One-pot, aqueous-phase synthesis of graphene oxide functionalized with heterocyclic groups to give increased solubility in organic solvents†
Wei
Ai
ab,
Ju-Qing
Liu
a,
Zhu-Zhu
Du
a,
Xiao-Xu
Liu
c,
Jing-Zhi
Shang
b,
Ming-Dong
Yi
a,
Ling-Hai
Xie
*a,
Jian-Jun
Zhang
a,
Hai-Feng
Lin
a,
Ting
Yu
*b and
Wei
Huang
*a
aKey Laboratory for Organic Electronics & Information Displays (KLOEID) and Institute of Advanced Materials (IAM), Nanjing University of Posts and Telecommunications, 9 Wenyuan Road, Nanjing 210046, P. R. China. E-mail: iamlhxie@njupt.edu.cn; wei-huang@njupt.edu.cn; Fax: +86 25 8349 2333; Tel: +86 25 8349 2333
bDivision of Physics and Applied Physics, School of Physical and Mathematical Sciences, Nanyang Technological University, 637371, Singapore. E-mail: yuting@ntu.edu.sg; Fax: +65 6316 7899; Tel: +65 6316 7899
cHeilongjiang Institute of Science and Technology, Harbin 150027, P. R. China
First published on 15th November 2012
Abstract
An easy and versatile method for the covalent functionalization of graphene oxide (GO) is reported. The functionalized GO is synthesized by a polyphosphoric acid-catalyzed cyclization reaction of the carboxylic groups on GO with the hydroxyl and amino groups on o-aminophenol and o-phenylenediamine, resulting in it being well dispersed in many organic solvents. The results may provide a way to extend the use of GO as a functional material in electronic devices and high performance structural materials.
Graphene and its derivatives, as ideal candidates for future functional and structural materials, have attracted great interest due to their outstanding mechanical, remarkable electrical, optical and stable thermal properties.1–3 Generally, pristine graphene with free defects is poorly soluble in water and most organic solvents because of its hydrophobic nature.4,5 To increase the solubility of graphene, two main approaches, covalent functionalization and non-covalent functionalization, have been developed.6 For non-covalent functionalization, it is usually achieved through the interaction of the delocalized π-bonds of graphene with the π-bonds of a molecule or polymer. However, π–π non-covalent binding is much weaker than the covalent one, and thus there is an inefficiency of the load transfer when graphene is used as an enhancement in structural materials. Furthermore, the electrical structure of graphene will not change too much after non-covalent functionalization, alternatively, it is hard to significantly tailor the electrical properties of graphene by non-covalent functionalization, which may limit the applications of graphene in electronics. Compared to non-covalent functionalization, covalent functionalization is particularly attractive because the chemical and physical properties of graphene can be tuned,7–10 for example by oxidizing graphene (abbreviated as GO), to attach carbonyl and carboxyl, hydroxyl, or epoxy groups to the edge and plane of graphene,11,12 and subsequently grafting additional specific functional groups onto GO.
GO is usually synthesized by oxidizing graphite, it is highly soluble in water and a few organic solvents, such as N-methyl pyrrolidone (NMP) and ethanol. Notably, the good solubility of GO in organic solvents is extremely desirable for device fabrication through the solution process.13,14 Nevertheless, the strong hydrophilic groups also make GO incompatible with most organic materials, especially as the filler for polymer nanocomposites, because most polymers are only soluble in organic solvents. This results in inevitable aggregation or the restacking of graphene sheets in the nanocomposites.15,16 To address those problems, several approaches have been explored to covalently functionalize GO and enable the functionalized GO to be soluble in most organic solvents,17 such as diazonium salts coupling with the sp2 area of GO,18 esterification reactions with the hydroxyl groups on GO,19,20 nucleophilic addition to the epoxy groups on GO,21,22 and the esterification and amidation of the carboxylic groups on GO.23,24 However, these reactions are usually carried out in organic solvents through a complex and non-environmentally friendly process.25 Hence, it is critical to explore alternative approaches to covalently functionalize GO with an easy and environmentally friendly process.
Herein, we present a novel and easy method for the covalent functionalization of GO through a one-step reaction in aqueous solvent. By using o-aminophenol (OAP) and o-phenylenediamine (OPD) as functionalizing reagents and polyphosphoric acid (PPA) as a catalyst, the carboxylic groups of GO were converted to benzoxazole and benzimidazole at room temperature (abbreviated as BO–GO and BI–GO, respectively). The obtained functionalized GO shows high solubility in most organic solvents, no aggregation or visible restacking is observed.
