DOI:
10.1039/C3PY00840A
(Paper)
Polym. Chem., 2014,
5, 220-226
Synthesis of well-defined poly(phenylcarbazole-alt-triphenylphosphine oxide) siloxane as a bipolar host material for solution-processed deep blue phosphorescent devices†
Received
28th June 2013
, Accepted 5th August 2013
First published on 8th August 2013
Abstract
Alternating copolymers with both hole and electron transporting side groups as bipolar hosts are of great interest for deep blue phosphorescent devices due to the uniform distribution of electrons and holes within the emitting layer. In this work, we synthesized an efficient alternating copolysiloxane-based host material poly(phenylcarbazole-alt-triphenylphosphine oxide) siloxane (PCzPOMSi) with phenylcarbazole and triphenylphosphine oxide moieties linked to the backbone of polysiloxane. PCzPOMSi exhibits a high decomposition temperature (Td = 437 °C) and glass transition temperature (Tg = 118 °C), and it can form a stable amorphous state. The silicon–oxygen linkage of PCzPOMSi disrupts its conjugation and results in a sufficiently high triplet energy level (ET = 3.0 eV). A bis((3,5-difluoro-4-cyanophenyl)pyridine) iridium picolinate (FCNIrpic)-based device using PCzPOMSi as a host shows a turn-on voltage of 7.7 V, a maximum external quantum efficiency of 4%, and a maximum current efficiency of 8.5 cd A−1. These results demonstrate that a well-designed alternating copolysiloxane-based host is a promising approach to realize high performance solution-processed deep blue PhPLEDs.
Introduction
Tremendous effort has been devoted to phosphorescent polymeric light emitting diodes (PhPLEDs) because of their capability of harvesting both singlet and triplet excitons, which can lead to an internal quantum efficiency of 100% by using phosphorescent dyes.1–5 In order to efficiently confine the electro-generated excitons in the emitting layer of typical PhPLEDs, various functional layers are required. Such sophisticated device configurations inevitably increase the manufacture complexity and the production cost.6 Therefore, polymeric hosts have attracted significant attention due to their simple device structures through solution processing technology. Over the last few decades, efforts to achieve efficient solution processed red and green PhPLEDs have led to considerable advances.2,4,7–10 However, the development of deep blue PhPLEDs is lagging behind, due to the lack of suitable polymeric host. Deep blue PhPLEDs play a crucial role in commercializing highly efficient full color flat panel displays and white general lighting with a high color rendering index (CRI).11 For this reason, it is vital to develop polymeric hosts for highly efficient and stable deep blue solution processed PhPLEDs.12 An ideal host for deep blue PhPLEDs should possess sufficiently high triplet energy to confine triplet excitons within the emitting layer and avoid reverse energy transfer from guest to host.13 In addition, the balanced hole and electron movement within an emitting layer is very important for the good charge balance.14–17 In particular, the highest occupied molecular orbital (HOMO) level and lowest unoccupied molecular orbital (LUMO) level should match the Fermi level of poly-(3,4-ethylenedioxythiophene): poly-(styrenesulfonate) (PEDOT:PSS) and the electron transporting layer.11,18 The polymeric host should also possess solubility, high thermal, electrochemical, morphological stability and good film-forming ability, no crystallization, for use in the light-emitting layer (EML).19
Recent research trends have, therefore, turned to the development of hosts possessing bipolar properties.20–26 Compared to the traditional hosts with either predominantly hole- or electron-transporting properties, bipolar hosts have potential advantages arising from more balanced injection and transportation of holes and electrons. Through the linking of both p- and n-type groups into the frame of the host, the carrier-injection/transport properties in the EML can achieve balanced charge fluxes with a concomitant reduction of the local density of phosphor excitons and the rates of triplet–triplet annihilation and other triplet quenching processes. The balanced charge fluxes will lead to a broader recombination zone within the emissive layer.
Herein, we have designed and successfully synthesized a new bipolar alternating copolysiloxane derivative (PCzPOMSi) with a sufficiently high triplet energy level (3.0 eV) by incorporating carbazole and phosphine oxide units into a polysiloxane backbone to effectively interrupt the conjugation of π electrons. Carbazole and phosphine oxide are well-known hole and electron transporting materials, respectively. They are the most widely used core structure of host materials due to their high triplet energy, charge-transporting functionality and versatile chemical modification.27,28 Additionally, the flexible polysiloxane main chain with methyl groups would enhance the likelihood that the material forms morphologically stable amorphous films and improved solubility.29 Therefore, it is expected that the well-defined alternating n- and p-type side groups will be beneficial for the uniform distribution of electrons and holes within the emitting layer, and also be useful for simplifying and fabricating highly efficient, deep-blue PhPLED devices. Meanwhile, it is facile to control the optical and electronic properties through the choice of side groups. Taking these into account, a deep blue phosphorescent polymeric light emitting device has been fabricated using copolysiloxane as a host and FCNIrpic as a guest phosphor, and its thermal and mechanical properties were also studied.
Results and discussion
Synthesis and characterization
The synthetic route and chemical structure of the new host material PCzPOMSi (7) are presented in Scheme 1. The starting material 9-(4-bromophenyl)-9-carbazole (1) was synthesized by a modified Ullmann condensation of carbazole with 1-bromo-4-iodobenzene in the presence of copper(I) iodide as a catalyst and 18-crown-6 as a ligand in DMF. Methyldiethyoxychlorosilane (2) was obtained by the alcoholysis of methyl trichlorosilane to give a colorless liquid. The transformation of bromide (1) and (5) to 9-(4-(diethoxy(methyl)silyl)phenyl)-9-carbazole (3) and (4-(diethoxy(methyl)silyl)phenyl)diphenylphosphine oxide (6) was achieved by Barbier–Grignard reaction to form monomers. The 1H NMR spectra of (3) and (6) show new peaks at 3.8–3.9 ppm, assigned to the proton of the Si–OCH2 groups, suggesting the successful preparation of (3) and (6). Compound (4) was obtained by the hydrolysis of (3) in a dilute THF–HCl solution at 0 °C. Low temperature could suppress the undesired condensation between the formed silanols. The 1H-NMRspectrum of (4) shows a new peak at 2.69 ppm, assigned to the proton of the Si–OH groups, while the peak at 3.83 ppm assigned to the protons of Si–OCH2 disappears, indicating the complete hydrolysis of (3). The desired polymer was then synthesized from the corresponding diethoxy monomer and dihydroxy monomer under tetrabutyl titanate catalyzed polycondensation. The end groups were blocked with trimethylchlorosilane to stabilize the resultant polysiloxane. After the end of polymerization, the solution was concentrated and precipitated in methanol to give the product as a white powder. Thus, the synthetic routes have obvious advantages over common noble metal catalyzed hydrosilylation reactions for the preparation of functional polysiloxanes. 1H, 13C and 29Si NMR spectroscopy and elemental analysis were employed to confirm the chemical structures of the newly synthesized compounds and the polymers, as described in the Experimental section of the ESI.† The number average molecular weight was determined to be 5428 g mol−1 for PCzPOMSi with a polydispersity index of 1.54 by gel permeation chromatography. The PCzPOMSi is highly soluble in common organic solvents such as chloroform, tetrahydrofuran, toluene and chlorobenzene. Therefore, it can be easily fabricated into thin films by casting, spin-coating, and dipping techniques.
