Amperometric detection of hydrogen peroxide utilizing synergistic action of cobalt hexacyanoferrate and carbon nanotubes chemically modified with platinum nanoparticles
Lijuan
Han
a,
Qiang
Wang
a,
Simon
Tricard
c,
Jianxin
Liu
a,
Jian
Fang
a,
Jihua
Zhao
*ac and
Weiguo
Shen
b
aCollege of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou, 730000, PR China. E-mail: zhaojihua@lzu.edu.cn; Fax: +869318912582; Tel: +86931891254 1
bCollege of Chemistry and Chemical Engineering, East China University of Science and Technology, Shanghai, 200237, PR China
cLaboratoire de Chimie de Coordination, CNRS UPR-8241 and Université de Toulouse, UPS INPT 31077 Toulouse Cedex 4, France
First published on 30th October 2012
Abstract
In this study, a novel composite of cobalt hexacyanoferrate nanoparticles (CoNP) and platinum nanoparticles (Pt) on carbon nanotubes (CNTs) is obtained by ultrasonically mixing CoNP synthesized in a microemulsion with CNTs chemically modified with platinum nanoparticles (Pt/CNTs). Cyclic voltammetric and amperometric measurements on a glass carbon electrode showed that the composite (called CoNP–Pt/CNTs) exhibits a well-defined pair of redox peaks and a prominent electrocatalytic activity toward hydrogen peroxide (H2O2) reduction. Besides, the current response of CoNP–Pt/CNTs is 2 orders of magnitude higher than the response of CoNP alone and 1 order of magnitude higher than the response of Pt/CNTs or CoNP/CNTs alone. This higher efficiency can be attributed to a remarkable synergistic effect between CoNP, Pt and CNTs. This sensor shows a linear response to H2O2 concentrations ranging from 0.2 μM to 1.25 mM with a detection limit of 0.1 μM, a maximum sensitivity of 0.744 A·M−1 and a fast response time below 2 s.
1. Introduction
Transition metal hexacyanoferrates (MHCF), an important class of mixed-valence compounds, have shown particular promise as chemical sensors due to their electrocatalytic properties.1,2 Among them, cobalt hexacyanoferrate (CoHCF) was intensively studied because of its perfectly reversible redox centres, very similar to those of Prussian blue.3–5 These materials have been widely used to electrocatalyze the oxidation of hydrazine,3 DNA,4 thiosulfate,5 dopamine,6 nicotinamide adenine dinucleotide,7p-chlorophenol8 and the reduction of H2O2.9–12 CoHCF was also combined with glucose oxidase for the fabrication of glucose biosensors.13,14 Cobalt hexacyanoferrate nanoparticles (CoNP) can improve the analytical performance of modified electrodes, showing very interesting attributes unmatched by their bulk counterparts. CoNP have been prepared using microemulsions,15,16 chemical depositions9 and reversed micelle synthesis.17 The microemulsion technique, a type of “soft chemistry”, required neither special nor expensive equipment. Moreover, the microemulsion technique allows control of the shape of the particles and their size distributions by varying the experimental parameters.16,18On the other hand, due to their excellent electrical properties, their high chemical stability and their high surface-to-volume ratio, carbon nanotubes (CNTs) provide a new avenue for designing different types of modified electrodes that can be used for electrocatalytic and sensing applications.19 Most CNTs-sensing research has focused on the functionalization of surface-confined CNTs with organic or inorganic molecules. The inorganic compound MHCF can interact and cooperate with CNTs due to the molecular interactions between the sidewall of CNTs and MHCF.20 Therefore, MHCF/CNTs composite was expected to be widely used in electrochemical fields. To the best of our knowledge, studies on the combination of MHCF and CNTs without other materials were relatively scarce in the literature.20–22
As a matter of fact, composite materials based on the combination of MHCF/CNTs composite and some other materials have received significant interest. Noncovalent and covalent modifications of the CNTs with polymers13,23,24 or metal nanoparticles9 were usually used to improve efficient direct electron communication between the redox centres of MHCF and the electrode. The combination of MHCF/CNTs composite with noble metal nanoparticles (NMNP) has especially led to the development of various electrochemical sensors, because certain NMNP exhibit strong surface plasmon resonance, facilitating the electron-transfer process and showing outstanding catalytic activities.25,26 Different ways have been explored to integrate the MHCF/CNTs with NMNP. One possibility consisted of immersing a NMNP/CNTs electrode, which was prepared by physical adsorption of NMNP on CNTs with or without a binder, in a solution of hexacyanoferrates and transition metal ions through electrochemical or chemical methods.9,27,28 Another possibility consisted of dispersing the MHCF/CNTs composite, which was synthesized by the deposition of MHCF on CNTs as a template, in NMNP colloids.29 More recently, MHCF nanoparticles modified with a NMNP hybrid have been created with a fine degree of control,30–32 and their electrochemical behavior was highly improved through integration in CNTs wrapped with polymers.26b,33
However, the methods mentioned above suffered from poor contact between CNTs and NMNP or weak electron-transfer reactions between CNTs and MHCF resulting from peculiar binder molecules. It has been suggested that the formation of Pt-deposited nanotube composites through a chemical modification method would be beneficial to the intimate contact between CNTs and Pt.19e,34 Recently, electrochemical biosensors of glucose were fabricated based on the synergic action of CNTs and platinum.34,35 On the other hand, due to the π–π stacking interaction coupled with an ionic interaction between the sidewall of CNTs and MHCF, the ultrasonical mixture of CoNP and Pt/CNTs without peculiar binder molecules exhibited effective molecular interactions between them. According to our knowledge, the integration of CoNP on CNTs chemically modified with PtNP has never been explored thus far.
