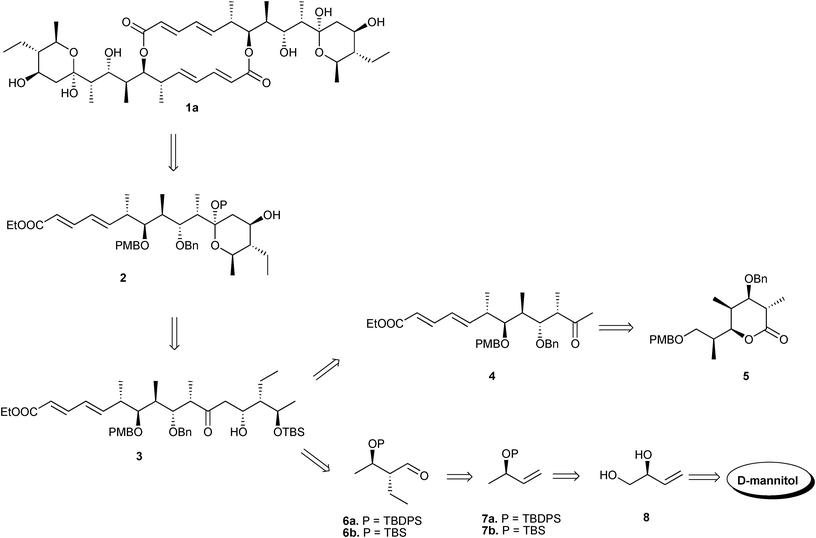 | ||
| Scheme 1 Retrosynthetic analysis. | ||
Results and discussion
The synthesis of keto ester 3 started with the known Prelog-Djerassi type lactone 5,10 which on treatment with AlMe3OMeNHMe·HCl at room temperature provided Weinreb amide 9 (98%).11 Oxidative acetalization of alcohol 9 with DDQ12 afforded acetal 10 (87%) (Scheme 2). Displacement of the amide functionality with 3.0 equiv. MeLi furnished methyl ketone 11 (99%).13 Treatment of keto-acetal 11 with 3.0 equiv. DIBAL-H led to the concomitant regioselective reductive opening 14 of the PMB acetal and reduction of the keto group to give 1,7-diol 12 (89%). 1,7-diol 12 was oxidized with IBX to furnish the keto aldehyde 13 (87%). Keto aldehyde 13 upon chemoselective HWE olefination15 with 2.5 equiv. crotyl phosphonate formed the aldol partner, keto ester 4 [>19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (E:Z), 85%].
:1 (E:Z), 85%].
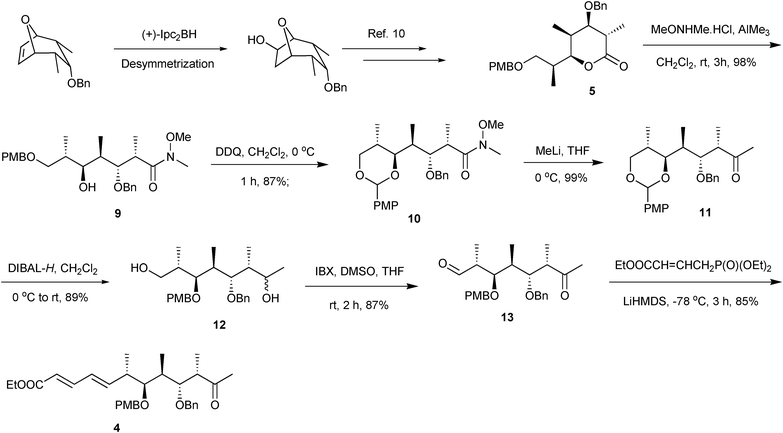 | ||
| Scheme 2 Synthesis of keto ester 4. | ||
The synthesis of aldehyde 6a commenced with the known diol 8,16 whose primary and secondary alcohols were masked successively as a tosyl ester (NEt3, nBu2SnO and Ts–Cl) and silyl ether (ImH and TBDPS-Cl) to give 15 (86% over two steps). Attempted reductive elimination of the tosyl group in 15 using LiAlH4 and superhydride for the synthesis of the carbo-metallation precursor resulted in the formation of a silyl deprotected, volatile and optically active methyl vinyl carbinol. This was circumvented by refluxing the tosyl ester 15 with NaBH4 in DMSO17 at 80 °C for 3 h to afford allylic silyl ether 7a (77%).
An ethyl stereocenter was introduced into the olefin 7a by zirconium assisted carbomagnesation18 with Cp2ZrCl2–EtMgBr to provide the desired anti isomer 16 (70%) with a 9:1 ds ratio (from NMR). Treatment of primary alcohol 16 with Dess–Martin periodinane furnished aldehyde 6a (95%) (Scheme 3).