The one-pot, aqueous-phase synthesis of BO–GO and BI–GO is schematically shown in Fig. 1 (also refer to the Experimental Section for details). Briefly, BO–GO and BI–GO were achieved through cyclization between the carboxylic groups on GO and the hydroxyl and amino groups on OAP and OPD, which is similar to the reported mechanism in conventional synthetic organic chemistry.26 Since the reaction mechanisms of BO–GO and BI–GO are similar, BO–GO is used as a model to study the mechanism of benzoxazole formation. At the initial step, GO is activated by PPA through the formation of GO-phosphoric anhydride and has reached a state of dynamic equilibrium. Then, the amino groups of OAP are protonated and some hydroxyl groups are converted to phosphate ester, which are also in dynamic equilibrium. The free hydroxyl portion reacts with the GO-phosphoric anhydride to generate an ester. It then undergoes rapid acyl migration, along with ring closure to form benzoxazole on GO with the aid of acid.
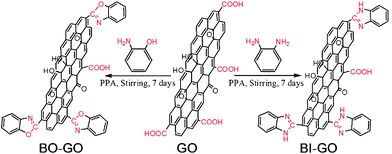 | ||
| Fig. 1 Schematic representation of the synthesis of BO–GO and BI–GO. | ||
Both BO–GO and BI–GO exhibit a high solubility in most organic solvents (Fig. 2), such as acetonitrile, acetone, NMP and N,N-dimethylformamide (DMF) for BO–GO, and NMP, DMF and tetrahydrofuran (THF) for BI–GO.
 | ||
| Fig. 2 Photographs of BO–GO (a) and BI–GO (b) dispersed in different organic solvents (1 mg ml−1) three days after preparation. | ||
Fig. 3 shows the FT-IR spectra of GO, BO–GO and BI–GO. The most characteristic feature in the FT-IR spectrum of GO is the adsorption band corresponding to the carboxyl stretching at 1730 cm−1.27 In the spectrum of BO–GO, a weaker absorption band appears at 1728 cm−1, indicating a reduced content of the carboxyl groups after functionalization. Furthermore, another two typical peaks at 1544 and 1359 cm−1 are also observed in BO–GO, which can be assigned to the stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and C–N, respectively.28 In comparison to BO–GO, the peak at 1730 cm−1 has disappeared in the spectrum of BI–GO, indicating the complete functionalization of the carboxyl groups. The new peak at 1520 cm−1 is attributed to the skeletal vibration of the benzene ring.29 Moreover, the two peaks at 1574 and 1362 cm−1 can be attributed to the stretching of CN and C–N, respectively.30 These results suggest that GO has been successfully functionalized through the formation of benzoxazole and benzimidazole by using OAP and OPD as the functionalizing reagents, respectively.
N and C–N, respectively.28 In comparison to BO–GO, the peak at 1730 cm−1 has disappeared in the spectrum of BI–GO, indicating the complete functionalization of the carboxyl groups. The new peak at 1520 cm−1 is attributed to the skeletal vibration of the benzene ring.29 Moreover, the two peaks at 1574 and 1362 cm−1 can be attributed to the stretching of CN and C–N, respectively.30 These results suggest that GO has been successfully functionalized through the formation of benzoxazole and benzimidazole by using OAP and OPD as the functionalizing reagents, respectively.
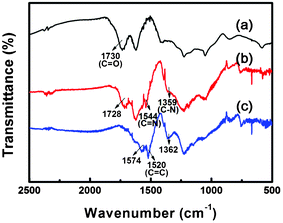 | ||
| Fig. 3 FT-IR spectra of GO (a), BO–GO (b) and BI–GO (c). | ||
Detailed elemental composition analysis in BO–GO and BI–GO was carried out by XPS spectroscopy, and the results were compared with that for GO, as shown in Fig. 4 (a). GO has four peaks at 284.6, 286.7, 288 and 289 eV, which represent the carbon bonds of C–C, C–O, CO and O–CO, respectively. No nitrogen signal is observed in the XPS spectrum of GO. For the C 1s spectra of BO–GO and BI–GO (see Fig. 4 (b) and 4 (c)), there is no difference in the binding energy of C–C, C–O, CO and O–CO, which is similar to that of GO. Notably, a new peak at 285.6 eV, corresponding to the C–N bonds, is observed in the spectra of both BO–GO and BI–GO.31 Additionally, distinct C 1s, N 1s and O 1s peaks can be seen in the XPS spectra of BO–GO and BI–GO. The amount of nitrogen in BO–GO and BI–GO is about 1.4 at% and 3.4 at%, respectively (Fig. 5 (c)).