In this paper, we report the integration of CoNP and CNTs chemically modified with Pt (Pt/CNTs) for constructing electrochemical sensors with improved sensitivity by using the synergistic effects of the superior electron-transfer ability of Pt/CNTs and the excellent reversible redox centers of CoNP. The products obtained were characterized by various techniques. The electrochemistry of the CoNP–Pt/CNTs composite was also investigated by electrochemical techniques, including cyclic voltammetry and amperometric detection of H2O2, which is a chemical threat to the environment36 and could be reduced effectively by CoHCF.
2. Experimental
2.1. Apparatus
Transmission electron microscope (TEM) images were obtained from a Hitachi H-600 microscope. Scanning electron microscopy (SEM) experiments were performed with a JEOL JSM-6701F. Infrared spectra (IR) of dried particles pressed into KBr pellets were obtained on a NICOLET NEXUS-670-FTIR spectrometer. Powder X-ray diffraction patterns were collected on a Rigaku D/MAX-2400 X-ray diffractometer with graphite monochromatized Cu-Kα radiation (λ = 0.15406 nm). Cyclic voltammetric and amperometric measurements were carried out on a CHI 660B electrochemical workstation (CH Instruments). A three-electrode cell was used with a modified glass carbon electrode (GCE, 3.0 mm in diameter) as the working electrode, a saturated calomel electrode (SCE) as the reference electrode and a platinum wire electrode as the counter electrode. All experiments were carried out at room temperature.2.2. Chemicals and materials
All the solutions were freshly prepared before use. Sodium bis(2-ethylhexyl) sulfosuccinate (AOT, 96% mass fraction) was purchased from Sigma-Aldrich and dried by P2O5 in a desiccator for two weeks. H2PtCl6·6H2O was purchased from Shenyang Chemical Reagents Company. 30% H2O2 solution was purchased from Shanghai Taopu Chemical Factory, and H2O2 aqueous solutions were prepared freshly before use. Other reagents were of analytical grade and used as received from Tianjin Guangfu Fine Reagent Factory. Phosphate buffer solution (PBS) (0.1 M K2HPO4 − KH2PO4 + 0.5 M KCl) was used as a supporting electrolyte, and its pH value was adjusted to 5.8 unless otherwise specified. Double distilled water was used throughout the measurement.2.3. Treatment of carbon nanotubes
The CNTs (95% 20–60 nm in diameter) were obtained from Shenzhen Nanotech Port Co. Ltd. Before use, CNTs were purified using a well-established method.37 Briefly, CNTs were purified under strong sonication in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) mixture of concentrated sulfuric and nitric acid for 1 h and then were refluxed at 80 °C for 8 h to form a dark brown suspension. The resultant mixture was diluted with distilled water and stirred for several hours. A black solid was obtained after filtration. It was subsequently washed several times with double distilled water until the filtrate became neutral, collected on a 200-nm-pore membrane filter and finally dried under a vacuum.
:1 (v/v) mixture of concentrated sulfuric and nitric acid for 1 h and then were refluxed at 80 °C for 8 h to form a dark brown suspension. The resultant mixture was diluted with distilled water and stirred for several hours. A black solid was obtained after filtration. It was subsequently washed several times with double distilled water until the filtrate became neutral, collected on a 200-nm-pore membrane filter and finally dried under a vacuum.