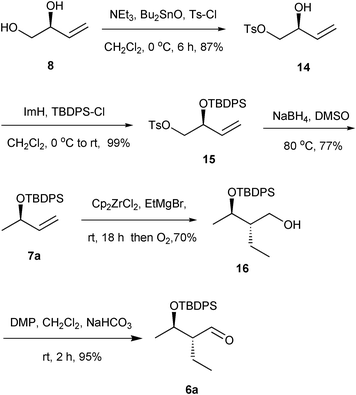 | ||
| Scheme 3 Synthesis of aldehyde 6a. | ||
Having both the fragments in hand, the aldol reaction performed between keto ester 4 and aldehyde 6a under TiCl4–iPr2NEt enolating conditions gave a 6:4 mixture of aldol adducts. Treatment of the lithium enolate of keto ester 4 with aldehyde 6a at −78 °C led to only 5–10% of the aldol adduct after a prolonged time. Assuming that the sterically encumbered TBDPS silyl protecting group may be the cause for the formation of the aldol adduct in a poor diastereomeric ratio and yield, we planned to study the aldol reaction with the TBS version of aldehyde 6a. Following similar reaction conditions as demonstrated in Scheme 3 and starting from known diol 8, TBS protected aldehyde 6b was obtained in five steps with 46% overall yield. Making our assumption true, the aldol reaction19 performed between keto ester 4 and aldehyde 6b surprisingly gave a single diastereomer of 3 (Scheme 4, 85%), whose formation can be ascribed to the preferential facial bias exerted by the conjugated dienoate and the protecting groups at the C3 and C5 positions from the ketone during the Felkin-Anh mode of addition on the aldehyde in the transition state.20
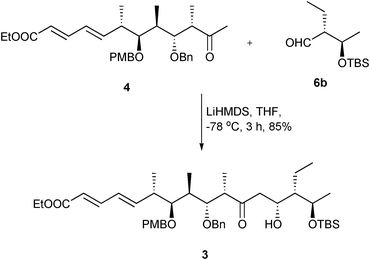 | ||
| Scheme 4 Synthesis of the C1–C15 unit 3. | ||
Conclusions
In conclusion, we have demonstrated the utilization of a desymmetrization strategy for the synthesis of intricate natural products comprised of polyketide domains by applying it to the C1–C15 unit of elaiolide approaching its total synthesis. Hoveyda's zirconium assisted carbometallation reaction has been promising to install the ethyl stereocenter at C14.References
- (a) F. M. Arcamone, C. Bertazzoli, M. Ghione and T. G. Scotti, G. Microbiol., 1959, 7, 207 CAS; (b) Azalomycin B, as reported by Arai, is identical with elaiophylin: M. Arai, J. Antibiot., Ser. A, 1960, 13(46), 51 CAS.
- (a) P. Hammann and G. Kretzschmar, Tetrahedron, 1990, 46, 5603 CrossRef CAS; (b) P. Hammann, G. Kretzschmar and G. Seibert, J. Antibiot., 1990, 43, 1431 CrossRef CAS; (c) C.-M. Liu, L. Jensen, J. W. Westley and D. Siegel, J. Antibiot., 1993, 46, 350 CrossRef CAS.
- S. Takahashi, M. Arai and E. Ohki, Chem. Pharm. Bull., 1967, 15, 1651 CrossRef CAS.
- S. Drose, K. U. Bindseil, E. J. Bowman, A. Siebers, A. Zeek and K. Altendorf, Biochemistry, 1993, 32, 3902 CrossRef CAS.
- (a) S. Takahashi, M. Arai and E. Ohki, Chem. Pharm. Bull., 1967, 15, 1651 CrossRef CAS; (b) S. Takahashi, M. Kurabayashi and E. Ohki, Chem. Pharm. Bull., 1967, 15, 1657 CrossRef CAS; (c) S. Takahashi and E. Ohki, Chem. Pharm. Bull., 1967, 15, 1726 CrossRef CAS.
- H. Kaiser and W. Keller-Schierlein, Helv. Chim. Acta, 1981, 64, 407 CrossRef CAS.
- (a) K. Neupert-Laves and M. Dobler, Helv. Chim. Acta, 1982, 65, 262 CrossRef CAS; (b) S. V. Ley, D. Neuhaus and D. J. Williams, Tetrahedron Lett., 1982, 23, 1207 CrossRef CAS.
