Photoactivatable RNA derivatives as tools for studying the structural and functional organization of complex cellular ribonucleoprotein machineries†
Dmitri
Graifer
ab and
Galina
Karpova
*a
aLaboratory of Ribosome Structure and Functions, Institute of Chemical Biology and Fundamental Medicine, Siberian Branch of the Russian Academy of Sciences, Novosibirsk, 630090, Russia. E-mail: karpova@niboch.nsc.ru
bDepartment of Molecular Biology, Novosibirsk State University, Novosibirsk, 630090, Russia
First published on 13th November 2012
Abstract
Ribonucleic acids (RNAs) are biopolymers that play key roles in many processes crucial for the life of cells. Functions of RNAs are generally mediated by their specific interactions with proteins and ribonucleoproteins (supramolecular RNA-protein complexes) that take place within complexes of RNA ligands with proteins or ribonucleoproteins. These interactions are defined by the molecular environment of the RNA ligand in the complex. A beneficial approach for studying such complexes involves site-directed cross-linking: RNA derivatives that contain chemically reactive groups form covalent bonds (cross-links) with those structural elements of the protein or ribonucleoprotein that neighbor the RNA ligand. Such RNA derivatives have been successfully used to study the interaction of messenger RNA (mRNA) with the protein synthesis machinery. By applying this approach to studies of the protein translation machinery of higher organisms, chemically reactive RNA derivatives have provided unique information that could not have been obtained by other approaches to date. This article presents a brief review of the types of RNA derivatives used as mRNA analogs, and their structures and synthesis, together with methods for identification of cross-linking targets. Furthermore, we summarize current data on mRNA interactions occurring during the course of protein synthesis in higher organisms.
 Dmitri Graifer | Dmitri Graifer is a leading scientist at the laboratory of Ribosome Structure and Functions headed by Prof. Galina Karpova, at the Novosibirsk Institute of Chemical Biology and Fundamental Medicine SB RAS. He obtained his Ph.D. degree in 1985 at the Novosibirsk Institute of Organic Chemistry, Biochemical Dept. In 1999 he was awarded the Russian State Prize in fields of science and technique. Since 2004 he has been an Associate Professor, and in 2008 he obtained the degree of Doctor of Chemical Sciences. The main topic of D. Graifer's research concerns studying structural and functional organization of human ribosomes with the use of site-directed cross-linking. |
 Galina Karpova | Galina Karpova obtained her Ph.D. degree in 1976 at the Novosibirsk Institute of Organic Chemistry, Biochemical Dept. Since 1980, she has headed the group (and the lab since 1990) of Ribosome Structure and Functions at the Novosibirsk Institute of Bioorganic Chemistry SB RAS (since 2003 Novosibirsk Institute of Chemical Biology and Fundamental Medicine). In 1990 she obtained the degree of Doctor of Chemical Sciences, and since 1993 she has been a Professor. In 1999 she was awarded the Russian State Prize in fields of science and technique. The main research field of G. Karpova's lab during the last 20 years has concerned studying structural and functional organization of human translational machinery. |
1 Introduction
Approximately half a century has passed since a crucial breakthrough in understanding the biological role of ribonucleic acids (RNAs), when Har Gobind Khorana, together with R.W. Holley and M.W. Nirenberg, showed that RNAs act as messengers that carry genetic information and are then translated into proteins. It is, of course, now well accepted that RNAs are essential biopolymers of the cells involved in the most important events of life, from the translation of genetic information from DNA to protein, to the regulation of various cellular processes. Key RNA players in protein translation include mRNAs, ribosomal RNAs (rRNAs) and transfer RNAs (tRNAs). rRNAs are the main components of supramolecular ribonucleoprotein cellular machines responsible for protein synthesis (ribosomes). tRNAs carry specific amino acid residues on their 3′-ends for the synthesis of proteins by the ribosome. Other RNAs contribute to gene expression regulation at earlier stages. These small RNAs (typically, 18–40 nucleotides) regulate histone modification, transcription, and mRNA maturation and degradation. Finally, the genomes of many viruses consist of RNAs. All of these examples highlight the fundamental role of RNA in biology, and therefore the importance of studying structural aspects of RNA interactions. Such approaches are crucial in order to understand how RNAs interact with other components of the cell and how these interactions relate to RNA functions.Various approaches have been used to study cellular processes involving RNAs, the protein synthesis apparatus being the subject of the majority of studies. As early as the 1970s, such studies on the interaction of aminoacyl-tRNA1 and mRNA2 with the ribosome were pioneered by D.G. Knorre with the use of site-directed cross-linking (affinity labeling). This approach is based on chemically active analogs of the ribosomal RNA ligands, which contain reactive groups that cross-link to ribosomal components in contact with or within a close proximity to the ligand. The affinity labeling is performed with mRNA and/or tRNA analogs bound within ribosomal complexes that imitate defined steps of translation, which is possible if the cross-linker does not interfere with the formation of such complexes. After the formation of cross-links, the identity of the covalently bound ribosomal components can be determined, and conclusions drawn concerning the structure of the ribosomal ligand binding centers.
Systematic studies of bacterial ribosomes were performed in the 1980s with the use of tRNA derivatives containing aromatic azides scattered throughout the molecule3 and mRNA derivatives consisting of short (3–12 nucleotides) oligoribonucleotides bearing a cross-linker (generally an alkylating 2-chloroethyl arylamine moiety) at the 3′ or 5′-end.4 These studies revealed for the first time ribosomal proteins located in tRNA binding sites3 and the mRNA binding center4 of the bacterial ribosome. Subsequently, the ribosomal mRNA binding center was studied by various groups using long (30–50 nucleotides) synthetic mRNA analogs bearing photoactivatable 4-thiouridine residues (e.g., see ref. 5–8). These cross-linking studies enabled the X-ray crystallographic data that followed to be correctly interpreted in order to reveal the structure of the bacterial ribosome at the atomic level (for a review, see ref. 9,10) and to make fundamental conclusions regarding the molecular mechanisms of translation.
Despite their general similarity to bacterial ribosomes, ribosomes from higher organisms (particularly human) are much more complicated and less well studied. This can be explained, in part, by the fact that several approaches used with success for studying bacterial ribosomes are not applicable yet to mammalian ribosomes. For example, approaches based on the reconstitution of active ribosomes from total ribosomal protein and rRNA are not possible with ribosomes of higher organisms. Furthermore, X-ray crystallography has only been successfully applied to lower eukaryotic ribosomes free from mRNA and tRNAs.11–13 This is why RNA analogs bearing cross-linkers at specific locations are the most suitable tools currently available for interrogating the interactions of RNA ligands with the mammalian translational machinery.
Here, we give an overview of the protein synthesis apparatus and describe current approaches for the synthesis of RNA analogs used for studying the mammalian translation system. These RNA analogs include synthetic mRNAs containing 4-thiouridines and derivatives of short oligoribonucleotides that bear photoactivatable perfluorophenyl azide cross-linkers at specific locations. We also describe an original method for site-specific introduction of a cross-linker into long, highly structured RNAs, and their application for studying the human translation system using the example of the hepatitis C virus internal ribosome entry site (IRES). Finally, we review the use of site-directed cross-linking in advancing our understanding of the fine structure of the ribosomal mRNA binding site, the arrangement of the hepatitis C virus IRES binding site on the small ribosomal subunit, and the molecular basis for recognition of mRNA stop codons by the translational termination factor eRF1.
2 An outline of the protein synthesis apparatus and model complexes used for studying it by site-directed cross-linking
Ribosomes translate genetic information in all organisms, from bacteria to humans. During translation, the ribosome interacts with two types of specific RNA ligands, mRNA and tRNAs. Each species of tRNA carries a specific amino acid (aa) residue for the synthesis of a polypeptide chain. Insertion of the correct aa residue into the growing polypeptide chain is achieved via base-pairing between an mRNA codon at the aminoacyl (A) site of the ribosome and the anticodon of the cognate aminoacyl-tRNA. This recognition (decoding) leads to binding of the cognate aminoacyl-tRNA at the A site, followed by the formation of a peptide bond between the aa residue of the A site aminoacyl-tRNA and the polypeptide moiety of the peptidyl-tRNA bound at the neighboring peptidyl (P) site (see Fig. 1 for a simplified scheme showing how the ribosome elongates the peptide chain).Each tRNA is specific to only one aa; the correspondence of mRNA codons to aa residues is governed by a genetic code that is practically universal for cytoplasmic ribosomes from all kingdoms of life. Translation is terminated when one of three stop codons (UAA, UAG or UGA) falls at the A site. These codons are recognized by specialised ‘class 1’ termination factors (in eukaryotes, eRF1) that trigger hydrolysis of the complex ester bond between the polypeptide and the final tRNA, thus releasing the newly synthesized protein from the ribosome.