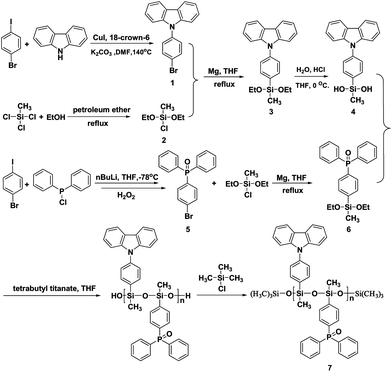 |
| | Scheme 1 Synthetic route for PCzPOMSi. | |
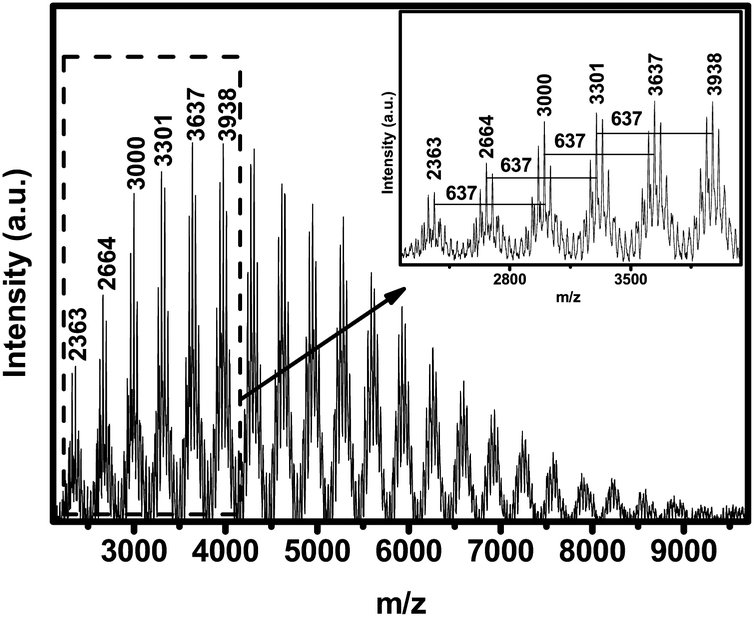
The block monomers (3) and (6) display similar chemical shifts in the 29Si-NMR spectra, which are −18.2 and −17.9 ppm, respectively. Thus PCzPOMSi also displays two single adjacent peaks at −33.1 and −33.7 ppm. To confirm the alternate polymerization of carbazole and phosphine oxide units in PCzPOMSi, matrix-assisted laser desorption ionizationmass spectroscopy (MALDI-TOF MS) of PCzPOMSi was performed, as shown in Fig. 1. We could obtain convincing structural information from it. The spectrum bears the characteristic shape of an alternating condensation polymer, which is made up of clusters of isotopic peaks. The nominal separation between these alternate major clusters, 637 Daltons, is exactly equal to the repeat unit of the hole transporting and electron transporting moieties, indicating that the synthesis proceeded as expected to give an alternating structure without a side reaction. What's more, the adjacent major clusters attribute to the Me3SiO- and HO-capped PCzPOMSi respectively.
Thermal analysis
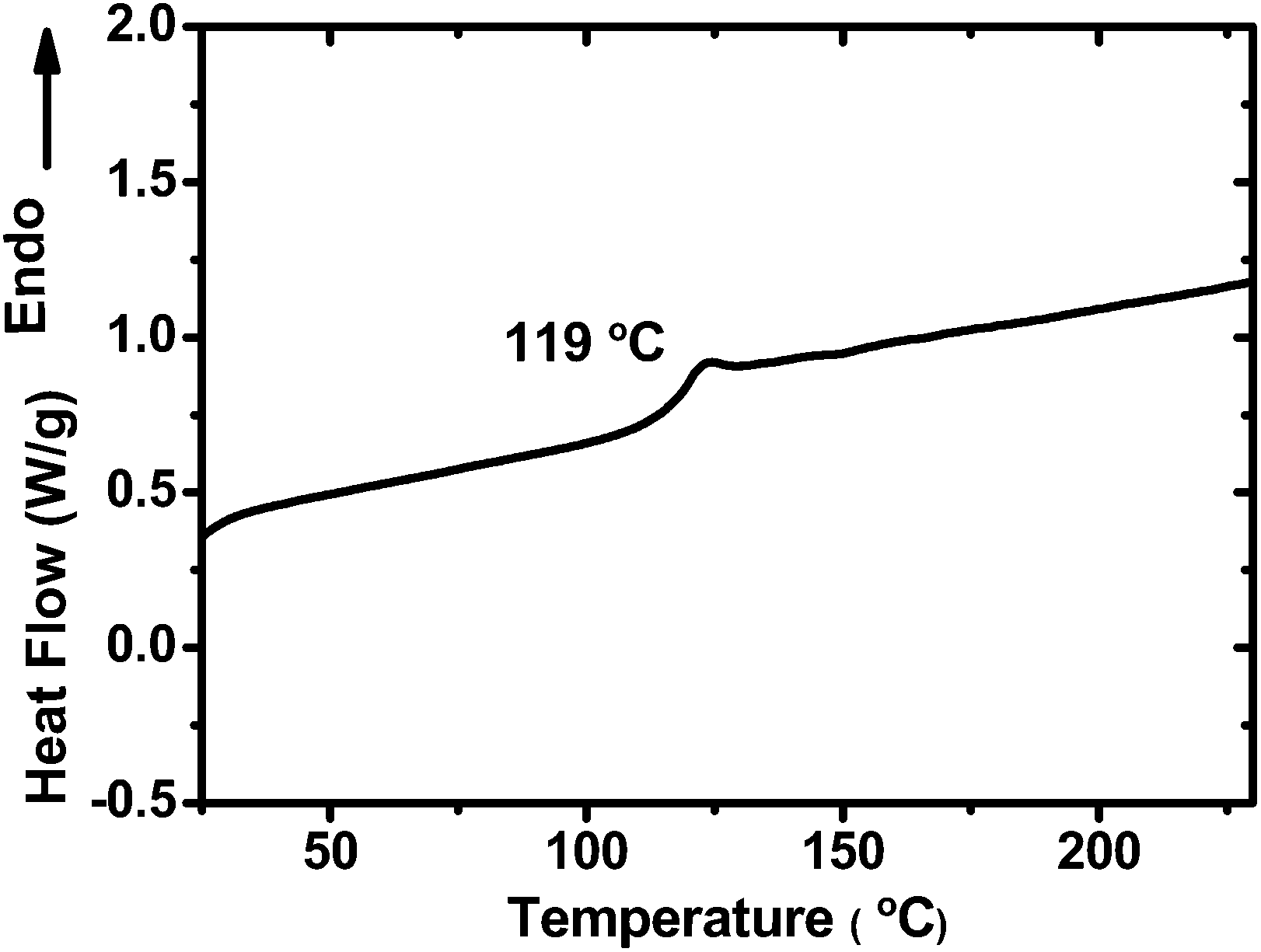
The thermal stability of PCzPOMSi was determined by thermal gravimetric analysis (TGA) and differential scanning calorimetry (DSC) under a nitrogen atmosphere. As shown in Fig. S1,† the decomposition temperature (Td), which corresponds to a weight loss of 5% at a heating rate of 10 °C min−1, was measured as 437 °C for PCzPOMSi, showing good thermal stability. Meanwhile, compared with traditional polysiloxanes, a relatively high glass transition temperature (Tg) of 119 °C was determined by DSC during its second heating cycle, as shown in Fig. 2. The relatively high Tg value is essential for stability of the luminance. In addition, no crystallization exotherm was observed. This obviously indicates that the presence of the carbazole and triphenylphosphine oxide units in the copolymer effectively suppresses the crystallization (or chain aggregation) of the polymer chain. The excellent thermal stability of PCzPOMSi appears to be a result of its polysiloxane backbone, which should be capable of enduring the inevitable Joule heating that occurs during device operation.