2.4. Preparation of cobalt hexacyanoferrate nanoparticles
CoNP was prepared in a microemulsion.15 In brief, 3.6 g of AOT, 4.5 g of 30 mM CoCl2 aqueous solution and 20 mL of isooctane were mixed and stirred until the formation of a transparent mixture emulsion (ME). 1.9 g of aqueous solution of K3Fe(CN)6 (14 mM) and CsCl (6 mM) was added dropwise to the ME under stirring. The mixture was continuously stirred for 11 h hours at 27 °C and then subjected to age for 11 h. The microemulsion was centrifuged and the precipitate was washed five times with ethanol and H2O to remove the contaminated oil, surfactant and other inorganic substances from the particles. The retentate was dried overnight in a vacuum at room temperature to give a quantitative powder compound.2.5. Synthesis of Pt/CNTs
Deposition of Pt on CNTs was prepared using a well-established method with a slight modification.38 Briefly, (i) 40 mg of pretreated CNTs were dispersed in 8 mL of deionized water by ultrasonic agitation for 20 min and then mechanically stirred for 4 h to get a black suspension. (ii) This black suspension was added dropwise to 2.66 mL of a 10 g L−1 aqueous H2PtCl6·6H2O solution. Then the pH value of the mixture was adjusted to 11 with the addition of an aqueous NaOH solution (2.5 M) under vigorous stirring. (iii) A formaldehyde/water solution (2 mL, 37%) was added immediately and the solution was stirred at 85 °C for 1.5 h under nitrogen. Finally, the products were filtered, washed with an excess of deionized water and dried at 70 °C. For comparison, Pt without CNTs was synthesized in a similar manner.2.6. Fabrication of the modified electrode
The working electrodes were prepared as follows. The pre-synthesized Pt/CNTs were ultrasonically dispersed in ethanol for 5 min to give a black suspension. Subsequently, CoNP was mixed with the black suspension by ultrasonic agitation for 10 min to obtain a CoNP doped Pt/CNTs homogeneous composite. 8 μL of the prepared black suspension of CoNP–Pt/CNTs composite was dropped on the cleaned GCE surface and dried upon heating below 30 °C under an infrared lamp. Other electrodes, such as CoNP–CNTs, Pt/CNTs, CoNP–Pt and CoNP, were prepared in the analogous manner by casting relevant solutions on the surface of glassy carbon electrodes. These modified electrodes were used for the electrochemical measurements.3. Results and discussion
3.1 Morphology analysis
As shown in Fig. 1A and Fig. 1B with different magnifications, uniform Pt attach to the sidewall of CNTs; the diameters of the Pt range from 5 to 10 nm. The dispersion of the Pt is crucial to improve the electronic and ionic transport capacity and hence the electrochemical and electrocatalytic properties of CNTs.38,39Fig. 1F demonstrates the EDS of the Pt/CNTs, in which the presence of Pt can be verified. Fig. 1D shows a typical SEM micrograph, which conforms the formation of a typical cubic CoNP with a diameter of about 50–100 nm, in accordance with the result of the representative TEM image in Fig. 1C. The SEM micrograph of the CoNP–Pt/CNTs sample is shown in Fig. 1E. It is worth noting that the composite dispersions in ethanol are homogeneous and stable for a couple of months, which is caused by the intrinsic interaction between CoNP and CNTs.20 | ||
| Fig. 1 TEM images of the Pt/CNTs ((A) low and (B) high magnifications) and CoNP (C). SEM micrographs of CoNP (D) and the CoNP–Pt/CNTs composite (E). The EDS pattern of Pt/CNTs (F). | ||
3.2 XRD analysis and FT-IR spectroscopy characterization
The structure of the CoNP is confirmed with XRD. In Fig. 2A, the sharp intense peaks at 2θ values equal to 17.18°, 24.40°, 34.76°, 39.05°, 43.04° correspond to the (200), (220), (400), (420) and (422) reflections, respectively, which demonstrates the face-centered cubic (fcc) structure of the synthesized CoNP with a cell parameter a = 10.31 Å.40 The XRD pattern for Pt/CNTs is shown in Fig. 2B. The four main characteristic peaks of the face-centered cubic crystalline Pt at 2θ values equal to 39.43°, 46.54°, 68.12° and 81.62° correspond to the (111), (200), (220), (311) reflections,19e,38 indicating that the Pt are composed of pure crystalline Pt. The X-ray diffraction peak at 25.99° can be attributed to the hexagonal graphite structure (002).38 According to the Scherrer formula,41 as shown below:| L = 0.9λ/Bcosθ, |
where L is the average particle size, λ the X-ray wavelength (1.54056 Å for Cu-Kα1 radiation), B the half-peak broadening and θ the angle corresponding to the peak maximum. The mean particle size of PtNP is calculated to be 7.2 nm, which is in good agreement with the TEM results. Besides, the main characteristic peaks of the face-centered cubic crystalline CoNP and Pt were confirmed with the XRD pattern of the final hybrid product (Fig. 2C).