The ribosome is a highly complex molecular machine consisting of two subunits, one small and one large, each of which contains rRNA and several dozen ribosomal proteins. The structures of ribosomes from bacteria and eukaryotes share significant similarity. In particular, the secondary structures of their respective rRNAs have a universally conserved core. Furthermore, approximately 2/3 of ribosomal proteins in eukaryotic ribosomes have a bacterial homologue. However, eukaryotic rRNAs are longer than their bacterial counterparts and contain additional fragments that do not belong to the conserved core, and eukaryotic ribosomal proteins are generally larger than their bacterial homologues. Eukaryotic ribosomal subunits also contain additional proteins (about 1/3 of the total number) that have no homologues among bacterial ribosomal proteins.
The small ribosomal subunit (30S in prokaryotes and 40S in eukaryotes) contains an mRNA binding centre, including the decoding site where an aminoacyl-tRNA is selected that is cognate to the A-site mRNA codon. The large subunit contains the peptidyl transferase center responsible for catalysis of peptide bond formation (Fig. 1). Three tRNA binding sites, namely A, P and E (E is the site at which the discharged tRNA binds before it exits the ribosome), are formed by the interfacial surfaces of both subunits. During translation, the ribosomal mRNA binding center not only interacts with mRNA codons and tRNAs at the A and the P sites, but also binds a 30–50 nucleotide mRNA fragment that contains these codons.14,15 The most common approach used to obtain model complexes for studying the interaction of mRNA and tRNAs with the ribosome has been the direct binding of the ligands to the ribosome in the absence of translation factors and at Mg2+concentrations of 10–20 mM (which are approximately one order of magnitude higher than that in the cell). Under these conditions, stable complexes are formed in which the tRNA is bound to the P site (for which it has the highest affinity relative to the A and E sites), where it interacts with a cognate mRNA codon.16–18 Application of short oligoribonucleotide derivatives (up to 12 nucleotides long) as mRNA analogs is advantageous for the successful study of mammalian ribosomes, since they have very low intrinsic affinity for the ribosome, and their binding is hardly detectable in the absence of tRNA.19–22 However, in the presence of a tRNA cognate to one of the mRNA codons, the analog becomes tightly bound to the ribosome and is fixed in a defined position by interaction with the tRNA anticodon at the P site.19–22 Thus, the position of the derivatized mRNA analog nucleotide in the ribosome can be unambiguously governed by a tRNA.
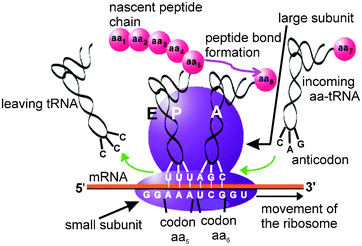 | ||
| Fig. 1 Simplified scheme of the protein synthesis on the ribosome. The tRNA carrying the nascent peptide chain is at the P site, and the mRNA codon for the next amino acid (aa) residue to be incorporated is at the A site, where it is recognized by the anticodon of tRNA carrying the respective aa. When the ribosome performs the catalysis of the peptide bond formation, the peptide chain elongated by one aa residue becomes attached to the A site tRNA, and the tRNA at the P site is deacylated. Then the ribosome moves towards the 3′ end of the mRNA, so that the peptidyl-tRNA, together with the respective mRNA codon, moves from the A to the P site. The deacylated tRNA moves from the P to the E site, at which it can leave the ribosome, and the A site programmed with the next mRNA codon becomes vacant for the new, incoming aa-tRNA. | ||
Although eukaryotic ribosomes are also able to translate viral RNAs, the mechanisms of the translation initiation are generally different to cellular RNAs. Cellular mRNAs contain a special 7-methylguanosine cap structure at their 5′-terminus that is recognized by the initiation factor eIF4E (“e” refers to “eukaryotic”), which, together with other initiation factors, promotes the binding of 40S subunits in complex with the initiator Met-tRNAiMet. Subsequently, the small ribosomal subunit ‘scans’ along the mRNA until it is positioned at the first AUG codon at the P site, where the first codon interacts with the initiator tRNAiMet (for a review, see ref. 23): This is considered the key event of translational initiation. An alternative mechanism is utilized by certain uncapped viral RNAs that contain highly-structured fragments in their 5′-untranslated regions (upstream of the start AUG codon), called IRESs (internal ribosome entry sites), that positions the 40S subunit on the mRNA in a manner that directly places the AUG codon in the P site region (reviewed in ref. 24). In particular, IRES-mediated translation initiation is inherent to the RNA of the hepatitis C virus (HCV), one of the most dangerous human pathogens.
3 Site-directed modification and identification of cross-linked components of the translational machinery
A general scheme of the cross-linking approach is shown in Fig. 2. In order to follow the ribosomal components cross-linked to mRNA analogs, the mRNA analog is labeled with 32P prior to the experiment. The label can be introduced to the 5′-terminus of the RNA by transfer from [γ-32P]ATP to the 5′-OH of the RNA 5′-terminal ribose using polynucleotide kinase. This is the generally accepted approach for chemically synthesized oligoribonucleotides and their derivatives. In some specific cases,25 the label can alternatively be introduced at the 3′-terminal ribose by its ligating to a cytidine-3′,5′ [32P]diphosphate with RNA ligase. Longer RNAs (obtained by transcription from specially synthesized DNA templates using T7 RNA polymerase) are generally uniformly labeled by using [γ-32P]nucleoside-triphosphates as substrates for the synthesis. Finally, a single 32P label can be introduced internally, 3′ of the nucleotide that bears the cross-linker by a multistep synthesis.26,27 To do this, two “halves” of the RNA are synthesized, one of them contains 4-thiouridine at the 3′-terminus, and another is 5′-32P labeled, as described above; then they are ligated together using RNA ligase so that the label occurs 3′ to the modified nucleotide.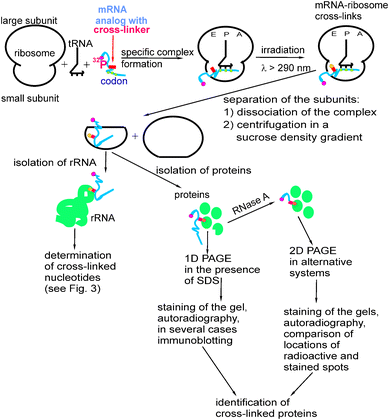 | ||
| Fig. 2 Basic scheme of studying the molecular environment of mRNA in the ribosome by site-directed cross-linking with the use of mRNA analogs bearing photoactivatable groups at the designed locations. 1D PAGE and 2D PAGE indicate one-dimensional and two-dimensional polyacrylamide gel electrophoresis, respectively. | ||
Identification of small subunit rRNA nucleotides cross-linked to labeled mRNA analogs is performed as follows (Fig. 3). rRNA is easily isolated from the modified ribosomes (or small subunits if the ribosomes were separated into subunits after cross-linking) by a standard procedure including extraction of proteins into water-saturated phenol with subsequent ethanol precipitation of the RNA. Regions of rRNA containing cross-linked mRNA analogs are then identified by hydrolysing the rRNA with RNase H in the presence of DNA oligomers that are complementary to specific rRNA sequences,21,28,29 thereby enabling rRNA cleavage at the designed sites. The resulting RNA fragments are separated by denaturing polyacrylamide gel electrophoresis (PAGE). Staining of the gels makes it possible to visualize all the RNA fragments, which can then be easily identified as their lengths correspond to the rRNA sites chosen for the RNase H cleavage; autoradiography of the gels shows which fragment contains the label. Identification of the cross-linked rRNA nucleotide is performed by reverse transcription using the modified rRNA as a template,30 and taking into account that as reverse transcriptase synthesizes DNA copies from the RNA template, it stops or pauses at the cross-linked RNA nucleotide. Primers for the reverse transcription (typically deoxy-20-mers), are designed based on the results of the RNase H experiments. The cross-linking site is generally assumed to be the nucleotide at 5′ to the primer extension end (pause) site, the position of which is determined by PAGE analysis of the primer extension products in parallel with the sequencing of unmodified rRNA, using each of the dideoxynucleoside triphosphates. This analysis allows identification of the exact sequence of the DNA transcript synthesized on the cross-linked rRNA and, consequently, the position of the cross-linked nucleotide in the rRNA. Control experiments are carried out with the rRNA isolated from unmodified subunits and processed in an identical manner, but in the absence of chemically reactive ribosomal ligands.
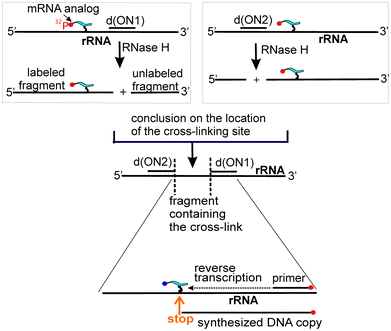 | ||
| Fig. 3 Principal steps of identification of cross-linked nucleotides of the target RNA. | ||
Identification of the ribosomal proteins cross-linked to mRNA analogs is more complicated than that of the rRNA nucleotides, with the choice of methodology depending on the identity of the cross-linked protein.22,25,31 To identify cross-linked proteins, modified ribosomes are first dissociated into subunits by centrifugation in a sucrose density gradient, before isolating total ribosomal protein from small subunits. In studies using short mRNA analogs, labeled proteins were identified using one- and two-dimensional PAGE in a variety of electrophoresis systems.32 Staining of the gels made it possible to visualize all unmodified proteins based on their known electrophoretic position, with autoradiography, then identifying the locations of the cross-linked proteins. Although the locations of radioactive spots did not exactly correspond to those of the respective unmodified proteins (due to the retarding effect of the cross-linked oligoribonucleotide on electrophoretic mobility), it has been possible to identify cross-linked proteins by comparing mobilities in different electrophoretic systems.22,25 In some cases, the identity of cross-linked ribosomal proteins has been confirmed with the use of antibodies against specific mammalian ribosomal proteins,22 mass spectrometry27 and other approaches (e.g., ref. 22). An example illustrating the identification of the ribosomal protein S26e (rpS26e) cross-linked to mRNA analogs is presented in Fig. 4.
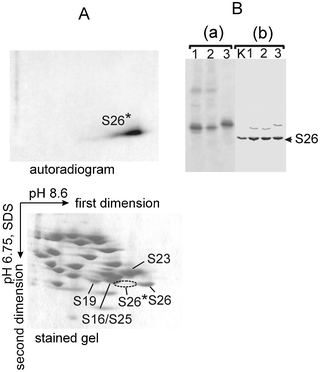 | ||
| Fig. 4 Identification of the rpS26e cross-linked to mRNA analogs whose ATB-derivatized uridine (U*) was placed in position −4, −6 or −9 (the respective mRNA analogs could be found in Fig. 10). (A) Analysis of the proteins cross-linked to one of the mRNA analogs by a two-dimensional PAGE after hydrolysis of the cross-linked RNA with RNase A. The cross-linked protein is marked with an asterisk (*). Spots of several unmodified proteins in the stained gel are indicated according to ref. 32. The location of the radioactive spot is indicated by the dotted line. (B) Identification of the cross-linked rpS26e protein by immunoblotting after 1D PAGE separation of 40S proteins untreated with RNase. Lanes 1, 2 and 3 correspond to the results with the complexes where U* was in the position −4, −6 or −9, respectively. Lane K corresponds to a control experiment with proteins isolated from unmodified 40S subunits. Panel (a) is an autoradiogram, panel (b) presents the identification of rpS26e with specific antibodies. Note that only a minor extent of the ribosomal rpS26e underwent the cross-linking, so the major part of it remained unmodified; so, immunoblotting developed strong bands of the unmodified rpS26e, and weaker bands of the protein cross-linked to mRNA analogs. | ||
With 30–50 nucleotide long mRNA analogs, direct identification of the cross-linked proteins by PAGE is highly ambiguous since the effect of the cross-linked RNA on protein mobilities is too strong. In these cases, two alternative approaches have been applied to identify the cross-linked proteins. The first utilized mRNA analogs carrying a single internal 32P label in the 3′-phosphate of the nucleotide bearing the cross-linker. This approach enabled hydrolysis of the cross-linked RNA without loss of the label followed by PAGE analysis, since modified proteins were only cross-linked with mono- or dinucleotide residues.27 This approach was used to identify amino acids of the translational termination factor eRF1 that cross-linked with the first nucleotide of the A site stop codon in an mRNA analog.26 The second approach is based on the application of uniformly labeled mRNA analogs. This allows the removal of the majority of RNA that is cross-linked using RNase, leaving a very short labeled oligonucleotide fragment.33 Both approaches have their specific disadvantages. Preparation of RNAs containing a single internal label in amounts sufficient for cross-linking experiments is sometimes problematic because of the low yield of a product obtained as a result of ligation of the two “halves” (see above). When using uniformly labeled RNAs, >95% of the label is lost in the course of the exhaustive RNase hydrolysis, which requires large amounts of labeled RNAs and other components that may be not easily available.
Site-directed cross-linking allows not only identification of the cross-linked proteins, but even the specific peptides that have been modified. A special methodology for the identification of cross-linked peptides of proteins was initially suggested,26 and then developed and successfully applied.34–37 The approach is based on the specific cleavage of the cross-linked protein with various proteolytic agents and subsequent separation of the resulting labeled and modified peptides by PAGE. These peptides can be identified by comparing their electrophoretic mobilities with those predicted from the known cleavage specificity of the proteolytic agent used. In the example given on the study of the termination factor eRF1, mutant forms of the factor were also used that contained single or double amino acid substitutions in the regions of interest that allowed fine mapping of protein fragments containing cross-links. These substitutions either provided new artificial methionine residues or eliminated natural methionines in the factor (methionine is a target for specific cleavage with cyanogen bromide).26,35,36
4 Types of RNA derivatives used as mRNA analogs
4.1 Oligoribonucleotides with an alkylating group at the 5′ or 3′-end
Oligoribonucleotide derivatives bearing an alkylating 4-(N-2-chloroethyl-N-methylamino)benzyl moiety at either the 5′-terminal phosphate or the 3′-terminal cis-diol group of ribose are shown in Fig. 5. These derivatives were used in systematic studies of bacterial ribosomes (e.g., see ref. 4 and references therein) and in early works on rat liver38,39 and human19,20,40 ribosomes. These studies exemplified the use of the site-directed cross-linking approach for studying the ribosomal mRNA binding center, and made it possible to obtain the first data on ribosomal components involved in the formation of this functional site, both in bacterial and mammalian ribosomes. However, these derivatives were not used for further detailed studies of mammalian ribosomes because of their substantial limitations. In particular, the alkylation of ribosomes required long incubation periods (5 to 30 h) at room temperature that led to the partial dissociation of the model complexes and inactivation of the ribosomes (discussed in ref. 21). Also, the alkylating group could only be introduced at the 5′ or the 3′-end of the oligoribonucleotide, which significantly limits the information that can be derived from the experiment.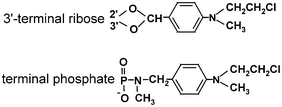 | ||
| Fig. 5 Alkylating aromatic 2-chloroethylamine groups introduced at the terminal phosphate or ribose of oligoribonucleotides. | ||
4.2 Synthetic long RNAs carrying 4-thiouridine residues
In cross-linking experiments using synthetic mRNA analogs that are 30–50 nucleotides in length, 4-thiouridine (s4U) residues are generally used as the cross-linker; a sulfur atom in s4U replaces the oxygen atom that naturally occurs in uridine (Fig. 6A). These residues can be activated by mild UV light (approximately 330 nm), which does not affect natural RNA bases (whose maximum absorbance is about 260 nm), and induces specific s4U-mediated zero-length cross-links that reflect intimate contacts between the mRNA analog and the ribosome. mRNA analogs with s4U residues at defined locations can be easily obtained by transcription with T7 polymerase from DNA templates containing a single A-residue at an appropriate position in the transcribed region. Transcription is performed in the presence of 4-thio-UTP instead of UTP, resulting in the incorporation of s4U at the designed mRNA position.5 Using DNA templates containing several A residues, one can obtain mRNA analogs containing s4U residues scattered throughout the RNA molecule.41 In either case, performing the transcription reaction in the presence of [γ-32P]ATP (or other nucleoside triphosphate) allows the preparation of the radioactively labeled mRNA analog for detection of cross-linked ribosomal components.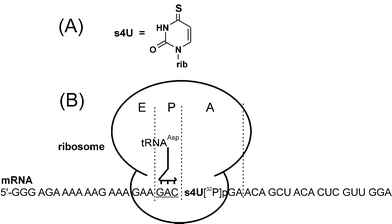 | ||
| Fig. 6 Structure of 4-thiouridine (s4U) (A) and a complex of the ribosome with a purine-rich s4U-containing mRNA analog applied for studying the ribosomal decoding site and the stop codon recognition site on the eRF1, (B). The mRNA analog is positioned on the ribosome by its GAC triplet that interacts with the cognate tRNAAsp at the P site. The subsequently modified stop codon s4UGA is at the A site where it can interact with the eRF1. The radioactive labeled phosphate is between the s4U and G of the stop codon, which makes hydrolysis of the cross-linked mRNA analog possible, without the loss of the label. | ||
s4U-containing mRNA analogs were extensively used in studies of bacterial ribosomes by groups led by R. Brimacombe and A. Bogdanov (e.g., see ref. 5,6), and P.Wollenzien.7,8 Initial reports on similar studies of human ribosomes30,41 revealed that it was difficult to achieve the unambiguous position of mRNA (and therefore, its s4U residues) on the ribosome by tRNA cognate to one of mRNA codons targeted to the P site. The problem was related to the conditions used for the in vitro assembly of the model ribosomal complexes, i.e. the absence of translation factors, and incubation with 10 mM of Mg2+ (which is 5-fold over that in the cell) at room temperature. Under these conditions the mRNA analogs had a large intrinsic affinity for the ribosomes in the absence of tRNA, which allowed them to bind the ribosome at ambiguous positions. Since the binding level and cross-linking pattern of mRNA analogs did not depend on the presence of tRNA, this implied that tRNA caused no significant change in the location of the mRNA on the ribosome.30,41 To overcome these difficulties, special purine-rich mRNA analogs were designed whose intrinsic affinities to 80S ribosomes were relatively low (an example is presented in Fig. 6B).27,42,43 Using these analogs, addition of tRNA cognate to the selected codon resulted in an increase in the level of mRNA binding, and the appearance of new cross-links that could be unambiguously assigned to complexes containing an mRNA analog fixed to the ribosome as a result of a codon–anticodon interaction of tRNA and mRNA at the P site (Fig. 6B). It should be noted that the problem concerning ambiguous binding of long mRNA analogs to the ribosome only arose under conditions of supraphysiological Mg2+ concentrations, but not more physiological levels in the presence of translation factors.33,44 However, in the majority of studies, model complexes were not assembled under these more physiological conditions, presumably because the respective mammalian translation factors were difficult to obtain at this time.26,27,30,41–43 Thus, a disadvantage of the relatively long s4U-containing mRNA analogs was that only special adenine-rich derivatives were suitable under the described ‘factor-free’ conditions, which in turn significantly restricts the sequence possibilities of the mRNA. Finally, a method for preparation of mRNA analogs containing a single 32P 3′ to the s4U residue (which is most suitable for precise identification of cross-linking products) often gives low yields of the final RNA that are insufficient for identification of the cross-linked products.
It is worth mentioning here that approaches based on enzymatic incorporation of photoactivatable nucleotides into RNA are also used to study RNA–protein interactions that do not directly relate to the translational machinery. So, an RNA containing 8-azido-adenosine residues obtained by T7 transcription from the appropriate DNA template was used in a cross-linking study with the coat protein of the Escherichia coli phage MS2.45 Besides, a method utilizing in vivo incorporation of s4U into RNAs by incubation of the living cells with s4U with subsequent UV-irradiation was elaborated and applied for studying RNA–protein interactions between s4U-containing RNA transcripts and RNA-binding proteins (RBPs) or microRNA-containing ribonucleoprotein complexes (RNPs) that regulate maturation, stability, transport, editing and translation of these transcripts.46 This method called PAR CLIP (photoactivatable-ribonucleoside-enhanced cross-linking and immunoprecipitation) is based on the immunoprecipitation of the cross-linked protein, the conversion of the cross-linked RNA fragments into a cDNA library and its sequencing; cross-linking sites are revealed by thymidine to cytidine transitions in the cDNAs. Photoactivatable nucleosides other than s4U (6-thioguanosine, 5-bromouridine and 5-iodouridine) are not good for PAR CLIP, since they provide too low yield of RNA–protein cross-links.46 The use of PAR CLIP with s4U allowed the authors to directly identify transcriptome-wide RNA sites for binding of several regulatory RBPs and RNPs in live cells.
4.3 Derivatives of oligoribonucleotides modified with perfluorophenyl azides
A cross-linker, whose application turned out to be the most fruitful in systematic studies of the structure of the mRNA binding center of the human ribosome, is a p-azidotetrafluorobenzoyl (ATB) group, which has two advantages. Firstly, it can be excited by relatively mild UV light (λ > 290 nm), which leads to the loss of nitrogen and the formation of biradical nitrenes attached to the perfluorinated benzol ring (Fig. 7). This is an active intermediate which is able to provide a high yield of cross-links with both nucleic acids and proteins under a short (less than 1 min) irradiation, using commercially available UV-lamps.47–49 The excellent cross-linking properties of perfluorated aryl azides relates to the higher reactivity of the respective nitrenes as compared to those of the unfluorinated aryl azides.50 Secondly, the ATB group can be introduced at various nucleotide positions not involved in classical Watson–Crick base-pairing, namely, at the C5 of uridines, N7 of guanosines or C8 of adenosines, in addition to the 5′- or 3′-terminal phosphates (Fig. 7). To obtain an ATB-derivatized oligoribonucleotide, a spacer bearing an aliphatic amino group should first be specifically introduced at the designed position. Then, the ATB group can be selectively introduced at this amino group by following its benzoylation with the N-oxysuccinimide ester of p-azidotetrafluorobenzoic acid. The resulting ATB-derivatized oligoribonucleotides can then be easily purified by reverse-phased high performance liquid chromatography (HPLC) (e.g., see ref. 51).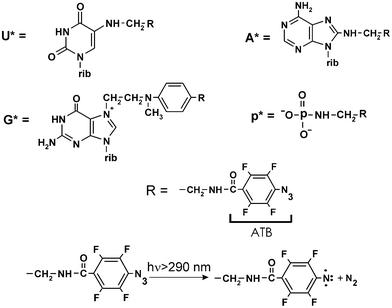 | ||
| Fig. 7 Structures of the ATB-modified nucleosides and a terminal phosphate, which were present in short mRNA analogs, derivatives of oligoribonucleotides and the formation of biradical nitrene upon UV-irradiation of the ATB group. | ||
A condensation reaction with aliphatic diamines (ethylene diamine or isopropylene diamine) is used to introduce an amino-spacer at the terminal phosphate of an oligoribonucleotide, with the help of a basic catalyst (N-methyl imidazole or dimethylaminopyridine) and a combination of triphenylphosphine with 2,2′-dipiridyl disulfide as a condensing agent (e.g., see ref. 19). In order to obtain an oligoribonucleotide derivative bearing an amino spacer at the N7 atom of the guanine, alkylation with 4-(N-2-chloroethyl-N-methylamino)benzylamine is employed under specific conditions that practically exclude the modification of other nucleotides.29 Evidently, this approach can only be applied to oligoribonucleotides that contain a single guanosine residue at a defined position. Lastly, the introduction of an amino spacer into a selected uridine or adenine is performed during the solid phase chemical synthesis of the oligoribonucleotide. 5-Br-uridine or 8-Br-adenosine monomers that contain all the groups necessary for oligoribonucleotide synthesis are used at the appropriate step in order to incorporate the modified nucleotide at the specified position.51 Following synthesis, the Br within the oligomer is replaced by ethylene diamine or isopropylene diamine residues using a nucleophilic substitution reaction performed with the respective diamine.51
When analyzing cross-linking results obtained using these ATB-derivatized RNA ligands, one should take into account that the maximum distance separating the derivatized RNA nucleotide and the reactive nitrogen of the cross-linker could be up to 11–14 angstroms, depending on the spacer structure. Therefore, the cross-linked ribosomal components could be those that either make direct contacts with the RNA ligand, or those that are located in close vicinity of, but not in direct contact with, the RNA.
4.4 Derivatives of long, highly structured RNAs bearing a perfluorophenyl azide-modified nucleotide in a defined location
In order to insert an aliphatic amine group spacer in long, highly structured RNAs for subsequent ATB coupling, a special strategy has been developed based on a so-called ‘complementary-addressed’ (i.e., sequence-specific) modification of nucleic acids, an approach originally proposed by N.I. Grineva in the 1960s.52 Specifically, the RNA is alkylated with a [4-(N-2-chloroethyl-N-methylamino)benzyl]phosphoramide derivative of an oligodeoxyribonucleotide that is complementary to a sequence adjacent to the target site. RNA sequences are chosen based on their predicted accessibility (i.e., sequences mainly not involved in the formation of secondary and/or tertiary RNA structures), to allow complementary base-pairing with the oligonucleotide moiety of the derivative. Any RNA nucleotide (except for U) that is located close to the modified terminus of the bound complementary deoxy-oligomer can then be alkylated at its nucleophilic center, leading to the formation of a covalent adduct (e.g., see ref. 53). The exact location of the modified nucleotide in the alkylated RNA is verified by a reverse transcription analysis (e.g., see ref. 54, 55) as described above in section 3. If the alkylated RNA is relatively short (about 100 nucleotides long), the modified nucleotide can be easily identified by hydrolysis with base-specific ribonucleases.53 The phosphoramide bond in the covalent adduct becomes labile under relatively mild acidic conditions (pH < 4, T° > 40 °C), thus allowing hydrolysis of the bond and liberation of a benzylamine group, without damage of the RNA. As a result, the specified RNA nucleotide is attached to a benzylamine spacer, which is identical to that used for the derivatization of guanines in oligoribonucleotides, as outlined above. The general scheme for the sequence-specific introduction of ATB groups in RNA is presented in Fig. 8.![Scheme of the site-specific introduction of a photoactivatable group into a selected RNA site, based on the complementary-addressed alkylation of the RNA with [4-(N-2-chloroethyl-N-methylamino)benzyl]-phosphoramides of oligodeoxyribonucleotides.](/image/article/2013/RA/c2ra22095d/c2ra22095d-f8.gif) | ||
| Fig. 8 Scheme of the site-specific introduction of a photoactivatable group into a selected RNA site, based on the complementary-addressed alkylation of the RNA with [4-(N-2-chloroethyl-N-methylamino)benzyl]-phosphoramides of oligodeoxyribonucleotides. | ||
The approach outlined above was successfully used to introduce a cross-linker in defined positions of the HCV IRES, which could be considered as a peculiar mRNA analog consisting of nucleotides 40–372 in the genomic RNA of HCV.56 To introduce a cross-linker into the IRES, the respective 32P-labeled RNA fragment was obtained by transcription with T7 RNA polymerase from a specially synthesized DNA template,57 an approach similar to that used for preparation of the 30–50 nucleotides long mRNA analogs described above. RNA containing biotin residues were obtained by transcription with biotinylated nucleoside triphosphates in several studies. Application of biotinylated RNAs made it possible to isolate cross-linked products containing the IRES derivatives by affinity chromatography on avidin-agarose.54 The RNA sites used to introduce the cross-linker were chosen based on available data regarding the domains and subdomains of HCV IRES involved in the binding to the small ribosomal subunit. To increase the yield of the IRES alkylation with deoxyoligomer derivatives, “helper oligomers” were employed that promoted the disruption of the IRES secondary structure, thereby allowing binding of oligomers containing the alkylating group to the structured RNA. The set of HCV IRES derivatives that were obtained by this approach54,55 (Fig. 9) helped to reveal ribosomal components that take part in HCV IRES binding to the human small ribosomal subunit that plays a key role in the initiation of the viral RNA translation (reviewed in ref. 24).
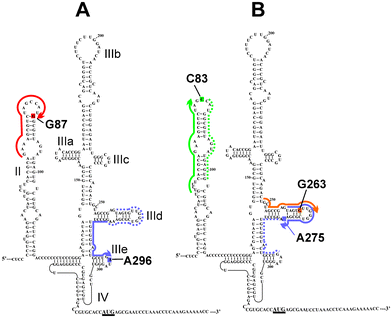 | ||
| Fig. 9 The secondary structure of the HCV IRES24 (the initiation AUG codon is underlined). RNA sequences complementary to the deoxy-oligomers used for site-specific modification of the HCV IRES used in ref. 54 (A) and in ref. 55 (B) are shown with thick lines with arrows indicating the 5'-phosphates derivatized with alkylating groups. RNA sequences complementary to “helper oligomers” used together with the alkylating derivatives are marked with dotted lines. Nucleotides of the HCV IRES modified with the oligonucleotide derivatives are shaded. | ||
5 Organisation of the mammalian translation machinery as revealed by site-directed cross-linking with RNA derivatives
This section contains a brief review of the structural elements of the translation machinery that contact mRNAs during the course of protein synthesis. Particular attention will be paid to those involved in forming the canonical ribosomal mRNA binding center, the site for binding of hepatitis C IRES on the small ribosomal subunit, and the mRNA stop codon recognition site on the termination translation factor eRF1.5.1. Structure of the mRNA binding center of the human ribosome
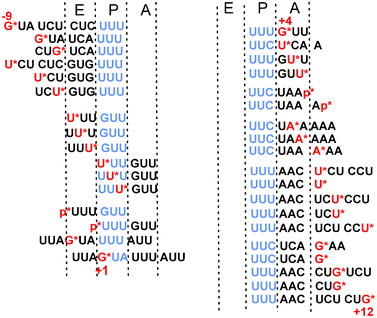 | ||
| Fig. 10 ATB-derivatized mRNA analogs applied for studying human translational machinery. The derivatized nucleotides or phosphates are in red and marked with asterisks. mRNA codons targeted to the ribosomal P site to fix the mRNA at the designed position on the ribosome are in blue. Position +1 conventionally corresponds to the first nucleotide of the P site codon. Positions of mRNA nucleotides upstream and downstream of this nucleotide are marked “−“ and “+”, respectively. | ||
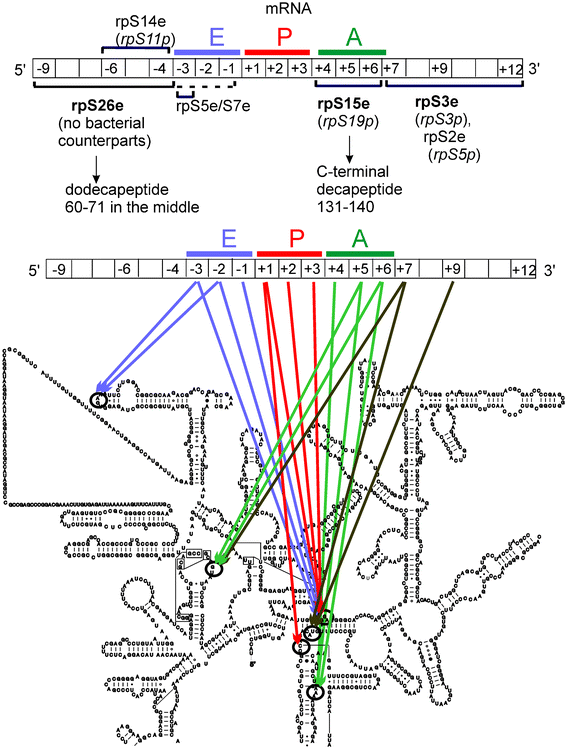 | ||
| Fig. 11 Summary of the structural elements of the human ribosome that neighbor particular positions of mRNA, according to cross-linking data with ATB-derivatized mRNA analogs. mRNA is shown as diagrams; positions of the mRNA nucleotides on the ribosome are indicated and the codons interacting with tRNAs are marked with colored lines. Upper diagram shows the protein environment of mRNA (the main targets of cross-linking are in bold).22,25,31,34,37 The bacterial counterparts are shown in brackets. Below, the mRNA–rRNA neighborhood21,22,28,29,59 is presented on the secondary structure of the human rRNA from the small ribosomal subunit taken from www.rna.ccbb.utexas.edu. | ||
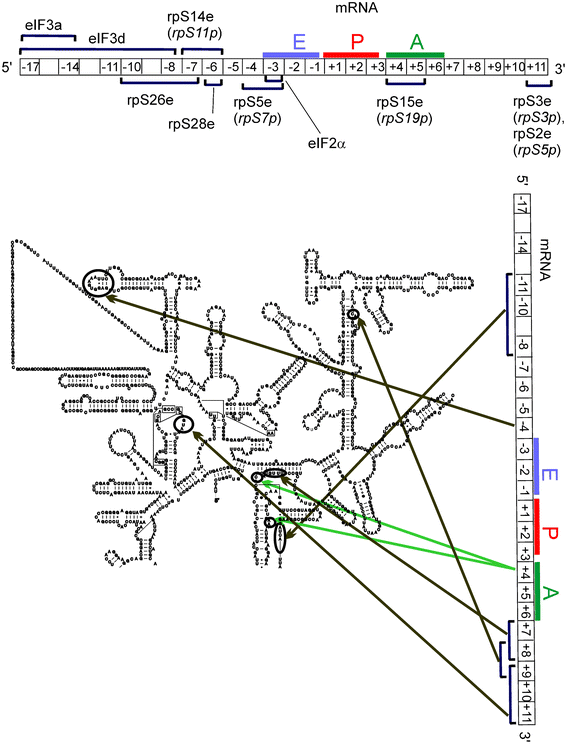 | ||
| Fig. 12 Components of mammalian translational machinery contacting mRNA during the translation initiation.33,44 For details, see the Fig. 11 legend. eIFs are subunits of the initiation factors. | ||
Comparison of the early data obtained from the application of short ATB-derivatized mRNA analogs (Fig. 11) with later data using longer synthetic s4U-containing mRNAs (Fig. 12) indicates that both tools gave similar results, despite the model complexes being formed in different ways. Discrepancies between the assigned environments of particular mRNA positions observed with these different mRNA analogs are expected, however, mainly due to differences in the chemical and stereochemical properties of the cross-linkers (s4U and ATB-modified nucleotides). Some dissimilarity in the cross-linking patterns obtained with these mRNA analogs could also be due to differences in how the mRNA analogs form their respective complexes. Interestingly, the direct interaction of the translation initiation factor (eIF) subunits with mRNA was also identified using this approach.33 These data, together with the finding that a eukaryote-specific motif in rpS26e cross-links to derivatized mRNA nucleotides in the same positions as those contacting eIF3, led to new ideas regarding how mRNA is recruited to the small ribosomal subunit during translation initiation in eukaryotes.37
5.2 Structure of the stop codon-recognition site in eRF1
Attempts to identify regions of the translation termination factor eRF1 responsible for the mRNA stop codon recognition at the ribosomal A site were conducted with a synthetic s4U-containing mRNA analog, in which the photoactivatable nucleotide was in the stop codon targeted to the A site.26,42 Since all three stop codons only contain U in the first position, these s4U-containing mRNA analogs were used to identify the eRF1 stop codon recognition site that interacts with the first nucleotide of the stop codons. First, it was shown that s4U-containing mRNA analogs are able to form cross-links with eRF1 within mammalian translation termination complexes, and that these cross-links are specific, that is, they were only observed when s4U was located in a stop codon and not in a sense codon targeted to the A site.42 Subsequently, it was found that s4U of the A site stop codon cross-links to the evolutionary conserved amino acid NIKS motif in the N-domain of eRF1 (Fig. 13).26 Thus, it was demonstrated for the first time that eRF1 participates in the termination process by making direct contacts with the mRNA stop codon.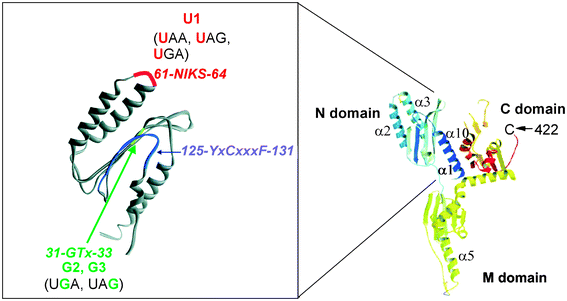 | ||
| Fig. 13 Spatial structure of the human translation termination factor eRF1 according to ref. 69 (right) and a magnified view of its N-domain (left), where conserved amino acid motifs are shown that were found to cross-link to derivatized U, A or G in the A site bound stop codon of mRNA analogs within the ribosomal termination complexes.26,35,36 | ||
To examine the position of the mRNA stop codon purines relative to eRF1 in translation termination complexes, short mRNA analogs were used that bore an ATB-derivatized guanine35 or adenine36 in a stop codon targeted to the A site. As with the s4U-containing mRNA analog described above, cross-linking to eRF1 was specific for those analogs containing the ATB-derivatized nucleotides in a stop codon, since it did not occur when control mRNA analogs were used that bore the derivatized A or G within a sense codon.35,36 Surprisingly, these experiments indicated that different evolutionary conserved motifs of the eRF1 N-domain cross-linked to modified guanines and adenines (Fig. 13). These results were then used to guide molecular modeling of eRF1 in the human ribosomal termination complex, using available structural data and software. This indicated that the peptide environments of the stop codon guanines and adenines are compatible with two alternative conformations of the eRF1 N-domain in the mammalian ribosomal termination complex. The capacity of the ribosome-bound eRF1 to adopt alternative conformations was suggested to be essential for recognition of all stop signals by a single termination factor.36 This recognition mode is principally different from that in bacteria, where two termination factors, RF1 and RF2, decode UAA/UAG and UAA/UGA, respectively. Thus, data obtained with the ATB-derivatized short mRNA analogs provided new insights into the mechanism of the stop codon decoding in the course of translation, and are in an agreement with NMR studies that also suggested rearrangement of the eRF1 N-domain during stop codon recognition.63
5.3 Structure of the HCV IRES binding site on the small ribosomal subunit
The first study of the HCV IRES binding site on the small ribosomal subunit was carried out with a HCV IRES derivative containing s4U residues scattered throughout the molecule.64 Although this approach identified a set of ribosomal proteins that contacted the IRES, it did not provide information on the interactions of these proteins with specific IRES domains and subdomains. This information was subsequently obtained using a set of HCV IRES derivatives, each bearing an ATB group in a defined location,54,55 within one of the IRES subdomains (Fig. 9). Similarly to the previous report, it was found that ribosomal proteins, but not rRNA, formed cross-links with the IRES. It should be noted here that the absence of the rRNA cross-links does not mean that the IRES does not contact rRNA, since an unfavorable orientation of the ATB group and target could impede cross-link formation. The cross-linking results54,55 were then interpreted in light of cryo-EM derived models of the mammalian small ribosomal subunit, with known locations of the ribosomal proteins.65 These analyses indicated that proteins cross-linked to the IRES derivatives are located on the back side of the small ribosomal subunit (Fig. 14), in a region homologous to the mRNA exit site in the bacterial ribosome, and that the HCV IRES binding site does not overlap well with the mRNA binding center (compare the data presented in Fig. 11 and 14). Remarkably, the positioning of the IRES on the small ribosomal subunit by cross-linking was in excellent agreement with results from the cryo-EM studies.66 Cross-linking results obtained with ATB-modified HCV IRES has provided substantial new information regarding the organization of the HCV IRES-binding site on the small ribosomal subunit, highlighting the key role played by specific ribosomal proteins in the organization of this site. It is worth mentioning that the set of cross-linked ribosomal proteins found in ref. 64 (S2e, S3e, S10e, S15e, S16e/S18e and S27e) only partially overlaps with those identified with ATB derivatives (rpS16e) (Fig. 14). However, it has become clear that the data obtained with different types of HCV IRES derivatives are not contradictory; rather, the differences observed could be explained by the proteins found in ref. 64 cross-linking to s4U residues that were located in positions not derivatized with ATB groups in ref. 54, 55 (Fig. 9). Indeed, it was recently found that rpS27e participates in the binding of the HCV IRES region, formed by the junction of the helices of subdomains IIIa, IIIb and IIIc with the central stem of domain III, and that rpS10e is involved in binding the region downstream of the nucleotide in position +12, with respect to the first nucleotide of the start AUG codon (Fig. 9).67 As for rpS2e, rpS3e and rpS15e, it is evident that cross-linking with these proteins occurred due to s4U residues substituting for U in the part of the IRES that corresponds to the coding sequence (after the AUG codon) in domain IV, because these proteins belong to the mRNA binding center as judged by cross-linking data with mRNA analogs (Fig. 11 and 12).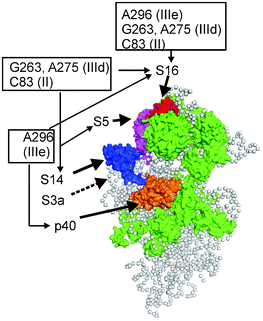 | ||
| Fig. 14 Structural model of the mammalian small ribosomal subunit obtained from cryo-electron microscopy data (adapted from ref. 65, Protein Data Bank accession number 2ZKQ, a view from the solvent side). Locations of ribosomal proteins cross-linked to HCV IRES nucleotides are shown in various colors (except for rpS3a, whose location became known from the later X-ray study,11 where this protein is designated as rpS1e, shown by the dotted line). All other proteins that have bacterial counterparts are shown in green, and proteins specific to eukaryotes are not shown. The gray balls represent the 18S rRNA. | ||
6 Conclusions and outlook
Studies using various RNA derivatives that bear photoactivatable cross-linkers at defined locations have provided an abundance of data concerning structural elements of the ribosome and of translation factors that mediate interactions with canonical mRNAs and a viral IRES. Methodologies have been developed that make it possible to identify these structural elements at the level of specific nucleotides of rRNA and of peptides in proteins. Such information could not be obtained to date by other means used for studying mRNA and IRES interactions in translation systems of higher eukaryotes. Although data obtained using different types of RNA derivatives bound at the same site are generally in a good agreement, they are not identical. Hence, using RNA derivatives that vary in the nature of their cross-linker and its location in the RNA chain provides comprehensive information on the structure of the RNA ligand binding center. Progress in developing methodologies for identification of cross-linking sites in proteins recently enabled the identification of the contacts between an RNA ligand and the ribosome at the level of a specific amino acid residue. So, by using a set of tRNA derivatives with their 3′-terminal ribose oxidized to a dialdehyde group (which is reactive towards aliphatic amino groups of proteins), it was established that the CCA-end of the P-site tRNA interacts with Lys53 of eukaryote/archaeal-specific rpL36AL.68 Evidently, the potential value of applying synthetic RNAs containing reactive nucleotides to studies of various biopolymers goes beyond the translation machinery. These derivatives could be used as tools for investigating RNA interactions occurring in the course of any cellular processes, in which RNAs are key players, particularly those involving an RNA ligand within a protein or ribonucleoprotein that is not amenable to X-ray crystallography analysis.Acknowledgements
The authors gratefully thank Mathew Coleman for the critical reading of this article. The work on this article was supported by the Presidium of Russian Academy of Sciences (Program on Molecular and Cell Biology, grant to G.K.) and by the Ministry of Education and Science of Russia, project 14.B37.21.0188.References
- E. S. Bochkareva, V. G. Budker, A. S. Girshovich, D. G. Knorre and N. M. Teplova, FEBS Lett., 1971, 19, 121–124 Search PubMed.
- V. G. Budker, A. S. Girshovich, N. I. Grineva, G. G. Karpova and D. G. Knorre, Dokl Akad Nauk SSSR (in Russian), 1973, 211, 725–728 Search PubMed.
- D. M. Graifer, G. T. Babkina, N. B. Matasova, S. N. Vladimirov, G. G. Karpova and V. V. Vlassov, Biochim. Biophys. Acta, Gene Struct. Expression, 1989, 1008, 146–156 Search PubMed.
- S. N. Vladimirov, G. T. Babkina, A. G. Venijaminova, O. I. Gimautdinova, M. A. Zenkova and G. G. Karpova, Biochim. Biophys. Acta, Gene Struct. Expression, 1990, 1048, 245–256 Search PubMed.
- K. Stade, J. Rinke-Appel and R. Brimacombe, Nucleic Acids Res., 17, 9889–9908 Search PubMed.
- J. Rinke-Appel, N. Junke, R. Brimacombe, S. Dokudovskaya, O. Dontsova and A. Bogdanov, Nucleic Acids Res., 1993, 21, 2853–2859 Search PubMed.
- R. Bhangu and P. L. Wollenzien, Biochemistry, 1992, 31, 5937–5944 Search PubMed.
- R. Bhangu, D. Juzumiene and P. Wollenzien, Biochemistry, 1994, 33, 3063–3070 Search PubMed.
- V. Ramakrishnan, Cell, 2002, 108, 557–72 CrossRef CAS.
- T. A. Steitz, Nat. Rev. Mol. Cell Biol., 2008, 9, 242–253 Search PubMed.
- J. Rabl, M. Leibundgut, S. F. Ataide, A. Haag and N. Ban, Science, 2011, 331, 730–736 Search PubMed.
- S. Klinge, F. Voigts-Hoffmann, M. Leibundgut, S. Arpagaus and N. Ban, Science, 2011, 334, 941–948 Search PubMed.
- A. Ben-Shem, N. Garreau de Loubresse, S. Melnikov, L. Jenner, G. Yusupova G. and M. Yusupov, Science, 2011, 334, 1524–1529 Search PubMed.
- D. Beyer, E. Skripkin, J. Wadzack and K. H. Nierhaus, J. Biol. Chem., 1994, 269, 30713–30717 Search PubMed.
- A. Hüttenhofer and H. F. Noller, EMBO J., 1994, 13, 3892–3901 Search PubMed.
- S. Watanabe, J. Mol. Biol., 1972, 67, 443–457 Search PubMed.
- R. Lill, J. M. Robertson and W. Wintermeyer, Biochemistry, 1986, 25, 3245–3255 Search PubMed.
- D. M. Graifer, S. Yu. Nekhai, D. A. Mundus, O. S. Fedorova and G. G. Karpova, Biochim. Biophys. Acta, Gene Struct. Expression, 1992, 1171, 56–64 Search PubMed.
- D. M. Graifer, M. A. Zenkova, A. A. Malygin, S. V. Mamaev, D. A. Mundus and G. G. Karpova, J. Mol. Biol., 1990, 214, 121–128 Search PubMed.
- A. A. Malygin, D. M. Graifer, K. N. Bulygin, M. A. Zenkova, V. I. Yamkovoy, J. Stahl and G. G. Karpova, Eur. J. Biochem., 1994, 226, 715–723 Search PubMed.
- N. Demeshkina, M. Repkova, A. Ven'yaminova, D. Graifer and G. Karpova, RNA, 2000, 6, 1727–1736 Search PubMed.
- D. Graifer, M. Molotkov, V. Styazhkina, N. Demeshkina, K. Bulygin, A. Eremina, A. Ivanov, E. Laletina, A. Ven'yaminova and G. Karpova, Nucleic Acids Res., 2004, 32, 3282–3293 Search PubMed.
- M. Rodnina and W. Wintermeyer, Curr. Opin. Cell Biol., 2009, 21, 435–443 Search PubMed.
- C. U. T. Hellen and P. Sarnow, Genes Dev., 2001, 15, 1593–1612 CrossRef CAS.
- M. V. Molotkov, D. M. Graifer, E. A. Popugaeva, K. N. Bulygin, M. I. Meschaninova, A. G. Ven'yaminova and G. G. Karpova, RNA Biol., 2006, 3, 122–129 Search PubMed.
- L. Chavatte, A. Seit-Nebi, V. Dubovaya and A. Favre, EMBO J., 2002, 21, 5302–5311 Search PubMed.
- K. Bulygin, L. Chavatte, L. Frolova, G. Karpova and A. Favre, Biochemistry, 2005, 44, 2153–2162 Search PubMed.
- K. N. Bulygin, D. M. Graifer, M. N. Repkova, I. A. Smolenskaya, A. G. Veniyaminova and G. G. Karpova, RNA, 1997, 3, 1480–1485 Search PubMed.
- N. Demeshkina, E. Laletina, M. Meschaninova, A. Ven'yaminova, D. Graifer and G. Karpova, Biochim. Biophys. Acta, Gene Struct. Expression, 2003, 1627, 39–46 Search PubMed.
- D. M. Graifer, D. I. Juzumiene, P. Wollenzien and G. Karpova, Biochemistry, 1994, 33, 3878–3884 Search PubMed.
- N. A. Demeshkina, E. S. Laletina, M. I. Meschaninova, M. N. Repkova, A. G. Ven'yaminova, D. M. Graifer and G. G. Karpova, Mol. Biol., 2003, 37, 132–139 Search PubMed.
- J. J. Madjar, M. Arpin, M. Buisson and J.P. Reboud, Mol. Gen. Genet., 1979, 171, 121–134 Search PubMed.
- A. V. Pisarev, V. G. Kolupaeva, M. M. Yusupov, C. U. T. Hellen and T. V. Pestova, EMBO J., 2008, 27, 1609–1621 Search PubMed.
- J. Khairulina, D. Graifer, K. Bulygin, A. Ven'yaminova, L. Frolova and G. Karpova, Biochimie, 2010, 92, 820–825 Search PubMed.
- K. N. Bulygin, Yu. S. Khairulina, P. M. Kolosov, A. G. Ven'yaminova, D. M. Graifer, Yu. N. Vorobjev, L. Yu. Frolova, L. L. Kisselev and G. G. Karpova, RNA, 2010, 16, 1902–1914 Search PubMed.
- K. N. Bulygin, Yu. S. Khairulina, P. M. Kolosov, A. G. Ven'yaminova, D. M. Graifer, Yu. N. Vorobjev, L. Yu. Frolova and G. G. Karpova, Nucleic Acids Res., 2011, 39, 7134–7146 Search PubMed.
- D. Sharifulin, Yu. Khairulina, A. Ivanov, M. Meschaninova, A. Ven'yaminova, D. Graifer and G. Karpova, Nucleic Acids Res., 2012, 40, 3056–3065 Search PubMed.
- J. Stahl and N. D. Kobetz, Mol. Biol. Rep., 1984, 9, 219–222 Search PubMed.
- J. Stahl and G. G. Karpova, Biomed. Biochim. Acta, 1985, 44, 1057–1064 Search PubMed.
- K. N. Bulygin, N. B. Matasova, D. M. Graifer, A. G. Veniyaminova, V. I. Yamkovoy, J. Stahl and G. G. Karpova, Biochim. Biophys. Acta, Gene Struct. Expression, 1997, 1351, 325–332 Search PubMed.
- D. M. Graifer, D. I. Juzumiene, G. G. Karpova and P. Wollenzien, Biochemistry, 1994, 33, 6201–6206 Search PubMed.
- L. Chavatte, L. Frolova, L. Kisselev and A. Favre, Eur. J. Biochem., 2001, 268, 2896–2904 Search PubMed.
- K. Bulygin, S. Baouz-Drahy, D. Graifer, A. Favre and G. Karpova, Biochim. Biophys. Acta, Gene Regul. Mech., 2009, 1789, 167–174 Search PubMed.
- A. V. Pisarev, V. G. Kolupaeva, V. P. Pisareva, W. C. Merrick, C. U. T. Hellen and T. V. Pestova, Genes Dev., 2006, 20, 624–636 Search PubMed.
- R. K. Gaur, in Methods in Molecular Biology, ed. Ren-Jang Lin, Humana Press Inc., Totowa, NJ, 2008, 488 Search PubMed.
- M. Hafner, M. Landthaler and L. Burger, et al. , Cell, 2010, 141, 129–141 CrossRef CAS.
- A. S. Levina, D. R. Tabatadze, L. M. Khalimskaya, N. A. Prihodko, G. V. Shishkin, L. A. Alexandrova and V. F. Zarytova, Bioconjugate Chem., 1993, 4, 319–325 Search PubMed.
- A. S. Levina, D. R. Tabatadze, M. I. Dobrikov, G. V. Shishkin, L. M. Khalimskaya and V. F. Zarytova, Antisense Nucleic Acid Drug Dev., 1996, 6, 119–126 Search PubMed.
- E. L. Chernolovskaya, P. P. Cherepanov, A. V. Gorozhankin, M. I. Dobrikov, V. V. Vlassov and N. D. Kobetz, Bioorgan. Khim. (in Russian), 1993, 19, 889–896 Search PubMed.
- R. P. Haugland in Molecular Probes Handbook: A Guide to Fluorescent Probes and Labeling Technologies, ed. I. Johnson and M. T. Z. Spence, 11th edn, 2010, section 5 Search PubMed.
- M. N. Repkova, T. M. Ivanova, N. I. Komarova, M. I. Meschaninova, M. A. Kuznetsova and A. G. Ven'yaminova, Russian J. Bioorgan. Chem., 1999, 25, 612–622 Search PubMed.
- A. M. Belikova, V. F. Zarytova and N. I. Grineva, Tetrahedron Lett., 1967, 37, 3557–3562 Search PubMed.
- K. Bulygin, A. Malygin, G. Karpova and P. Westermann, Eur. J. Biochem., 1998, 251, 175–180 Search PubMed.
- E. Laletina, D. Graifer, A. Malygin, A. Ivanov, I. Shatsky and G. Karpova, Nucleic Acids Res., 2006, 34, 2027–2036 Search PubMed.
- E. Babaylova, D. Graifer, A. Malygin, J. Stahl, I. Shatsky and G. Karpova, Nucleic Acids Res., 2009, 37, 1141–1151 Search PubMed.
- R. J. Jackson, C. U. T. Hellen and T. V. Pestova, Nat. Rev. Mol. Cell Biol., 2010, 11, 113–127 Search PubMed.
- A. A. Malygin, D. M. Graifer, E. S. Laletina, I. N. Shatsky and G. G. Karpova, Molekul. Biol. (in Russian), 2003, 37, 873–879 Search PubMed.
- N. A. Demeshkina, M. N. Repkova, A. G. Ven'yaminova, D. M. Graifer and G. G. Karpova, Mol. Biol., 2004, 38, 421–426 Search PubMed.
- M. Molotkov, D. Graifer, N. Demeshkina, M. Repkova, A. Ven'yaminova and G. Karpova, RNA Biol., 2005, 2, 63–69 Search PubMed.
- A. R. Morgan, R. D. Wells and H. G. Khorana, J. Mol. Biol., 1967, 26, 477–497 Search PubMed.
- A. P. Potapov, F. J. Triana-Alonso and K. H. Nierhaus, J. Biol. Chem., 1995, 270, 17680–17684 Search PubMed.
- D. Graifer, A. Zhigailov, A. Ven'yaminova, A. Malygin, B. Iskakov and G. Karpova, FEBS Lett., 2012, 586, 3731–3736 Search PubMed.
- L. E. Wong, Y. Li, S. Pillay, L. Frolova and K. Pervushin, Nucleic Acids Res., 2012, 40, 5751–5765 Search PubMed.
- G. A. Otto, P. J. Lukavsky, A. M. Lancaster, P. Sarnow and J. D. Puglisi, RNA, 2002, 8, 913–923 Search PubMed.
- P. Chandramouli, M. Topf, J.-F. Menetret, N. Eswar, J. J. Cannone, R. R. Gutell, A. Sali and C. W. Akey, Structure, 2008, 16, 535–548 Search PubMed.
- C. M. T. Spahn, J. S. Kieft, R. A. Grassucci, P. A. Penczek, K. Zhou, J. A. Doudna and J. Frank, Science, 2001, 291, 1959–1962 Search PubMed.
- A. A. Malygin, I. N. Shatsky and G. G. Karpova, Biochemistry (Moscow), 2012 Search PubMed , in press.
- C. Hountondji, K. Bulygin, A. Woisard, P. Tuffery, J.-B. Créchet, M. Pech, K. H. Nierhaus, G. Karpova and S. Baouz, ChemBioChem, 2012, 13, 1791–1797 Search PubMed.
- H. Song, P. Mugnier, H. M. Webb, D. R. Evans, M. F. Tuite, B. A. Hemmings and D. Barford, Cell, 2000, 100, 311–321 Search PubMed.
Footnote |
| † This review article is dedicated to the memory of Har Gobind Khorana, a scientist whose contribution to understanding the functions of nucleic acids is invaluable. |
| This journal is © The Royal Society of Chemistry 2013 |