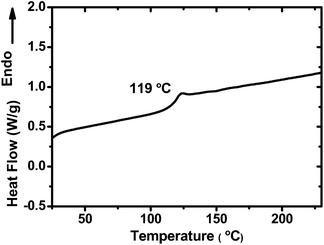 |
| | Fig. 2 DSC curves of PCzPOMSi recorded at a heating rate of 10 °C min−1. | |
Photophysical properties
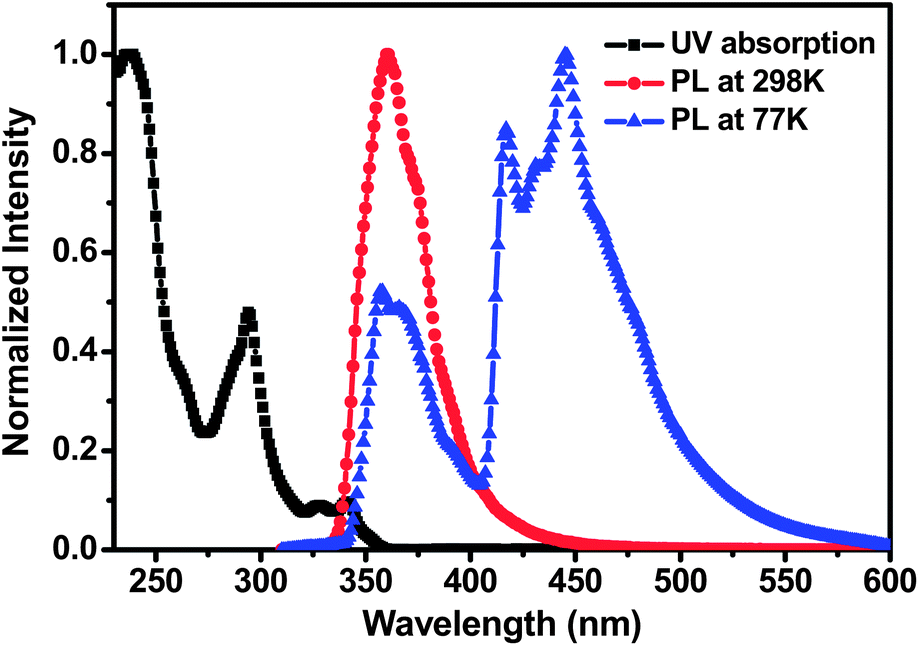
The UV-vis absorption and photoluminescence (PL) spectra of PCzPOMSi in CH2Cl2 solution at a concentration of 5 × 10−6 mol mL−1 are presented in Fig. 3. The absorption peaks in the UV-vis spectrum at around 294 can be assigned to the π → π* transitions of the carbazole moiety and weaker absorption peaks at longer wavelengths of 324 and 344 nm could be attributed to the n → π* transitions of extended conjugation of the carbazole moiety.30 In addition, a relatively wide energy gap (Eg) of 3.46 eV is obtained from the onset of absorption. The photoluminescence (PL) spectrum of PCzPOMSi displays intense UV emission peaks at 361 nm. An ET of 3.0 eV was calculated from the highest-energy vibronic sub-band of the phosphorescence spectrum, which is substantially higher than that of the deep-blue phosphor FCNIrpic. Compared to the conjugated polymeric host, the higher ET of PCzPOMSi can be attributed to the disruption of π conjugation caused by the insertion of a silicon–oxygen bond between each side group.
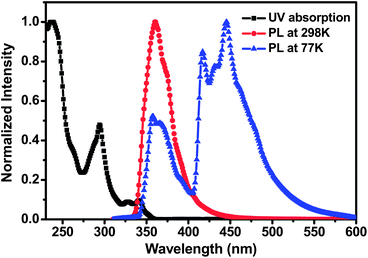 |
| | Fig. 3 The absorption and emission spectra of PCzPOMSi in dichloromethane with a concentration of 5 × 10−6 mol L−1 at room temperature and the phosphorescence spectra at 77 K. | |
Electrochemical analysis
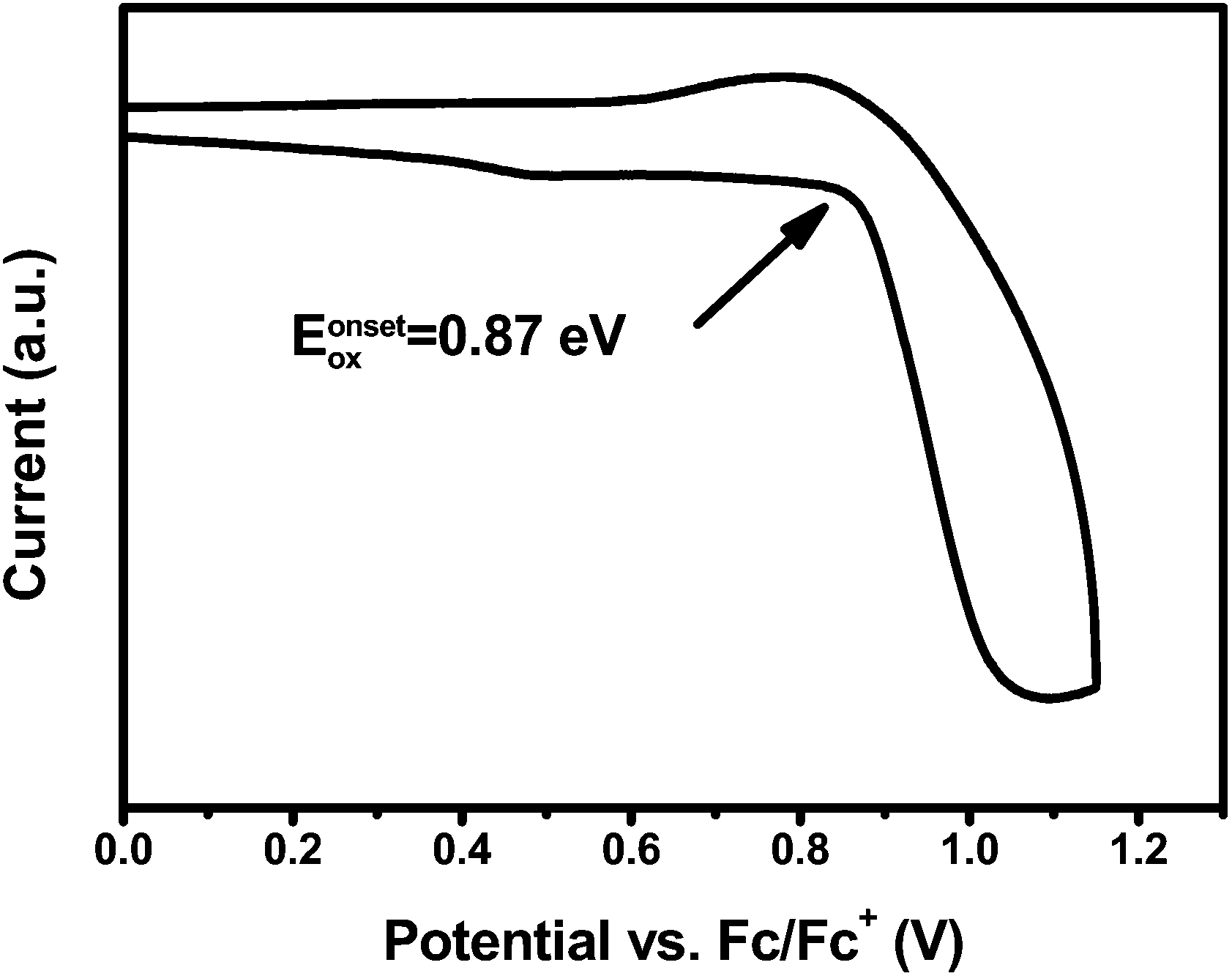
The electrochemical behavior of PCzPOMSi was investigated by cyclic voltammetry (CV) using 0.1 M tetra (n-butyl) ammonium hexafluorophosphate (Bu4NPF6) as a supporting electrolyte in anhydrous CH2Cl2 under a nitrogen atmosphere. The CV shows an irreversible oxidation feature, similar to those of other carbazole derivatives with unprotected 3, 6-positions, as shown in Fig. 4.