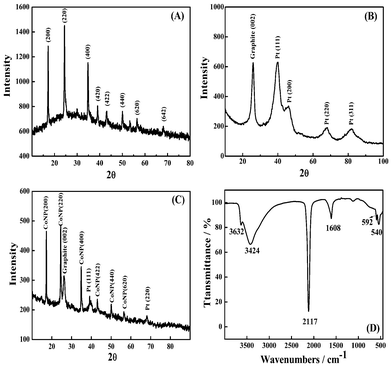 | ||
| Fig. 2 XRD patterns of CoNP (A), Pt/CNTs (B) and the CoNP–Pt/CNTs composite (C). The FTIR spectrum of CoNP (D). | ||
Fig. 2D shows the IR spectrum of CoNP in the region of 450–4000 cm−1. The sharp band at 2117 cm−1 is assigned to the CN stretching frequencies of the FeII–CN–CoIII states, according to our previous study.42,43 No characteristic band of the surfactant AOT is detected in the spectra. In addition, the peaks at 3633 cm−1 and 3424 cm−1 are attributed to the stretching vibrations of the O–H bond in the CoHCF·xH2O complex. The frequency at 1608 cm−1 corresponds to the bending vibration of H–O–H, which confirms the presence of crystal water in the structure of CoHCF.
3.3 Electrochemical characterization
Films of CoNP–Pt/CNTs (trace a), CoNP (trace b), CoNP–Pt (trace c), Pt/CNTs (trace d) and CoNP–CNTs (trace e) were studied by cyclic voltammetric (CV) in order to ascertain the role of individual components and possible synergy among them. Fig. 3A shows CVs recorded at film modified electrodes in PBS at the scan rate of 50 mV s−1. The CoNP–CNTs film (trace e) exhibits one set of reversible redox peaks with formal potential (E° = [(Ep,a + Ep,c)/2]) of about 0.53 V, which has been attributed to the transformations between Fe(II) and Fe(III) in cobalt hexacyanoferrate. Therefore, the redox couple can be expected as Co1.5IIFeIII(CN)6/KCo1.5IIFeII(CN)6.4 For the CoNP–Pt/CNTs film, the peak potential separations increase with deposition of Pt on CNTs, revealing that both oxidation and reduction processes become difficult. We notice a strong increase of the peak current with the formal potential unchanged of about 0.53 V. In the inset, a pair of redox peaks can be observed for CoNP (curve b) with a formal potential of 0.53 V, while no obvious voltammetric peak is observed in the solution for the Pt/CNTs film (curve d) because no Fe is present. In addition, the peak current of the CoNP–Pt/CNTs modified electrode (curve a) was much larger than that of the CoNP–Pt modified electrode (trace c), for Pt nanoparticles deposited on CNTs do not aggregate with each other to form larger clusters. In particular, the peak current of the CoNP–Pt/CNTs composite is about 17 times that of the CoNP–CNTs, 32 times that of the CoNP–Pt and 50 times that of the CoNP (no peak for Pt/CNTs). The increased peak current on the GCE modified by CoNP–Pt/CNTs composite film is due apparently to the synergistic effects among constitents.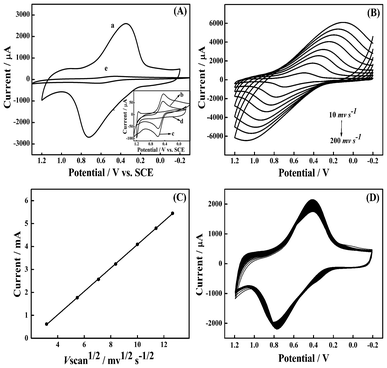 | ||
| Fig. 3 (A) CV of CoNP–Pt/CNTs (trace a), CoNP (trace b), CoNP–Pt (trace c), Pt/CNTs (trace d) and CoNP–CNTs (trace e) films modified glassy carbon electrodes at a scan rate of 50 mV s−1. (B) CV of the CoNP–Pt/CNTs films modified electrode at different scan rates: 10, 30, 50, 70, 100, 130, 160 and 200 mV s−1. (C) A plot of the reduction peak current versus the square root of scan rate in the range 10–200 mV s−1. (D) CV of the CoNP–Pt/CNTs film during the first 100 cycles at a scan rate of 50 mV s−1. Each CV was conducted in PBS (pH 5.8). | ||
We investigated the effect of Pt/CNTs on the improvement of the redox properties of CoNP in detail by varying the content of CoNP in the CoNP–Pt/CNTs film. Fig. 4A shows the CV of the film with different amounts of CoNP, as the content of Pt/CNTs is kept constant at 2.0 mg mL−1. With the amount of CoNP increasing, the peak current rose because of the increasing number of electrocatalytic sites in the film. The well-defined peaks indicate that the film is highly homogeneous. We fixed the concentration of CoNP in the film at 2.0 mg mL−1. Fig. 4B shows the CV of the CoNP–Pt/CNTs film in PBS with an increasing amount of Pt/CNTs at the scan rate of 50 mV s−1. It can be seen that the peak current increased almost linearly with the increase of the content of Pt/CNTs, whereas the quantity of the redox active species – the hexacyanoferrate – is kept constant, again indicating the synergy effect between CoNP and Pt/CNTs.
