Photoelectrochemically active species and photoelectrochemical biosensors
Xiaoru
Zhang
a,
Yingshu
Guo
a,
Mingshuai
Liu
a and
Shusheng
Zhang
*ab
aKey Laboratory of Biochemical Analysis, Ministry of Education, College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, P. R. China. E-mail: shushzhang@126.com
bCollege of Chemistry and Chemical Engineering, Linyi University, Linyi 276000, P. R. China
First published on 13th November 2012
Abstract
Photoelectrochemical biosensors developed from solar cells have received increasing attention due to their desirable sensitivity and better analytical performance. In recent decades, much attention has been paid to the preparation of photoelectrochemical biosensors. This paper aims to provide an overview on the development of photosensitizers, including the sensing principle of photoelectrochemical biosensors and a special kind of photoelectrochemical biosensor without external irradiation. The latest developments and applications are also included.
 Xiaoru Zhang | Dr Xiaoru Zhang was born in Heilongjiang Province, China. She received her MS degree from Tianjin University in 2000 and her PhD from the Ocean University of China in 2003. She is currently working at Qingdao University of Science and Technology. Her scientific interests focus on photoelectrochemistry biosensors, carbon and metal nanomaterials for electrochemical applications. |
Introduction
The principle of photoelectrochemistry has been applied to solar energy conversion for decades.1 During this time, the major components of photoelectrochemical solar cells have been optimized in a stepwise way. The most successful system in terms of quantum yield was developed by O′Regan and Gratzel.2 The photoelectrochemical (PEC) detection method is a newly developed and promising analytical method for biological assays.3–5 In photoelectrochemical detection, light is used to excite active species on the electrode, and current is obtained as the detection signal, which is just the reverse process of electrochemiluminescence (ECL). Coupling photoirradiation with electrochemical detection, photoelectrochemical sensors have the advantages of both optical methods and electrochemical sensors. Due to its separate source for excitation and detection, the sensitivity of the photoelectrochemistry-based analytical method could potentially match that of the ECL with low background signals. At the same time, compared with the optical detection methods such as fluorescence, chemiluminescence (CL), and ECL, which have to use complex and expensive optical imaging devices and sophisticated image recognition software, the use of electronic detection makes the photoelectrochemical instrument simple and low-cost.6–8 However, given the advantages mentioned above, only rather limited PEC analysis on the determination of DNA damage,6,8 DNA,9–11 proteins12–14 and small molecules4,15 have been reported in the past decade. With the emergence of novel photoelectrochemically active species and new detection methods, this PEC analysis shows great potential in the field of biosensors. Here we provide a comprehensive review from the view point of photosensitizers and the sensing principle of PEC biosensors often used.1 Photoelectrochemically active species
1.1 Semiconducting nanoparticles or quantum dots
Recent advances in semiconducting nanoparticles (NPs), or quantum dots (QDs), have shown great promise due to their high fluorescence quantum yields, stability against photobleaching, and size-controlled luminescence properties.15–22 The organization of functional semiconductor nanoparticles on surfaces, and specifically electrodes, has attracted substantial research efforts.23–25 Semiconductor NPs architectures on surfaces have been used to assemble photoelectrochemical cells,26–28 to tailor light-emitting diodes,29–31 to fabricate electrochromic devices,32 and to organize sensor systems.33,34 Photoexcitation of semiconductor QDs results in the transfer of electrons from the valence band to the conduction band, thus yielding electron–hole pairs, which is the primary event for the photocurrent generation. The ejection of the conduction-band electrons to the electrode, with the concomitant transfer of electrons from a solution-solubilized electron donor D, yields an anodic photocurrent (Fig. 1A). In contrast, transfer of the conduction-band electrons to a solution-solubilized electron acceptor D, followed by the supply of electrons from the electrode to neutralize the valence-band holes, yields a cathodic photocurrent (Fig. 1B).35 | ||
| Fig. 1 Photocurrents generated by semiconductor NPs associated with electrodes: A) anodic photocurrent, B) cathodic photocurrent. Reprinted from ref. 35 with permission from Wiley–VCH. | ||
Willner et al. prepared assembly of the DNA-cross-linked CdS particles on an Au electrode.11 The array of CdS NP layers was constructed by a layer-by-layer hybridization process using CdS QDs functionalized with nucleic acids. The photocurrents were generated by the DNA-cross-linked CdS arrays that contained two and four generations of nanoparticles. In the presence of [Ru(NH3)6]3+ which were electrostatically bound to the DNA, the photocurrent was about two fold higher. This method can be used for the quantitative detection of DNA.
Later, Pardo-Yissar et al. prepared acetylcholine esterase (AChE) labeled CdS nanoparticles on electrodes.36 CdS NPs were assembled on a gold electrode, and the NPs were further modified with AChE. The photocurrent generation in the system is attributed to the AChE-catalyzed hydrolysis of acetylthiocholine to thiocholine, which acted as an electron donor for the photogenerated holes in the valence band of the CdS NPs (Fig. 2). The study demonstrated that enzyme inhibitors decrease the photocurrents, and thus the nanoparticle/AChE system could act as a biosensor for the respective inhibitor.
 | ||
| Fig. 2 Assembly of the CdS nanoparticle/AChE hybrid system used for photoelectrochemical detection of enzyme activity. Reprinted from ref. 36 with permission from the American Chemical Society. | ||
The photoelectrochemical detection of tyrosinase (TR) activity was also reported by Willner et al., using CdS NPs as photoelectrochemical reporter units.14 CdS NPs were modified with amidated L-tyrosine methyl ester, and then reacted with tyrosinase/O2. The resulting dihydroxyphenyl methyl ester was linked to a phenylboronic acid monolayer-functionalized electrode (Fig. 3). Illumination of the CdS-NPs-functionalized electrode generated photocurrents in the presence of TEOA as a sacrificial electron donor. The detection limit for analysing tyrosinase was 0.2 U.
 | ||
| Fig. 3 Photoelectrochemical analysis of tyrosinase by a boronic acid monolayer-functionalized electrode and tyramine-labeled CdS NPs. Reprinted from ref. 14 with permission from the American Chemical Society. | ||
Recently, many label-free photoelectrochemical biosensors were developed based on semiconducting nanoparticles modified electrode. Wang et al. developed a label-free photoelectrochemical immunosensor using a CdS quantum dots (QDs) multilayer film coupled with a biospecific interaction.13 The CdS QDs multilayer film was prepared by layer-by-layer assembling positively charged poly(dimethyldiallylammonium chloride) (PDDA) and thioglycolic acid (TGA)-capped water-soluble CdS QDs on the surface of an indium-tin oxide (ITO) electrode. Ascorbic acid (AA) was exploited as an electron donor for scavenging photogenerated holes and mouse IgG was used as a model analyte (Fig. 4). On the basis of the photocurrent decrease due to the formation of the immunocomplex, a label-free photo-electrochemical immunosensor was achieved. Similarly, our group prepared a photoelectrochemical cytosensor by integrating the LBL assembly of CdSe NPs and DNA aptamer (a single-stranded DNA/RNA oligonucleotide, which has been selected for binding to a broad range of targets). The formed aptamer–cell complex due to the recognition of aptamer can block the diffusion of AA to the surface of electrode and lead to the decrease of photocurrent. The developed cytosensor exhibited excellent sensitivity and selectivity. Under optimal conditions, a detection limit of 84 cells mL−1 was achieved.37
 | ||
| Fig. 4 Schematic diagram of the stepwise immunosensor fabrication process. Reprinted from ref. 13 with permission from the Royal Society of Chemistry. | ||
As for semiconductor nanoparticles, the study showed that large photoanodic currents were obtained when the CdSe particles were replaced by CdSe@CdS NPs38 or CdSe/ZnS NPs39 (core/shell). As these core/shell QDs have a unique surface chemistry for coupling to biomolecules, they can be used as labeling agents in photoelectrochemical biosensor systems.
1.2 Small organic molecules
Some small organic molecules can also serve as photoelectrochemically active species to be used in photoelectrochemical sensors. Ikeda et al. prepared 5, 10, 15, 20-tetra(4-pyridyl)porphyrin (TPyP)-deposited ITO electrodes, and the electrode operated as a photoelectrochemical sensor for nucleotides.40 When 5′-monophosphate (AMP), adenosine 5′-diphosphate (ADP), and adenosine 5′-triphosphate (ATP) were added, the photocurrent was decreased due to the phosphate ions of nucleotides prevented the electron transfer from photoactivated TPyP to the ITO electrode (Fig. 5). The order of photocurrent changes was AMP > ADP = ATP. The photoelectrochemical sensor can be used several times after successive washing with water and ethanol. | ||
| Fig. 5 Schematic illustration of a TPyP-deposited ITO electrode as a sensor of nucleotides. Reprinted from ref. 40 with permission from the American Chemical Society. | ||
Anthraquinone (AQ) is an effective photosensitizer41–45 as well as a common redox-active material for electroanalytical chemistry.46–50 Okamoto et al. reported for the first time on the photostimulated hole transport through DNA duplexes immobilized on gold electrodes.51 By modifying the surface of a gold electrode with a DNA duplex containing anthraquinone, which was the molecule responsible for the generation of the photocurrent, a sequence-dependent cathodic photocurrent was accomplished (Fig. 6). The result suggested that the efficiency of photostimulated hole transport through the DNA duplexes on the gold electrodes was strongly affected by the duplex sequences and the hole transport efficiency was lowered by elongating the hole-hopping distance.
 | ||
| Fig. 6 Schematic illustration of the measurement of a photocurrent using a gold electrode modified with an anthraquinone-modified DNA duplex. Reprinted from ref. 51 with permission from the American Chemical Society. | ||
Yamada et al. prepared AQ photosensitizer-tethered oligodeoxynucleotide (ODN) duplexes bearing 5-methylcytosine (mC) or the corresponding cytosine (C) at a restriction site of the ODN strand immobilized on gold electrodes, and measured their photocurrent responses arising from hole transport after enzymatic digestion.52 After being treated with HapII or HhaI, a significant suppression of photocurrent response was observed for duplexes with a normal C at the restriction site strand due to the cleavage at the C target site, by which the AQ photosensitizer on the duplex was eliminated from the electrode. In contrast, the duplex containing mC at the target site did not undergo such an enzymatic digestion and therefore the photocurrent response was preserved (Fig. 7). This system has a strong potential for the photoelectrochemical identification of methylation status. Similarly, Haruna et al. studied the pH effect on the hole transport through AQ photosensitizer-tethered DNA duplex possessing a partial triplex region using a photoelectrochemical method.53
 | ||
| Fig. 7 The protocol for photoelectrochemical discrimination of C and mC in DNA immobilized on a gold electrode. Reprinted from ref. 52 with permission from the Royal Society of Chemistry. | ||
1.3 Metal complex
A ruthenium tris(2,2-bipyridine) (Ru-bipy) derivative was usually employed as the photoelectrochemical signal-generating molecule with porous SnO2 film as semiconductor electrode. Oxalate54,6,8 or guanine7 served as an electron donor to recycle the signal reporter and amplify the signal. Gao et al. synthesized a bis-intercalator consisting of two N,N′-bis(3-propyl-imidazole)-1,4,5,8-naphthalene diimides (PIND) linked by a Ru(bpy)22+ (bpy = 2,2′-bipyridine) complex (PIND–Ru–PIND).9 The extremely low dissociation rate of the adduct and unique photoelectrochemical properties of the nucleic acid/PIND–Ru–PIND provided a PCR-free ultrasensitive rout for the detection of nucleic acids with a sensitivity enhancement of four orders of magnitude over voltammetry.Haddour et al. synthesized ruthenium complex functionalized with electropolymerizable pyrrole groups and biotin as affinity binding groups (Fig. 8a).12 After electrochemical polymerization, biotinylated poly(pyrrole-ruthenium) film was obtained as a photosensitive and anchoring polymeric layer. Then, a layer of biotinylated cholera toxin was firmly bound to the functionalized polypyrrole film via avidin bridges (Fig. 8b). Such successive binding of avidin and biotinylated toxin induces a marked decrease in the photocurrent response, which was ascribed to the increase in steric hindrances toward the diffusion of quencher molecules to the underlying poly Ru film with the increase in protein layers. The resulting modified electrodes were used as immunosensors for the detection of the corresponding antibody from 0 to 200 μgmL−1.
![a, Structure of the [Ru(L2)2(L1)]2+complex. b, Assembly of polypyrrole ruthenium(ii) film used for photoelectrochemical label-free detection of anti-cholera toxin. Reprinted from ref. 12 with permission from the American Chemical Society.](/image/article/2013/RA/c2ra22238h/c2ra22238h-f8.gif) | ||
| Fig. 8 a, Structure of the [Ru(L2)2(L1)]2+complex. b, Assembly of polypyrrole ruthenium(II) film used for photoelectrochemical label-free detection of anti-cholera toxin. Reprinted from ref. 12 with permission from the American Chemical Society. | ||
Two photoelectrochemical methods for the detection of DNA damage were presented by Lang's group for the detection of DNA-damage. One was based on photoelectrochemically catalyzed base oxidation (Fig. 9A), and the other employed a photoelectrochemical indicator (Fig. 9B). Of the two approaches investigated, the first approach was more convenient to use, as it was a true sensor configuration with all the reagents immobilized on the sensor surface. The intercalator approach was less demanding on the photocurrent detector due to its high signal.6 The results demonstrated for the first time that the photoelectrochemical DNA sensor could detect both DNA adduct formation and DNA oxidation. However, both methods require the presence of H2O2 in the case of transition metals for the Fenton reaction to occur and can only detect reactive organic chemicals which form covalent adducts with DNA directly. Their utility as a screening tool for genetic toxicity is therefore limited. So, the same group reported rapid detection of in situ DNA damage induced by enzyme-catalyzed Fenton reaction using photoelectrochemical method later. Enzymes catalyze the formation of H2O2 in the presence of glucose, which then reacts with Fe2+ and generates hydroxyl radicals by the Fenton reaction.8
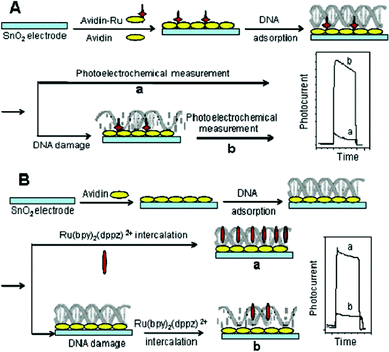 | ||
| Fig. 9 Schematic diagram illustrating the DNA damage detection method based on (A) photoelectrochemically catalyzed base oxidation and a (B) photoelectrochemical indicator. Reprinted from ref. 6 with permission from the American Chemical Society. | ||
Using a high-affinity DNA intercalator, Ru(bpy)2dppz (bpy = 2,2-bipyridine, dppz = dipyrido[3,2-a:2′,3′-c]phenazine) as the signal indicator, Liu's group developed a photoelectrochemical method for double-stranded DNA detection.10 When double-stranded DNA was added into the solution of Ru(bpy)2dppz, photocurrent dropped substantially due to the intercalation of Ru(bpy)2dppz into DNA and, consequently, the reduced mass diffusion of the indicator to the electrode, as well as electrostatic repulsion between oxalate anion and negative charges on DNA.
Natural as well as synthetic lipids have long been employed as general building blocks in many research areas owing to their ability to assemble into stable, well-defined microscopic structures. Jiang et al. reported a new photocurrent-generation system based on Ru(bpy)32+ tethered on phospholipid/alkanethiol hybrid bilayers in aqueous media.55 The construction of such a system comprises two steps (Fig. 10). First, a self-assembled monolayer (SAM) of alkanethiol was formed on gold, and separately, liposomes containing Ru(bpy)32+-conjugated dioleoylphosphoethanolamine (DOPE) were prepared by extrusion. Subsequent exposure of the Ru(bpy)32+-containing liposome solution to the preformed SAM induced the addition of a monolayer of phospholipids on top of the SAM and thereby the immobilization of a Ru(bpy)32+ layer on the gold electrode. Either anodic or cathodic photocurrent generation could be realized in this system, when ascorbate (anodic) and methyl violgen/oxygen (cathodic) were used as the sacrificial electron donor and acceptor, respectively. This study demonstrated a new photocurrent-generation system constructed on phospholipid/alkanethiol hybrid bilayers.
 | ||
| Fig. 10 Formation of Ru(bpy)32+ conjugated phospholipid/alkanethiol hybrid bilayers. Reprinted from ref. 55 with permission from the American Chemical Society. | ||
In addition to (Ru-bipy) derivatives, metal-porphyrin complexes are often used as photoelectrochemically active species.56,57 Tu et al. synthesized [meso-tetrakis(4-sulfonatophenyl) porphyrin] iron(III) monochloride (FeTPPS)-TiO2 nanoparticles, which were prepared by the dentate binding of TiO2 nanoparticles with sulfonic groups of FeTPPS.58 FeTPPS could efficiently improve the photocurrent conversion efficiency of the TiO2 and the nanoparticles showed a stable photoelectrochemical response. Using glutathione (GSH) as a model, a novel method for the detection of GSH was thus developed (Fig. 11). Later, this group prepared free-base-porphyrin- functionalized ZnO nanoparticles.59 This nanohybrid can sensitize the photoelectrochemical efficiency of ZnO nanoparticles, which produced a photoelectrochemical biosensing platform for the detection of cysteine. The detection limit was 0.2 mmolL−1 at a signal-to-noise ratio of 3.
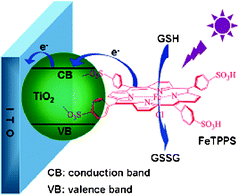 | ||
| Fig. 11 Schematic illustration of photoelectrochemical process for oxidation of GSH at FeTPPS-TiO2-modified ITO electrode. Reprinted from ref. 58 with permission from the American Chemical Society. | ||
1.4 Nanocomposites

Bare TiO2 is a wide band gap semiconductor material and is photoelectrochemically active under UV irradiation. The destructive effect of the UV light and the strong oxidation power of the photo holes of TiO2 limit its application as a photoelectrochemical biosensor. It was reported that coupling of TiO2 with CdS could greatly improve the photocurrent intensity of CdS due to the enhanced charge separation.68–70 Hence, an improved approach was developed for obtaining TiO2/CdS hybrid modified electrode by Wang et al.71 The CdS deposition on TiO2 modified electrode from [Cd(NH3)4]2+ solution showed much higher photocurrent intensity with fewer coating times than that from Cd2+ solution. After the ITO/TiO2/CdS electrode was coated with chitosan (CS), α-fetoprotein (AFP) antibodies were covalently conjugated on the surface of the electrode (Fig. 13). After the as obtained immunosensor was incubated with the corresponding antigen, the photocurrent intensity further decreased. The decrease in the photocurrent was ascribed to the increase in steric hindrances for AA to the surface of CdS due to the formation of the immuno-complex. The immunosensor displayed a linear response to AFP in the ranges from 50 pg mL−1 to 50 ng mL−1 with a relatively low detection limit of 40 pg mL−1. The photoelectrochemical results for the detection of AFP in five human sera showed acceptable accuracy, indicating the attractive perspective of the immunosensor in clinical immunoassay for AFP in serum.
 | ||
| Fig. 13 Schematic diagram of the immunosensor construction process. Reprinted from ref. 71 with permission from Elsevier Limited. | ||
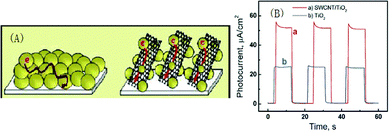 | ||
| Fig. 14 (A) Electron transport across nanostructured semiconductor films in the absence and in the presence of a nanotube support architecture. (B) Photocurrent response vs. time profiles of carbon fiber electrode/SWCNT/TiO2 (a) and carbon fiber electrode/TiO2 (b) electrodes at 0 V vs. SCE. Light intensity was 50 mW cm−2 (λ > 300 nm). Reprinted from ref. 76 with permission from the American Chemical Society. | ||
Willner et al. have demonstrated unprecedented high quantum yields for the generation of photocurrents by using semiconductor nanoparticles/carbon nanotubes as a hybrid system. As shown in Fig. 15, the Au electrode was functionalized with a primary monolayer of cysteamine to which the carboxylic acid functionalized CNTs were then coupled. The CdS NPs (protected by a capping monolayer of cysteamine and 2-thio-ethanesulfonic acid) were then coupled to the ends of the CNTs. The results suggest that the length of the CNTs plays a major role in the formation of photocurrents and probably the defects in the CNT affect the extent of charge separation that follows the photoexcitation of the CdS NPs.25
 | ||
| Fig. 15 Assembly of the CdS NP/CNT system on an Au electrode, and the photoinduced charge transport in the system. TEOA = triethanolamine, h+ = hole. Reprinted from ref. 25 with permission from Wiley–VCH. | ||
Graphene, an atomic-layer-thick 2D system, has drawn great attention owing to its outstanding electronic, optical, thermal and mechanical properties.83–88 Due to its unique structure and excellent optical and electronic properties,86 much attention has been focused on its application in the field of photoelectrochemistry. As a good candidate for the acceptor material in OPV applications, graphene has large donor/acceptor interfaces for charge generation and a continuous pathway for electron transfer. Recently, Chen et al. have reviewed applications of graphene in organic photovoltaic (OPV) cells, including transparent electrodes, active layers and interfaces layer in OPV.89
Guo et al. reported the preparation of a layered graphene/QDs device by electrophoretic deposition of chemically reduced graphene, followed by direct synthesis of CdS QDs on predeposited graphene layer through sequential chemical bath deposition from their salt aqueous solutions (Fig. 16a).81 The significantly improved photo-responses confirm that graphene is a good candidate for the collection and transport of photogenerated charges. The performance of {graphene/QDs}8 samples was further compared with that of {SWNT/QDs}8 samples of the same thickness (Fig. 16b). Reproducible responses to ON–OFF light cycles on both {graphene/QDs}8 and {SWNT/QDs}8 showed that the photocurrent response of {graphene/QDs}8 is more than 2.5 times that of {SWNT/QDs}8. This can be explained from two aspects. Firstly, from the energy-level diagrams of graphene/QDs and SWNT/QDs photoelectrodes shown in Fig. 16c, we can see that ITO with a work function of around 4.8 eV can facilitate the fast capture of electrons from graphene, whereas this does not occur to SWNTs. Secondly, the good distribution of QDs on graphene together with the favorable work function of graphene could make it effective for separation of the photogenerated electron–hole pairs and transfer of the electrons to the electrode surface.
 | ||
| Fig. 16 a) Fabrication of the layered graphene/QDs on ITO glass; b) photocurrent responses of {graphene/QDs}8 and {SWNT/QDs}8versus time profiles; c) Energy-level diagram of the bilayer system. Reprinted from ref. 81 with permission from Wiley–VCH. | ||
Geng et al. fabricated composite films via π–π stacking interaction between graphene and pyridine-capped QDs in aqueous solutions.90 The flexible and transparent optoelectronic films fabricated from the composites show improved photosensitivity with increasing loadings of QDs. Chang's group developed another rout for the preparation of QD functioned graphene.91 Graphene was first functionalized with pyrenebutyrate (PB) to form PB functionalized graphene (PB-graphene) with negative charges. PB-graphene absorbed positive Cd2+ on graphene through electrostatic interaction between negative PB and Cd2+. The absorbed Cd2+ ions reacted with S2− ions, resulting in the form of CdS QDs on graphene and photoelectrodes on ITO substrate. These QDs sensitized graphene photoelectrodes showed enhanced photocurrent generation capability and incident IPCE at visible light, and could also be an efficient platform for other optoelectronic applications.
Our group demonstrated the first photoelectrochemical biosensor sensitized by functionalized graphene.92 In this work, pyrene grafted poly(acrylic acid) (PAA) was synthesized first. After being coupled with graphene through π–π stacking between the surface of graphenes and pyrenes, graphene became negatively charged. Then CdSe sensitized graphene multilayers (PAA–G/CdSe)n were assembled on the modified ITO electrode based on the electrostatic interactions. Finally, –PO3H groups modified with the DNA aptamer was covalently bound. When putting such a modified electrode into the solution containing thrombin, the formed aptamer–thrombin complex could block the diffusion of sacrificial electron donor AA to the surface of the electrode and led to the decrease of photocurrent (Fig. 17).
 | ||
| Fig. 17 Schematic diagram of the construction of graphene and CdSe multilayers and the fabrication of a thrombin biosensor. Reprinted from ref. 92 with permission from the Royal Society of Chemistry. | ||
Zhao et al. report a one-step synthetic route for the fabrication of GR–CdS nanocomposites. GO was simultaneously reduced to GR during the deposition of CdS.93 Thioacetamide (TAA) served not only as a gentle reducing agent to reduce GO, but also as a sulfur source to form CdS. As a capping agent, poly(vinyl pyrrolidone) (PVP) prevented the aggregation of GR–CdS nanocomposites (Fig. 18a). Then a layer of folic acid (FA) was conjugated to the GR–CdS film. The modified electrode was studied as a sensor for cell detection through affinity interactions between FA and folate receptor on the cell surface (Fig. 18b). The cytosensor showed a good photoelectronic effect and cell-capture ability, and had a wide linear range and low detection limit for Hela cells.
 | ||
| Fig. 18 a) The one-step synthesis of GR–CdS nanocomposites. b) The fabrication of a photoelectrochemical cytosensor. Reprinted from ref. 93 with permission from Wiley–VCH. | ||
After further improving the above work, the same group reported a novel route for the fabrication of GR-CdS nano-composite.94 The introduction of hydroxylamine (NH2OH) has two advantages: firstly, it can facilitate the coordination with cadmium, which made a higher coverage of CdS nanocrystals on the GR sheets. Secondly, NH2OH also contributed to the assistant reduction of GO and the stabilization of CdS NCs via attachment with the negatively charged –COO− and –OH− groups on GO (Fig. 19).95–97 The size and crystalline phase of CdS NCs in this work were different from previous work, and the uniform and small size led to a higher coverage of CdS on GR sheets so that an enhanced photocurrent compared to previous work.93 Using GSH as a hole scavenger, the fast photoelectronic communication among GSH, CdS, GR sheet and ITO electrode led to a novel strategy for the photoelectrochemical detection of GSH with a detection limit of 0.003 mmol L−1. This method also shows an excellent specificity against anticancer drugs and can be successfully applied for GSH detection in real samples.
 | ||
| Fig. 19 Schematic illustration of the one step synthesis of GR–CdS nanocomposites. Reprinted from ref. 94 with permission from the Royal Society of Chemistry. | ||
2 Sensing principle of photoelectrochemical biosensors
Compared to the large amount of research on photosensitizers, research on the sensing principle of photoelectrochemical biosensors is much more limited. The signalling mechanism is mainly focused on using the altered electron donor concentration or its diffusion efficiency caused by enzyme/affinity reaction to achieve the signal diminution/enhancement. Namely, the signalling strategy was established on the direct interfacial electron communication between the photoactive material and ambient environment.35,98 Recently, Xu's group conducted research on this topic from various angles.They explored the energy transfer between CdS QDs and Au NPs in a PEC system to search for an advanced energy transfer-based PEC bioassay protocol.99 The emission peak of CdS QDs centered at ca. 510 nm is observed in the PL spectrum, however the absorption spectra of the Au NPs with the maximum plasmon absorption peak is observed at ca. 520 nm. Apparently, the PL emission of the CdS QDs has a large spectral overlap with the surface plasmon resonance (SPR) absorption of the Au NPs (Fig. 20). This would be beneficial to the inducement of the SPR of the Au NPs and thus affect the subsequent interparticle interaction. Then the sensitive biosensing of DNA was realized using this method.
 | ||
| Fig. 20 Schematic diagram of the process to fabricate the energy transfer-controlled PEC system. Reprinted from ref. 99 with permission from the Royal Society of Chemistry. | ||
Very recently, this group investigated the exciton-plasmon interactions (EPI) between CdS quantum dots and Ag nanoparticles in a photoelectrochemical system (Fig. 21).98 Due to the natural overlap of their absorption spectra, the exciton and the plasmon could be induced simultaneously, resulting in the photoresponse of the QDs greatly attenuated by the stimulated EPI. The EPI resonant nature enabled manipulating photoresponse of the QDs via tuning interparticle distances and complete PEC signal quenching could be realized. This work provides a new protocol for DNA sensing with the linear range of target DNA from 2.0 × 10−15 to 2.0 × 10−11 M.
 | ||
| Fig. 21 EPI between Ag NP and CdS QD. Reprinted from ref. 98 with permission from the American Chemical Society. | ||
QDs-based amplified PEC immunoassay with the integration of horseradish peroxidase (HRP) catalyzed biocatalytic precipitation (BCP) was also study by the same group aiming to search for an advanced immunoassay format.100 Compared to the conventional label-free PEC immunoassays with enhanced steric hindrances or donor concentration as the exclusive cause for signal variation, the bioconjugates of HRP-secondary antibodies (Ab2) used in this strategy not only can enhance steric hindrance, but also stimulate the BCP onto the electrode surface for signal amplification (Fig. 22). As a result of the multisignal amplification in this HRP catalyzed BCP-based PEC immunoassay, the antigen could be detected from 0.5 pg mL−1 to 5.0 ng mL−1 with a detection limit of 0.5 pg mL−1.
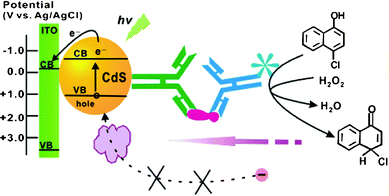 | ||
| Fig. 22 Development of the amplified QD-based sandwich PEC immunoassay with HRP-catalyzed BCP format. Reprinted from ref. 100 with permission from the American Chemical Society. | ||
3 Photoelectrochemical biosensors without external irradiation
In all the work mentioned above, in order to produce photocurrents, a physical light source and monochromator are required, which makes the volume of the instrument bigger and contradicts the trend for portable biosensors. Consequently, a strategy for the substitution of physical light source is highly deserved. CL emission is a promising substitute for the physical light source.101 Adjusting the reaction conditions of CL systems can bring light emission with different wavelengths to realize photoelectrochemical detection without the need of a physical light source and monochromator. Most recently, several works have focused on this aspect.Our group reported PEC analysis using CL emission of the isoluminol–H2O2–Co2+ system as a light source for the determination of physiological thiols in cancer cells.102 As shown in Fig. 23, isoluminol and thiolated signal DNA were labeled on the surface of the polystyrene microspheres (PSMs), and at the same time, the amino group on the surface of the magnetic beads (MBs) were converted into pyridyldisulfide groups by treatment with N-succinimidyl-3-(2-pyridyldithio)-propionate (SPDP). After treating the SPDP activated MBs with the modified PSMs, isoluminol molecules on the surface of the PSMs were attached to the surface of the MBs through disulfide bonds to form a CL probe. In the presence of thiols, the disulfide bonds could be cleaved readily and PSMs modified with the isoluminol molecules and signal DNA were released. The released PSMs were captured by CdS modified electrode through DNA hybridization and transferred to the dark cell for PEC detection.
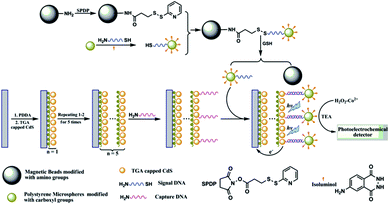 | ||
| Fig. 23 Schematic diagram of the stepwise fabrication and detection process for determination of the thiols. Reprinted from ref. 102 with permission from the Royal Society of Chemistry. | ||
Willner’s group also reported a photoelectrochemical biosensor without external irradiation (Fig. 24). The sensing systems were based on the hemin/G-quadruplex-catalyzed generation of chemiluminescence in the presence of H2O2/luminol. The subsequent internal chemiluminescence resonance energy transfer (CRET) stimulated excitation of the semiconductor QDs that led to the generation of electron–hole pairs.103 The transfer of the conduction-band electrons to the electrode, and the concomitant scavenging of the valence-band holes by the triethanolamine electron donor resulted in the generation of photocurrents. This CRET-stimulated generation of photocurrents was applied to sense DNA and glucose.
 | ||
| Fig. 24 Generation of photocurrents by CdS QDs associated with an Au electrode and functionalized by a hemin/G-quadruplex tethered to a BSA layer associated with the QDs. Reprinted from ref. 103 with permission from the American Chemical Society. | ||
In addition to the study on chemiluminescence excited photoelectrochemistry, our group also investigated an electrochemiluminescent (ECL)-induced PEC system (EPE) based on DNA-linked CdS NPs superstructure with methylene blue as the intercalator molecule.104 In order to reduce background current, EPE synthesis containing a six-electrode system was divided into two parts, the outer cell (ECL cell) and the inner cell (PEC cell). The excitation light source was generated from luminol (ECL substrate) in the ECL cell. CdS NPs as photovoltaic sensitive substrates were modified on the Au electrode in order to accept light energy. The photocurrent from CdS NPs was detected by an electrochemistry detector (Fig. 25). The detection limit for target DNA was 1 fM.
 | ||
| Fig. 25 (A) EPE cell synthesis. (B) The schematic diagrams of the stepwise process assembled on Au electrode 2. Reprinted from ref. 104 with permission from the Royal Society of Chemistry. | ||
Conclusions
Developed from solar cells, photoelectrochemical detection is a newly emerging yet dynamically developing technique for biological sensing. The examples covered in this article highlight the recent important advances in the species of photosensitizers and the different sensing principle of photoelectrochemical biosensors. Despite many important achievements mentioned in the article, such technique is still in its infancy, with many challenges and opportunities open to researchers in the field. The most challenging aspect of working with CdS-based probes in biological systems, however, is related to toxicity that can result from the decomposition and release of heavy metal ions. So, they might be toxic to cells if used for in vitro characterization of cell cultures, and if used for diagnostic tests, the particles might be a hazard to the technician carrying out an assay. With the further development of materials science, new photoactive materials will emerge constantly. Preparing photoelectrochemically active species with high IPCE can not only promote the development of solar cells, but also improve the sensitivity of photoelectrochemical biosensors. Moreover, compared with its flourishing counterparts, such as electrochemical sensors and optical sensors, the signalling mechanism of photoelectrochemical biosensors deserves further study. Finally, owing to significant advantages, the detection range of photoelectrochemical biosensors is highly limited. Therefore, it is necessary to expand range of object type and realize the detection for multiple targets simultaneously.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 21275003, 21275086), Excellent Young Scientists Encouragement Foundation of Shandong Province (No. BS2011CL015), and the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT).References
- A. C. Arias, J. D. MacKenzie, I. McCulloch, J. Rivnay and A. Salleo, Chem. Rev., 2010, 110, 3 CrossRef CAS.
- B. O'Regan and M. Gratzel, Nature, 1991, 353, 737 CrossRef CAS.
- G. N. Brown, J. W. Birks and C. A. Koval, Anal. Chem., 1992, 64, 427 CrossRef CAS.
- J. A. Cooper, M. Wu and R. G. Compton, Anal. Chem., 1998, 70, 2922 CrossRef CAS.
- H. Tokudome, Y. Yamada, S. Sonezaki, H. Ishikawa, M. Bekki, K. Kanehira and M. Miyauchi, Appl. Phys. Lett., 2005, 87, 213901 CrossRef.
- M. Liang and L. Guo, Environ. Sci. Technol., 2007, 41, 658 CrossRef CAS.
- M. Liang, S. Liu, M. We and L. Guo, Anal. Chem., 2006, 78, 621 CrossRef CAS.
- M. Liang, S. Jia, A. Zhu and L. Guo, Environ. Sci. Technol., 2008, 42, 635 CrossRef CAS.
- Z. Q. Gao and N. C. Tansil, Nucleic Acids Res., 2005, 33, e123 CrossRef.
- S. L. Liu, C. Li, J. Cheng and Y. X. Zhou, Anal. Chem., 2006, 78, 4722 CrossRef CAS.
- I. Willner, F. Patolsky and J. Wasserman, Angew. Chem., Int. Ed., 2001, 40, 1861 CrossRef CAS.
- N. Haddour, J. Chauvin, C. Gondran and S. Cosnier, J. Am. Chem. Soc., 2006, 128, 9693 CrossRef CAS.
- G. L. Wang, P. P. Yu, J. J. Xu and H. Y. Chen, J. Phys. Chem. C, 2009, 113, 11142 CAS.
- H. B. Yildiz, R. Freeman, R. Gill and I. Willner, Anal. Chem., 2008, 80, 2811 CrossRef CAS.
- M. Hojeij, B. Su, S. X. Tan, G. Mériguet and H. H. Girault, ACS Nano, 2008, 2, 984 CrossRef CAS.
- A. P. Alivisatos, Science, 1996, 271, 933 CAS.
- G. F. Jie, L. Wang, J. X Yuan and S. S. Zhang, Anal. Chem., 2011, 83, 3873 CrossRef CAS.
- M. Nirmal and L. Brus, Acc. Chem. Res., 1999, 32, 407 CrossRef CAS.
- C. J. Murphy and J. L. Coffer, Appl. Spectrosc., 2002, 56, 16A CrossRef CAS.
- V. I. Klimov, Los Alamos Science, 2003, 28, 214 CAS.
- B. Su, D. J. Fermin, J. P. Abid, N. Eugster and H. H. Girault, J. Electroanal. Chem., 2005, 583, 241 CrossRef CAS.
- K. Cottingham, Anal. Chem., 2005, 77, 354A CrossRef CAS.
- A. N. Shipway, E. Katz and I. Willner, ChemPhysChem, 2000, 1, 18 CrossRef CAS.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS.
- L. Sheeney-Haj-Ichia, B. Basnar and I. Willner, Angew. Chem., Int. Ed., 2005, 44, 78 CrossRef CAS.
- S. G. Hickey, D. J. Riley and E. J. Tull, J. Phys. Chem. B, 2000, 104, 7623 CrossRef CAS.
- M. Miyake, T. Torimoto, T. Sakata, H. Mori and H. Yoneyama, Langmuir, 1999, 15, 1503 CrossRef CAS.
- H. Jensen, D. J. Fermin, J. E. Moser and H. H. Girault, J. Phys. Chem. B, 2002, 106, 10908 CrossRef CAS.
- B. O. Dabbousi, M. G. Bawendi, O. Onitsuka and M. F. Rubner, Appl. Phys. Lett., 1995, 66, 1316 CrossRef CAS.
- H. Mattoussi, L. H. Radzilowski, B. O. Dabbousi, E. L. Thomas, M. G. Bawendi and M. F. Rubner, J. Appl. Phys., 1998, 83, 7965 CrossRef CAS.
- N. Tessler, V. Medvedev, M. Kazes, S. H. Kan and U. Banin, Science, 2002, 295, 1506 CrossRef.
- C. Bechinger, S. Ferrer, A. Zaban, J. Sprague and B. A. Gregg, Nature, 1996, 383, 608 CrossRef CAS.
- F. Patolsky, R. Gill, Y. Weizmann, T. Mokari, U. Banin and I. Willner, J. Am. Chem. Soc., 2003, 125, 13918 CrossRef CAS.
- I. Willner, F. Patolsky and J. Wasserman, Angew. Chem., 2001, 113, 1913 CrossRef.
- R. Gill, M. Zayats and I. Willner, Angew. Chem., Int. Ed., 2008, 47, 7602 CrossRef CAS.
- V. Pardo-Yissar, E. Katz, J. Wasserman and I. Willner, J. Am. Chem. Soc., 2003, 125, 622 CrossRef CAS.
- X. R. Zhang, S. G. Li, X. Jin and X. M. Li, Biosens. Bioelectron., 2011, 26, 3674 CrossRef CAS.
- M. Hojeij, B. Su, S. X. Tan, G. Mériguet and H. H. Girault, ACS Nano, 2008, 2, 984 CrossRef CAS.
- D. Baş and İ. H. Boyacι, Electroanalysis, 2009, 21, 1829 CrossRef.
- A. Ikeda, M. Nakasu, S. Ogasawara, H. Nakanishi, M. Nakamura and Jun-ichi Kikuchi, Org. Lett., 2009, 11, 1163 CrossRef CAS.
- G. B. Schuster, Acc. Chem. Res., 2000, 33, 253 CrossRef CAS.
- B. Armitage, Chem. Rev., 1998, 98, 1171 CrossRef CAS.
- A. E. Alegŕıa, A. Ferrer, G. Santigo, E. Sepúlveda and W. J. Flores, J. Photochem. Photobiol., A, 1999, 127, 57 CrossRef.
- K. K. Mothilal, J. J. Inbaraj, R. Gandhidasan and R. J. Murugesan, J. Photochem. Photobiol., A, 2004, 162, 9 CrossRef CAS.
- P. C. Pandeyt and H. H. Weetall, Anal. Chem., 1994, 66, 1236 CrossRef.
- P. Tissot and A. Huissoud, Electrochim. Acta, 1996, 41, 2451 CrossRef CAS.
- S. W. Han, S. W. Joo, T. H. Ha, Y. Kim and K. J. Kim, J. Phys. Chem. B, 2000, 104, 11987 CrossRef CAS.
- T. Ossowski, P. Pipka, A. Liwo and D. Jeziorek, Electrochim. Acta, 2000, 45, 3581 CrossRef CAS.
- A. Salimi, C. E. Banks and R. G. Compton, Phys. Chem. Chem. Phys., 2003, 5, 3988 RSC.
- K. Yamana, N. Kawakami, T. Ohtsuka, Y. Sugie, H. Nakano and I. Saito, Nucleic Acids Symp. Ser., 2003, 3, 89 CrossRef CAS.
- A. Okamoto, T. Kamei, K. Tanaka and I. Saito, J. Am. Chem. Soc., 2004, 126, 14732 CrossRef CAS.
- H. Yamada, K. Tanabe and S. Nishimoto, Org. Biomol. Chem., 2008, 6, 272 CAS.
- K. Haruna, H. Iida, K. Tanabe and S. Nishimoto, Org. Biomol. Chem., 2008, 6, 1613 CAS.
- D. Dong, D. Zheng, F. Q. Wang, X. Q. Yang, N. Wang, Y. G. Li, L. H. Guo and J. Cheng, Anal. Chem., 2004, 76, 499 CrossRef CAS.
- K. Jiang, H. Xie and W. Zhan, Langmuir, 2009, 25, 11129 CrossRef CAS.
- A. Kira, T. Umeyama, Y. Matano, K. Yoshida, S. Isoda, J. K. Park, D. Kim and H. Imahori, J. Am. Chem. Soc., 2009, 131, 3198 CrossRef CAS.
- K. Kaunisto, T. Vuorinen, H. Vahasalo, V. Chukharev, N. V. Tkachenko, A. Efimov, A. Tolkki, H. Lehtivuori and H. Lemmetyinen, J. Phys. Chem. C, 2008, 112, 10256 CAS.
- W. W. Tu, Y. T. Dong, J. P. Lei and H. G. Ju, Anal. Chem., 2010, 82, 8711 CrossRef CAS.
- W. W. Tu, J. P. Lei, P. Wang and H. G. Ju, Chem.–Eur. J., 2011, 17, 9440 CrossRef CAS.
- W. Lu, G. Wang, Y. Jin, X. Yao, J. Q. Hu and J. H. Li, Appl. Phys. Lett., 2006, 89, 263902 CrossRef.
- H. J. Zhao, D. L. Jiang, S. Q. Zhang, K. Catterall and R. John, Anal. Chem., 2004, 76, 155 CrossRef CAS.
- N. Chandrasekharan and P. V. Kamat, J. Phys. Chem. B, 2000, 104, 10851 CrossRef CAS.
- H. X. Li, Z. F. Bian, J. Zhu, Y. N. Huo, H. Li and Y. F. Lu, J. Am. Chem. Soc., 2007, 129, 4538 CrossRef CAS.
- N. Zhou, J. Wang, T. Chen, Z. G. Yu and G. X. Li, Anal. Chem., 2006, 78, 5227 CrossRef CAS.
- S. Phadtare, A. Kumar, V. P. Vinod, C. Dash, D. V. Palaskar, M. Rao, P. G. Shukla, S. Sivaram and M. Sastry, Chem. Mater., 2003, 15, 1944 CrossRef CAS.
- W. Zhu, Y. R. An, X. M. Luo, F. Wang, J. H. Zheng, L. L. Tang, Q. J. Wang, Z. H. Zhang, W. Zhang and L. T. Jin, Chem. Commun., 2009, 19, 2682 RSC.
- Y. R. An, L. L. Tang, X. L. Jiang, H. Chen, M. C. Yang, L. T. Jin, S. P. Zhang, C. G. Wang and W. Zhang, Chem.–Eur. J., 2010, 16, 14439 CrossRef CAS.
- R. Vogel, P. Hoyer and H. Weller, J. Phys. Chem., 1994, 98, 3183 CrossRef CAS.
- P. A. Sant and P.V. Kamat, Phys. Chem. Chem. Phys., 2002, 4, 198 RSC.
- C. Nasr, P. V. Kamat and S. Hotchandani, J. Phys. Chem. B, 1998, 102, 10047 CrossRef CAS.
- G. L. Wang, J. J. Xu, H. Y. Chen and S. Z. Fu, Biosens. Bioelectron., 2009, 25, 791 CrossRef CAS.
- L. Sheeney-Haj-Ichia, S. Pogorelova, Y. Gofer and I. Willner, Adv. Funct. Mater., 2004, 14(5), 416 CrossRef CAS.
- H. B. Yildiz, R. Tel-Vered and I. Willner, Adv. Funct. Mater., 2008, 18, 3497 CrossRef CAS.
- G. Centi and S. Perathoner, Eur. J. Inorg. Chem., 2009, 26, 3851 CrossRef.
- N. M. Gabor, Z. H. Zhong, K. Bosnick, J. Park and P. L. McEuen, Science, 2009, 325, 1367 CrossRef CAS.
- A. Kongkanand, R. M. Dominguez and P. V. Kamat, Nano Lett., 2007, 7, 676 CrossRef CAS.
- D. M. Guldi, G. M. A. Rahman, V. Sgobba, N. A. Kotov, D. Bonifazi and M. Prato, J. Am. Chem. Soc., 2006, 128, 2315 CrossRef CAS.
- B. Farrow and P. V. Kamat, J. Am. Chem. Soc., 2009, 131, 11124 CrossRef CAS.
- I. Robel, B. A. Bunker and P. V. Kamat, Adv. Mater., 2005, 17, 2458 CrossRef CAS.
- L. Hu, Y. L. Zhao, K. Ryu, C. Zhou, J. F. Stoddart and G. Gruner, Adv. Mater., 2008, 20, 939 CrossRef CAS.
- C. X. Guo, H. B. Yang, Z. M. Sheng, Z. S. Lu, Q. L. Song and C. M. Li, Angew. Chem., Int. Ed., 2010, 49, 3014 CrossRef CAS.
- P. V. Kamat, J. Phys. Chem. C, 2008, 112, 18737 CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguye and R. S. Ruoff, Nature, 2006, 442, 282 CrossRef CAS.
- M. J. Alle, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132 CrossRef.
- F. Bonaccorso, Z. Sun, T. Hasan and A. C. Ferrari, Nat. Photonics, 2010, 4, 611 CrossRef CAS.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752 CrossRef CAS.
- M. I. Katsnelson, Mater. Today, 2007, 10, 20 CrossRef CAS.
- X. Wan, G. Long, L. Huang and Y. Chen, Adv. Mater., 2011, 23, 5342 CrossRef CAS.
- X. M. Geng, L. Niu, Z. Y. Xing, R. S. Song, G. T. Liu, M. T. Sun, G. S. Cheng, H. J. Zhong, Z. H. Liu, Z. J. Zhang, L. F. Sun, H. X. Xu, L. Lu and L. W. Liu, Adv. Mater., 2010, 22, 638 CrossRef CAS.
- H. X. Chang, X. J. Lv, H. Zhang and J. H. Li, Electrochem. Commun., 2010, 12, 483 CrossRef CAS.
- X. R. Zhang, S. G. Li, X. Jin and S. S. Zhang, Chem. Commun., 2011, 47, 4929 RSC.
- X. M. Zhao, S. W. Zhou, L. P. Jiang, W. H. Hou, Q. M. Shen and J. J. Zhu, Chem.–Eur. J., 2012, 18, 4974 CrossRef CAS.
- X. M. Zhao, S. W. Zhou, Q. M. Shen, L. P. Jiang and J. J. Zhu, Analyst, 2012, 137, 3697 RSC.
- K. Jasuja and V. Berry, ACS Nano, 2009, 3, 2358 CrossRef CAS.
- X. J. Zhou, J. L. Zhang, H. X. Wu, H. J. Yang, J. Y. Zhang and S. W. Guo, J. Phys. Chem. C, 2011, 115, 11957 CAS.
- J. L. Lyon, D. A. Fleming, M. B. Stone, P. Schiffer and M. E. Williams, Nano Lett., 2004, 4, 719 CrossRef CAS.
- W. W. Zhao, P. P. Yu, Y. Shan, J. Wang, J. J. Xu and H. Y. Chen, Anal. Chem., 2012, 84, 5892 CrossRef CAS.
- W. W. Zhao, J. Wang, J. J. Xu and H. Y. Chen, Chem. Commun., 2011, 47, 10990 RSC.
- W. W. Zhao, Z. Y. Ma, P. P. Yu, X. Y. Dong, J. J. Xu and H. Y. Chen, Anal. Chem., 2012, 84, 917 CrossRef CAS.
- X. R. Zhang, Y. Q. Zhao, H. R. Zhou and B. Qu, Biosens. Bioelectron., 2011, 26, 2737 CrossRef CAS.
- C. F. Ding, H. Li, X. L. Li and S. S. Zhang, Chem. Commun., 2010, 46, 7990 RSC.
- E. Golub, A. Niazov, R. Freeman, M. Zatsepin and I. Willner, J. Phys. Chem. C, 2012, 116, 13827 CAS.
- Y. S. Guo, Y. S. Sun and S. S. Zhang, Chem. Commun., 2011, 47, 1595 RSC.
| This journal is © The Royal Society of Chemistry 2013 |