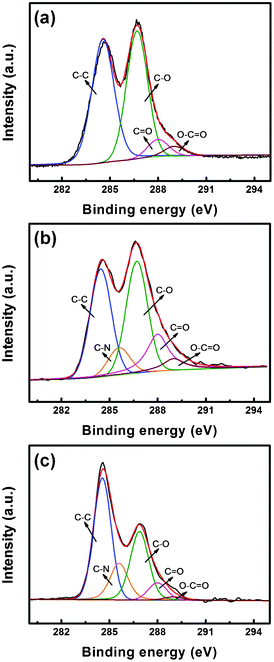 | ||
| Fig. 4 XPS spectra of GO (a), BO–GO (b) and BI–GO (c). | ||
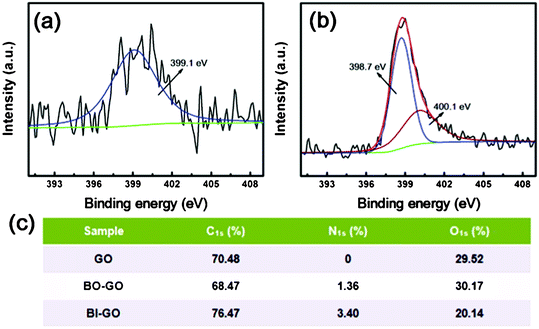 | ||
| Fig. 5 XPS N 1s spectra of BO–GO (a) and BI–GO (b). The element content data of GO, BO–GO and BI–GO (c). | ||
Moreover, the bonding configurations of the nitrogen atoms in BO–GO and BI–GO were further analyzed by N 1s spectra (Fig. 5 (a) and 5 (b)). The N 1s peak of BO–GO at 399.1 eV corresponds to the pyridine-like N from the benzoxazole rings.32 Whereas the N 1s peak of BI–GO could be fitted in to two sets of peaks, 398.9 eV for the pyridine-like N and 400.1 eV for the pyrrole-like N from the benzimidazole rings.33,34 These results confirm that the OAP and OPD had been successfully grafted to GO through the formation of benzoxazole and benzimidazole rings. Meanwhile, the structures of the as-synthesized BO–GO and BI–GO were further confirmed by Raman spectroscopy and TGA (Fig. S1 and Fig. S2†).
The XRD patterns of GO, BO–GO, and BI–GO were used to study the changes in the interlayer spacings of these graphene derivatives. As shown in Fig. 6, the XRD profile of GO shows a strong 2θ peak at 9.98°, which corresponds to an interlayer distance of 8.85 Å, which is comparable with the value in a previous report.35 In contrast, BO–GO and BI–GO show a slightly higher angle of reflection at 10.7° and 10.8°, respectively, with a reduced interlayer distance of 8.26 Å for BO–GO and 8.18 Å for BI–GO. This decreased interlayer distance can be explained by the formation of benzoxazole and benzimidazole rings on the edges of GO after covalent functionalization, because the introduced aromatic molecules will weaken the electrostatic repulsion between the GO sheets and thus induce a smaller interlayer distance. Additionally, a peak at 25.8°, corresponding to the chemically converted graphene,36 is observed in BI–GO, indicating the partial reduction and re-stacking of GO occurring during the functionalization process. This phenomenon is consistent with our XPS data mentioned above and the previous results by Ma and coworkers,37 where p-phenylenediamine was used as a reducing reagent to reduce GO.