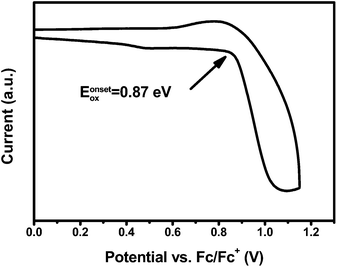 |
| | Fig. 4
CV of PCzPOMSi in CH2Cl2. | |
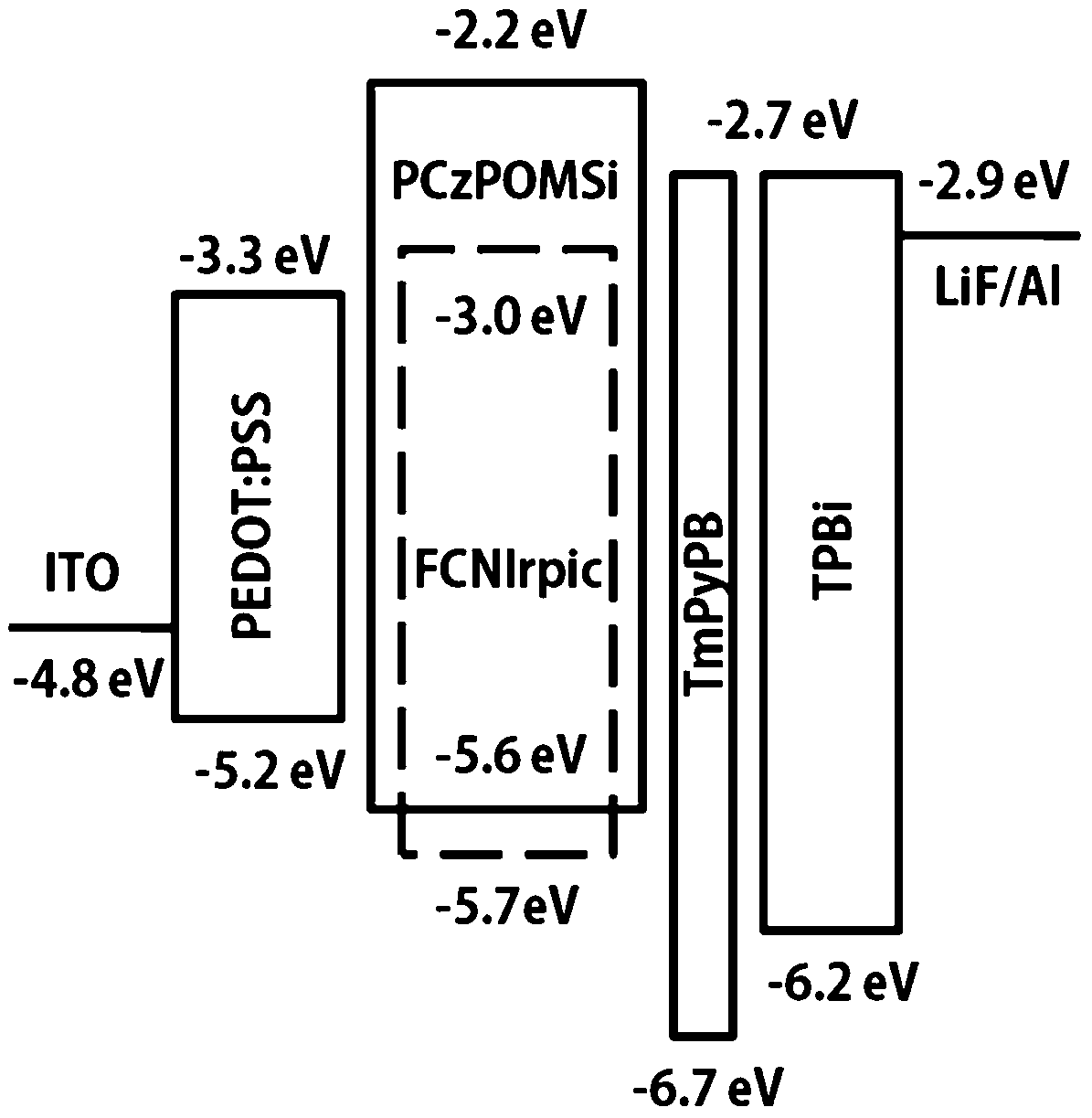
The HOMO energy level of PCzPOMSi was calculated according to the formula EHOMO = −(4.8 + Eonox − EFc) eV. The LUMO energy level was obtained by adding the optical Eg to the HOMO level (as shown in Table 1). In this way, the HOMO and LUMO levels of PCzPOMSi were estimated to be −5.6 eV and −2.2 eV respectively. The HOMO level of PCzPOMSi was significantly higher than that of carbazole derivatives, such as CBP (−6.3 eV), mCP (−5.9 eV) and CzSi (−6.0 eV), these results indicate that the incorporation of the carbazole unit and phosphine oxide unit would facilitate both hole injection and electron injection into PCzPOMSi.
Table 1 Physical properties of PCzPOMSi
|
E
T
a (eV) |
HOMOb (eV) |
LUMOc (eV) |
T
g
d (°C) |
T
d
e (°C) |
|
Triplet energy level.
Highest occupied molecular orbital.
Lowest unoccupied molecular orbital.
Glass-transition temperature determined by DSC.
Decomposition temperature determined by TGA at 5% weight loss.
|
| 3.0 |
−5.60 |
−2.2 |
118 |
437 |
Electroluminescent properties
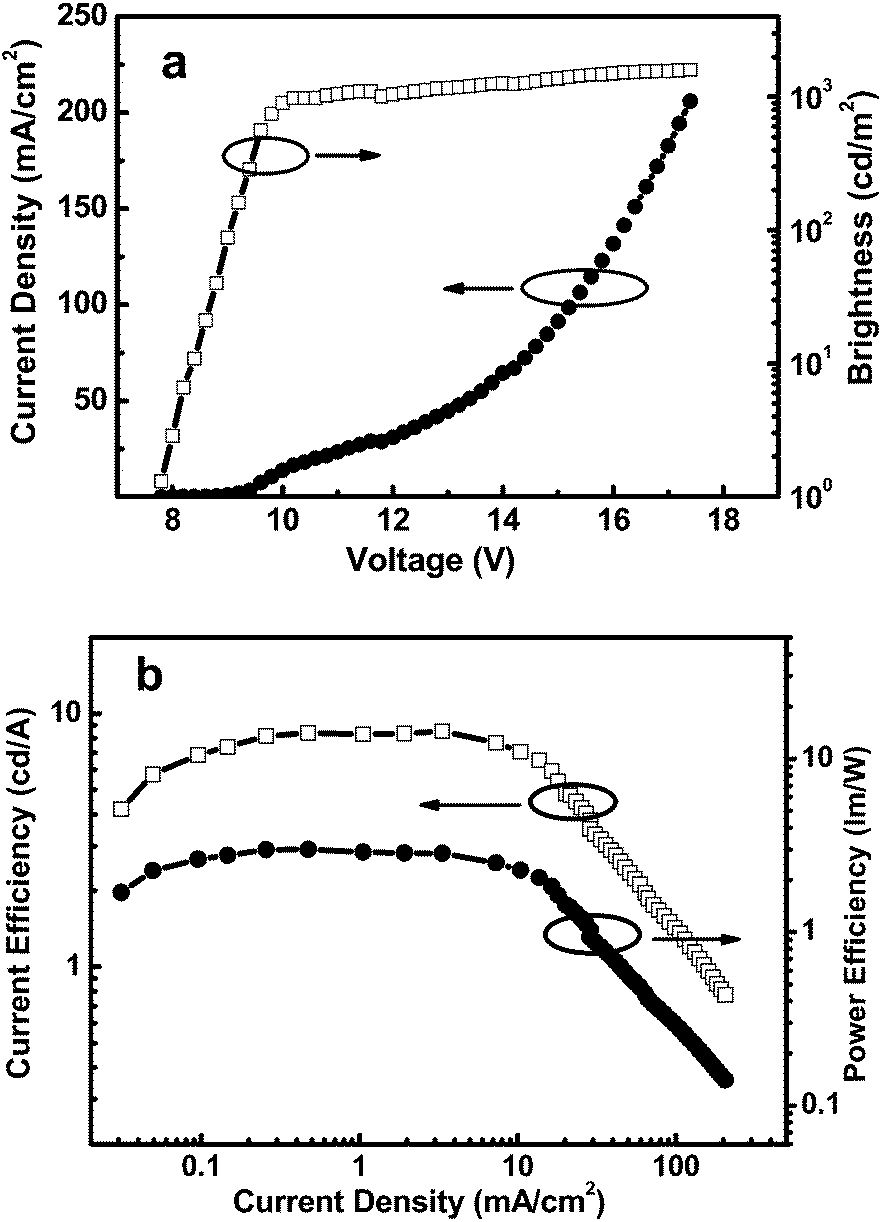
The high triplet energy, the high-lying HOMO level, the bipolar character, and the good film-forming ability of the new polymer make it a good candidate as a solution processed host for deep-blue PhPLEDs. We fabricated the FCNIrpic-based blue phosphorescent device with a configuration of indium tin oxide (ITO)/PEDOT:PSS (40 nm)/host: 10 wt% FCNIrpic (40–45 nm)/1,3,5-tri(m-pyrid-3-yl-phenyl)benzene (Tm3PyPB, 5 nm)/1,3,5-tris(1-phenyl-1H-benzimidazol-2-yl)benzene (TPBI, 30 nm)/LiF (1 nm)/Al (100 nm). In this architecture, PEDOT:PSS acts as a hole injection layer, host doped with 10 wt% FCNIrpic is spin-coated to form an emitting layer, and Tm3PyPB and TPBI were used as the hole/exciton-blocking and electron-transporting layers, respectively. The relative energy levels of each layer are presented in Fig. 5. As shown in Table 2, the ET of PCzPOMSi is sufficient to confine triplet excitons in the emitting layer. The current density–voltage–brightness (J–V–L) curves of the deep blue PhOLED are presented in Fig. 6a. The device exhibits a turn-on voltage of 7.7 V (corresponding to 1 cd m−2). Meanwhile, a maximum brightness of 1598 cd m−2 was achieved. Power and current efficiencies of the device plotted with respect to current density are depicted in Fig. 6b. The device shows maximum power and current efficiencies of 3 lm W−1 and 8.5 cd A−1 at 286 cd m−2, respectively, which are observed over a wide range of high luminance. It is noted that the highest external quantum efficiency for the device was 4% at a practical luminance of 286 cd m−2.
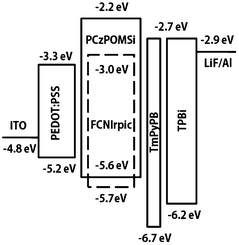 |
| | Fig. 5 Relative energy levels in a PhPLED containing deep blue emitter FCNIrpic doped in PCzPOMSi as a host. | |
Table 2 Performance of the FCNIrpic-based device
|
V
on
a (V) |
L
max
b (cd m−2) |
η
c,max
c (cd A−1) |
η
p,maxd (lm W−1) |
η
ext,max
e (100%) |
CIEf (x, y) |
|
Turn-on voltage.
Maximum luminance.
Maximum luminous efficiency.
Maximum power efficiency.
External quantum efficiency.
Commission Internationale deI'Eclairage coordinates.
|
| 7.7 |
1598 |
8.5 |
3 |
4 |
0.14, 0.22 |
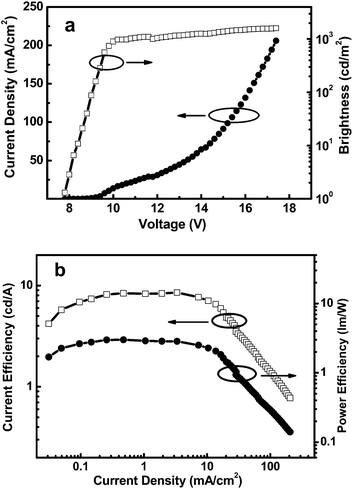 |
| | Fig. 6 (a) J–V–L characteristics of a 10 wt% FCNIrpic-doped PhPLED. (b) Power efficiency and current efficiency versus the luminance of a deep blue PhPLED containing FCNIrpic doped in PCzPOMSi. | |
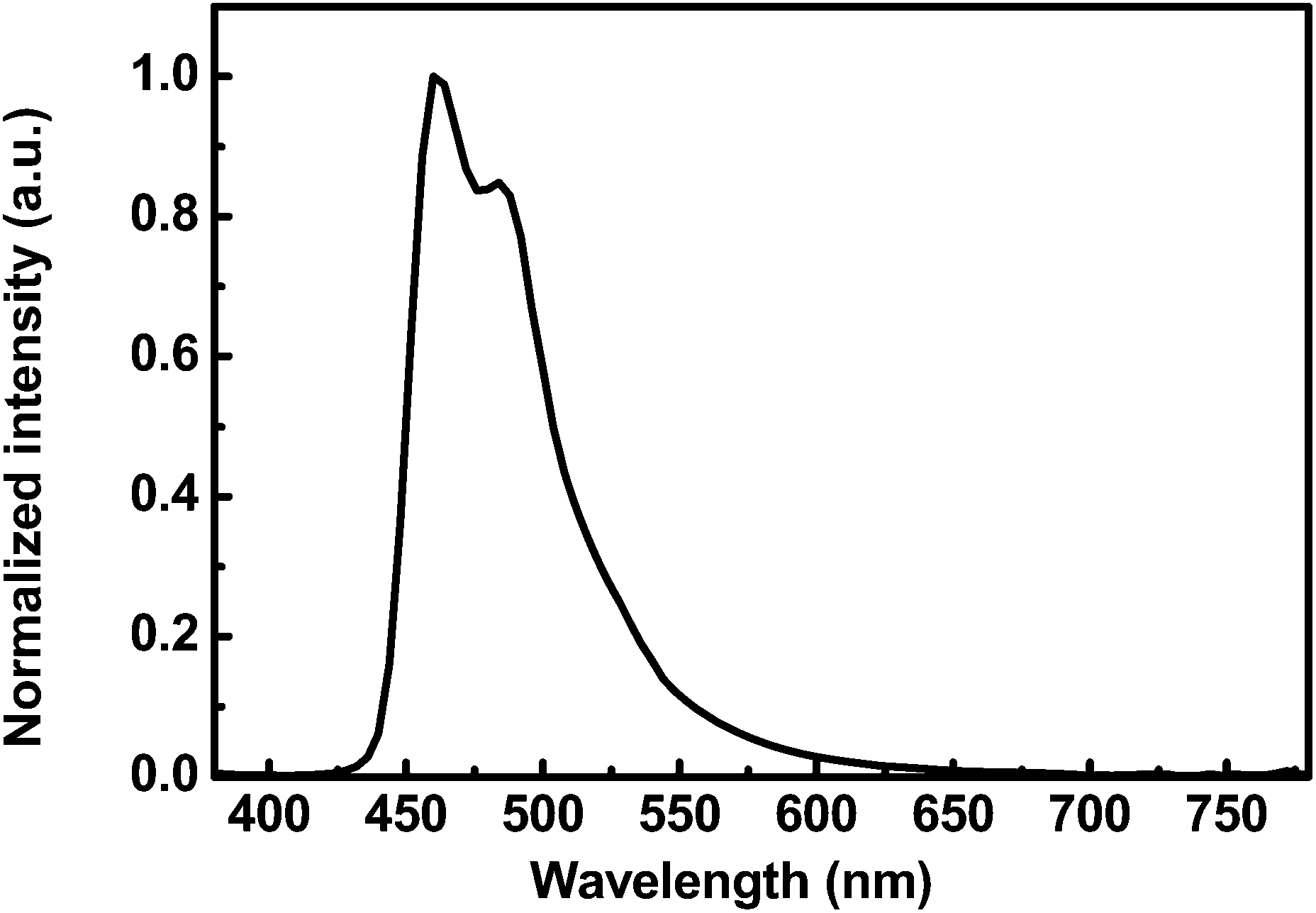
The electroluminescence (EL) spectrum of the deep blue PhPLED is shown in Fig. 7. The EL spectrum was normalized, and the device exhibited a peak centered at 460 nm and a vibrational peak centered at 484 nm with CIE coordinates (0.14, 0.22). The EL spectrum was affected by the doping concentration and red-shift of the CIE coordinates was observed due to significant intermolecular interactions between the dopant molecules. The FCNIrpic has strong polar F and CN functional groups in the ligands, resulting in strong intermolecular interactions at a high doping concentration.31
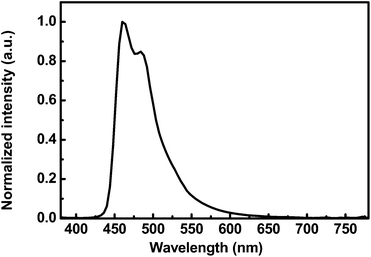 |
| | Fig. 7 Normalized EL spectrum of deep blue PhPLEDs with respect to luminance at 100 cd m−2. | |
Mechanical properties
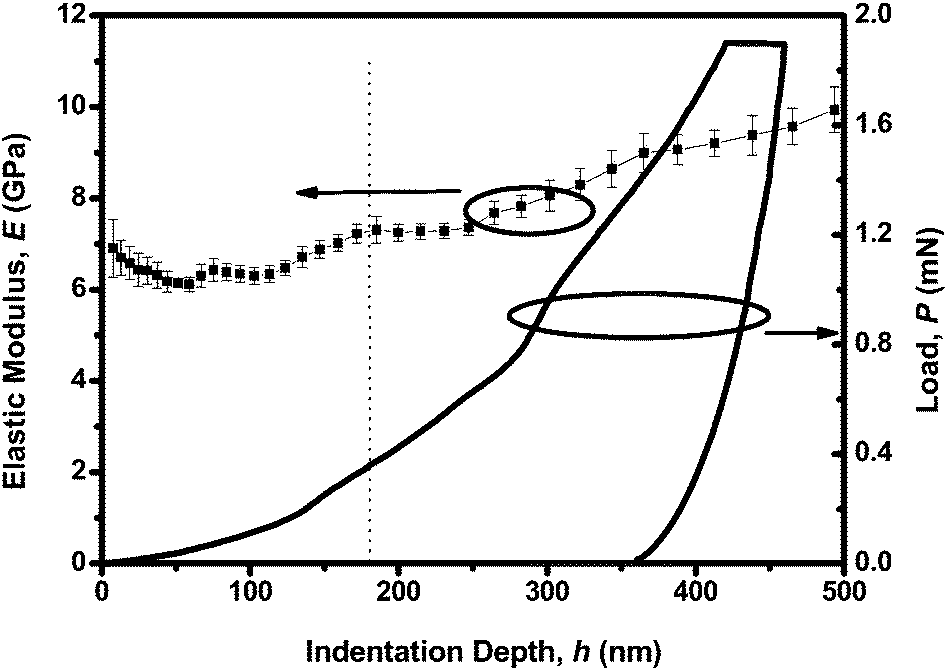
Although PCzPOMSi is a highly efficient host material, for practical application good mechanical performance is of importance. Therefore, the mechanical properties of the host PCzPOMSi film were determined by the nanoindentation technique. Nanoindentation, using applied load and depth data obtained from the experiment, has been extensively applied to study the surface and near-surface mechanical properties of the thin films.32–34 A three sided pyramidal Berkovich tip (tip radius ∼ 100 nm) was used to determine the Young's modulus (E). As shown in Fig. 8, the Young's modulus of the PCzPOMSi films is 8.424(287) GPa, which was calculated from the depth of 100–500 nm. The Young's modulus of the PCzPOMSi films is a little higher than the conventional polymers which have a Young's modulus in the range of 0.2 GPa to 5 GPa.35 The higher Young's modulus of PCzPOMSi could be attributed to the ionic properties of the silicon–oxygen bond. Therefore the mechanical properties of the host PCzPOMSi films could meet the needs of practical applications.
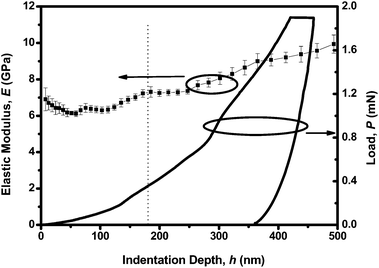 |
| | Fig. 8 Young's modulus and load of PCzPOMSi as a function of indentation depth, wherein each error bar represents the standard deviation from 20 indents. | |
Experimental section
Materials
All reactants (Adamas-beta) were purchased from Adamas Reagent, Ltd. without further purification and all solvents were supplied by the Beijing Chemical Reagent Co., Ltd. Anhydrous and deoxygenated solvents were obtained by distillation over sodium benzophenone complex.
Device fabrication and characterization
The hole-injection material PEDOT:PSS (8000), and electron-transporting and hole-blocking material TPBI, were used from commercial sources. ITO-coated glass with a sheet resistance of 10 Ω per square was used as the substrate. Before device fabrication, the ITO-coated glass substrate was precleaned carefully and exposed to UV-ozone for 2 min. After that, PEDOT:PSS was spin-coated to the clean ITO substrate as a hole-injection layer. Then, the host material doped with 10% (by weight) FCNIrpic was spin-coated to form a 40 nm thick emissive layer (EML) and annealed at 80 °C for 30 min to remove residual solvent. Finally a 5 nm-thick exaction block layer of Tm3PyPB was vacuum deposited followed by a 30 nm-thick electron-transporting layer (ETL) of TPBI, and a cathode composed of a 1 nm-thick layer of lithium fluoride (LiF) and aluminum (100 nm) which were sequentially deposited onto the substrate through shadow masking with a pressure of 10−6 Torr. The current density–voltage–luminance (J–V–L) characteristics of the device were measured using a Keithley 2400 Source meter and a Keithley 2000 Source multimeter equipped with a calibrated Si-photodiode in a glove box. The EL spectra were measured using a JY SPEX CCD3000 spectrometer. The EQE values were calculated from the luminance, current density, and electroluminescence spectrum according to previously reported methods.36 All measurements were performed at room temperature under ambient conditions.
Synthetic procedures
9-(4-Bromophenyl)-9-carbazole37.
This was synthesized according to the general Ullmann procedure and the characterization data can be found in the literature.
Methyldiethyoxylclorosilane.
To boiling petroleum ether (30–60 boiling range) (200 mL), methyltrichlorosilane (150 mL, 127 mmol) and alcohol (146 mL, 254 mmol) were slowly dropped under the same velocity. Afterwards, the mixture was distilled to give a colorless liquid product (127–128 °C) with 52% yield (111 g). 1H NMR (400 MHz, CDCl3, δ): 0.45 (s, 3H, Si–CH3), 1.25 (t, 6H, J = 7.3 Hz; –CH2CH3), 3.87 (q, 4H, J = 7.2 Hz; –OCH2CH3); 29Si-NMR (400 MHz, CDCl3, δ): 29.32.
9-(4-(Diethoxy(methyl)silyl)phenyl)-9-carbazole.
Under argon atmosphere, 9-(4-bromophenyl)-9-carbazole (1.93 g, 6 mmol) was dissolved in 200 mL anhydrous THF. The reaction flask was cooled to −78 °C in a Dewar flask and n-butyllithium (2.2 M in hexane, 3.3 mL) was added dropwise slowly within 15 minutes. Stirring was continued for half an hour with keeping this low temperature, followed by the addition of chlorodiethoxy(methyl)silane (1.5 g, 8.9 mmol) under argon atmosphere. After the addition was completed, the resulting mixture was allowed to warm to ambient temperature gradually and quenched with 20 mL water. Then it was extracted with Et2O, the combined organic layer was washed with distilled water 3 times and dried over anhydrous magnesium sulfate. The solvent was evaporated in vacuo to give the crude product, which was purified by column chromatography on silica gel using (petroleum ether![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dichloromethane = 6:1) as the eluent to obtain the product as a colorless oil (0.76 g, 33.9%). 1H NMR (400 MHz, CDCl3, δ): 0.48 (s, 3H, –CH3), 1.34 (t, J = 7 Hz, 6H; –OCH2CH3), 3.93 (q, J = 7 Hz, 4H; –OCH2), 7.32 (t, J = 7 Hz, 2H; Ar H), 7.43 (t, J = 8 Hz, 2H; Ar H), 7.48 (d, J = 8 Hz, 2H; Ar H), 7.62 (d, J = 8 Hz, 2H; Ar H), 7.90 (d, J = 8 Hz, 2H; Ar H), 8.17 (d, J = 8 Hz, 2H; Ar H). 13C NMR (400 MHz, CDCl3, δ): −4.00, 18.51, 58.78, 109.94, 120.22, 123.58, 126.16, 134.28, 135.71, 139.37, 140.74. 29Si-NMR (400 MHz, CDCl3, δ): −18.20. Anal. calcd for C23H25NO2Si: C, 73.56; H, 6.71; N, 3.73. Found: C, 73.63; H, 6.65; N, 3.67%.
:dichloromethane = 6:1) as the eluent to obtain the product as a colorless oil (0.76 g, 33.9%). 1H NMR (400 MHz, CDCl3, δ): 0.48 (s, 3H, –CH3), 1.34 (t, J = 7 Hz, 6H; –OCH2CH3), 3.93 (q, J = 7 Hz, 4H; –OCH2), 7.32 (t, J = 7 Hz, 2H; Ar H), 7.43 (t, J = 8 Hz, 2H; Ar H), 7.48 (d, J = 8 Hz, 2H; Ar H), 7.62 (d, J = 8 Hz, 2H; Ar H), 7.90 (d, J = 8 Hz, 2H; Ar H), 8.17 (d, J = 8 Hz, 2H; Ar H). 13C NMR (400 MHz, CDCl3, δ): −4.00, 18.51, 58.78, 109.94, 120.22, 123.58, 126.16, 134.28, 135.71, 139.37, 140.74. 29Si-NMR (400 MHz, CDCl3, δ): −18.20. Anal. calcd for C23H25NO2Si: C, 73.56; H, 6.71; N, 3.73. Found: C, 73.63; H, 6.65; N, 3.67%.
(4-(9-Carbazol-9-yl)phenyl)(methyl)silanediol.
To a mixture of 9-(4-(diethoxy(methyl)silyl)phenyl)-9-carbazole (0.65 g, 1.7 mmol) and THF 100 mL, water (2 mL) and two drops of 1 M HCl was added. The mixture was stirred at 0 °C for 24 h. After the reaction was completed, the solvent was extracted with Et2O and washed with distilled water 3 times and dried over anhydrous magnesium sulfate. The filtrate was condensed by reduced pressure and precipitated with toluene to give a crude product which was purified by column chromatography on silica gel using petroleum ether:ethyl acetate (4:1) as the eluent to obtain the product as a colorless oil (0.26 g, 46.2%). 1H NMR (400 MHz, CDCl3, δ): 0.61(s, 3H; –CH3), 2.69(s; –OH), 7.35 (d, J = 8 Hz, 2H; ArH), 7.39 (t, J = 8 Hz, 2H; ArH), 7.42 (t, J = 8 Hz, 2H; ArH), 7.66 (d, J = 8 Hz, 2H; ArH), 7.96 (d, J = 8 Hz, 2H; ArH), 8.17 (d, J = 8 Hz, 2H; ArH). 13C NMR (400 MHz, CDCl3, δ): −0.01, 110.48, 120.95, 124.26, 126.8, 135.82, 136.56, 140.20, 141.31. 29Si-NMR (400 MHz, CDCl3, δ): −23.01.
(4-Bromophenyl)diphenylphosphine oxide.
n-Butyllithium (1.6 M in hexane, 13.8 mL, 22 mmol) was added dropwise to a solution of 1,4-dibromobenzene (4.72 g, 20 mmol) in anhydrous THF (160 mL) at −78 °C. The reaction mixture was kept at this temperature for half an hour, and then chlorodiphenylphosphine (3.95 mL, 22 mmol) was added. The resulting mixture was stirred for a further 3 h at −78 °C before quenching with water. Then the mixture was extracted with CH2Cl2, washed with water, and dried over anhydrous Mg2SO4. After the solvent was completely removed, 30% hydrogen peroxide (50 mL) and THF (100 mL) were added to the obtained residue and the mixture was stirred overnight at room temperature. The organic layer was separated and washed with water and then brine. The extract was evaporated to dryness, and the residue was purified by column chromatography on silica gel with dichloromethane/ethyl acetate as eluent to give a white solid (7.14 g, 86%). 1H NMR (400 MHz, CDCl3, δ): 7.18 (t, J = 8 Hz, 2H; Ar H), 7.32 (t, J = 8 Hz, 4H; Ar H), 7.37 (d, J = 8 Hz, 6H; Ar H), 7.48 (d, 2H; Ar H). 13C NMR (400 MHz, CDCl3, δ): 123.5, 128.7, 131.7, 133.7, 135.2, 136.6.
(4-(Diethoxy(methyl)silyl)phenyl)diphenylphosphine oxide.
Under an argon atmosphere, methyldiethyoxylclorosilane (3.8 mL, 20 mmol) and magnesium powder (0.24 g, 10 mmol) were added in 30 mL anhydrous THF. The reaction flask was heated to 70 °C and a solution of (4-bromophenyl)diphenylphosphine oxide (3.57 g, 10 mmol) in 50 mL of THF was added dropwise slowly within 30 minutes. After the addition was completed, the mixture was stirred for 3 h keeping this temperature. Then it was cooled to room temperature and extracted with Et2O. The solvent was evaporated in vacuo to give the crude product, which was purified by column chromatography on silica gel using (petroleum ether:dichloromethane = 4:1) as the eluent to obtain the product as a colorless oil (2.58 g, 63%). 1H NMR (400 MHz, CDCl3, δ): 0.38 (s, 3H, –CH3), 1.26 (t, J = 7 Hz, 6H; –OCH2CH3), 3.83 (q, J = 7 Hz, 4H; –OCH2), 7.32 (brs,12H; Ar H), 7.62 (d, J = 8 Hz, 2H; Ar H). 13C NMR (400 MHz, CDCl3, δ): −2.1, 20.4, 60.6, 130.6, 134.8, 135.8, 137.3, 138.9, 141.4. 29Si-NMR (400 MHz, CDCl3, δ): −17.9.
PCzPOMSi.
To a mixture of (4-(diethoxy(methyl)silyl)phenyl)diphenylphosphine oxide (6) (5 mmol), (4-(9-carbazol-9-yl)phenyl)(methyl)silanediol (4) (5 mmol) and 50 mL anhydrous THF, two drops of tetrabutyl titanate (TBOT) were added as catalyst. The mixture was stirred at 80 °C for four days by tracking the degree of the reaction with a Fourier Transform Infrared (FTIR) Spectrometer. After the reaction was completed, 1 mL trimethylchlorosilane was added and the mixture was stirred for another 12 h. Afterwards, the solvent was evaporated in vacuo to give a sticky liquid, which was dissolved in toluene and precipitated with methanol to obtain the product as a white powder. 1H NMR (400 MHz, CDCl3, δ): 0.38 (brs, 3H, –CH3), 7.21 (brs, 6H; Ar H), 7.52 (brs, 6H; Ar H), 8.09 (brs, 2H; Ar H). 29Si-NMR (400 MHz, CDCl3, δ): −33.1, −33.7.
Conclusions
In summary, a well-defined alternating copolysiloxane bipolar host was designed and synthesized successfully through incorporating hole and electron transporting units into a polysiloxane backbone. This host exhibits excellent thermal and morphological stability as a deep blue phosphorescent emitter FCNIrpic. Because the phenylcarbazole and triphenylphosphine oxide segments are connected through silicon–oxygen bonds that disrupt the π conjugation, PCzPOMSi possesses a high ET of 3.0 eV. Electrochemical studies revealed that the carbazole unit increases the HOMO level and the phosphine oxide unit reduces the LUMO of PCzPOMSi, facilitating hole and electron injection. A device containing PCzPOMSi as a host exhibits both high luminance and desirable efficiency. It is believed that the synthetic strategy used to produce PCzPOMSi can be extended to other polysiloxane-based bipolar hosts by changing the side groups and through further design of the device structure alternating copolysiloxane hosts can be developed to fabricate efficient solution-processed PhPLEDs.
Acknowledgements
The financial support of the NSFC (no. 21104002) and the fundamental research funds for the central university (ZY1204) are gratefully acknowledged. Zhongjie Ren is grateful to Dr Wei Li for the nanoindentation tests.
Notes and references
- S. Shao, J. Ding, T. Ye, Z. Xie, L. Wang, X. Jing and F. Wang, Adv. Mater., 2011, 23, 3570–3574 CrossRef CAS PubMed
 .
.
- J. Huang, T. Watanabe, K. Ueno and Y. Yang, Adv. Mater., 2007, 19, 739–743 CrossRef CAS .
- A. van Dijken, J. J. Bastiaansen, N. M. Kiggen, B. M. Langeveld, C. Rothe, A. Monkman, I. Bach, P. Stössel and K. Brunner, J. Am. Chem. Soc., 2004, 126, 7718–7727 CrossRef CAS PubMed .
- C. Jiang, W. Yang, J. Peng, S. Xiao and Y. Cao, Adv. Mater., 2004, 16, 537–541 CrossRef CAS .
- P. T. Furuta, L. Deng, S. Garon, M. E. Thompson and J. M. Fréchet, J. Am. Chem. Soc., 2004, 126, 15388–15389 CrossRef CAS PubMed .
- S. Gong, C. Yang and J. Qin, Chem. Soc. Rev., 2012, 41, 4797–4807 RSC .
- B. S. Du, J. L. Liao, M. H. Huang, C. H. Lin, H. W. Lin, Y. Chi, H. A. Pan, G. L. Fan, K. T. Wong, G. H. Lee and P. T. Chou, Adv. Funct. Mater., 2012, 22, 3491–3499 CrossRef CAS .
- L. Chen, B. Zhang, Y. Cheng, Z. Xie, L. Wang, X. Jing and F. Wang, Adv. Funct. Mater., 2010, 20, 3143–3153 CrossRef CAS .
- X. Chen, J. L. Liao, Y. Liang, M. O. Ahmed, H. E. Tseng and S. A. Chen, J. Am. Chem. Soc., 2003, 125, 636–637 CrossRef CAS PubMed .
- M. Zhu, T. Ye, X. He, X. Cao, C. Zhong, D. Ma, J. Qin and C. Yang, J. Mater. Chem., 2011, 21, 9326–9331 RSC .
- K. S. Yook and J. Y. Lee, Adv. Mater., 2012, 24, 3169–3190 CrossRef CAS PubMed .
- D. Kim, L. Zhu and J. L. Brédas, Chem. Mater., 2012, 24, 2604–2610 CrossRef CAS .
- M. Sudhakar, P. I. Djurovich, T. E. Hogen-Esch and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7796–7797 CrossRef CAS PubMed .
- C. T. Chen, Y. Wei, J. S. Lin, M. V. Moturu, W. S. Chao, Y. T. Tao and C. H. Chien, J. Am. Chem. Soc., 2006, 128, 10992–10993 CrossRef CAS PubMed .
- S. Gong, Q. Fu, Q. Wang, C. Yang, C. Zhong, J. Qin and D. Ma, Adv. Mater., 2011, 23, 4956–4959 CrossRef CAS PubMed .
- C. J. Zheng, J. Ye, M. F. Lo, M. K. Fung, X. M. Ou, X. H. Zhang and C. S. Lee, Chem. Mater., 2012, 24, 643–650 CrossRef CAS .
- A. Chaskar, H. F. Chen and K. T. Wong, Adv. Mater., 2011, 23, 3876–3895 CrossRef CAS PubMed .
- Y. Tao, C. Yang and J. Qin, Chem. Soc. Rev., 2011, 40, 2943–2970 RSC .
- Z. Ge, T. Hayakawa, S. Ando, M. Ueda, T. Akiike, H. Miyamoto, T. Kajita and M. Kakimoto, Adv. Funct. Mater., 2008, 18, 584–590 CrossRef CAS .
- Y. Tao, Q. Wang, C. Yang, J. Qin and D. Ma, ACS Appl. Mater. Interfaces, 2010, 2, 2813–2818 CAS .
- N. Lin, J. Qiao, L. Duan, H. Li, L. Wang and Y. Qiu, J. Phys. Chem. C, 2012, 116, 19451–19457 CAS .
- C. W. Lee and J. Y. Lee, Adv. Mater., 2013, 25, 596–600 CrossRef CAS PubMed .
- X. H. Zhao, Z. S. Zhang, Y. Qian, M. D. Yi, L. Xie, C. P. Hu, G. Xie, H. Xu, C. Han and Y. Zhao, J. Mater. Chem. C, 2013, 1, 3482–3490 RSC .
- E. Mondal, W. Y. Hung, H. C. Dai and K. T. Wong, Adv. Funct. Mater., 2013, 23, 3096–3105 CrossRef CAS .
- C. W. Lee, J. Kim, S. Joo and J. Y. Lee, ACS Appl. Mater. Interfaces, 2013, 5, 2169–2173 CAS .
- D. Hu, F. Shen, H. Liu, P. Lu, Y. Lv, D. Liu and Y. Ma, Chem. Commun., 2012, 48, 3015–3017 RSC .
- M. H. Tsai, T. H. Ke, H. W. Lin, C. C. Wu, S. F. Chiu, F. C. Fang, Y. L. Liao, K. T. Wong, Y. H. Chen and C. I. Wu, ACS Appl. Mater. Interfaces, 2009, 1, 567–574 CAS .
- L. Fu, G. Xiao and D. Yan, ACS Appl. Mater. Interfaces, 2010, 2, 1601–1607 CAS .
- Z. Ren, Y. Tian, P. Xie, S. Yan and R. Zhang, Polym. Chem., 2010, 1, 1095–1101 RSC .
- M. Lin, S. Yang, H. Chang, Y. Huang, Y. Tsai, C. Wu, S. Chou, E. Mondal and K. Wong, J. Mater. Chem., 2012, 22, 16114–16120 RSC .
- S. O. Jeon, K. S. Yook, C. W. Joo and J. Y. Lee, Adv. Funct. Mater., 2009, 19, 3644–3649 CrossRef CAS .
- K. Geng, F. Yang, T. Druffel and E. A. Grulke, Polymer, 2005, 46, 11768–11772 CrossRef CAS PubMed .
- T. H. Fang and W. J. Chang, Microelectron. J., 2004, 35, 595–599 CrossRef CAS PubMed .
- K. H. T. Raman, M. S. R. N. Kiran, U. Ramamurty and G. Mohan Rao, Appl. Surf. Sci., 2012, 258, 8629–8635 CrossRef CAS PubMed .
- S. Bundschuh, O. Kraft, H. K. Arslan, H. Gliemann, P. G. Weidler and C. Woll, Appl. Phys. Lett., 2012, 101, 10190 CrossRef .
- S. Forrest, D. Bradley and M. Thompson, Adv. Mater., 2003, 15, 1043–1048 CrossRef CAS .
- J. Ding, Q. Wang, L. Zhao, D. Ma, L. Wang, X. Jing and F. Wang, J. Mater. Chem., 2010, 20, 8126–8133 RSC .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3py00840a |
|
| This journal is © The Royal Society of Chemistry 2014 |