Wavelength-tunable luminescent gold nanoparticles generated by cooperation ligand exchange and their potential application in cellular imaging†
Zhengyong
Zhang
,
Lingjia
Xu
,
Huixiang
Li
and
Jilie
Kong
*
Department of Chemistry and Institutes of Biomedical Sciences, Fudan University, Shanghai, 200433, P. R. China. E-mail: jlkong@fudan.edu.cn; Fax: 86 21 65641740; Tel: 86 21 65642138
First published on 30th October 2012
Abstract
This work reports a simple and novel method for the synthesis of wavelength-tunable luminescent gold nanoparticles (NPs) based on a cooperation ligand exchange using 11-mercaptoundecanoic acid (11-MUA) and D-penicillamine (DPA). Next, the obtained luminescent gold NPs were used for cellular imaging, and the observations indicated that the wavelength-tunable luminescent gold NPs have potential applications in cellular luminescence labeling and imaging.
Noble metal NPs have gained tremendous interest in the fields of chemistry, biology, materials and some new hybrid fields due to their molecule-like properties, such as luminescence and unique charging.1–3 There has been a great deal of research work on the luminescence of metal NPs, especially gold NPs and silver NPs, in the past decade.4–6 Compared with common commercial fluorophores, such as organic dyes and semiconductor quantum dots (QDs), luminescent gold NPs have been demonstrated to be promising practical bioimaging probes in single molecule optoelectronics and bioassays7–9 because of their ultrafine size, great photostability, and low biological toxicity.10,11
The synthesis of luminescent gold NPs reported so far could be broadly classified into two categories. The first method is the direct synthesis of gold NPs by the reduction of gold ions in the presence of suitably selected ligands12 or by template-assisted synthesis. Biomolecules (e.g., proteins),13 dendrimers (poly(amidoamine) (PAMAM))14,15etc., have been used as a template to guide the formation of luminescent gold NPs. The second method is the synthesis of gold NPs by the core “etching” of metallic NPs using etching molecules (e.g., alkylated thiols).16–18
However, the preparation of wavelength-tunable luminescent gold NPs is still a challenge. Chang et al. have reported that by controlling the molar ratios of tetrakis(hydroxymethyl)phosphonium chloride (THPC) to gold/silver ions, luminescent gold and gold/silver NPs with tunable size and emission wavelength could be obtained.19 However, it seems that the starting size of the seed gold NPs should be carefully chosen. Kawasaki and co-workers have developed a pH-dependent synthesis of gold nanoclusters (NCs) with blue, green and red luminescent emission using porcine pepsin.20 Here, a novel and simple strategy has been proposed to prepare wavelength-tunable luminescent gold NPs, where the molar ratio of THPC to gold ions and the pH no longer require intense adjustments.
Firstly, we prepared a gold NP solution through the reduction of HAuCl4·4H2O with THPC, which acted as both a reducing and capping agent.21 Using transmission electron microscopy (TEM, Fig. S1†), we determined that the size of the spherical gold NPs were ca. 2.5 (± 0.4) nm. The as-prepared gold NPs were then mixed with 11-MUA to form very strong Au–S bonds on the gold NPs surface, where a bright luminescence was observed.22,23 In this experiment, DPA is used as a cooperation ligand exchange reagent for the first time, and as shown in Scheme 1, by adjusting the concentration ratio of 11-MUA to DPA, luminescent gold NPs with a tunable emission wavelength could be generated on demand. To the best of our knowledge, this is the first time wavelength-tunable gold NPs were synthesised using cooperation ligand exchange.
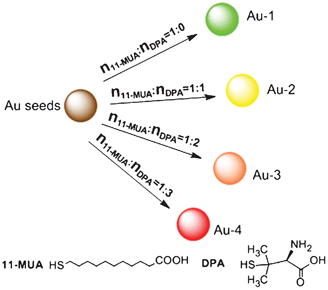 | ||
| Scheme 1 Schematic depiction of the synthesis route of wavelength-tunable luminescent gold NPs via controlling the ratio of 11-MUA to DPA. | ||
In this study, the obtained THPC-gold NPs do not have a visible luminescence signal (Fig. S2†). After the as-prepared gold NPs were mixed with different concentration ratios of 11-MUA to DPA (for more details see the ESI)† and then left to react for several days in the dark, the luminescence colors of the four solutions upon excitation at 365 nm with a UV-lamp were green, yellow, orange and red, respectively, as shown in Fig. 1A .
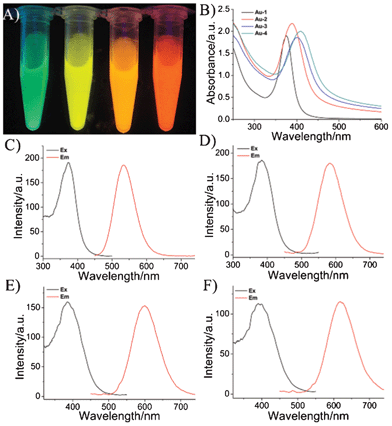 | ||
Fig. 1 (A) Photographs of the obtained wavelength-tunable gold NPs under a UV-light source with excitation at 365 nm. (The ratio of 11-MUA to DPA is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 1:1, 1:2, and 1:3 from left to right.) (B) The UV-Vis absorption spectra. (C–F) The luminescence spectra of the wavelength-tunable gold NPs; (C) Au-1, (D) Au-2, (E) Au-3, and (F) Au-4. :0, 1:1, 1:2, and 1:3 from left to right.) (B) The UV-Vis absorption spectra. (C–F) The luminescence spectra of the wavelength-tunable gold NPs; (C) Au-1, (D) Au-2, (E) Au-3, and (F) Au-4. | ||
Fig. 1B and Fig. S3† display the UV-Vis absorption spectra of the wavelength-tunable luminescent gold NPs solutions. The molar ratios are over the range of 1:0–1:3 (11-MUA:DPA), and the names of the obtained products are abbreviated as Au-1, Au-2, Au-3, Au-4 for 1:0, 1:1, 1:2, 1:3, respectively. The Au-1, Au-2, Au-3, Au-4 from left to right exhibited absorption-band maxima at wavelengths of 375 nm, 389 nm, 399 nm and 407 nm, respectively. According to the literature, the absorption bands for the thiol luminescent gold NPs originate from the metal center (Au 5d10 to 6sp interband transitions) and/or ligand–metal charge-transfer transitions.24,25 Plus, the UV absorption peak is significantly red shifted with increasing DPA, maybe due to the core in the gold NPs interacting with the solvent (water) more closely, owing to the DPA shorter chain length.26
Fig. 1C-F and Fig. S4† reveal luminescence spectra ranging from 530–630 nm of the wavelength-tunable luminescent gold NPs solutions. The quantum yields (QYs) of the obtained Au-1, Au-2, Au-3 and Au-4 luminescent gold NPs are estimated, with results of 1.4%, 1.6%, 1.2% and 1.0%, respectively (see Fig. S5†), in comparison to rhodamine 6G (which has a standard QY of 95% in ethanol).27 Plus, the obtained luminescent gold NPs have a large Stokes shift (more than 100 nm), which indicates many potential attractive properties, such as being easily detected above the background, and usefulness in detecting multiple analytes using a single excitation wavelength, etc. Additionally, as the control experiment, the DPA was also used alone as the ligand exchange reagent to prepare gold NPs (Fig. S6†), but no obvious luminescence signal could be observed in aqueous solution at room temperature. A possible reason is that the luminescence was quenched by the water,28 and 11-MUA was better for stabilizing luminescent gold NPs.
We further studied the emission behavior of the wavelength-tunable luminescent gold NPs through measurements of their luminescence lifetimes. By fitting to a biexponential luminescence decay, we obtained lifetimes (t1/t2) for Au-1, Au-2, Au-3 and Au-4 of 512.9/149.1, 283.4/986.4, 363.9/1229.1, and 393.6/1241.3 ns, respectively (Fig. S7†). The long lifetime is a general characteristic of thiol–Au(I) complexes that display ligand–metal charge transfer and metal(I)–metal(I) interactions.29,30 Plus, the increase of the lifetime upon increasing the ratio of DPA could be attributed to the effect of DPA. Zeta potentials of the obtained luminescent gold NPs were tested in an aqueous solution, as shown in Fig. S8† and Fig. S9†, which were −39.8 ± 4.7 mV, −29.4 ± 5.4 mV, −26.3 ± 1.8 mV, −22.5 ± 3.1 mV for Au-1, Au-2, Au-3 and Au-4, respectively. The positive shift could be attributed to the increasing number of amine groups which resulted from increasing the ratio of DPA. Fig. S10† shows typical Fourier transform infrared spectroscopy (FTIR) spectra of 11-MUA alone, DPA alone and the obtained luminescent gold NPs. It can be found that S–H stretching bands display at around 2554 cm−1 and 2597 cm−1 for 11-MUA and DPA, respectively, but disappear in the spectra of the obtained luminescent gold NPs, resulting from the formation of the gold–sulfur bond.
Fig. 2 displays the high resolution transmission electron microscopy (HRTEM) images of the prepared luminescent gold NPs. The corresponding HRTEM images indicated that the Au-1, Au-2, Au-3 and Au-4 are spherical, narrowly distributed and have average diameters of ca. 1.8 (± 0.3), 2.2 (± 0.4), 2.2 (± 0.3) and 2.3 (± 0.4) nm, respectively. The sizes of these NPs decreased after treatment with 11-MUA because of the production of significant fragmentation energies. Once the 11-MUA molecules adsorbed onto the gold NPs surface, they tended to dissociate the particles into smaller gold and gold-thiolate clusters, as addressed in previous work.31 After being mixed with the different concentrations of DPA, the size of the obtained gold NPs appears to grow a little. A possible explanation for this phenomenon is mainly due to the strong combination of DPA with the surface of the gold NPs, so as to effectively prevent the over etching by 11-MUA, and thus result in different sizes of luminescent gold NPs. For further research, glutathione (GSH), 3-mercaptopropionic acid (MPA) and L-cysteine (L-Cys) were employed as the cooperation reagents, replacing DPA, and reacting in the same manner. The results revealed that only L-cysteine could cause a similar phenomenon, which suggests that the amino and thiol groups for the cooperation reagents played important roles in this system, as well as the distance between the amino and thiol groups (Fig. S11-S13†).
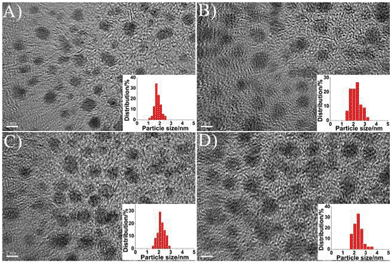 | ||
| Fig. 2 HRTEM images of the wavelength-tunable luminescent gold NPs. (A) Au-1, (B) Au-2, (C) Au-3, and (D) Au-4. (Scale bar: 2 nm.) | ||
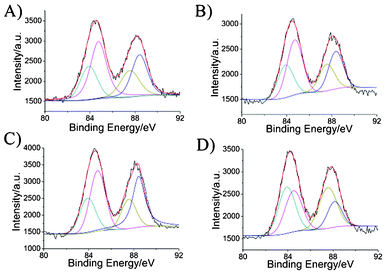 | ||
| Fig. 3 XPS spectra of the wavelength-tunable luminescent gold NPs. (A) Au-1, (B) Au-2, (C) Au-3, and (D) Au-4. | ||
To analyze the valence states of gold in the obtained wavelength-tunable gold NPs, we carried out X-ray photoelectron spectroscopy (XPS) measurements to investigate the oxidation states of their surfaces. The Au 4f XPS spectra shows that the binding energy (BE) of the Au 4f7/2 BE of the resulting gold NPs is located between the Au(0) BE (84 eV) of a metallic gold film and the Au(I) BE (86 eV) of gold thiolate, which proves that Au(0) and Au(I) exist in the luminescent gold NPs (Fig. 3).32,33 The further calculated amounts of Au(I) present were 65.8%, 63.1%, 62.9% and 48.0% for Au-1, Au-2, Au-3 and Au-4, respectively. This phenomenon supports the hypothesis that a fraction of the gold atoms in luminescent gold NPs exist in the Au(I) oxidation state, consistent with previously reported literature.9,34–36
As addressed by previous work, the luminescence of the obtained wavelength-tunable luminescent gold NPs could be dependent on many parameters, including the nature and size of the NPs, the surface ligands, temperature, solution composition and so on.19,37–42 It has been known that gold NCs, composed of a few to a hundred atoms, are comparable to the Fermi wavelength of electrons (ca. 0.7 nm), resulting in molecule-like properties including discrete and size-tunable electronic transitions and unique optical properties due to the strong quantum-confinement effect.43,44 However, Zheng et al. recently suggested that this particle size effect only applies to the few-atom NCs (<1 nm), and for larger gold NPs (1–3 nm), their emission energy is slightly deviated from the perfect free-electron model. For the few nm gold nanoparticles, the surface ligand effect and valence state effect have had significant impacts on the luminescence properties.37 Although the emission mechanisms of these luminescent gold NPs are still not fully understood, the differing ratios of the Au(I) state in the luminescent gold NPs, would be responsible for their unique optical properties.36,45
To further investigate the potential application for the presented gold NPs as luminescent probes in biological imaging, we then tested the imaging capability of the obtained luminescent gold NPs by using human cervical carcinoma (HeLa) cells. After 3 h of incubation with the wavelength-tunable luminescent gold NPs, cells were observed using a laser scanning confocal microscope (LSCM) (Fig. 4). The emission luminescence from the gold NPs can be clearly observed, and the luminescence signals suggest that the luminescent gold NPs were bonding to the cell membrane and that some of them had been ingested by the HeLa cells. Plus, to make sure that the observed luminescence was indeed from the luminescent gold NPs, we also performed a control experiment in the same experimental conditions without the addition of gold NPs (Fig. S14†). No obvious luminescence signal from the cells without incubation with the gold NPs was collected. Hence, this observation indicated that the wavelength-tunable luminescent gold NPs could be used for cellular luminescence imaging.
 | ||
| Fig. 4 Confocal microscopy images of the HeLa cells treated with the wavelength-tunable gold NPs (excitation wavelength: 405 nm). (A) Au-1, (B) Au-2, (C) Au-3, and (D) Au-4. (Scale bar: 75 μm.) | ||
The cytotoxicity of the obtained luminescent gold NPs was assessed via MTT assay after incubation for 24 h with HeLa cells. Fig. 5 shows the viability of the cancer cells incubated with eight different concentrations of the luminescent gold NPs. No apparent reduction in viability was observed after incubation with Au-1, Au-2, Au-3 and Au-4, even at the concentration of 200 μg mL−1. These results suggest that these luminescent gold NPs have good biocompatibility, and will be applicable for the imaging of living cells.
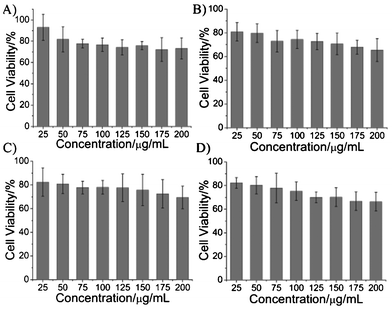 | ||
| Fig. 5 The viability results after incubation of the HeLa cells with various concentrations of (A) Au-1, (B) Au-2, (C) Au-3 and (D) Au-4, respectively. | ||
In summary, we have demonstrated that by adjusting the molar ratio of 11-MUA to DPA, the luminescence emission of prepared gold NPs was tunable within the region from 530 to 630 nm. The luminescent gold NPs exhibited long lifetimes and a high resolution of the luminescence. The imaging of the experimental results also suggests that the luminescent gold NPs could be potentially employed for cellular imaging.
Acknowledgements
This work was supported by The National Natural Science Foundation of China (20890022, 21175029) and the Shanghai Leading Academic Discipline Project (B109).References
- W. Chen, G. Lan and H. Chang, Anal. Chem., 2011, 83, 9450–9455 CrossRef CAS
.
- Y. Lin and W. Tseng, Anal. Chem., 2010, 82, 9194–9200 CrossRef CAS
- H. Liu, X. Zhang, X. Wu, L. Jiang, C. Burda and J. Zhu, Chem. Commun., 2011, 47, 4237–4239 RSC
- H. Tsunoyama, P. Nickut, Y. Negishi, K. Al-Shamery, Y. Matsumoto and T. Tsukuda, J. Phys. Chem. C, 2007, 111, 4153–4158 CAS
- X. Yang, M. Shi, R. Zhou, X. Chen and H. Chen, Nanoscale, 2011, 3, 2596–2601 RSC
- Y. Guo, Z. Wang, H. Shao and X. Jiang, Analyst, 2012, 137, 301–304 RSC
- C. Huang, C. Chen, Y. Shiang, Z. Lin and H. Chang, Anal. Chem., 2009, 81, 875–882 CrossRef CAS
- Y. Tseng, H. Chang, C. Chen, C. Chen and C. Huang, Biosens. Bioelectron., 2011, 27, 95–100 CrossRef CAS
- L. Shang, R. M. Dörlich, S. Brandholt, R. Schneider, V. Trouillet, M. Bruns, D. Gerthsen and G. U. Nienhaus, Nanoscale, 2011, 3, 2009–2014 RSC
- X. Yuan, Z. Luo, Q. Zhang, X. Zhang, Y. Zheng, J. Y. Lee and J. Xie, ACS Nano, 2011, 5, 8800–8808 CrossRef CAS
- Y. Wang, Y. Cui, Y. Zhao, R. Liu, Z. Sun, W. Li and X. Gao, Chem. Commun., 2012, 48, 871–873 RSC
- Y. Shichibu, Y. Negishi, H. Tsunoyama, M. Kanehara, T. Teranishi and T. Tsukuda, Small, 2007, 3, 835–839 CrossRef CAS
- J. Xie, Y. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888–889 CrossRef CAS
- S. Lin, N. Chen, S. Sum, L. Lo and C. Yang, Chem. Commun., 2008, 4762–4764 RSC
- M. L. Tran, A. V. Zvyagin and T. Plakhotnik, Chem. Commun., 2006, 2400–2401 RSC
- R. Zhou, M. Shi, X. Chen, M. Wang and H. Chen, Chem.–Eur. J., 2009, 15, 4944–4951 CrossRef CAS
- L. Shang, S. Dong and G. U. Nienhaus, Nano Today, 2011, 6, 401–418 CrossRef CAS
- C. Huang, Y. Hung, Y. Shiang, T. Lin, Y. Lin, C. Chen and H. Chang, Chem. Asian J., 2010, 5, 334–341 CrossRef CAS
- C. Huang, H. Liao, Y. Shiang, Z. Lin, Z. Yang and H. Chang, J. Mater. Chem., 2009, 19, 755–759 RSC
- H. Kawasaki, K. Hamaguchi, I. Osaka and R. Arakawa, Adv. Funct. Mater., 2011, 21, 3508–3515 CrossRef CAS
- D. G. Duff, A. Baiker and P. P. Edwards, Langmuir, 1993, 9, 2301–2309 CrossRef CAS
- Z. Wu and R. Jin, ACS Nano, 2009, 3, 2036–2042 CrossRef CAS
- Z. Yuan, M. Peng, Y. He and E. S. Yeung, Chem. Commun., 2011, 47, 11981–11983 RSC
- Z. Zhang, P. Zhang, K. Guo, G. Liang, H. Chen, B. Liu and J. Kong, Talanta, 2011, 85, 2695–2699 CrossRef CAS
- C. Liu, M. Ho, Y. Chen, C. Hsieh, Y. Lin, Y. Wang, M. Yang, H. Duan, B. Chen, J. Lee, J. Hsiao and P. Chou, J. Phys. Chem. C, 2009, 113, 21082–21089 CAS
- E. S. Shibu, M. A. H. Muhammed, T. Tsukuda and T. Pradeep, J. Phys. Chem. C, 2008, 112, 12168–12176 CAS
- C. J. Lin, T. Yang, C. Lee, S. H. Huang, R. A. Sperling, M. Zanella, J. K. Li, J. Shen, H. Wang, H. Yeh, W. J. Parak and W. H. Chang, ACS Nano, 2009, 3, 395–401 CrossRef CAS
- K. Lam, P. Chan and K. Wong, J. Nanosci. Nanotechnol., 2009, 9, 2337–2345 CrossRef CAS
- Y. Shiang, C. Huang and H. Chang, Chem. Commun., 2009, 3437–3439 RSC
- L. Shang, N. Azadfar, F. Stockmar, W. Send, V. Trouillet, M. Bruns, D. Gerthsen and G. U. Nienhaus, Small, 2011, 7, 2614–2620 CrossRef CAS
- C. Huang, Z. Yang, K. Lee and H. Chang, Angew. Chem., Int. Ed., 2007, 46, 6824–6828 CrossRef CAS
- M. A. Habeeb Muhammed, P. K. Verma, S. K. Pal, A. Retnakumari, M. Koyakutty, S. Nair and T. Pradeep, Chem.–Eur. J., 2010, 16, 10103–10112 CrossRef CAS
- E. S. Shibu and T. Pradeep, Chem. Mater., 2011, 23, 989–999 CrossRef CAS
- Z. Wang, W. Cai and J. Sui, ChemPhysChem, 2009, 10, 2012–2015 CrossRef CAS
- T. Udayabhaskararao, Y. Sun, N. Goswami, S. K. Pal, K. Balasubramanian and T. Pradeep, Angew. Chem., Int. Ed., 2012, 51, 2155–2159 CrossRef CAS
- C. Zhou, C. Sun, M. Yu, Y. Qin, J. Wang, M. Kim and J. Zheng, J. Phys. Chem. C, 2010, 114, 7727–7732 CAS
- J. Zheng, C. Zhou, M. Yu and J. Liu, Nanoscale, 2012, 4, 4073–4083 RSC
- Y. Shiang, C. Huang, W. Chen, P. Chen and H. Chang, J. Mater. Chem., 2012, 22, 12972–12982 RSC
- R. Jin, Nanoscale, 2010, 2, 343–362 RSC
- J. Zheng, C. Zhang and R. M. Dickson, Phys. Rev. Lett., 2004, 93, 77402 CrossRef
- H. Qian, Y. Zhu and R. Jin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 696–700 CrossRef CAS
- G. Schmid, Y. Liu, M. Schumann, T. Raschke and C. Radehaus, Nano Lett., 2001, 1, 405–407 CrossRef CAS
- W. Guo, J. Yuan and E. Wang, Chem. Commun., 2012, 48, 3076–3078 RSC
- O. Varnavski, G. Ramakrishna, J. Kim, D. Lee and T. Goodson III, ACS Nano, 2010, 4, 3406–3412 CrossRef CAS
- X. Tu, W. Chen and X. Guo, Nanotechnology, 2011, 22, 095701 CrossRef
Footnote |
| † Electronic Supplementary Information (ESI) available: experimental details, more characterization and other supporting data. See DOI: 10.1039/c2ra21785f |
| This journal is © The Royal Society of Chemistry 2013 |