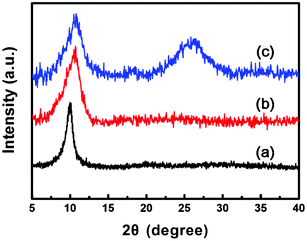 | ||
| Fig. 6 XRD patterns of GO (a), BO–GO (b) and BI–GO (c). | ||
The morphology of BO–GO and BI–GO was studied by tapping-mode AFM. Fig. 7 shows the AFM images and the cross-section analyses of a single-layer GO, BO–GO and BI–GO deposited on Si/SiO2 substrates. The measured thickness of a single-layer GO sheet was about 1.1 nm, which is comparable with values in previous studies.38,39 In contrast, single-layer functionalized GO shows a larger thickness, which is 1.6 nm for BO–GO and 1.4 nm for BI–GO, respectively. The increased thickness is attributed to the functional groups grafted on the GO sheets.40,41 Notably, the surface morphology of both BO–GO and BI–GO are relatively coarse, indicating many defects has been induced by the covalent modification of OAP and OPD, which is also confirmed by the Raman spectra results (Fig. S1†).
 | ||
| Fig. 7 Tapping-mode AFM images of GO, BO–GO and BI–GO on Si/SiO2 substrates. | ||
In summary, we have developed a one-pot and aqueous-phase method for the synthesis of covalently functionalized GO with the aid of PPA at room temperature. The resulting functionalized GO exhibits good solubility in many organic solvents. This strategy provides a new avenue for the covalent functionalization of GO, with the features of one-pot and aqueous-phase synthesis, possessing the advantage of being simple and environmentally friendly. The details of the effects of the covalent functionalization of GO on its electrical properties in semiconductor devices and interfacial properties in structural materials are under investigation.
This work was supported by the “973” project (No. 2009CB930600, No. 2012CB933301), NNSFC (Grants No. 21144004, No. 20974046, No. 21101095, No. 21003076, No. 20774043, No. 51173081, No. 50428303, No. 61136003, No. 50428303), the Ministry of Education of China (No. IRT1148), the NSF of Jiangsu Province (Grants No. SBK201122680, No. 11KJB510017, No. BK2008053, No. 11KJB510017, No. BK2009025, No. 10KJB510013 and No. BZ2010043) and NUPT (No. NY210030 and NY211022). L. H. Xie thanks the Jiangsu Overseas Research & Training Program for University Prominent Young & Middle-aged Teachers and Presidents.
References
- C. Biswas and Y. H. Lee, Adv. Funct. Mater., 2011, 21, 3806 CrossRef CAS.
- N. A. Kotov, Nature, 2006, 442, 254 CrossRef CAS.
- H. Kim, A. A. Abdala and C. W. Macosko, Macromolecules, 2010, 43, 6515 CrossRef CAS.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. GunKo, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563 CrossRef CAS.
- M. Lotya, Y. Hernandez, P. J. King, R. J. Smith, V. Nicolosi, L. S. Karlsson, F. M. Blighe, S. De, Z. M. Wang, I. T. McGovern, G. S. Duesberg and J. N. Coleman, J. Am. Chem. Soc., 2009, 131, 3611 CrossRef CAS.
- J. M. Englert, C. Dotzer, G. Yang, M. Schmid, C. Papp, J. M. Gottfried, H. P. Steinruck, E. Spiecker, F. Hauke and A. Hirsch, Nat. Chem., 2011, 3, 279 CrossRef CAS.
- I. Y. Jeon, D. Yu, S. Y. Bae, H. J. Choi, D. W. Chang, L. Dai and J. B. Baek, Chem. Mater., 2011, 23, 3987 CrossRef CAS.
- D. Yu, K. Park, M. Durstock and L. Dai, J. Phys. Chem. Lett., 2011, 2, 1113 CrossRef CAS.
- S. Niyogi, E. Bekyarova, M. E. Itkis, H. Zhang, K. Shepperd, J. Hicks, M. Sprinkle, C. Berger, C. N. Lau, W. A. deHeer, E. H. Conrad and R. C. Haddon, Nano Lett., 2010, 10, 4061 CrossRef CAS.
- A. Sinitskii, A. Dimiev, D. A. Corley, A. A. Fursina, D. V. Kosynkin and J. M. Tour, ACS Nano, 2010, 4, 1949 CrossRef CAS.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477 CrossRef CAS.
- T. Szabo, O. Berkesi, P. Forgo, K. Josepovits, Y. Sanakis, D. Petridis and I. Dekany, Chem. Mater., 2006, 18, 2740 CrossRef CAS.
- J. Liu, Z. Yin, X. Cao, F. Zhao, A. Lin, L. Xie, Q. Fan, F. Boey, H. Zhang and W. Huang, ACS Nano, 2010, 4, 3987 CrossRef CAS.
- Q. He, S. Wu, S. Gao, X. Cao, Z. Yin, H. Li, P. Chen and H. Zhang, ACS Nano, 2011, 5, 5038 CrossRef CAS.
- V. Eswaraiah, K. Balasubramaniam and S. Ramaprabhu, Nanoscale, 2012, 4, 1258 RSC.
- H. J. Salavagione, G. Martinez and G. Ellis, Macromol. Rapid Commun., 2011, 32, 1771 CrossRef CAS.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228 RSC.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679 CrossRef CAS.
- S. H. Lee, D. R. Dreyer, J. An, A. Velamakanni, R. D. Piner, S. Park, Y. Zhu, S. O. Kim, C. W. Bielawski and R. S. Ruoff, Macromol. Rapid Commun., 2010, 31, 281 CrossRef CAS.
- X. Sun, Z. Liu, K. Welsher, J. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203 CrossRef CAS.
- W. R. Collins, E. Schmois and T. M. Swager, Chem. Commun., 2011, 47, 8790 RSC.
- C. Zeng, Z. Tang, B. Guo and L. Zhang, Phys. Chem. Chem. Phys., 2012, 14, 9838 RSC.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282 CrossRef CAS.
- X. D. Zhuang, Y. Chen, G. Liu, P. P. Li, C. X. Zhu, E. T. Kang, K. G. Noeh, B. Zhang, J. H. Zhu and Y. X. Li, Adv. Mater., 2010, 22, 1731 CrossRef CAS.
- R. Salvio, S. Krabbenborg, W. J. M. Naber, A. H. Velders, D. N. Reinhoudt and W. G. van der Wiel, Chem.–Eur. J., 2009, 15, 8235 CrossRef CAS.
- Y. H. So and J. P. Heeschen, J. Org. Chem., 1997, 62, 3552 CrossRef CAS.
- S. Stankovich, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Carbon, 2006, 44, 3342 CrossRef CAS.
- H. Reyes, H. I. Beltran and E. Rivera-Becerril, Tetrahedron Lett., 2011, 52, 308 CrossRef CAS.
- T. J. Lane, I. Nakagawa, A. j. Kandathi and J. L. Walter, Inorg. Chem., 1962, 1, 267 CrossRef CAS.
- Y. Guo, Z. Lu, L. Yao and Z. Shi, Chin. J. Chem., 2011, 29, 489 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS.
- A. L. M. Reddy, A. Srivastava, S. R. Gowda, H. Gullapalli, M. Dubey and P. M. Ajayan, ACS Nano, 2010, 4, 6337 CrossRef CAS.
- D. Wei, Y. Liu, Y. Wang, H. Zhang, L. Huang and G. Yu, Nano Lett., 2009, 9, 1752 CrossRef CAS.
- Z. Luo, S. Lim, Z. Tian, J. Shang, L. Lai, B. MacDonald, C. Fu, Z. Shen, T. Yu and J. Lin, J. Mater. Chem., 2011, 21, 8038 RSC.
- Y. Zhu, M. D. Stoller, W. Cai, A. Velamakanni, R. D. Piner, D. Chen and R. S. Ruoff, ACS Nano, 2010, 4, 1227 CrossRef CAS.
- H. J. Shin, K. K. Kim, A. Benayad, S. M. Yoon, H. K. Park, I. S. Jung, M. H. Jin, H. K. Jeong, J. M. Kim, J. Y. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987 CrossRef CAS.
- Y. Chen, X. Zhang, P. Yu and Y. Ma, Chem. Commun., 2009, 4527 RSC.
- F. Zhao, J. Liu, X. Huang, X. Zou, G. Lu, P. Sun, S. Wu, W. Ai, M. Yi, X. Qi, L. Xie, J. Wang, H. Zhang and W. Huang, ACS Nano, 2012, 6, 3027 CrossRef CAS.
- C. X. Cong, T. Yu, H. M. Wang, K. H. Zheng, P. Q. Gao, X. D. Chen and Q. Zhang, Small, 2010, 6, 2837 CrossRef CAS.
- H. He and C. Gao, Chem. Mater., 2010, 22, 5054 CrossRef CAS.
- J. R. Lomeda, C. D. Doyle, D. V. Kosynkin, W. F. Hwang and J. M. Tour, J. Am. Chem. Soc., 2008, 130, 16201 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: experimental section, Raman spectra of GO, BO–GO and BI–GO, thermogravimetric curves of GO, BO–GO and BI–GO. See DOI: 10.1039/c2ra22009a |
| This journal is © The Royal Society of Chemistry 2013 |