- (a) D. Seebach, H.-F. Chow, R. F. W. Jackson, K. Lawson, M. A. Sutter, S. Thaisrivongs and J. Zimmermann, J. Am. Chem. Soc., 1985, 107, 5292 CrossRef CAS; (b) K. Toshima, K. Tatsuta and M. Kinoshita, Bull. Chem. Soc. Jpn., 1988, 61, 2369 CrossRef CAS; (c) K. Toshima, K. Tatsuta and M. Kinoshita, Tetrahedron Lett., 1986, 27, 4741 CrossRef CAS; (d) D. Seebach, H.-F. Chow, R. F. W. Jackson, M. A. Sutter, S. Thaisrivongs and J. Zimmermann, Liebigs Ann. Chem., 1986, 1281 CrossRef CAS; (e) T. Wakamatsu, H. Nakamura, E. Naka and Y. Ban, Tetrahedron Lett., 1986, 27, 3895 CrossRef CAS; (f) T. Wakamatsu, S. Yamada, H. Nakamura and Y. Ban, Heterocycles, 1987, 25, 43 CrossRef CAS; (g) Formal total synthesis of elaiophylin: H. Nakamura, K. Arata, T. Wakamatsu, Y. Ban and M. Shibasaki, Chem. Pharm. Bull., 1990, 38, 2435 CrossRef CAS; (h) K. U. Bindseil and A. Zeeck, J. Org. Chem., 1993, 58, 5487 CrossRef CAS; (i) D. A. Evans and D. M. Fitch, J. Org. Chem., 1997, 62, 454 CrossRef CAS; (j) F. E. Ziegler and J. S. Tung, J. Org. Chem., 1991, 56, 6530 CrossRef CAS; (k) I. Paterson, H.-G. Lombart and C. Allerton, Org. Lett., 1999, 1, 19 CrossRef CAS; (l) I. Paterson and J. Man, Tetrahedron Lett., 1997, 38, 695 CrossRef CAS; (m) R. Barth and J. Mulzer, Tetrahedron, 2008, 64, 4718 CrossRef CAS.
- (a) J. S. Yadav, C. Srinivas Rao, S. Chandrasekhar and A. V. Rama Rao, Tetrahedron Lett., 1995, 36, 7717 CrossRef CAS; (b) J. S. Yadav, S. Abraham, M. Muralidhar Reddy, G. Sabitha, A. Ravi Sanker and A. C. Kunwar, Tetrahedron Lett., 2001, 42, 4713 CrossRef CAS; (c) J. S. Yadav and M. M. Ahmed, Tetrahedron Lett., 2002, 43, 7147 CrossRef CAS; (d) J. S. Yadav, R. Srinivas and K. Sathaiah, Tetrahedron Lett., 2006, 47, 1603 CrossRef CAS; (e) J. S. Yadav, T. V. Pratap and V. Rajender, J. Org. Chem., 2007, 72, 5882 CrossRef CAS; (f) J. S. Yadav and V. Rajender, Eur. J. Org. Chem., 2010, 2148 CrossRef CAS.
- J. S. Yadav, V. Rajender and Y. G. Rao, Org. Lett., 2010, 12, 348 CrossRef CAS.
- (a) J. L. Levin, E. Turos and S. M. Weinreb, Synth. Commun., 1982, 12, 989 CrossRef CAS; (b) T. Shimizu, K. Osako and T. Nakata, Tetrahedron Lett., 1997, 38, 2685 CrossRef CAS; (c) M. Miyashita, Y. Toshimitu, T. Shiratani and H. Lrie, Tetrahedron: Asymmetry, 1993, 4, 1573 CrossRef CAS; (d) T. Shimizu, R. Kobayashi, K. Osako, H. Osada and T. Nakata, Tetrahedron Lett., 1996, 37, 6755 CrossRef CAS.
- (a) Y. Oikawa, T. Yoshioka and O. Yonemitsu, Tetrahedron Lett., 1982, 23, 889 CrossRef CAS; (b) J. S. Yadav, M. C. Chander and B. V. Joshi, Tetrahedron Lett., 1988, 29, 2737 CrossRef CAS.
- (a) S. Nakamura and M. Shibasaki, Tetrahedron Lett., 1994, 35, 4145 CrossRef CAS; (b) L. C. Dias, R. J. Baú, M. A. de Sousa and J. Z. Schpectorb, Org. Lett., 2002, 4, 4325 CrossRef CAS.
- S. L. Shreiber, Z. Wang and G. Schulte, Tetrahedron Lett., 1988, 29, 4085 CrossRef.
- B. Bennacer, D. Trubuil, C. Rivalle and D. S. Grierson, Eur. J. Org. Chem., 2003, 4561 CrossRef CAS.
- S. C. Bergmeier and D. M. Stanchina, J. Org. Chem., 1999, 64, 2852 CrossRef CAS.
- T. Hiyama, K. Kobayashi and K. Nishide, Bull. Chem. Soc. Jpn., 1987, 60, 2127 CrossRef CAS.
- A. F. Houri, M. T. Didiuk, Zhongmin Xu, N. R. Horan and A. H. Hoveyda, J. Am. Chem. Soc., 1993, 115, 6614 CrossRef CAS.
- I. Paterson, E. A. Anderson, A. D. Findlay and C. S. Knappy, Tetrahedron, 2008, 64, 4768 CrossRef CAS.
- (a) W.R. Roush, T. D. Bannister, M. D. Wendt, M. S. VanNieuwenhze, D. J. Gustin, G. J. Dilley, G. C. Lane, K. A. Scheidt and W. J. Smith III, J. Org. Chem., 2002, 67, 4284 CrossRef CAS; (b) C. M. Liu, W. J. Smith III, D. J. Gustin and W.R. Roush, J. Am. Chem. Soc., 2005, 127, 5770 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, 1H and 13C NMR spectra for all the new compounds are available. See DOI: 10.1039/c2ra22122e |
| This journal is © The Royal Society of Chemistry 2013 |