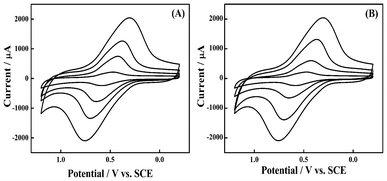 | ||
| Fig. 4 CV of CoNP–Pt/CNTs film modified GCE in PBS (A) with 2.0 mg mL−1 of Pt/CNTs and different CoNP content (from inner to outer, 0.5, 1.0, 1.5, 2.0 mg mL−1) and (B) with 2.0 mg mL−1 CoNP and different Pt/CNTs content (from inner to outer, 0.5, 1.0, 1.5, 2.0 mg mL−1). Scan rate, 50 mV s−1. | ||
The effect of the potential scan rate on the reduction current of the CV peak is investigated in the range of 10–200 mV s−1, as shown in Fig. 3B. With the increase of the scan rate, the peak current of the redox wave increases simultaneously, and the peak separation widens as well. As expected, the current is found to be proportional to the square root of the scan rate between 10 and 200 mV s−1 (Fig. 3C), suggesting that the reaction kinetics is the diffusion-controlled process of the modified electrode.9 As shown in Fig. 3D, the stability of the modified electrodes is satisfactory. Sweeping the potential from −0.2 to 1.2 V at a scan rate of 50 mV s−1 shows less than 15% decrease of peak current after 100 cycles for the modified electrodes; this result indicates a relatively stable film in PBS.
3.4 Amperometric response of the modified electrodes to hydrogen peroxide
The above synergistic effect of the CoNP–Pt/CNTs composite film modified electrode can also be observed in potentiostatic measurements. The incorporation of Pt into CoNP/CNTs film facilitates the electroreduction of H2O2. Fig. 5A displays a typical steady-state response of the CoNP–Pt/CNTs film modified electrode on successive addition of H2O2 to the stirring of PBS (pH 5.8) at −0.2 V. A subsequent addition of H2O2 results in a significant increase in the reduction current intensity, showing the catalytic property of the modified electrode to the reduction of H2O2. The time required to reach the 95% steady-state response is within 2 s, and the detection limit of the sensor with a signal-to-noise ratio of 3 is determined to be 0.1 μM. The electrochemical response to H2O2 shows a linear range from 0.2 μM to 1.25 mM with a high sensitivity of 0.744 A·M−1 (R2 = 0.9979) (inset of Fig. 5A, up right). The inset (down left) shows the magnified image of the current-time curve at the CoNP–Pt/CNTs modified electrodes from 50 s to 200 s. Varying concentrations of hydrogen peroxide solution were added dropwise to a stirring PBS solution (pH 5.8) every 30 s.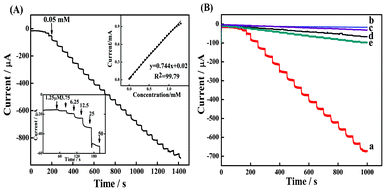 | ||
| Fig. 5 (A) The performance of the CoNP–Pt/CNTs sensor in amperometric detection of H2O2 operated at −0.2 V in a stirred PBS (pH 5.8). The inset (down left) shows the magnified image of injection every 30 s from 50 s to 200 s. The other inset (up right) shows the calibration curve for H2O2 concentration with the modified electrode. (B) A comparison of the current–time graph of CoNP–Pt/CNTs (trace a), CoNP (trace b), CoNP–Pt (trace c), Pt/CNTs (trace d) and CoNP–CNTs (trace e) films modified electrodes. | ||
To discern the contribution of individual components and the possible synergistic effect among them, control experiments on CoNP (trace b), CoNP–Pt (trace c), Pt/CNTs (trace d) and CoNP–CNTs (trace e) composite film electrodes under the same conditions were also carried, as shown in Fig. 5B. The CoNP and CoNP–Pt films yield a detectable, but very small response to H2O2 in the order of 10−7 A. The presence of current at the Pt/CNTs and CoNP/CNTs film electrodes was due to the interaction between Pt and CNTs or between CoNP and CNTs.21,34,35 When PtNP is incorporated into the CoNP–Pt/CNTs composite film, the response current of the CoNP–Pt/CNTs electrode is about 94 times higher than the response current of CoNP, 27 times than the one of CoNP–Pt, 14 times than the one of Pt/CNTs and 8 times than the one of CoNP/CNTs. This result shows a synergistic effect among them because the current response of the CoNP–Pt/CNTs electrode is much larger than the sum of any of the other two electrodes. According to the literature,44,45 such synergy has been recently surveyed in a study of the oxidation of the enzyme cofactor on the electrode system integrated with CNTs and redox mediators. The observed synergy is explained by the hypothesis that the introduction of CNTs into the polymer matrix containing redox mediators improved its electronic and ionic transport capacity and electron self-exchange in the polymer film. This hypothesis can also be applied to our system of a CoNP–Pt/CNTs-modified GC electrode. The CNTs and Pt in the composite film improve the electronic transport capacity, resulting in a prominent increase in the enhancement of the electrocatalytic activity toward H2O2 reduction. In addition, the introduction of Pt in CNTs can not only facilitate the electron transfer, but also have high catalytic activities for H2O2.34,35 Another important aspect is that the intimate contact between Pt and CNTs surface would also be helpful to electron transfer; herein, both promotions contribute to an improvement of electrocatalytic activity toward the reduction of H2O2.35 The faster response and the better stability of the CoNP–Pt/CNTs film are primarily due to the introduction of PtNP. To reveal the possible advantage of the present sensors, the performance of the sensors is compared with previously reported sensors based on CoHCF, as presented in Table 1. To our knowledge, the CoNP–Pt/CNTs sensor displays the better combination of sensitivity and linear range. A set of 10 different amperometric measurements for 0.05 mM H2O2 with the modified electrode yielded a relative standard deviation (R.S.D.) of 5.1%, indicating a good repeatability of the sensing for H2O2. Moreover, we checked the reproducibility of the sensor. Seven sensors were prepared in the same batch of CoNP–Pt/CNTs, and a R.S.D. of 6.9% was observed toward 0.05 mM H2O2, indicating the reliability of the method.
| Type of electrode | E (V) | Linear range (mM) | Sensitivity (A M−1 cm−2) | Refs. |
|---|---|---|---|---|
| a Potential vs. SCE. | ||||
| CoNP/Pt/CNTs/GCE | −0.2 | 0.2 × 10−3–1.25 | 0.744 | Present |
| CoNP–CNTs–CHIT/GCE | −0.2 | 13 | ||
| CoNP/Au/CNTs/GCE | −0.1 | 5 × 10−3–10 | 18.65 × 10−3 | 9 |
| CoNP/CNTs/GCE | −0.1 | 0.01–8 | 3.57 × 10−3 | 9 |
| CoHCF/aluminum electrode | 0.0 | 0.5 × 10−3–1.7 | 0.225 × 10−3 | 11 |
| Hb/POAP/FeCoHCF/AuE | −0.25 | 1.73 × 10−2–4.03 | 0.16 × 10−3 | 46 |
| CuCoHCF/GCE | +0.02 | 0.81 × 10−3–2.3 | 47 | |
Conclusions
In this paper, a simple chemical approach for fabrication of CoNP–Pt/CNTs sensors is described based on the combination of CoNP and CNTs chemically modified with Pt. Compared to the CoNP–CNTs film, incorporation of PtNP into CoNP–Pt/CNTs results in a more than eight-fold increase in the amperometric response to H2O2. We show that the increased amperometric response is due to a synergistic effect of three kinds of nanomaterials: CoNP, Pt and CNTs. An advantage of this approach is the ease of synthesis and modification because of the absence of any peculiar binder molecules or entrapment reagents. The high stability, high sensitivity and high reproducibility of the sensor make it promising for reliable detection of H2O2. This system represents a new electrochemical platform that can provide operational access to a large number of oxidase-based enzymes towards the fabrication biosensors.Acknowledgements
Financial support was provided by the National Natural Science Foundation of China (Projects 20603014, 20673059, 20973061 and 20903051), the Chinese Ministry of Education (Key project 105074), the Committee of Science and Technology of Shanghai (Projects nos. 0652nm010 and 08JC1408100), and the Fundamental Research Funds for the Central Universities (Project lzujbky-2011-116 and lzujbky-2010-36). Jihua Zhao thanks Simon Tricard, Gabor Molnar, Lionel Salmon and Azzedine Bousseksou for their generous help.References
- A. A. Karyakin, E. A. Puganova, I. A. Budashov, I. N. Kurochkin and E. Karyakina, Anal. Chem., 2004, 76, 474 CrossRef CAS.
- A. Eftekhari, J. Electroanal. Chem., 2002, 537, 59 CrossRef CAS.
- S. M. Chen, Electrochim. Acta, 1998, 43, 3359 CrossRef CAS.
- A. Abbaspour and M. A. Mehrgardi, Anal. Chem., 2004, 76, 5690 CrossRef CAS.
- W. Tao, D. Pan, Y. Liu, L. Nie and S. Yao, J. Electroanal. Chem., 2004, 572, 109 CrossRef CAS.
- Z. Xun, C. Cai, W. Xing and T. Lu, J. Electroanal. Chem., 2003, 545, 19 CrossRef CAS.
- C. X. Cai, H. X. Ju and H. Y. Chen, J. Electroanal. Chem., 1995, 397, 185 CrossRef.
- T. R. I. Cataldi, G. D. Benedetto and A. Bianchini, J. Electroanal. Chem., 1999, 471, 42 CrossRef CAS.
- S. Wang, L. Lu, M. Yang, Y. Lei, G. Shen and R. Yu, Anal. Chim. Acta, 2009, 651, 220 CrossRef CAS.
- R. Garjonyte and A. Malinauskas, Sens. Actuators, B, 1998, 46, 236 CrossRef.
- A. Eftekhari, Microchim. Acta, 2003, 141, 15 CrossRef CAS.
- X. Cui, L. Hong and X. Lin, J. Electroanal. Chem., 2002, 526, 115 CrossRef CAS.
- M. Yang, J. Jiang, Y. Yang, X. Chen, G. Shen and R. Yu, Biosens. Bioelectron., 2006, 21, 1791 CrossRef CAS.
- M. Florescu, M. Barsan, R. Pauliukaite and C. M. A. Brett, Electroanalysis, 2007, 19, 220 CrossRef CAS.
- J. F. Xu, H. Liu, P. Liu, C. H. Liang, Q. Wang, J. Fang, J. H. Zhao and W. G. Shen, J. Iran. Chem. Soc., 2010, 7, 123 CrossRef.
- S. B. Vaucher, J. Fielden, M. Li, E. Dujardin and S. Mann, Nano Lett., 2002, 2, 225 CrossRef CAS.
- M. Yamada, T. Sato, M. Miyake and Y. Kobayashi, J. Colloid Interface Sci., 2007, 315, 369 CrossRef CAS.
- S. Vaucher, M. Li and S. Mann, Angew. Chem., Int. Ed., 2000, 39, 1793 CrossRef CAS.
- (a) D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105 CrossRef CAS; (b) K. Gong, M. Zhang, Y. Yan, L. Su, L. Mao, S. Xiong and Y. Chen, Anal. Chem., 2004, 76, 6500 CrossRef CAS; (c) T. W. Ebbesen, H. J. Lezec, H. Hiura, J. W. Bennett, H. F. Ghaemi and T. Thio, Nature, 1996, 382, 54 CrossRef CAS; (d) R. H. Baughman, Science, 2002, 297, 787 CrossRef CAS; (e) Y. Mu, H. Liang, J. Hu, L. Jiang and L. Wan, J. Phys. Chem. B, 2005, 109, 22212 CrossRef CAS.
- Y. Zhang, Y. Wen, Y. Liu, D. Li and D. Li, Electrochem. Commun., 2004, 6, 1180 CrossRef CAS.
- E. Nossol and A. J. G. Zarbin, J. Mater. Chem., 2012, 22, 1824 RSC.
- (a) S. Han, Y. Chen, R. Pang and P. Wan, Ind. Eng. Chem. Res., 2007, 46, 6847 CrossRef CAS; (b) L. Qian and X. Yang, Talanta, 2006, 69, 957 CrossRef CAS; (c) Q. Tang, W. Xiong and L. Long, Anal. Lett., 2007, 40, 1610 CrossRef CAS.
- F. Qu, M. Yang, Y. Lu, G. Shen and R. Yu, Anal. Bioanal. Chem., 2006, 386, 228 CrossRef CAS.
- (a) J. Li, J. D. Qiu, J. J. Xu, H. Y. Chen and X. H. Xia, Adv. Funct. Mater., 2007, 17, 1574 CrossRef CAS; (b) Y. Lin and X. Cui, Chem. Commun., 2006, 2226 Search PubMed.
- B. M. Quinn, C. Dekker and S. G. Lemay, J. Am. Chem. Soc., 2005, 127, 6146 CrossRef CAS.
- (a) M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS; (b) J. Zhang, J. Li, F. Yang, B. Zhang and X. Yang, Sens. Actuators, B, 2009, 143, 373 CrossRef.
- X. Che, R. Yuan, Y. Chai, J. Li, Z. Song, W. Li and X. Zhong, Colloids Surf., B, 2011, 84, 454 CrossRef CAS.
- J. Zhang, J. Li, F. Yang, B. Zhang and X. Yang, J. Electroanal. Chem., 2010, 638, 173 CrossRef CAS.
- H. Yang, R. Yuan, Y. Chai, H. Su, Y. Zhuo, W. Jiang and Z. Song, Electrochim. Acta, 2011, 56, 1973 CrossRef CAS.
- Y. Miao, J. Chen, X. Wu and J. Miao, Colloids Surf., A, 2007, 295, 135 CrossRef CAS.
- S. S. Kumar, J. Joseph and K. L. Phani, Chem. Mater., 2007, 19, 4722 CrossRef CAS.
- (a) J. Zhang, W. Yang, H. Zhu, J. Li, F. Yang, B. Zhang and X. Yang, J. Colloid Interface Sci., 2009, 338, 319 CrossRef CAS; (b) J. D. Qiu, H. Z. Peng, R. P. Liang, J. Li and X. H. Xia, Langmuir, 2007, 23, 2133 CrossRef CAS.
- C. Ou, S. Chen, R. Yuan, Y. Chai and X. Zhong, J. Electroanal. Chem., 2008, 624, 287 CrossRef CAS.
- S. Hrapovic, Y. Liu, K. B. Male and J. H. T. Luong, Anal. Chem., 2004, 76, 1083 CrossRef CAS.
- F. Qu, M. Yang, G. Shen and R. Yu, Biosens. Bioelectron., 2007, 22, 1749 CrossRef CAS.
- A. Salimi, R. Hallaj, S. Soltanian and H. Mamkhezri, Anal. Chim. Acta, 2007, 594, 24 CrossRef CAS.
- R. Yu, L. Chen, Q. Liu, J. Lin, K. Tan, S. C. Ng, H. S. O. Chan, G. Xu and T. S. A. Hor, Chem. Mater., 1998, 10, 718 CrossRef CAS.
- J. E. Huang, D. J. Guo, Y. G. Yao and H. L. Li, J. Electroanal. Chem., 2005, 577, 93 CrossRef CAS.
- J. M. Liu, H. Meng, J. L. Li, S. Liao and J. H. Bu, Fuel Cells, 2007, 7, 402 CrossRef CAS.
- (a) S. Choudhury, G. K. Dey and J. V. Yakhmi, J. Cryst. Growth, 2003, 258, 197 CrossRef CAS; (b) M. Berrettoni, M. Giorgetti, S. Zamponi, P. Conti, D. Ranganathan, A. Zanotto, M. L. Saladino and E. Caponetti, J. Phys. Chem. C, 2010, 114, 6401 CrossRef CAS; (c) A. Bleuzen, V. Escax, A. Ferrier, F. Villain, M. Verdaguer, P. Münsch and J. P. Itié, Angew. Chem., 2004, 116, 3814 CrossRef.
- V. Radmilovic, H. A. Gasteiger and P. N. R. Jr, J. Catal., 1995, 154, 98 CrossRef CAS.
- C. F. Yang, Q. Wang, C. Y. Yi, J. H. Zhao, J. Fang and W. G. Shen, J. Electroanal. Chem., 2012, 674, 30 CrossRef CAS.
- R. O. Lezna and R. Romagnoli, J. Phys. Chem. B, 2002, 106, 3612 CrossRef CAS.
- M. G. Zhang and W. Gorski, J. Am. Chem. Soc., 2005, 127, 2058 CrossRef CAS.
- M. G. Zhang and W. Gorski, Anal. Chem., 2005, 77, 3960 CrossRef CAS.
- W. Tao, D. Pan, Y. Liu, L. Nie and S. Yao, Anal. Biochem., 2005, 338, 332 CrossRef CAS.
- X. Cui, L. Hong and X. Lin, J. Electroanal. Chem., 2002, 526, 115 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |