Preparation of ceramic nanoparticles via cellulose-assisted glycine nitrate process: a review
Hansu
Birol
a,
Carlos
Renato Rambo
*b,
Marcela
Guiotoku
c and
Dachamir
Hotza
b
aCentro de Inovações CSEM Brasil, Pça. Carlos Chagas, 49, 30170-020, Belo Horizonte, MG, Brazil
bGroup of Ceramic and Glass Materials (CERMAT), Federal University of Santa Catarina (UFSC), 88040-900 Florianópolis, SC, Brazil
cEmpresa Brasileira de Pesquisa Agropecuária (EMPRAPA), Centro Nacional de Pequisa de Florestas, 83411-000 Colombo, PR, Brazil
First published on 15th November 2012
Abstract
Ceramics exhibit several interesting properties, which make them the material of choice for a broad range of applications. Their physical and chemical properties are significantly improved by sub-micrometer ceramic powders with narrow particle size distribution, high chemical purity and crystallinity and no/weak agglomeration. However, powders with such superior characteristics are mostly synthesized by complex and costly processes, which are usually not ideal for production at an industrial scale. Therefore, developing simple, efficient, inexpensive and environmentally-benign processes for the preparation of high quality ceramic powders is of great interest both for the research community and industry. In this regard, this article reviews the research efforts in the preparation of ceramic nanopowders from cotton-cellulose, which is used as a sacrificial bio-template, in a glycine–nitrate process. Low processing temperatures, self-propagating nature of the reactions, high reaction rates, no necessity for extra energy and special apparatus are the characteristics of this process yielding extremely fine, homogenous and non-agglomerated powders.
 Hansu Birol | Dr Hansu Birol completed his Ph.D. in fabrication of sensors and micro-fluidic structures using low temperature co-fired ceramic (LTCC) and thick film technologies at EPFL, Switzerland. Following his doctorate, he joined the Piezotechnology Division of EPCOS OHG (pka. Siemens-Matsushita Components AG) as process development engineer to optimize the yield and product quality of the piezoelectric stacks used for fuel injection in diesel engines. He is currently the leader of the LTCC Technology Group at CSEM Brasil in Belo Horizonte, where his current area of interest is the development of advanced microsystems for high reliability applications in Brazilian Industry. |
 Carlos Renato Rambo | Prof. Dr Carlos Renato Rambo graduated in Physics from the University of São Paulo, Brazil (1994) and as a doctor in Materials Science and Engineering (University of São Paulo, Brazil, 2001), and then worked as a post-doc at the University of Erlangen, Germany (2002–2005). He has worked at the Electrical Engineering Department of Federal University of Santa Catarina, Brazil as Adjunct Professor since 2010. He has been working on the development of nanostructured ceramics and composites for energy and electronic applications and has authored more than 60 publications. |
 Marcela Guiotoku | Dr Marcela Guiotoku received a Bachelor in Chemistry (1999) and Ph.D. in Materials Science and Engineering (2008) from the Federal University of Santa Catarina. She has been a chemical analyst at the Brazilian Agricultural Research Corporation since 2004. She has a post-doctoral fellowship at the Federal University of Santa Catarina with a research focus on carbon-based nanostructured materials for energy storage. Her research interests are in the development of hydrothermal processing of materials and carbon based nanomaterials for energy applications. |
 Dachamir Hotza | Prof. Dr Dachamir Hotza: Chemical Engineer, M.Sc. in Mechanical Engineering, Ph.D. in Materials Engineering from TUHH, Germany, post-docs at the University of Erlangen, Germany; OMTRI, Japan; and University of Queensland, Australia. Currently Associate Professor at the Department of Chemical Engineering of the Federal University of Santa Catarina - UFSC, Brazil. Main research interests: ceramic processing, nanotechnology, rheology. He has published more than 170 papers. |
1. Introduction
In the last twenty years, notably in the last decade, several research papers have focused on the synthesis of ceramic nanoparticles/nanopowders (NP) with superior characteristics. The great majority of this work reported on the synthesis of high quality NP by complex and costly processes, which are usually not useful for production at an industrial scale. Therefore, developing simple, efficient, inexpensive and environmentally-benign processes for the preparation of high quality ceramic NP is of great interest both for the research community and industry. This is a consequence of the dramatic alterations in the physical and chemical properties of NP,1–3 through which fascinating applications in electronics,4,5 photonics,6,7 catalysis8,9 and biomedicine10,11 have been realized. The unusual and attractive characteristics of NP are mainly due to their significantly-increased specific surface area to volume ratio, making NP more reactive than coarser particles.1,2 Particle size is a critical parameter in materials processing and fine crystallites from NP, in general, improve the mechanical, thermal, electrical and magnetic properties of ceramics, sintered metals and composites.1 Moreover, they reduce the sintering temperature of the powders and the shrinkage-related defects due to the increased reactivity and higher green-density, respectively.1,12–14There are two main approaches to prepare NP: top-down and bottom-up (Fig. 1). In the former approach, NP are prepared by reducing coarser structures in size by mechanical or chemical processes, whereas in the latter, NP are constructed by arrangements at the atomic-level via chemical or biological processes.2,15–17 Due to the improved homogeneity, superior control at the atomic-level and reduced number of defects, the bottom-up approach is more favourable for preparing high quality NP.2,15,16 However, most of the NP synthesis methods belonging to the bottom-up approach are costly and time consuming due to the complexity of the processes.12,18–20
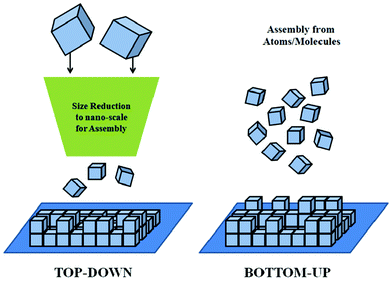 | ||
| Fig. 1 Top-down and bottom-up approaches in nanotechnology. | ||
Among these methods, the glycine–nitrate (GN) process has several distinctive features, which optimize the preparation of high-quality NP.20 The process is a sub-class of combustion synthesis techniques and, as will be detailed in the next chapters, is efficient, simple, self-sustaining, fast and feasible, requiring no special apparatus.18
Considering the aforementioned requirements and motives in NP synthesis, this article aims to review the literature on ceramic NP preparation using environmentally-friendly cellulose and the GN process. In the next section, ceramic NP processing routes will be briefly discussed, whereas the focus will be on combustion synthesis (CS) in section three. Finally, the cotton cellulose-assisted GN process for the preparation of ceramic NP will be reviewed.
2. History of ceramic NP synthesis routes
One of the most critical issues other than those discussed in the previous paragraph is the selection of an environmentally-benign process in NP synthesis.2,16 In this respect, biological templating attracts growing interest since the biotemplate (BT) is a natural substance that is abundant in nature and therefore renewable.21 Moreover, biological substances exhibit optimized characteristics such as high strength to density ratios, high stiffness and elasticity and high damage tolerance.21 However, what makes the BT exceptional in NP synthesis is its complex and unique hierarchic structure defined by elementary units and pores varying in size and distribution (2–100 nm).21–23 Bacteria,24,25 pollen,26 cornstarch,27 living cells28 and wood29 are some of the BTs used to develop NP. In spite of the potential and demonstrated success in the synthesis of NP,4–11,30–36 biological templating still has challenges to overcome such as a limited control of the size, shape and orientation of the material and incomplete replication of structural hierarchy in higher-order assemblies and over extended dimensions.37–40 Bulk ceramic materials with nanograins exhibit enhanced physical and chemical properties compared to traditional ceramics with grains on the micrometer scale.1,18,41–48 One of the most fundamental prerequisites in preparation of bulk nanoceramics is the synthesis of highly-pure, agglomerate and contaminant-free ceramic NP with narrow size distribution.1,43,45,49,50 Fine ceramic particles with increased specific surface area reduce the sintering temperatures and times, which in turn avoids the extremely undesirable problems related to high temperature sintering such as grain growth, volatilization of the material, and processing costs.1,18,19 Therefore, the preparation of ceramic NP is one of the most critical issues to be addressed to make high quality ceramic nanomaterials and products.Powder synthesis methods can be grouped in three main categories: i) solid phase reactant, ii) liquid phase reactant and iii) gas phase reactant. The most conventional technique for the preparation of ceramic powders is the solid-state reaction, which is widely employed in industry due to its simplicity and relatively low processing cost.14,18,44 In this technique, the initial powders, the sizes of which are in the order of a few microns, are calcined following mixing and milling to react chemically and form a homogenous phase. The calcination temperatures are usually high (>1100 °C) and carried out over long periods of time to facilitate diffusion during the solid-state reaction.18 The resulting powder is often coarse and agglomerated, exhibiting limited purity and homogeneity.44,51 Therefore, one of the biggest challenges to prepare ceramic NP relies on facilitating homogenous mixing of the constituent particles, which can be best achieved at the atomic level.18 However, this requires precise control on the process parameters making the whole process complicated, time-consuming and costly.43,44,51
A complete classification and description of ceramic powder processing methods is not within the interests of this review and the interested reader is advised to go through ref. 18–20 for a broad description of ceramic powder processing techniques. However, several critical factors such as the cost of production (initial materials, machinery, equipment, time and efficiency), simplicity and flexibility of the processes employed and the number and nature of the intermediate and post processing steps must be carefully considered in evaluating the efficiency of a technique to produce ceramic powder for a specific application. For particle sizes below 100 nm, high purity and crystallinity, fine crystalline size and a single phase are desired powder characteristics, which usually impose strict restrictions on the process parameters and thus increase the complexity and costs of the process. Wet-chemical syntheses such as sol–gel, although they facilitate mixing of the initial materials at the molecular level, are usually costly, complicated and time consuming.12,41 Therefore, new techniques, which facilitate the production of high quality powders at high yields, are constantly being researched.
3. Combustion synthesis
Combustion synthesis (CS) is a low-cost and energy-efficient method with a high production rate for the preparation of a broad range of materials such as ceramics, intermetallics and composites.52–57 The powder/ceramic synthesized by CS is extremely fine and of high chemical homogeneity and purity due to the distinctive features of the process, such as:• The mixing of constituents at the molecular level,
• Self-sustained, high combustion temperatures resulting in the direct formation of phase composition,
• Fast reaction rates (short exposure times to high temperatures, eliminating particle size growth, volatilization),
• The stabilization of metastable phases.14,57–61
Pigments and catalysts, solid oxide fuel cells (SOFCs) electrodes, batteries, oxygen sensors and novel dielectric and piezoelectric materials exhibit significantly-improved properties when prepared by this technique.41,56–62
In a typical CS process, the desired material is synthesized by igniting the mixed oxidizer, which is the raw material for the final powder, and the fuel. Upon ignition of the mixture at relatively low temperatures, intense thermal energy is released due to the highly exothermic nature of the chemical reactions, which lead to temperatures in the 2000–5000 K range. The reactants, however, are exposed to such temperatures at very short time intervals (seconds to a few minutes) so that the previously-mentioned desired powder characteristics are obtained.57,63,64 Heat generation is accompanied by the production of gas, which actually prevents the overheating of the particles by dissipating the heat.47
Before elucidating the details of the GN process, CS will be classified according to the type and physical state of the reactants and the reaction medium.57 Depending on this classification, CS is referred to as self-propagating, high temperature synthesis (SHS), volume combustion synthesis (VCS) and solution combustion synthesis (SCS), which can be further classified as gel-combustion, emulsion combustion, aqueous combustion synthesis and the glycine–nitrate process in literature57,71 (Table 1).
| Solid state combustion | Solution sombustion synthesis (SCS) |
|---|---|
| Self-propagating high-temperature synthesis (SHS) | Gel/emulsion/aqueous combustion synthesis |
| Bulk/volume combustion synthesis (VCS) | Glycine-nitrate process (GN) |
3.1. Solid state combustion
In solid state combustion, the reactants, intermediates and products are all in the solid state57 and the material is synthesized by igniting the reactants, which are initially prepared as pellets. Aluminides, borides, carbides, nitrides and oxides are some of the materials prepared by solid state combustion.60,65There are several critical and interdependent parameters to consider in SHS of NP such as the amount, composition, density, particle size and purity of the reactants, pressure of the gaseous phase and the type and amount of the fuel.63 Moreover, the critical reaction temperature (>1500 °C) and burning rate (0.6–2.5 mm s−1) are other conditions which need to be fulfilled in order to maintain the self-sustaining nature of the SHS reactions,64 which are, for instance, impeded by coarse reactant particle size and density.64
In order to understand the chemistry of SHS reactions, it is essential to understand the nature of redox reactions since SHS reactions are of a redox type.64 In a reduction reaction, the electrons are gained from donors, which are also known as reductants or reducing agents. Reductants such as Mg, Fe and Zn can reduce other species whilst themselves being oxidized.63 Therefore, the oxidizer or the oxidant in a SHS reaction is an electron acceptor, whereas the fuel is an electron donor.64
However, the chemistry of the reagents is not the only variable to consider in SHS; the decisive factor in the performance of the technique depends on the rates of heat evolution and heat transfer in the system,63 which are influenced by the velocity, form and structure of the combustion wave (CW). There are critical phases or subzones of a typical CW in SHS, each of which involves a series of processes.63,64 Initially, intense heat exchange occurs although no chemical reactions take place. This is followed by the main heat release phase, which is accompanied by the main reactions. These two subzones significantly influence the heat release and heat transfer of the wave front. Elevated heat losses in the initial stage may also lead to combustion quenching, which delays or annuls the proceeding subzones. After burning of subzones is a consequence of the unaltered heat propagation. Critical processes such as crystallization and phase transformation occur in the structure formation subzone determining the most critical structural and chemical properties of the products. Finally the reactions are completed and the product loses heat in the cool down subzone.
Although no post processes are required to prepare NP by SHS, the final powder may lack high specific surface area in the case of coarse initial powders, which are exposed to temperatures over 2000 K, are used.53 In such cases, SHS is usually followed by additional processes such as intensive milling67 or mechanical activation.68 Alternatively, SHS may be accompanied by a process known as chemical dispersion, in which the powder prepared by SHS is initially etched in dilute acid to remove impurities and is then milled to prepare fine NP.69 Carbon combustion synthesis is a derivative of SHS, which is based on the utilization of carbon instead of metal as the fuel. The high release rate of CO2 during the reaction leads to the formation of porous powders with sub-100 nm size.70
Fig. 2 shows La0.8Sr0.2CrO3 (LSC) perovskite nanoparticles produced by VCS (Fig. 2a) and SHS (Fig. 2b) modes after calcination.72
 | ||
| Fig. 2 SEM micrographs of calcined LSC powder produced in a) VCS and b) SHS combustion synthesis modes. Reprinted from S. Mukasyan, C. Costello, K. P. Sherlock, D. Lafarga, A. Varma, Perovskite membranes by aqueous combustion synthesis: synthesis and properties, Sep. Purif. Technol., 2001, 25, 117–126, Copyright (2001), with permission from Elsevier. | ||
The LSC nanoparticles are uniform in size with a well defined particle morphology of each particle. The powder synthesized in the SHS mode is more homogeneous and less agglomerated than the powder produced in the VCS mode.
3.2. Solution combustion synthesis (SCS)
SCS is an efficient method for the preparation of extremely fine, homogenous and crystalline oxide ceramics without the need for intermediate thermal treatments.74,75 The first step in the synthesis of NP by SCS is the preparation of the precursor, which is accomplished by mixing the oxidizer, which is typically a metal nitrate, with an organic fuel homogenously. Oxidizers are selected from metal nitrates since nitrates are water soluble and efficient oxidizers, whereas the fuel is ideally a substance containing carboxylic and amine groups such as urea and hydrazides. These fuels are rich in C and H and thus they facilitate the liberation of heat by the formation of CO2 and H2O during combustion. Moreover, they form complexes with metal ions efficiently so that the level of mixing is improved, avoiding segregation.53,57,60,74 Other fuel options are citric acid, poly vinyl alcohol (PVA) and glycine. The formation of a precursor that is either a viscous liquid or a gel is essential to mix the initial materials at the molecular level and to prevent random redox reactions between the oxidizer and the fuel.57 The fuel, on the other hand, is expected to initiate combustion with an oxidizer at low ignition temperatures.57,76 The type of fuel, the amount of fuel, the pH of the starting solution and the F/O ratio are some of the critical parameters influencing the characteristics of the precursor and hence the synthesized powder.75–77The second step in NP synthesis by SCS is auto or self ignition, which stems from the highly exothermic nature of the reactions taking place, which occur between the GN reactants. The heat generated during combustion facilitates the crystallization and formation of the desired phase, whereas an excess of it might lead to crystal size growth, hard agglomerates and a higher specific surface area.47 The increase in temperature is limited by simultaneously occurring gas evolution processes during combustion. Therefore, the powder characteristics are primarily governed by these two processes which are closely related to the previously mentioned parameters such as type and amount of fuel and the F/O ratio.47,75–79
Luminescent materials and catalysts are mostly prepared by SCS53,57 due to the superior characteristics of the NP. A detailed list of oxide materials prepared by SCS and their applications are listed in a review by Patil.57
4. GN process
4.1 Cotton cellulose-assisted GN process
The GN process is one of the SCS routes, which was introduced by Chick et al.80 in 1990 as an efficient method for the preparation of extremely fine mixed-metal oxides with a single phase.80–83Fig. 3 shows a bright-field transmission electron micrograph of La0.76Sr0.24CrO3 powder produced by glycine–nitrate combustion synthesis, displaying nanoparticles with uniform diameters of approximately 20 nm.80 | ||
| Fig. 3 Bright-field transmission electron micrograph of La0.76Sr0.24CrO3 particles prepared by the GN process. Reprinted from L. A. Chick, L. R. Pederson, G. D. Maupin, J. L. Bates, L. E. Thomas and G. J. Exarhos, Glycine-Nitrate combustion synthesis of oxide ceramic powders, Mater. Lett., 1990, 10, 6–12, Copyright (1990), with permission from Elsevier. | ||
Compared to other methods such as a solid-state reaction, a co-precipitation reaction, hydrothermal synthesis and sol–gel, GN process is inexpensive and efficient yielding desired powder characteristics.84–90 Moreover, GN yields LSC nanoparticles with smaller sizes and narrower size distribution when compared to SHS or VHS (see Fig. 2). The process is simple and realized by the preparation and decomposition of a precursor solution at a critical reaction temperature. The precursor solution is prepared by dissolving the metal nitrates, which are the raw materials for the final powder, in water and by mixing the solution with glycine. The mixture is gradually heated to remove the water until self-ignition occurs.83,91 The extremely rapid propagation of the combustion flame is caused by the strongly exothermic reaction between the oxidizer and fuel and yields ash, which is actually the desired product. The reasons for selecting glycine as the fuel rather than other alternatives are varied. First of all, it has a higher heat of combustion than other alternatives such as carbohydrazide, citric acid and urea.83 Moreover, it is an efficient complexing agent, which facilitates the intimate mixing of the powders, avoiding segregation.83,85,92 Another attractive feature is its cost, which is the lowest among amino acids.85
The GN process is capable of preparing a variety of powders from single metal oxides to multi-metal complex oxides.93–99 Zhang et al.97 reported the synthesis of a nanostructured LiMn2O4 spinel using a glycine–nitrate combustion process followed by short-time calcination. In this article it was shown that the glycine–nitrate process is an attractive method for the fabrication of cost-effective nanostructured electrodes for lithium secondary batteries. Fig. 4 shows TEM (Fig. 4a) and scanning electron microscopy (SEM) (Fig. 4b) images of the as-synthesized and calcined LiMn2O4 nanopowders.97
 | ||
| Fig. 4 (a) TEM image of as-synthesized LiMn2O4 powder; (b) SEM image of calcined LiMn2O4 powder. Reprinted from 171, Y. Zhang, H. C. Shin, J. Dong, M. Liu, Nanostructured LiMn2O4 prepared by a glycine–nitrate process for Lithium-ion batteries, Solid State Ionics, 2004, 171, 25–31, Copyright (2004), with permission from Elsevier. | ||
The particle size of the as-synthesized powders (Fig. 4a) is approximately 10 nm with visible agglomerations. Fig. 4b displays the typical morphology of calcined LiMn2O4 powder, which exhibits a macroporous structure with uniformly distributed interconnected pores of about 500 nm in diameter. This high porosity is attributed to the gas evolution (H2O, N2 and CO2) during the air combustion synthesis, represented by the following reaction (eqn (1)):
| 2Mn(CH3COO)2·4H2O + 4HNO3 + LiNO3 + 2NH2CH2COOH → LiMn2O4 + 17H2O↑ + (7/2)N2↑ + 12CO2↑ | (1) |
The high reactivity of the powders, which makes them an ideal choice for catalyzing electrochemical reactions in SOFCs as well as in other fields such as gas sensors, oxygen separation membranes,100–103 is easily obtained by the GN process. In spite of the attractive features of the process and the superior characteristics of the powders prepared, there are certain concerns regarding the environmental impact of the GN process.87,91,104 Although the process is referred to as being environmentally benign,80,92 combustion processes might well end up with uncompleted reactions because the reactions occur at extremely fast rates, which imposes restrictions on the kinetics of the transformation process.88 Actually in a relatively recent work by Pine,91 in which the emission levels of polluting products in a GN process were measured by an exhaust gas analyzer for one F/O ratio, a high emission level of NOx and CO is clearly demonstrated. Another problem is the production of a large amount of gases blowing away the NP and thus reducing the yield and becoming a threat to environment.87,104
Although these disadvantages do not change the fact that the NP prepared by the GN process are of ideal uniformity, purity and perfect stoichiometry,91 constant efforts are being made to improve the process. In this regard, a cellulose-assisted GN process is one of the very recent methods used in the preparation of high quality NP, tending to resolve the aforementioned difficulties and problems encountered by the traditional GN process.104 In this method, cotton cellulose is used as a micro-bioreactor and integrated into the traditional process.104
4.2. The structure and chemistry of cellulose for the GN process
Cellulose is the most abundant, renewable and oxygen-rich polysaccharide, which has been used recently as a sacrificial BT for the synthesis of different materials.104–106 It exhibits very attractive structural and chemical properties, which makes it an ideal template for the GN process.First of all it has a distinctive fibre morphology that is composed of microfibrils, which are 10–30 nm in width and have a surface area of 30–55 m2 g−1.107–111 These fibrils are three-dimensionally interconnected and they have nanopores on them varying in size from a few nm to a few hundred nm.51,104,105 These pores are efficient in binding metal salts easily and firmly by an electrostatic interaction that occurs between the electron rich oxygen atoms of the polar hydroxyl (R–OH) and ether groups (R–O–R) of cellulose and electropositive metal cations following the reduction of the metal salt.51,105 This interaction, in addition to the random arrangement of cellulose molecular chains, prevents the segregation of the metals and crystallization during the evaporation of water and hence increases the level of mixing and the homogeneity.51,105,112–114 Consequently, the firing temperature of the mixture is reduced as a result of the lower diffusion barrier due to enhanced mixing.51 Binding the metals is not the only feature of the nanopores on the three-dimensionally connected fibrils. The structure itself functions as an escape-channel for the gas products, thereby avoiding the spreading out of fine powders during combustion.104
In addition to the advantages brought by its environmentally friendly nature, cellulose is not soluble in water and thus it remains as a solid reagent during the evaporation process.104 Moreover, it is fully burned out during the combustion process, yielding even finer particles than those prepared by the traditional GN process.104
4.3. Cellulose-assisted GN process
A cellulose-assisted GN route is a new approach for preparing ceramic NP. The concept was first introduced by Shao et al. in 2008 and most of the work centres on his group's efforts in developing electrodes and electrolytes of SOFCs by using this process,41,104,115–117 which will be reviewed in this section. For convenience ceramic NP prepared by this method are shown in Table 2 prior to a detailed description of the powder synthesis and its characteristics. The application-related properties of synthesized powders such as electrical and electrochemical characteristics and tests will not be discussed. Comparison of the cellulose-assisted GN process with the traditional GN process as well as other routes such as a solid state reaction can be found in ref. 118–120.The first step in the synthesis of ceramic NP by the cellulose-assisted GN process is the preparation of activated cellulose, by immersing dewaxed cotton into nitric acid (HNO3) followed by washing to remove excess HNO3. The template is then dried at 100 °C to finally form the activated cellulose fibre. In the second step, the precursor is prepared by mixing the metal nitrates, which are previously blended at a stoichiometric ratio to yield the desired material composition, with glycine at a selected F/O ratio. This is further followed by heating the mixture to 80–100 °C to form the sol, which is then poured over the activated cellulose prepared in the first step and kept at this temperature for several h. In the third step, metal-deposited cotton fibre is finally exposed to a temperature of ca. 250 °C to trigger auto-combustion, which occurs upon the reaction of glycine with nitrates, releasing large amounts of heat. The combustion takes place within a few seconds to minutes and the temperature reached, which is a result of the cellulose amount used and F/O ratio selected, can be calculated using thermodynamic calculations. A very attractive function of cellulose in the process as cited in ref. 104 is its blocking effect, which prevents the contact of oxide particles and hence suppresses coarsening and agglomeration, yielding extremely fine particles. This is facilitated by the high amount of gas formation as well as the full burnout of cellulose fibre at a temperature higher than the combustion or the GN reaction temperature, which consequently allows the fibre to maintain its morphology and thus the reacted particles be trapped in the hierarchical cellulose structure with no/loose contact.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio of the total sum of metal ions to glycine. The mixture is heated at 80 °C to form the viscous sol, which is impregnated slowly into the activated cellulose. Soaking of the activated cellulose fibre in GN solution is followed by drying at 80 °C for several h to form the precursor, which is put in an electric oven at 250 °C to initiate self-combustion. The resulting powder, which is around 20 nm in size, is reported to be weakly agglomerated and to resemble the morphology of the cellulose fibre.
:2 ratio of the total sum of metal ions to glycine. The mixture is heated at 80 °C to form the viscous sol, which is impregnated slowly into the activated cellulose. Soaking of the activated cellulose fibre in GN solution is followed by drying at 80 °C for several h to form the precursor, which is put in an electric oven at 250 °C to initiate self-combustion. The resulting powder, which is around 20 nm in size, is reported to be weakly agglomerated and to resemble the morphology of the cellulose fibre.
Compared to the traditional GN process, the cellulose-assisted GN process is shown to be much milder and without smog or ash at a higher cellulose to GN-precursor weight ratio. Ref. 104 demonstrates the reactions taking place during heating the activated-cellulose, the conventional GN precursor and the cellulose-GN precursor measured by differential thermal analysis (DTA) and thermo gravimetric analysis (TGA), which are summarized in Table 3. An important conclusion that can be drawn from these results is that the cellulose is immediately burned out at 175 °C, right after the combustion process of the GN precursor, which takes place at 168 °C. This suggests that the cellulose structure is efficient in limiting the contact of metals at the GN-precursor combustion temperature and hence in preventing particle coarsening. The resemblance of the morphological features of the synthesized powders to that of the cellulose fibre is also related to the same effect.
| Material analyzed | Reactions (temp./weight loss) | Nature of reaction and peak temp. | Observations |
|---|---|---|---|
| Activated cellulose | 200–300 °C/30% | Exothermal/236 °C | Slight weight loss at higher GN to cellulose ratios |
| 300–350 °C/50% | Exothermal/330 °C | ||
| 350–500 °C/20% | Exothermal/459 °C | ||
| Conventional GN precursor | Data not recorded due to sudden loss of powder by vigorous combustion reaction at 128 °C | ||
| Cellulose-GN precursor | 168 °C | Extremely exothermal | GN combustion reaction |
| 175 °C | Exothermal | Combustion of cellulose | |
The influence of cellulose on the process and powder characteristics is discussed in the light of thermodynamic calculations and X-ray diffraction (XRD) spectra. First of all, it is demonstrated, as calculated from the adiabatic combustion models, that the crystalline size is efficiently decreased by an increase in cellulose amount. The combustion temperature, which is calculated to reach ca. 4000 K in the conventional GN process, is shown to decrease to 2000 K by the addition of cellulose. Moreover, 1.5 g of cellulose is found to be the critical amount of cellulose to absorb the GN precursor solution completely and hence limit the crystalline size in preparation of 0.01 mol LSCF. The as-prepared powders, which are found to lack phase purity, are also shown to be totally pure for a cellulose amount above 1.5 g when calcined at 900 °C; which otherwise is still impure for a smaller amount of cellulose.
The main difference between the two articles by the same group discussing the LSCF powder preparation via cellulose-assisted GN process is the ca. 60 and 30 °C temperature differences (higher for Zhou et al.104) in GN-precursor combustion and cellulose burn out temperatures, respectively (Table 3 and 4). However, neither the origin nor the significance of this difference is discussed in the articles.
| Material analyzed | Critical reactions (temp.) | Nature of reaction | Observations |
|---|---|---|---|
| Activated cellulose (heated in air) | Identical results to those in ref. 41 | ||
| Conventional GN precursor | Data not recorded due to sudden loss of powder due to a vigorous combustion reaction at 180 °C | ||
| Cellulose-GN precursor | 195 °C | Extremely exothermal | GN combustion reaction |
| 203 °C | Exothermal | Combustion of cellulose | |
It is shown that the as-prepared SDC and GDC powders exhibit pure phases regardless of the cellulose amount, whereas their crystalline size continuously decreases with increasing cellulose addition. There is, however, no explanation for this behaviour. Therefore, the highlight of the article is the as-prepared SDC and GDC powders, which are shown to be pure and 10–20 nm in size making them ideal materials for further processing and application.
The first step in preparation of the GN precursor is slowly immersing tetrabutyl titanate (Ti(C4H9O)4) in de-ionized water in an ice-water bath and stirring, which yields TiO(OH)2. This is followed by the addition of HNO3 and stirring the mixture, which results in a transparent titanyl nitrate solution. After the stoichiometric addition of LiNO3 to the product, glycine is added at a ratio of 1:3 (total metal ions to glycine ratio). In the next step, the GN-precursor is slowly soaked into the activated cellulose and the mixture is dried at 80 °C for several h. The cellulose-GN precursor is finally heated to 250 °C, causing self-combustion upon the reaction of glycine with the nitrates. The fluffy powder, which is observed to contain organics, is calcined at different temperatures between 500–900 °C.
XRD results prove that the powders calcined at 700 °C are pure and highly crystalline in comparison to pure powders obtained via the solid state reaction route at 850 °C and for 17 h. The process is reported to yield almost-spherical powders with an average crystalline size of 28 and 60 nm at 700 and 800 °C, respectively, and to be highly pure as the evaporation of Li is prevented.
:3:1.
| Material | Sequence 1 | Sequence 2 | Sequence 3 |
|---|---|---|---|
| a Gly = Glycine; AC = Activated cellulose CF = Cellulose fiber | |||
| Activated cellulose | Identical to the procedure explained in ref. 104 | ||
| Cellulose-GN precursor | Stoichiometric HNO3, LiNO3, TiO2(s), Gly | TiO2, H2O dropped onto CF | LiNO3, Gly soaked onto CF |
| Mixture dropped on AC | Dried (80 °C/3 h) | Dried (no details) | |
| Solid precursor (SP-I) after drying (80 °C) | CF immersed in HNO3, LiNO3, Gly | CF immersed in HNO3, TiO2 | |
| Dried solid precursor (SP-II) (80 °C/3 h) | Dried solid precursor (SP-III) (80 °C/3 h) | ||
The characteristics of the materials and/or processes prepared by three different routes shown in Table 5 are demonstrated in Table 6 according to the observations in ref. 116. The reason for the absence of combustion in SP-II is ascribed to the decomposition of LiNO3 and hence the loss of NO3− that is necessary for combustion, due to the solid TiO2 closing the pores of the cellulose fibre. At this point no information is given regarding the particle size and soaking extent of TiO2 in the cellulose fibre. The main criticism could probably be made on the phase purity of the powder prepared by SP-III, since this process is cited as a highly smog and gas releasing process, which is expected to lead to the escape of NP. Considering the direct influence of the cellulose amount on the progress of the combustion process, which is mentioned explicitly in ref. 41 and 104 a more experiment-based argument seems to be necessary in this article to make a conclusion about the purity of LTO produced by SP-III.
| Characteristics of materials/processes | Sequence 1 | Sequence 2 | Sequence 3 |
|---|---|---|---|
| Ignition | Sparks | Temp. up to 300 °C | Sparks |
| First combustion flame at | 62 °C | No flame, sparks | 61 °C |
| Increase of temperature to | 450 °C | 500 °C | |
| XRD (as-prepared powder) | Max. LTO phase pure, crystalline | ||
| XRD after calcination (750 °C for 5 h) | Pure, crystalline |
The route referred to in the article starts with dispersion of the YSZ powder (less than 2 g) in 100 ml of distilled-water by stirring. This step is followed by the addition of La(NO3)3·6H2O, Sr(NO3)2, and Mn(NO3)2·6H2O to the primary solution. Following the addition of glycine, the mixture is dried at 80 °C and, upon transforming into a sol, mixed with activated cellulose . Ratios of reactants are manipulated so that two moles of glycine and 75 g of activated cellulose are used for one mole of total metal cations. The mixture is heated at 250 °C and calcined at 850 °C for 3 h.
Transmission electron microscopy (TEM) images revealed that the produced LSM nanoparticles exhibit uniform size of 50 nm (Fig. 5a), which are smaller than particles of commercial powder, with sizes ranging from 70 to 150 nm (Fig. 5b).
 | ||
| Fig. 5 TEM images of: (a) LSM nano particles, (b) YSZ particles. Reprinted from J. Park, J. Zou and J. Chung, Synthesis and evaluation of nano-size Lanthanum Strontium Manganite–Yttria-stablized zirconia composite powders as cathodes for solid oxide fuel cells, J. Power Sources, 195, 2010, 4593–4599, Copyright (2010), with permission from Elsevier. | ||
The rest of the article details the electrical characteristics of the mixed cathode for eight different LSM:YSZ wt% ratios, which is not going to be discussed here.
Table 7 summarizes the particle size and specific surface area of LSC nanopowders produced by different CS routes, from selected references.
Despite the similarity in the processes, glycine–nitrate combustion synthesis of complex oxide ceramics leads to a finer powder with a higher specific surface area among nitrate methods.
Wet-chemical methods, on the other hand, facilitate preparation of homogenous and highly pure ceramic NP, but the processes employed are complex and the production rates are low. Compared to these routes, cellulose-assisted GN process facilitates the preparation of mixed-oxide ceramic NP and materials with high purity and crystallinity by optimizing the yield and production cost as well as energy efficiency. The significant advantages of the cellulose-assisted GN process over other routes in light of the available references can be summarized as:
• The use of relatively simple equipment requiring almost no investment;
• The estimation of process parameters with ease by thermodynamic calculations;
• The formation of high purity NP without intermediate processes
• The possibility to synthesize multi-metal oxide powders including those requiring high temperatures for phase formation;
• Extremely short combustion intervals preventing particle coarsening;
• Suppressed particle growth and agglomeration by the cellulose fibre that burns out following combustion.
In spite of these positive perspectives, certain arguments about the process still remain empirical. Compared to some of the process parameters such as the combustion temperature or the quantity of released thermal energy, which can be directly calculated by thermodynamics, there is a lack of evidence or limited quantitative data to estimate the amount of GN-precursor that is not deposited on cellulose and the significance of this. Moreover, as in the case of LSCF, as-prepared powders might lack purity requiring calcination at high temperatures. However, the interest in the GN process for the production of extremely pure and fine ceramic NP is expected to increase and resolve the gaps of conventional, as well as other CS processes.
Conclusions
The conventional method to prepare ceramic powders is a solid state reaction of oxide and carbonate powders. Although this method is widely preferred in industry due to its simplicity, it is both costly and time consuming due to its intermediate processes such as calcination. Moreover, it results in undesirable powder characteristics such as low purity due to the lack of atomic-level mixing and shifted stoichiometry resulting from high temperature sintering over long periods.In light of the literature reviewed here, developing simple, efficient, inexpensive and environmentally-benign processes for the preparation of high quality ceramic powders is a great interest both for the research community and industry.
Acknowledgements
The authors kindly acknowledge the support of the Brazilian Council for Scientific and Technological Development (CNPq).References
- A. Franks, J. Phys. E: Sci. Instrum., 1987, 20, 1442 CrossRef.
- K. N. Thakkar, S. S. Mhatre and R. Y. Parikh, Nanomed.: Nanotechnol., Biol. Med., 2010, 6, 257 CrossRef CAS.
- J. Blackman, Metallic Nanoparticles, Elsevier, New York, 2008 Search PubMed.
- W. Lu and C. M. Lieber, Nat. Mater., 2007, 6, 841 CrossRef CAS.
- J. Y. Park, C. W. Oh, C. W. Choi and S. S. Kim, J. Am. Ceram. Soc., 2009, 92, 2982 CrossRef CAS.
- A. Mitra, B. Deutsch, F. Ignatovich, C. Dykes and L. Novotny, ACS Nano, 2010, 4, 1305 CrossRef CAS.
- K. L. Kelly, L. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668 CrossRef CAS.
- N. Narayanan and M. A. Sayed, J. Phys. Chem. B, 2005, 109, 12663 CrossRef.
- B. R. Cuenya, Thin Solid Films, 2010, 518, 3127 CrossRef CAS.
- H. C. Wu, X. Chang, L. Fiu, F. Zhao and Y. Zhao, J. Mater. Chem., 2010, 20, 1036 RSC.
- M. Epple, K. Ganesan, R. Heumann, J. Klesing, A. Kovtun, S. Neumann and V. Sokolova, J. Mater. Chem., 2010, 20, 18 RSC.
- D. W. Richerson, Modern Ceramic Engineering: Properties, Processing and Use in Design, 2nd ed., Marcel-Dekker, New York, 1992 Search PubMed.
- S. J. Kang, Sintering, Densification, Grain-growth and Microstructure, Elsevier Butterworth-Heinemann, New York, 2005 Search PubMed.
- M. N. Rahaman, Ceramic Processing and Sintering, 2nd ed., New York, Marcel & Dekker, 1995 Search PubMed.
- F. Sanchez and K. Sobolev, Constr. Build. Mater., 2010, 24, 2061 Search PubMed.
- K. E. Drexler, C. Peterson, G. Pergamit, Unbounding the Future: The Nanotechnology Revolution, William Morrow and Company Inc., New York, 1991. Chapter 3 Search PubMed.
- W. D. Callister, Materials Science and Engineering: An Introduction, 7th ed., Wiley, New York, 2007 Search PubMed.
- B. Lee and S. Komarneni (editors), Chemical Processing of Ceramics, Taylor and Francis, New York, 2005 Search PubMed.
- T. A. Ring, Fundamentals of Ceramic Powder Processing and Synthesis, Academic Press, New York, 1996 Search PubMed.
- M. N. Rahaman, Ceramic Processing and Sintering, 2nd ed., New York, Marcel & Dekker, 1995, Chapter 2 Search PubMed.
- Y. Shin and G. J. Exarhos, Cellulose, 2007, 14, 269 CrossRef CAS.
- M. Sarikaya, Microsc. Res. Tech., 1994, 27, 360 CrossRef CAS.
- S. A. Davis, M. Breulmann, K. H. Rhodes, B. Zhang and S. Mann, Chem. Mater., 2001, 13, 3218 CrossRef CAS.
- S. A. Davis, S. L. Burkett, N. H. Mendelson and S. Mann, Nature, 1997, 385, 420 CrossRef CAS.
- B. Zhang, S. A. Davis, S. Mann and N. H. Mendelson, Chem. Commun., 2000, 781 RSC.
- S. R. Hall, H. Bolger and S. Mann, Chem. Commun., 2003, 2784 RSC.
- B. Zhang, S. A. Davis and S. Mann, Chem. Mater., 2002, 14, 1369 CrossRef CAS.
- S. Chia, J. Urano, F. Tananoi, B. Dunn and J. I. Zink, J. Am. Chem. Soc., 2000, 122, 6488 CrossRef CAS.
- Y. Shin, J. Liu, J. H. Chang, Z. Nie and G. J. Exarhos, Adv. Mater., 2001, 13, 728 CrossRef CAS.
- Y. Shin, I. Bae and G. J. Exarhos, Colloids Surf., A, 2009, 348, 191 CrossRef CAS.
- S. Ifuku, M. Tsuji, M. Morimoto, H. Saimoto and Hi. Yano, Biomacromolecules, 2009, 10, 2714 CrossRef CAS.
- K. Watanabe, D. Menzel, N. Nilius and H.-J. Freund, Chem. Rev., 2006, 106, 4301 CrossRef CAS.
- M. Wu, S. Kuga and Y. Huang, J. Am. Chem. Soc., 2008, 24, 10494 CAS.
- K. D. Hermanson, S. O. Lumsdon, J. P. Williams, E. W. Kaler and O. D. Velev, Science, 2001, 294, 1082 CrossRef CAS.
- Z. Zhou, S. Wang, W. Zhou, G. Wang, L. Jiang, W. Li, S. Song, J. Liu, G. Sun and Q. Xin, Chem. Commun., 2003, 394 RSC.
- I. Sondi and B. S. Sondi, J. Colloid Interface Sci., 2004, 275, 177 CrossRef CAS.
- R. Daw, Nature, 2002, 418, 491 CrossRef CAS.
- J. Huang and T. Kunitake, J. Am. Chem. Soc., 2003, 125, 11834 CrossRef CAS.
- A. T. Harris and A. R. Maddocks, Mater. Sci. Technol., 2010, 26, 375 CrossRef CAS.
- S. Sotiropoulou, Y. Sierra-Sastre, S. S. Mark and C. A. Batt, Chem. Mater., 2008, 20, 821 CrossRef CAS.
- W. Zhou, Z. Shao, R. Ran, H. Gu, W. Jin and N. Xu, J. Am. Ceram. Soc., 2008, 91, 1155 CrossRef CAS.
- S. Zhu, Y. Wu, Q. Chen, Z. Yu, C. Wang, S. Jin, Y. Dinga and G. Wuc, Green Chem., 2006, 8, 325 RSC.
- A. Mukhopadhyay and B. Basu, Int. Mater. Rev., 2007, 52, 257 CrossRef CAS.
- D. Segal, J. Mater. Chem., 1997, 7, 1297 RSC.
- V. V. Srdic, M. Winterer and H. Hahn, J. Am. Ceram. Soc., 2000, 83, 729 CrossRef CAS.
- D. Sarkar, M. Chu, S. J. Cho, Y. I. Kim and B. Basu, J. Am. Ceram. Soc., 2009, 92, 2877 CrossRef CAS.
- A. K. Tyagi, Barc. Newsletter, 2007, 285, 39 CAS.
- L. Zhang, Preparation of Multi-component Ceramic Nanoparticles, Ohio State University, Department of Materials Science and Engineering, Group of Inorganic Materials Science, 2004 Search PubMed.
- M. F. Yan, Solid State Sintering, Engineering Materials Handbook, Vol. 4, Ceramics and Glasses, ASM International, Amsterdam, 1991, 270 Search PubMed.
- W. M. Sigmund, N. S. Bell and L. Bergström, J. Am. Ceram. Soc., 2000, 83, 1557 CrossRef CAS.
- Z. Shao, G. Shiong, Y. Ren and Y. Cong, J. Mater. Sci., 2000, 35, 5639 CrossRef CAS.
- K. C. Patil, M. S. Hegde, R. Tanu and S. T. Aruna, Chemistry of nanocrystalline oxide materials: combustion synthesis, properties and applications. World Scientific, Singapore, 2008 Search PubMed.
- S. T. Aruna and A. S. Mukasyan, Curr. Opin. Solid State Mater. Sci., 2008, 12, 44 CrossRef CAS.
- A. M. Segadaes, Eur. Ceram. News Lett., 2006, 9, 1–5 Search PubMed.
- C. C. Hwang, T. Y. Wu, J. Wan and J. S. Tsai, Mater. Sci. Eng., B, 2004, 111, 49 CrossRef.
- M. Lackner (editor), Combustion Synthesis: Novel Routes to Novel Materials, Bentham e-Books, New York, 2010 Search PubMed.
- K. C. Patil, S. T. Aruna and T. Mimani, Curr. Opin. Solid State Mater. Sci., 2002, 6, 507 CrossRef CAS.
- A. Cincotti, R. Licheri, A. M. Locci, R. Orru and G. Cao, J. Chem. Technol. Biotechnol., 2003, 78, 122 CrossRef CAS.
- A. S. Mukasyan and P. Dinka, Adv. Eng. Mater., 2007, 1 Search PubMed.
- A. Makino and C. K. Law, J. Am. Ceram. Soc., 1994, 77, 778 CrossRef CAS.
- T. Mimani and T. C. Patil, Mater. Phys. Mech., 2001, 4, 134 CAS.
- T. P. Raming, A. J. A. Winnubst, W. E. van Zyl and H. Verweij, J. Eur. Ceram. Soc., 2003, 23, 1053 CrossRef CAS.
- A. A. Borisov, L. Luca and A. G. Merzhanov (editors), Combustion Science and Technology Book Series, Vol. 5, Self-Propagating High-Temperature Synthesis of Materials, Taylor and Francis, New York, 2002. Chapters 9 and 10 Search PubMed.
- A. G. Merzhanov, J. Mater. Chem., 2004, 14, 1779 RSC.
- A. Varma, A. S. Rogachev, A. S. Mukasyan and S. Hwang, Adv. Chem. Eng., 1998, 24, 79 CrossRef CAS.
- F. Maglia, U. Anselmi-Tamburini, G. Spinolo and Z. A. Munir, J. Am. Ceram. Soc., 2000, 83, 1935 CrossRef CAS.
- L. Stobierski, Z. Wegrzyn, J. Lis and M. Buck, Int. J. Self-Prop. High-Temp. Synth., 2001, 10, 217 CAS.
- F. Bernard and E. Gaffet, Int. J. Self-Prop. High-Temp. Synth., 2001, 10, 109 CAS.
- I. P. Borovinskaya, T. I. Ignat'eva, V. I. Vershinnikov, G. G. Khurtina and N. V. Sachkova, Inorg. Mater., 2003, 39, 588 CrossRef CAS.
- K. S. Martirosyan and D. Luss, AIChE J., 2005, 51, 2801 CrossRef CAS.
- T. Striker and J. A. Ruud, J. Am. Ceram. Soc., 2010, 93, 2622 CrossRef CAS.
- S. Mukasyan, C. Costello, K. P. Sherlock, D. Lafarga and A. Varma, Sep. Purif. Technol., 2001, 25, 117 CrossRef.
- K. Deshpande, A. Mukasyan and A. Varma, J. Am. Ceram. Soc., 2003, 86, 1149 CrossRef CAS.
- O. Burgos-Montes, R. Moreno, M. T. Colomer and J. C. Farinas, J. Am. Ceram. Soc., 2006, 89, 484 CrossRef CAS.
- Y. F. Tang, L. Feng, Y. F. Chen and A. D. Li, Adv. Eng. Mater., 2004, 6, 69 CrossRef CAS.
- R. D. Purhoit and A. K. Tyagi, J. Mater. Chem., 2002, 12, 1218 RSC.
- J. Schafer, W. Sigmund, S. Roy and F. Aldinger, J. Mater. Res., 1997, 12, 2518 CrossRef CAS.
- S. V. Chavan and A. K. Tyagi, J. Mater. Res., 2004, 19, 3181 CrossRef CAS.
- P. B. Avakyan, M. D. Nersesyan and A. G. Merzhanov, Am. Ceram. Soc. Bull., 1996, 75, 50 CAS.
- L. A. Chick, L. R. Pederson, G. D. Maupin, J. L. Bates, L. E. Thomas and G. J. Exarhos, Mater. Lett., 1990, 10, 6 CrossRef CAS.
- G. Zhu, X. Fang, C. Xia and X. Liu, Ceram. Int., 2005, 31, 115 CrossRef CAS.
- D. Lee, I. Lee, Y. Jeon and R. Song, Solid State Ionics, 2005, 176, 1021 CrossRef CAS.
- R. D. Purohit, S. Saha and A. K. Tyagi, J. Nucl. Mater., 2001, 288, 7 CrossRef CAS.
- J. Yuan, L. Jiang, H. Tianmin, C. Ligong, W. Jinxia and S. Wenhui, J. Alloys Compd., 2003, 353, 322 CrossRef.
- M. Valefi, C. Falamaki, T. Ebadzadeh and M. S. Hashjin, J. Am. Ceram. Soc., 2007, 90, 2008 CrossRef CAS.
- C. Xinliang, X. Qing, H. Duanping, Z. Feng and C. Wen, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2006, 21, 53 CrossRef.
- S. M. Maliyekkal, Anshup, K. R. Antony and T. Pradeep, Sci. Total Environ., 2010, 408, 2273 CrossRef CAS.
- L. Cong, T. He, Y. Ji, P. Guan, Y. Huang and W. Su, J. Alloys Compd., 2003, 348, 325 CrossRef CAS.
- Z. Lei, Q. Zhu and S. Zhang, J. Eur. Ceram. Soc., 2006, 26, 397 CrossRef CAS.
- H. S. Potdar, S. B. Deshpande, Y. B. Khollam, A. S. Deshpande and S. K. Date, Mater. Lett., 2003, 57, 1066 CrossRef CAS.
- T. Pine, X. Lu, D. R. Mumm, G. S. Samuelsen and J. Brouwer, J. Am. Ceram. Soc., 2007, 90, 3735 CAS.
- J. C. Toniolo, M. D. Lima, A. S. Takimi and C. P. Bergmann, Mater. Res. Bull., 2005, 40, 561 CrossRef CAS.
- T. Mimani, J. Alloys Compd., 2001, 315, 123 CrossRef CAS.
- D. G. Lamas, R. E. Juarez, G. E. Lascalea and N. E. Walsoe de Reca, J. Mater. Sci. Lett., 2001, 20, 1447 CrossRef CAS.
- T. V. Anuradha, S. Ranganathan, T. Mimani and K. C. Patil, Scr. Mater., 2001, 44, 2237 CrossRef CAS.
- G. Fagherazzi, S. Polizzi, M. Bettinelli and A. Speghini, J. Mater. Res., 2000, 15, 586 CrossRef CAS.
- Y. Zhang, H. C. Shin, J. Dong and M. Liu, Solid State Ionics, 2004, 171, 25 CrossRef CAS.
- N. P. Bansal and Z. Zhong, J. Power Sources, 2006, 158, 148 CrossRef CAS.
- Y. Jin, W. P. Qin, J. S. Zhang, Y. Wang and C. Y. Cao, J. Solid State Chem., 2008, 181, 724 CrossRef CAS.
- Z. P. Shao, S. M. Haile, J. M. Ahn and P. D. Ronney, Nature, 2004, 431, 170 CrossRef CAS.
- P. Y. Zeng, Z. H. Chen, W. Zhou, H. X. Gu, Z. P. Shao and S. M. Liu, J. Membr. Sci., 2007, 291, 148 CrossRef CAS.
- S. Tao, J. T. S. Irvine and S. M. Plint, J. Phys. Chem. B, 2006, 110, 21771 CrossRef CAS.
- H. Wang, C. Tablet, A. Feldhoff and J. Caro, Adv. Mater., 2005, 17, 1785 CrossRef CAS.
- W. Zhou, Z. P. Shao, R. Ran, W. Q. Jin and N. P. Xu, Mater. Res. Bull., 2008, 43, 2248 CrossRef CAS.
- J. He, T. Kunitake and A. Nakao, Chem. Mater., 2003, 15, 4401 CrossRef CAS.
- Z. Shao, G. Li, G. Xiong and W. Yang, Powder Technol., 2002, 122, 26 CrossRef CAS.
- C. Kaewprasit, E. Hequet, N. Abidi and J. P. Gourlot, J. Cotton Sci., 1998, 164 Search PubMed.
- J. G. Huang, I. Ichinose and T. Kunitake, Angew. Chem., Int. Ed., 2006, 45, 1 CrossRef.
- D. Zabetakis, M. Dinderman and P. Schoen, Adv. Mater., 2005, 17, 734 CrossRef CAS.
- Y. Shin, X. H. S. Li, C. M. Wang, J. R. Coleman and G. J. Exarhos, Adv. Mater., 2004, 16, 1212 CrossRef CAS.
- J. H. He, T. Kunitake and T. Watanabe, Chem. Commun., 2005, 795 RSC.
- H. Dong and J. P. Hinestroza, ACS Appl. Mater. Interfaces, 2009, 1, 797 CAS.
- N. Sharifi, F. Tajabadi and N. Taghavinia, Int. J. Hydrogen Energy, 2010, 35, 3258 CrossRef CAS.
- S. Zhu, Y. Wu, Q. Chen, Z. Yu, C. Wang, S. Jin, Y. Ding and G. Wu, Green Chem., 2006, 8, 325 RSC.
- T. Yuan, R. Cai, K. Wang, R. Ran, S. Liu and Z. Shao, Ceram. Int., 2009, 35, 1757 CrossRef CAS.
- T. Yuan, K. Wang, R. Cai, R. Ran and Z. Shao, J. Alloys Compd., 2009, 477, 665 CrossRef CAS.
- J. Park, J. Zou and J. Chung, J. Power Sources, 2010, 195, 4593 CrossRef CAS.
- T. Yuan, R. Cai, P. Gu and Z. Shao, J. Power Sources, 2010, 195, 2883 CrossRef CAS.
- T. Yuan, X. Yu, R. Cai, Y. Zhou and Z. Shao, J. Power Sources, 2010, 195, 4997 CrossRef CAS.
- R. Cai, T. Yuan, R. Ran, X. Liu and Z. Shao, Int. J. Energy Res., 2011, 35, 68 CrossRef CAS.
- H. U. Anderson, Solid State Ionics, 1992, 52, 33 CrossRef CAS.
- E. Maguire, B. Gharbage, F. M. B. Marques and J. A. Labrincha, Solid State Ionics, 2000, 127, 329 CrossRef CAS.
- B. C. H. Steele, Solid State Ionics, 2000, 129, 95 CrossRef CAS.
- R. Tian, F. Zhao, F. Chen and C. Xia, Solid State Ionics, 2011, 192, 580 CrossRef CAS.
- A. S. Prakash, P. Manikandan, K. Ramesha, M. Sathiya, J. M. Tarascon and A. K. Shukla, Chem. Mater., 2010, 22, 2857 CrossRef CAS.
- Y. H. Rho, K. Kanamura and T. Umegaki, J. Electrochem. Soc., 2003, 150, A107 CrossRef CAS.
- D. E. Vladikova, Z. B. Stoynov, A. Barbucci, M. Viviani, P. Carpanese, J. A. Kilner, S. J. Skinner and R. Rudkin, Electrochim. Acta, 2008, 53, 7491 CrossRef CAS.
- C. Lu, T. Z. Sholklapper, C. P. Jacobson, S. J. Visco and L. C. De Jonghe, J. Electrochem. Soc., 2006, 153, A1115 CrossRef CAS.
- K. Zupan, D. Kolar and M. Marinšek, J. Power Sources, 2000, 86, 417 CrossRef CAS.
- K. Prabhakaran, J. Lakra, M. O. Beigh, N. M. Gokhale, S. C. Sharma and R. Lal, J. Mater. Sci., 2006, 41, 6300 CrossRef CAS.
- S. R. Nair, R. D. Purohit, P. K. Sinha and A. K. Tyagi, J. Alloys Compd., 2009, 477, 644 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |