Synthesis of self-assembled prismatic iron oxide nanoparticles by a novel thermal decomposition route
Geetu
Sharma
and
Pethaiyan
Jeevanandam
*
Department of Chemistry Indian Institute of Technology Roorkee, Roorkee-247667, India. E-mail: jeevafcy@iitr.ernet.in; Fax: 91-1332-286202; Tel: 91-1332-285444
First published on 25th October 2012
Abstract
Magnetic nanostructured materials with novel morphologies are often required for interesting applications. In the present study, iron oxide nanoparticles with prismatic hexagonal morphology were synthesized using a novel low temperature thermal decomposition approach. The thermal decomposition of [Fe(CON2H4)6](NO3)3 in diphenyl ether at about 200 °C for 70 min leads to the formation of prismatic iron oxide nanoparticles. The prismatic particles were found to be made up of self-assembled iron oxide nanoparticles. The iron oxide nanoparticles were characterized using X-ray diffraction, thermal gravimetric analysis, elemental analysis, infrared spectroscopy, field emission-scanning electron microscopy coupled with energy dispersive X-ray analysis, and magnetic measurements. The mechanism of formation of prismatic iron oxide nanoparticles has also been investigated.
Introduction
Magnetic nanoparticles have gained considerable attention recently because of their potential technological applications.1,2 Among the magnetic nanoparticles, iron oxide has been widely investigated because of its applications in data storage,3 ferrofluids,4 biosensors,5 bioprobes,6 contrast agents for magnetic resonance imaging,7 catalysis,8etc. Iron oxide nanoparticles possess tunable magnetic properties according to their size,9,10 shape11,12 and phase.13 Two factors that dominate the magnetic behavior of nanoparticles are size and surface anisotropy.14 For example Hyeon et al. have reported an increase in the blocking temperature (TB) with an increase in particle size of γ-Fe2O3 nanoparticles.10 Chalasani et al. have reported differences in the blocking temperatures of spherical and cubic shaped iron oxide nanoparticles of comparable dimensions.11 Differences in surface anisotropy and exchange bias lead to differences in the blocking temperature between the spherical and cubic nanoparticles.11 α-Fe2O3 nanoparticles with truncated hexagonal bipyramidal morphology show weak ferromagnetism below the Morin transition at 260 K due to the high surface energy of the exposed {101} and {001} facets.15 γ-Fe2O3 nanowires, synthesized by flame vapor deposition, exhibit single domain structure and low coercivity at room temperature due to their superior crystallinity.16Self-assembly of magnetic nanoparticles into organized and designed structures is of great interest since the organized assemblies exhibit interesting magnetic properties resulting from structural organizations.17 The nanostructures possess wide range of applications in the field of magnetic and magneto-electronic devices.18,19 Various synthetic methods have been reported for the preparation of iron oxide nanoparticles which include co-precipitation,20 ultrasound irradiation,21 solvothermal method,22 hydrothermal synthesis,23 microemulsion,24 microwave synthesis25 and thermal decomposition.26–28 Among the reported methods, thermal decomposition of iron precursors in organic media has been widely used for the synthesis of iron oxide nanoparticles as their size and morphology can be easily controlled.29,30 The commonly used iron precursors in the thermal decomposition approach are Fe(acac)3,29 Fe(CO)5,31 Fe(Cup)3,32 iron carboxylates (e.g. oleates, myristates, palmitates),33 iron oxyhydroxide34 and [Fe(η5-C6H3Me4)2].35 Recently, different shapes of iron oxide nanoparticles such as star,33 sphere,9 cube,36 octahedron,37 worm,38 diamond, prism and hexagon,39 hollow sphere,40 and truncated triangle41 have been synthesized by the thermal decomposition approach and the shape control has been achieved using different ligands. Out of different reported methods for the synthesis of self-assembled nanostructures, thermal decomposition is the easiest one for the synthesis of monodisperse iron oxide nanoparticles which can self assemble into various organized structures.37,41–43
The reported thermal decomposition methods often require high temperatures (e.g. 250–350 °C), inert conditions (e.g. Ar and N2 atmospheres), ligands, and sometimes involve complicated precursor synthesis (e.g. iron-oleate). Simple and economical routes for the synthesis of iron oxide nanostructures with controlled morphologies are often explored. In the present study, iron oxide nanoparticles have been synthesized using the thermal decomposition of an iron–urea complex, iron(III) hexakisurea trinitrate ([Fe(CON2H4)6](NO3)3), in diphenyl ether without the need for the use of any capping agent and special conditions. The iron oxide nanoparticles self-assemble into prismatic structures depending on the synthetic conditions.
2. Results and discussion
The XRD patterns of as prepared and calcined iron oxide samples are shown in Fig. 1. The as prepared sample is X-ray amorphous. The sample, after calcination at 350 °C, shows weak reflections at 2θ values of 35.30° and 62.55° that can be attributed to either Fe3O4 (JCPDS file no. 85-1436) or γ-Fe2O3 (JCPDS file no. 25-1402); the lattice spacing (d) values of these two phases are similar. Since, in the present study, the thermal decomposition of the iron–urea complex is carried out in air and under refluxing conditions, the as prepared sample is proposed to be γ-Fe2O3. Further evidence for the presence of γ-Fe2O3 in the as prepared sample comes from magnetic measurement results discussed later. After calcination at 500 °C, well defined diffraction peaks due to α-Fe2O3 (JCPDS file no. 85-0987) are observed. Elemental analysis of the iron-urea complex indicated the weight percent of carbon, nitrogen and hydrogen as 11.7, 34.9 and 3.9, respectively which is close to the theoretical values (% C = 11.97; % N = 34.89; % H = 4.02).44 Elemental analysis of the as prepared iron oxide indicated the weight percent of C, N and H as 3.3, 3.7 and 2.2, respectively with a carbon to nitrogen ratio of close to unity (0.9), suggesting the presence of organic moieties on the surface of iron oxide nanoparticles.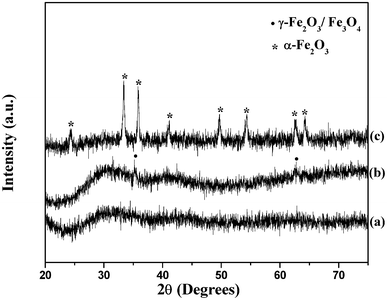 | ||
| Fig. 1 XRD patterns of iron oxide nanoparticles; (a) as prepared, (b) after calcination at 350 °C and (c) after calcination at 500 °C. | ||
Fig. 2a shows the TGA curve for the iron–urea complex along with that of as prepared iron oxide. The observed decomposition pattern of the iron–urea complex matches with the reported behavior.44 The iron–urea complex decomposes at about 210 °C to ferric nitrate and urea; the ferric nitrate further decomposes to iron oxide with the evolution of NO2, NO and O2. In the as prepared iron oxide, the total weight loss observed is about 25%. The first decomposition step at about 100 °C is attributed to the loss of adsorbed water molecules. The weight loss observed in the temperature range 200–330 °C is attributed to the removal of organic moieties on the surface of iron oxide nanoparticles. The organic moieties are proposed to be cyanuric acid molecules on the basis of elemental analysis, DTA and IR results discussed later.
 | ||
| Fig. 2 (a) TGA and (b) DTA curves of (i) iron–urea complex and (ii) as prepared iron oxide sample. | ||
The DTA curves for the iron–urea complex and the as prepared iron oxide are shown in Fig. 2b. An endothermic peak observed at 177 °C in the case of the iron–urea complex is due to the melting of the complex as reported.44 The exothermic peak at 210 °C is attributed to the decomposition of [Fe(CON2H4)6](NO3)3 to ferric nitrate and its subsequent decomposition to iron oxide. The broad exothermic hump between 400 °C and 600 °C is attributed to the phase transformation of γ-Fe2O3 to α-Fe2O3. The as prepared iron oxide shows two exothermic peaks at 317 °C and 489 °C. The exothermic peak at 317 °C is attributed to the decomposition of cyanuric acid present on the surface of iron oxide nanoparticles.45 The exothermic peak at 489 °C is attributed to the phase transformation of γ-Fe2O3 to α-Fe2O3.
In the present study, τhe γ-Fe2O3 to α-Fe2O3 transition occurs as a broad hump between 400 °C and 600 °C in the DTA pattern in the case of iron oxide nanoparticles prepared from the solid state thermal decomposition of the iron–urea complex (curve (i), Fig. 2b). The same transformation in the case of prismatic iron oxide nanoparticles occurs at 489 °C (curve (ii), Fig. 2b). The γ-Fe2O3 to α-Fe2O3 transition occurs when the size of the γ-Fe2O3 nanoparticles reaches a certain critical value between 10 and 25 nm.46,47 The broad exothermic hump in curve (i) is attributed to the broad size distribution of γ-Fe2O3 nanoparticles which undergoes the transition at different temperatures. On the other hand, the sharp exothermic peak for the prismatic iron oxide nanoparticles (curve (ii)) is attributed to the narrow size distribution of γ-Fe2O3 nanoparticles. The lower transition temperature (489 °C) in the case of prismatic iron oxide nanoparticles is attributed to the smaller particle size of iron oxide nanoparticles compared to the iron oxide nanoparticles produced by the direct solid state decomposition of the iron–urea complex. It is known that the γ-Fe2O3 to α-Fe2O3 transition temperature decreases when size of the nanoparticles is decreased.48,49
The IR spectra of the as prepared and calcined iron oxide samples are shown in Fig. 3. The band positions and their assignments are given in Table 1. All the samples show bands near 3400 cm−1 and 1630 cm−1 attributed to O–H stretching and bending, respectively due to physisorbed water molecules. The as prepared iron oxide shows a band at 3413 cm−1 due to N–H stretching.50 It also shows additional bands at 1715 cm−1, 1469 cm−1, 1375 cm−1 and 1015 cm−1. The band at 1715 cm−1 is attributed to carbonyl stretching of cyanuric acid.50 The band at 1469 cm−1 corresponds to C–N stretching.45 The bands around 1375 cm−1 and 1015 cm−1 are attributed to the bending vibrations of carbonyl and N–H groups, respectively.50 The FT-IR results give evidence for the presence of cyanuric acid in the as prepared iron oxide. The sample also shows bands at about 590 cm−1 and 430 cm−1 attributed to γ-Fe2O3.51 The bands at about 540 cm−1 and 460 cm−1 in the calcined iron oxide samples are the characteristic bands due to α-Fe2O3.52
 | ||
| Fig. 3 FT-IR spectra of as prepared and calcined samples of iron oxide nanoparticles. | ||
| Band positions (cm−1) | Assignment | ||
|---|---|---|---|
| As prepared | Calcined at 350 °C | Calcined at 500 °C | |
| 3413 | 3440 | 3445 | ν(N–H) and ν(O–H) |
| 1715 | — | — |
ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) O) |
| 1630 | 1630 | 1630 | δ(O–H) |
| 1469 | — | — | ν(C–N) |
| 1375 | — | — |
δ(CO) |
| 1015 | — | — | δ(N–H) |
| 590 | 548 | 537 | ν(Fe–O) |
| 430 | 457 | 460 | ν(Fe–O) |
FE-SEM images of as prepared iron oxide at different magnifications are shown in Fig. 4. The SEM images show the formation of self assembled prismatic structures on an iron oxide substrate; the evidence for the presence of iron oxide as the substrate comes from EDXA results discussed later. The image at higher magnification clearly shows that each prismatic structure is made up of small particles (Fig. 4c). To understand the formation of prismatic structures on the iron oxide substrate, SEM images of iron oxide samples prepared at different reaction time intervals (5, 15, 35 and 70 min) were recorded and are shown in Fig. 5. At a reaction time of 5 min, small particles are observed. With an increase in the reaction time (e.g. 15 min), bigger spherical aggregates (mean diameter = 199 ± 96 nm) are observed (Fig. 5b). The SEM image of an iron oxide sample synthesized at 35 min shows the formation of distorted prisms. At a reaction time of 70 min, the formation of hexagonal prisms is complete (Fig. 5d).
 | ||
| Fig. 4 FE-SEM images of the as prepared iron oxide at different magnifications. | ||
 | ||
| Fig. 5 FE-SEM images of the as prepared iron oxide samples prepared at different reaction intervals: (a) 5 min, (b) 15 min, (c) 35 min and (d) 70 min. | ||
TEM observations would have been more useful to look at the fine features of the prismatic iron oxide nanostructures. However, the attempts to do TEM measurements for the prisms were not fruitful. The self assembled prismatic structures of iron oxide nanoparticles are formed on big lumps of iron oxide substrate (see Fig. 4a). The substrate itself is made up of very small iron oxide particles (see Fig. 6b). Since the substrate on which prismatic iron oxide nanostructures are formed is thick (about 2.5 to 10.5 μm), the electron beam is not able to pass through and provide good image of the prisms. It may be noted that the thickness of the sample required for transmission electron microscopy measurements should be usually less than 200 nm.53
EDX analysis indicated the presence of O and Fe in the as prepared and calcined iron oxide samples. In addition to these elements, the analysis also indicated the presence of carbon and nitrogen in the as prepared iron oxide. The percentage of carbon and nitrogen was found to be more on the prismatic structures than on the non-prismatic structures, i.e. the substrate (Fig. 6). In the prismatic structures, the weight percentage of carbon varied from 21.1 to 34.0 and that of nitrogen varied from 8 to 14.8. In the non-prismatic structures (i.e. the substrate), the percentage of carbon varied from 9.4 to 16.3 and that of nitrogen varied from 3.3 to 3.8. The iron content varied from 46.4 to 59.3 in the prisms and 69.7 to 71.9 in the substrate. The EDXA results prove the presence of more organic content on the prismatic structures compared to that on the substrate. On calcination of the as prepared iron oxide, the carbon content decreased to about 5.6 to 6.0% at 350 °C and to 0% at 500 °C. The iron content in calcined iron oxide samples varied from 85 to 86% after calcination at 350 °C and it varied from 88 to 89% on calcination at 500 °C. To see the effect of heat treatment on the prismatic morphology of the as prepared iron oxide, SEM images were recorded after calcination at 350 °C and 500 °C. It can be observed from the SEM images (Fig. 7) that the prismatic morphology is lost on calcination.
 | ||
| Fig. 6 EDXA patterns of as prepared iron oxide sample: (a) analysis on prismatic structures, and (b) analysis on non-prismatic areas. | ||
 | ||
| Fig. 7 FE-SEM images of prismatic iron oxide particles after calcination at (a) 350 °C, and (b) 500 °C. | ||
The EDX analysis was also performed for the iron oxide samples in which spherical aggregates (reaction time = 15 min) and distorted prisms (reaction time = 35 min) were observed in the SEM analysis. The analysis on the spherical aggregates and the distorted prisms also indicated more weight percent of carbon (27.6%) and nitrogen (8.1%) compared to that on the substrate (% C = 12.2; % N= 1.6). This suggests that the chemical nature of the organic content is the same in the spherical aggregates, distorted prisms and completely formed prisms.
Field dependent magnetization (M–H) plots for the as prepared and calcined iron oxide samples at 300 K and 5 K are shown in Fig. 8. The observed magnetic parameters for the iron oxide nanoparticles such as coercivity (Hc) and remnant magnetization (Mr) have been summarized in Table 2. The as prepared iron oxide exhibits weak superparamagnetic behavior at 300 K, whereas at 5 K, ferromagnetic behavior is observed with a large coercivity of 2816 Oe (Fig. 8a). This indicates that the superparamagnetic blocking temperature is below room temperature for the iron oxide nanoparticles.54 The magnetization does not saturate at both the temperatures at a maximum field of 50 kOe. A careful observation of the hysteresis loop indicated that it is shifted towards the negative field at 5 K (see the inset in Fig. 8a). This indicates the presence of exchange anisotropy in γ-Fe2O3 nanoparticles.55,56 The observed magnetic behavior for the as prepared iron oxide is similar to that reported in the literature for γ-Fe2O3 nanoparticles.55–57
 | ||
| Fig. 8 M–H curves at 300 and 5 K for the iron oxide nanoparticles: (a) as prepared, (b) after calcination at 350 °C, and (c) after calcination at 500 °C. | ||
| Sample | H c (Oe) | M r (emu/g) | T B (K) | T M (K) | ||
|---|---|---|---|---|---|---|
| 5 K | 300 K | 5 K | 300 K | |||
| As-prepared iron oxide | 2816 | Negligible | 0.21 | Negligible | 42 | — |
| After calcination at 350 °C | 1813 | 1103 | 0.62 | 0.35 | >300 K | — |
| After calcination at 500 °C | 730 | 3076 | 0.002 | 0.016 | >300 K | 250 K |
After calcination at 350 °C, ferromagnetic behavior with hysteresis loops is observed at 300 K and 5 K with Hc values of 1103 Oe and 1813 Oe, respectively (Fig. 8b). The decrease in the Hc value after calcination at 350 °C is attributed to the increase in particle size of iron oxide.58 After calcination of the as prepared iron oxide at 500 °C, coercivity values of 3076 Oe at 300 K and 730 Oe at 5 K were observed (Fig. 8c). Bulk α-Fe2O3 is an antiferromagnet with a high Neel temperature (TN) of 960 K59 which undergoes magnetic phase transition from a canted state to an antiferromagnetically ordered one at about 260 K, called the Morin transition.59 At 300 K (i.e. above the Morin transition), α-Fe2O3 is known to exhibit ferromagnetic behavior.60 Because of this transition, the Hc value increases from 1103 Oe to 3076 Oe on increasing the calcination temperature from 350 °C to 500 °C. At T ≪ TM (e.g. 5 K), the Hc value decreases from 1813 Oe to 730 Oe and this is attributed to the antiferromagnetic behavior of α-Fe2O3.60
The zero field cooled (ZFC) and field cooled (FC) magnetization curves measured in a field of 100 Oe as a function of temperature are shown in Fig. 9. In the as prepared iron oxide, the ZFC curve (Fig. 9a) exhibits a sharp maximum at about 42 K indicating the superparamagnetic blocking transition. The decrease of magnetization in the FC curve below the blocking temperature is attributed to the strong interparticle interactions in γ-Fe2O3 nanoparticles.54,61,62 From the blocking temperature (TB), the average particle size of γ-Fe2O3 nanoparticles was estimated using the following formula:63
| TB = KeffV/25 kB |
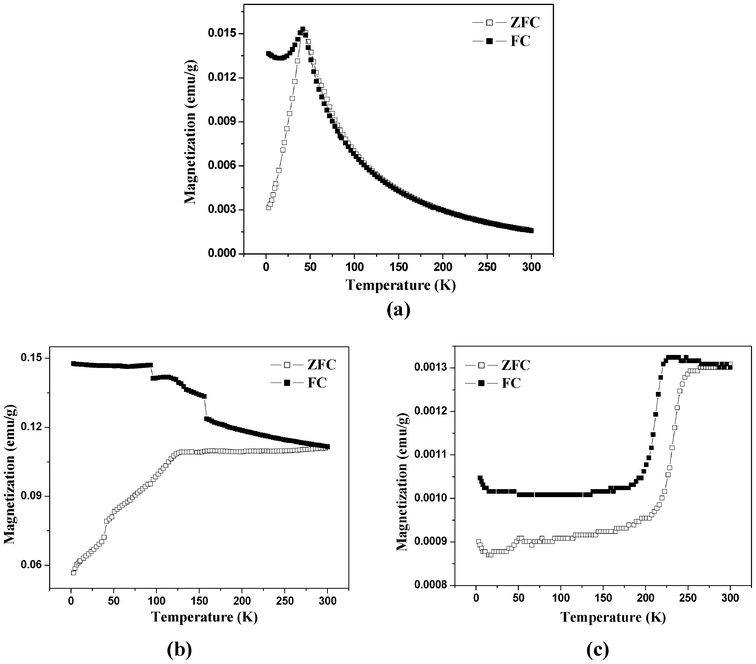 | ||
| Fig. 9 ZFC–FC curves under 100 Oe for the iron oxide nanoparticles: (a) as prepared, (b) after calcination at 350 °C, and (c) after calcination at 500 °C. | ||
Fig. 9c shows the ZFC and FC magnetization curves for the iron oxide after calcination at 500 °C. The ZFC and FC magnetization curves give clear evidence for the Morin transition at 250 K confirming the formation of α-Fe2O3 nanoparticles after calcination at 500 °C.
On the basis of observed TG-DTA, elemental and FT-IR results, the proposed sequence of the reactions taking place during the thermal decomposition of the iron–urea complex with the formation of iron oxide nanoparticles is depicted in Scheme 1.44,45,50 On heating the iron–urea complex ([Fe(CON2H4)6](NO3)3), melting occurs at 178 °C with a simultaneous decomposition to ferric nitrate and urea. Ferric nitrate decomposes to form iron oxide at about 200 °C with the evolution of NO, NO2 and O2. Conversion of urea to isocyanuric acid and ammonia occurs above 140 °C. The isocyanuric further reacts with urea at 190 °C to form biuret which converts to cyanuric acid. Thus, on thermal decomposition of the iron–urea complex, cyanuric acid is formed along with the iron oxide nanoparticles. The cyanuric acid acts as the capping agent for iron oxide nanoparticles and thus there is no need for the external addition of any capping agent (ligand) during the synthesis. The proposed mechanism of the formation of iron oxide prisms is given in Scheme 2. The iron oxide nanoparticles coated with cyanuric acid self assemble to form spherical aggregates (see Fig. 5b). With an increase in the reaction time, the spherical structures convert into prismatic structures. Aggregates of iron oxide nanoparticles act as the substrate for the self assembly.
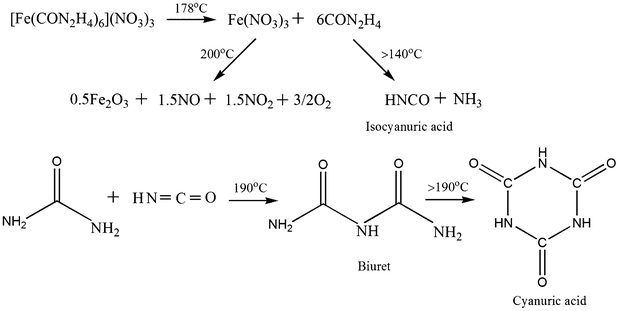 | ||
| Scheme 1 Possible reactions involved in the formation of iron oxide nanoparticles. | ||
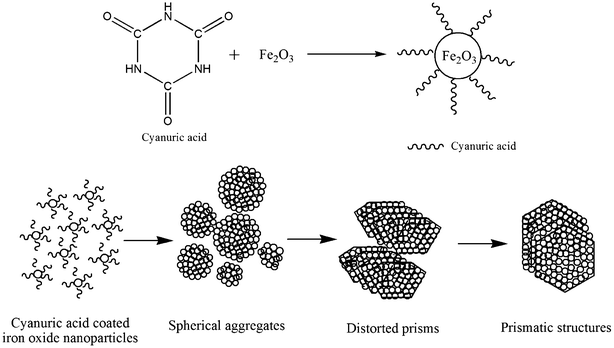 | ||
| Scheme 2 Illustration of the formation of prismatic iron oxide nanoparticles. | ||
In the present study, reaction time was found to have a great influence on the morphology of iron oxide. It was found that as the reaction time was increased from 5 min to 70 min, the iron oxide nanoparticles first self assemble to form spherical structures (see Fig. 5b) which later on convert to hexagonal prismatic structures. With regards to the size, the concentration (amount) of the iron–urea complex was found to be the key factor. For example, when 250 mg of the complex was employed during the thermal decomposition, the mean edge length of the hexagonal prisms was 110 ± 37 nm. On the other hand, when 500 mg of the iron–urea complex was used, the mean edge length of the prisms increased to 170 ± 38 nm.
Self assembly is a spontaneous process which can be directed through non-covalent interactions (e.g. hydrogen bonding, van der Waals, electrostatic, etc).66,67 In the present study, iron oxide nanoparticles self assemble to form prismatic structures because of the interparticle interactions. The cyanuric acid molecules present on the surface of iron oxide nanoparticles facilitate the self assembly. γ-Fe2O3 has an inverse spinel structure with three low-energy surfaces: {100}, {110}, and {111}.39 Particles with a prismatic morphology are formed when the ligand (cyanuric acid) is preferentially bound to the highest energy {111} surface among the above three surfaces.39 Cheon et al. have synthesized hexagonal shaped nanocrystals of γ-Fe2O3 using a time consuming thermolysis method (9 h) at 180 °C using dodecylamine as a ligand.39 Zhao et al. have used the same complex as that reported in the present study ([Fe(CON2H4)6](NO3)3) for the synthesis of γ-Fe2O3 nanoparticles by thermal decomposition at 200 °C.44 However, spherical iron oxide nanoparticles are observed and the decomposition was carried out in the solid state. In the present study, iron oxide nanostructures with prismatic morphologies are formed when the iron–urea complex is subjected to thermal decomposition at 200 °C for 70 min in diphenyl ether. The in situ formation of cyanuric acid and its effective capping action leads to the formation of prismatic structures due to the self-assembly of iron oxide nanoparticles. The synthesis does not require external addition of a capping agent (ligand).
Organized assemblies of magnetic nanoparticles exhibit interesting magnetic properties such as high saturation magnetization,68 high coercivity69 and enhanced electromagnetic loss.70 In the present study, the self assembled iron oxide nanostructures retain superparamagnetic behavior at room temperature. The advantages of such assembly are: (i) they can be easily separated from solutions by applying a moderate magnetic field which makes them suitable for biomedical applications such as drug delivery and bio-separation71 and (ii) the self-assembled iron oxide nanoparticles may improve contrast in magnetic resonance imaging compared to individual nanoparticles.72
3. Experimental methods
Reagents
All the reagents were of analytical grade and were used as received. Ferric nitrate nonahydrate was received from SD Fine Chemicals Ltd., urea was received from Rankem, and diphenyl ether was received from Sigma Aldrich.Synthesis
The synthesis of [Fe(CON2H4)6](NO3)3 was carried out as reported.44 Fe(NO3)3·9H2O and urea in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6.2 molar ratio were dissolved in about 25 mL of ethanol and vigorously stirred for 1 h. The light green colored precipitate obtained was centrifuged, washed repeatedly with ethanol and kept for drying under vacuum. The formation of [Fe(CON2H4)6](NO3)3 was confirmed by comparing the FT-IR, TGA and elemental analysis results for the complex with that reported.44
:6.2 molar ratio were dissolved in about 25 mL of ethanol and vigorously stirred for 1 h. The light green colored precipitate obtained was centrifuged, washed repeatedly with ethanol and kept for drying under vacuum. The formation of [Fe(CON2H4)6](NO3)3 was confirmed by comparing the FT-IR, TGA and elemental analysis results for the complex with that reported.44
For the preparation of iron oxide nanoparticles, typically, about 500 mg of the iron–urea complex was taken in 10 mL of diphenyl ether in a round bottom flask. Thereafter, the mixture was refluxed in air at 200 °C with stirring for about 70 min. After cooling the contents to room temperature, about 30 mL methanol was added. The precipitate was centrifuged, washed several times with methanol and kept for drying in air. The as prepared sample was calcined at 350 °C and 500 °C in air inside a muffle furnace (Nabertherm®, heating rate = 2 °C min−1).
Characterization
The as prepared and calcined iron oxide samples were characterized using different analytical techniques such as X-ray diffraction, infrared spectroscopy, elemental analysis, thermal gravimetric analysis coupled with differential thermal analysis, scanning electron microscopy coupled with energy dispersive X-ray analysis, and superconducting quantum interference device magnetic measurements. Powder X-ray diffraction patterns were recorded using a Brucker AXS D8 diffractometer with Cu-Kα radiation (λ = 1.5406 Å) with a scan speed of 1° min−1. Thermal gravimetric analysis was carried out using a Perkin Elmer (Pyris Diamond) instrument in the temperature range 25 °C to 800 °C, in air, at a heating rate of 10 °C min−1. The percentages of carbon, hydrogen and nitrogen, present in the iron–urea complex and the as prepared iron oxide, were measured using a Vario ELIII CHNS analyzer. IR spectra were recorded using a Thermo Nicolet Nexus Fourier Transform infrared spectrometer using KBr pellets. The morphology of the iron oxide samples was analyzed using a FEI Quanta 200F scanning electron microscope operating at an accelerating voltage of 20 kV coupled with an energy dispersive X-ray analysis facility. Magnetic measurements (M–H) on sample powders were done at 300 K and 5 K using a Quantum Design Magnetometer (MPMS XL-5) by varying the field up to 50 kOe. Zero field cooled and field cooled measurements were carried out at an applied field of 100 Oe with temperature varying from 300 K to 5 K.4. Conclusions
Prismatic iron oxide nanostructures formed by the self-assembly of iron oxide nanoparticles were successfully synthesized using a low temperature easy thermal decomposition approach. The TG-DTA, FT-IR and elemental analysis results indicate the in situ formation of cyanuric acid during the decomposition of the iron–urea complex which acts as the ligand for the formation of iron oxide nanoparticles. SEM studies indicate the formation of small iron oxide nanoparticles in the beginning which convert to bigger spherical structures, as the reaction proceeds, which ultimately convert into prisms. Magnetic studies reveal superparamagnetic behavior for the prismatic iron oxide nanostructures with a blocking temperature of about 42 K. The present thermal decomposition approach may be extended for the synthesis of other magnetic nanoparticles with novel morphology.Acknowledgements
Financial support from Council of Scientific and Industrial Research, Government of India (Project No: 01 (2311)/09/EMR-II) is gratefully acknowledged. We are thankful to the Institute Instrumentation Centre, Indian Institute of Technology, Roorkee for providing the XRD, TG-DTA and FE-SEM facilities. Thanks are also due to Prof. S. Vasudevan and Mr. Rajesh Chalasani, Department of Inorganic and Physical Chemistry, Indian Institute of Science, Bangalore for their help in carrying out the SQUID measurements.References
- A. Wu, P. Ou and L. Zeng, Nano, 2010, 5, 245 CrossRef CAS.
- J. S. Beveridge, J. R. Stephens and M. E. Williams, Annu. Rev. Anal. Chem., 2011, 4, 251 CrossRef CAS.
- Y. K. Sung, B. W. Ahn and T. J. Kang, J. Magn. Magn. Mater., 2012, 324, 916 CrossRef CAS.
- V. Lobaz, R. N. Klupp Taylor and W. Peukert, J. Colloid Interface Sci., 2012, 374, 102 CrossRef CAS.
- S. Ghoshal, A. A. M. Ansar, S. O. Raja, A. Jana, N. R. Bandyopadhyay, A. K. Dasgupta and M. Ray, Nanoscale Res. Lett., 2011, 6, 540 CrossRef.
- S. J. DeNardo, G. L. DeNardo, L. A. Miers, A. Natarajan, A. R. Foreman, C. Gruettner, G. N. Adamson and R. Ivkov, Clin. Cancer Res., 2005, 11, 7087s CrossRef CAS.
- J. Lu, S. Yang, K. M. Ng, C. H. Su, C. S. Yeh, Y. N. Wu and D. B. Shieh, Nanotechnology, 2006, 17, 5812 CrossRef CAS.
- C. Huang, H. Zhang, Z. Sun, Y. Zhao, S. Chen, R. Tao and Z. Liu, J. Colloid Interface Sci., 2011, 364, 298 CrossRef CAS.
- J. Park, E. Lee, N. M. Hwang, M. Kang, S. C. Kim, Y. Hwang, J. G. Park, H. J. Noh, J. Y. Kim, J. H. Park and T. Hyeon, Angew. Chem., Int. Ed., 2005, 44, 2872 CrossRef CAS.
- T. Hyeon, S. S. Lee, J. Park, Y. Chung and H. B. Na, J. Am. Chem. Soc., 2001, 123, 12798 CrossRef CAS.
- R. Chalasani and S. Vasudevan, J. Phys. Chem. C, 2011, 115, 18088 CAS.
- G. Salazar-Alvarez, J. Qin, V. Sepelak, I. Bergmann, M. Vasilakaki, K. N. Trohidou, J. D. Ardisson, W. A. A. Macedo, M. Mikhaylova, M. Muhammed, M. D. Baro and J. Nogues, J. Am. Chem. Soc., 2008, 130, 13234 CrossRef CAS.
- S. Liu, J. Zhou and L. Zhang, J. Phys. Chem. C, 2011, 115, 3602 CAS.
- A. H. Lu, E. L. Salabas and F. Schuth, Angew. Chem., Int. Ed., 2007, 46, 1222 CrossRef CAS.
- T. K. Van, H. G. Cha, C. K. Nguyen, S. W. Kim, M. H. Jung and Y. S. Kang, Cryst. Growth Des., 2012, 12, 862 CAS.
- P. M. Rao and X. Zheng, Nano Lett., 2011, 11, 2390 CrossRef CAS.
- M. J. Benitez, D. Mishra, P. Szary, G. A. Badini Confalonieri, M. Feyen, A. H. Lu, L. Agudo, G. Eggeler, O. Petracic and H. Zabel, J. Phys.: Condens. Matter, 2011, 23, 126003 CrossRef CAS.
- R. P. Tan, J. Carrey and M. Respaud, J. Appl. Phys., 2008, 104, 023908 CrossRef.
- P. Poddar, T. Fried and G. Markovich, Phys. Rev. B: Condens. Matter, 2002, 65, 172405 CrossRef.
- C. Blanco-Andujar, D. Ortega, Q. A. Pankhurst and N. T. K. Thanh, J. Mater. Chem., 2012, 22, 12498 RSC.
- S. Zhang, Y. Zhang, Y. Wang, S. Liu and Y. Deng, Phys. Chem. Chem. Phys., 2012, 14, 5132 RSC.
- I. M. Grabs, C. Bradtmoeller, D. Menzel and G. Garnweitner, Cryst. Growth Des., 2012, 12, 1469 CAS.
- L. Feng, M. Cao, X. Ma, Y. Zhu and C. Hu, J. Hazard. Mater., 2012, 217-218, 439 CrossRef CAS.
- M. Gotic, T. Jurkin and S. Music, Colloid Polym. Sci., 2007, 285, 793 CAS.
- K. Nadeem, H. Krenn, T. Traussnig, R. Wuerschum, D. V. Szabo and I. Letofsky-Papst, J. Appl. Phys., 2012, 111, 113911 CrossRef.
- F. Zhao, B. Zhang and L. Feng, Mater. Lett., 2012, 68, 112 CrossRef CAS.
- B. H. Kim, N. Lee, H. Kim, K. An, Y. I. Park, Y. Choi, K. Shin, Y. Lee, S. G. Kwon, H. B. Na, J. G. Park, T. Y. Ahn, Y. W. Kim, W. K. Moon, S. H. Choi and T. Hyeon, J. Am. Chem. Soc., 2011, 133, 12624 CrossRef CAS.
- Y. Zhu, F. Y. Jiang, K. Chen, F. Kang and Z. K. Tang, J. Alloys Compd., 2011, 509, 8549 CrossRef CAS.
- P. Guardia, N. Perez, A. Labarta and X. Batlle, Langmuir, 2010, 26, 5843 CrossRef CAS.
- A. Shavel, B. Rodriguez-Gonzalez, J. Pacifico, M. Spasova, M. Farle and L. M. Liz-Marzan, Chem. Mater., 2009, 21, 1326 CrossRef CAS.
- S. Peng, C. Wang, J. Xie and S. Sun, J. Am. Chem. Soc., 2006, 128, 10676 CrossRef CAS.
- J. Rockenberger, E. C. Scher and A. P. Alivisatos, J. Am. Chem. Soc., 1999, 121, 11595 CrossRef CAS.
- L. M. Bronstein, J. E. Atkinson, A. G. Malyutin, F. Kidwai, B. D. Stein, D. G. Morgan, J. M. Perry and J. A. Karty, Langmuir, 2011, 27, 3044 CrossRef CAS.
- C. R. Lin, R. K. Chiang, J. S. Wang and T. W. Sung, J. Appl. Phys., 2006, 99, 08N710 CrossRef.
- D. A. J. Herman, P. Ferguson, S. Cheong, I. F. Hermans, B. J. Ruck, K. M. Allan, S. Prabakar, J. L. Spencer, C. D. Lendrum and R. D. Tilley, Chem. Commun., 2011, 47, 9221 RSC.
- P. Guardia, J. Perez-Juste, A. Labarta, X. Batlle and L. M. Liz-Marzan, Chem. Commun., 2010, 46, 6108 RSC.
- L. Zhang, J. Wu, H. Liao, Y. Hou and S. Gao, Chem. Commun., 2009, 4378 RSC.
- S. Palchoudhury, Y. Xu, J. Goodwin and Y. Bao, J. Appl. Phys., 2011, 109, 07E314 CrossRef.
- J. Cheon, N. J. Kang, S. M. Lee, J. H. Lee, J. H. Yoon and S. J. Oh, J. Am. Chem. Soc., 2004, 126, 1950 CrossRef CAS.
- S. Peng and S. Sun, Angew. Chem., Int. Ed., 2007, 46, 4155 CrossRef CAS.
- X. Teng and H. Yang, J. Mater. Chem., 2004, 14, 774 RSC.
- M. Pauly, B. P. Pichon, P. A. Albouy, S. Fleutot, C. Leuvrey, M. Trassin, J. L. Gallani and S. Begin-Colin, J. Mater. Chem., 2011, 21, 16018 RSC.
- T. Harada and T. A. Hatton, Langmuir, 2009, 25, 6407 CrossRef CAS.
- S. Zhao, H. Y. Wu, L. Song, O. Tegus and S. Asuha, J. Mater. Sci., 2009, 44, 926 CrossRef CAS.
- S. Desilets, P. Brousseau, D. Chamberland, S. Singh, H. Feng, R. Turcotte, K. Armstrong and J. Anderson, Thermochim. Acta, 2011, 521, 59 CrossRef CAS.
- F. S. Yen, W. C. Chen, J. M. Yang and C. T. Hong, Nano Lett., 2002, 2, 245 CrossRef CAS.
- G. Schimanke and M. Martin, Solid State Ionics, 2000, 136–137, 1235 CrossRef CAS.
- G. Gnanaprakash, S. Ayyappan, T. Jayakumar, J. Philip and B. Raj, Nanotechnology, 2006, 17, 5851 CrossRef CAS.
- L. Machala, J. Tucek and R. Zboril, Chem. Mater., 2011, 23, 3255 CrossRef CAS.
- H. Rostkowska, L. Lapinski and M. J. Nowak, Vib. Spectrosc., 2009, 49, 43 CrossRef CAS.
- L. Hu, A. Percheron, D. Chaumont and C. H. Brachais, J. Sol-Gel Sci. Technol., 2011, 60, 198 CrossRef CAS.
- H. Wang and W. C. Geng, Res. Chem. Intermed., 2011, 37, 523 CrossRef CAS.
- A. R. West in Basic Solid State Chemistry, John-Wiley & Sons, Ltd., 2nd edn, 2000, ch. 4, p 172 Search PubMed.
- V. Sreeja and P. A. Joy, Mater. Res. Bull., 2007, 42, 1570 CrossRef CAS.
- T. Tsuzuki, F. Schaffel, M. Muroi and P. G. McCormick, J. Alloys Compd., 2011, 509, 5420 CrossRef CAS.
- D. Li, W. Y. Teoh, C. Selomulya, R. C. Woodward, P. Munroe and R. Amal, J. Mater. Chem., 2007, 17, 4876 RSC.
- M. P. Morales, S. Veintemillas-Verdaguer, M. I. Montero, C. J. Serna, A. Roig, L. Casas, B. Martinez and F. Sandiumenge, Chem. Mater., 1999, 11, 3058 CrossRef CAS.
- C. M. Sorensen in Nanoscale Materials in Chemistry, ed. K. J. Klabunde, John Wiley & Sons, Inc, 2001, ch. 6, pp 210–211 Search PubMed.
- F. Gronvold and E. J. Samuelsen, J. Phys. Chem. Solids, 1975, 36, 249 CrossRef.
- Y. Yang, J. B. Yi, X. L. Huang, J. M. Xue and J. Ding, IEEE Trans. Magn., 2011, 47, 3340 CrossRef CAS.
- E. S. Leite, J. A. H. Coaquira, W. R. Viali, P. P. C. Sartoratto, R. L. de Almeida, P. C. Morais and S. K. Malik, J. Phys.: Conf. Ser., 2010, 200, 072060 CrossRef.
- D. Parker, F. Ladieu, E. Vincent, G. Meriguet, E. Dubois, V. Dupuis and R. Perzynski, J. Appl. Phys., 2005, 97, 10A502 CrossRef.
- B. D. Cullity and C. D. Graham in Introduction to Magnetic Materials, John Wiley & Sons, Inc, 2nd edn, 2009, ch. 11, p 386 Search PubMed.
- V. A. M. Brabers in Handbook of Magnetic Materials, ed. K. H. J. Buschow, Elsevier, North-Holland, 1995, vol. 8, ch. 3, p 212 Search PubMed.
- P. Hajra, S. Basu, S. Dutta, P. Brahma and D. Chakravorty, J. Magn. Magn. Mater., 2009, 321, 2269 CrossRef CAS.
- J. Jin, T. Iyoda, C. Cao, Y. Song, L. Jiang, T. J. Li and D. B. Zhu, Angew. Chem., Int. Ed., 2001, 40, 2135 CrossRef CAS.
- M. E. Khosroshahi and L. Ghazanfari, Mater. Sci. Eng., C, 2012, 32, 1043 CrossRef CAS.
- J. Wang, T. Xia, C. Wu, J. Feng, F. Meng, Z. Shi and J. Meng, RSC Adv., 2012, 2, 4220 RSC.
- M. Tadic, N. Citakovic, M. Panjan, B. Stanojevic, D. Markovic, D. Jovanovic and V. Spasojevic, J. Alloys Compd., 2012, 543, 118 CrossRef CAS.
- P. F. Guan, X. F. Zhang and J. J. Guo, Appl. Phys. Lett., 2012, 101, 153108 CrossRef.
- J. Ge, Y. Hu, M. Biasini, W. P. Beyermann and Y. Yin, Angew. Chem., Int. Ed., 2007, 46, 4342 CrossRef CAS.
- J. F. Berret, N. Schonbeck, F. Gazeau, D. E. Kharrat, O. Sandre, A. Vacher and M. Airiau, J. Am. Chem. Soc., 2006, 128, 1755 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |