RAFT copolymerization of acid chloride-containing monomers†
Myungeun
Seo‡
and
Marc A.
Hillmyer
*
Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, USA. E-mail: hillmyer@umn.edu
First published on 27th August 2013
Abstract
Controlled copolymerization of acryloyl chloride (AC), methacryloyl chloride (MAC), and vinylbenzoyl chloride (VBC) with styrene via the reversible addition–fragmentation chain transfer (RAFT) process was investigated. Copolymerization was conducted in 1,4-dioxane at 60 °C using azobisisobutyronitrile as an initiator and S-1-dodecyl-S′-(R,R′-dimethyl-R′′-acetic acid) trithiocarbonate as a chain transfer agent (CTA). The reactive copolymer was obtained by precipitating in hexanes. Methyl ester analogues of the reactive polymers were obtained by precipitating in methanol for analytical purposes and their 1H nuclear magnetic resonance spectroscopy and size exclusion chromatography analyses indicated that the best control was achieved for P(S-co-VBC) produced by copolymerization of styrene and VBC. Kinetics of the copolymerization of styrene and VBC was consistent with the RAFT mechanism. Reactive block polymers consisting of the P(S-co-VBC) block were also readily prepared using a macromolecular chain transfer agent. P(S-co-VBC) was successfully functionalized by reaction with alcohols or amines to form ester or amide linkages demonstrating its utility for the postpolymerization modification approach.
Introduction
One great advantage that controlled radical polymerization offers is high functionality tolerance.1 In contrast to other controlled polymerization techniques where very few functional groups are inert under the polymerization conditions, controlled radical polymerization provides versatile opportunities to directly (co)polymerize reactive monomers possessing various functional groups to produce reactive polymers with well-defined chain length, narrow dispersity, and controlled amount of reactive groups. The resulting reactive polymer allows postpolymerization modification with high yield to introduce another functionality, and therefore enables construction of diverse libraries of various functional polymers with identical average chain length and distribution.2 Postpolymerization modification with multifunctional reactants will also provide a route toward crosslinked polymeric material with controlled crosslinking density.3–6A number of reactive monomers that can be directly (co)polymerized by controlled radical polymerization and used for postpolymerization modification have been developed and discussed in recent reviews.2,7 Vinyl monomers containing activated carbonyl leaving groups such as N-hydroxysuccinimide8–10 and pentafluorophenol ester11 have been shown to be highly useful for this purpose, since these functionalities do not interfere with the polymerization and readily react with nucleophiles such as alcohols and amines under mild conditions to produce ester or amide bonds with high yield. However, there has been surprisingly no literature reporting controlled (co)polymerization of acid chloride-containing monomers despite the fact that acid chloride functionality is probably the simplest, well-known, and industrially feasible activated carbonyl group.
Free radical polymerizations of acryloyl chloride (AC)12–18 and methacryloyl chloride (MAC)19–21 have been known as a means to produce poly(acryloyl chloride) (PAC) and poly(methacryloyl chloride) (PMAC), respectively.22 Since polymers with a smaller but controlled number of reactive groups are often more attractive for synthetic purposes,22 copolymerization of AC15–17,23–26 and MAC22,27,28 with non-reactive monomers such as methyl methacrylate has been also studied. These reactive polymers have been used to synthesize poly(metha)crylates with complex side groups such as azobenzene moieties via postpolymerization modification because those polymers were difficult to prepare by direct polymerization.13,18,23,24,29–35 Vinylbenzoyl chloride (VBC), another acid chloride-containing monomer, was developed in 1968 and copolymerized with styrene via the free radical polymerization mechanism.36
Controlled radical copolymerization of acid chloride-containing monomers will provide a simple and efficient methodology to incorporate a controlled amount of reactive acid chloride groups into well-defined macromolecular architectures such as block polymers with narrow dispersity. To this end, we explored copolymerization of AC, MAC, and VBC with styrene (S) via the reversible addition–fragmentation chain transfer (RAFT) polymerization process.1,37 Excellent functionality tolerance of RAFT polymerization38 makes it suitable for copolymerization of acid chloride-containing monomers. We focused on VBC which produced a relatively more controlled copolymer than others, and demonstrate that P(S-co-VBC) and reactive block polymers containing the P(S-co-VBC) block can be readily synthesized by the RAFT process. We also show successful postpolymerization modification of P(S-co-DVB) with various amines and alcohols.
Results and discussion
The copolymerization scheme is shown in Scheme 1. S-1-Dodecyl-S′-(R,R′-dimethyl-R′′-acetic acid) trithiocarbonate was used as a chain transfer agent (CTA) for RAFT polymerization because of its excellent control over styrene (co)polymerization.6,39 Copolymerization was conducted in 1,4-dioxane initiated by azobisisobutyronitrile (AIBN) at 60 °C for 23–25 h. Reactive polymers were obtained in a form of solid by precipitation in hexanes. For characterization, polymers were precipitated in methanol and stirred overnight to form methyl ester analogues which were analyzed by 1H nuclear magnetic resonance (NMR) spectroscopy and size exclusion chromatography (SEC). Table 1 summarizes the characterization data of the copolymers.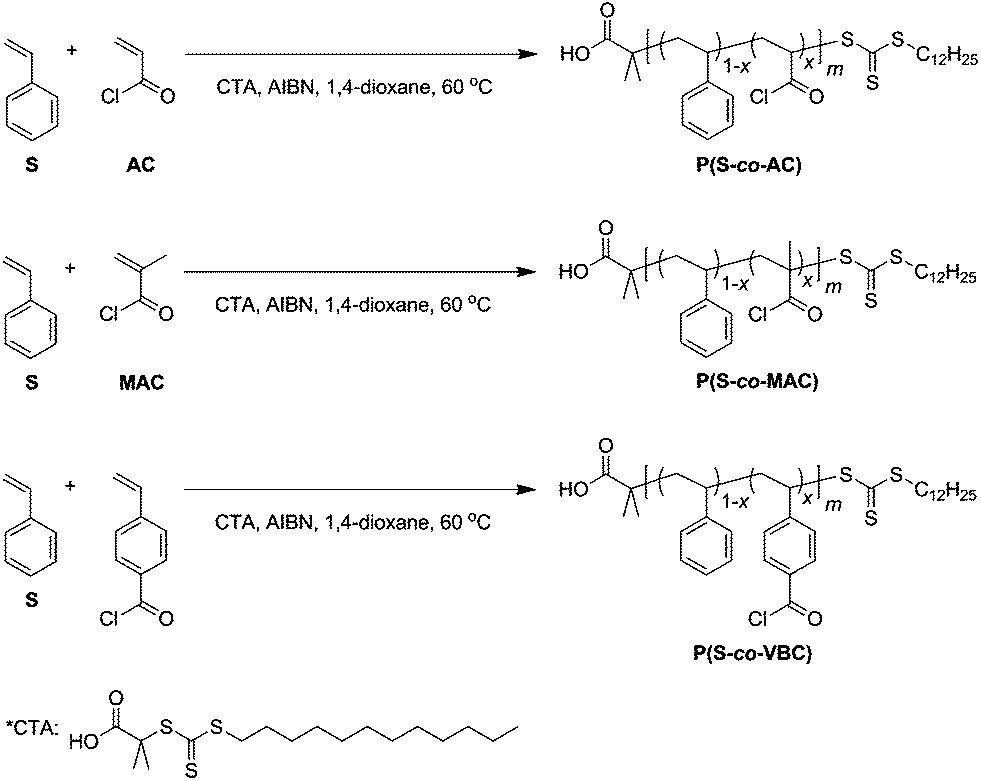 | ||
| Scheme 1 RAFT copolymerization of styrene with acid chloride-containing monomers. | ||
| Polymer | [M1]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[M2]:[CTA]a :[M2]:[CTA]a |
x M2,feed (%) | x M2,polymer (%) | M n,SEC (kg mol−1) | Đ |
|---|---|---|---|---|---|
| a Concentration of CTA in the mixture was 10 mM. b Mol%. c Calculated based on linear polystyrene standards. | |||||
| P(S-co-AC) | 610:32:1 |
5.0 | 39 | 13 | 1.39 |
| P(S-co-AC) | 540:61:1 |
10 | 37 | 16 | 1.48 |
| P(S-co-MAC) | 530:29:1 |
5.2 | 33 | 9.9 | 1.45 |
| P(S-co-MAC) | 530:60:1 |
10 | 38 | 11 | 1.47 |
| P(S-co-VBC) | 540:27:1 |
4.8 | 19 | 15 | 1.26 |
| P(S-co-VBC) | 560:62:1 |
10 | 30 | 16 | 1.34 |
Fig. 1 shows representative 1H NMR spectra of the methyl ester analogues of P(S-co-AC), P(S-co-MAC), and P(S-co-VBC), respectively. For P(S-co-AC) and P(S-co-MAC), the mol percent of acid chloride-containing monomers incorporated in the copolymer was calculated based on the integral of signals corresponding to aromatic protons of PS and that corresponding to –COOCH3 protons, assuming 100% modification of the acid chloride groups into the ester form. For P(S-co-VBC), the signal corresponding to two aromatic protons adjacent to the carbonyl (8.0–7.5 ppm) was separated from those corresponding to other aromatic protons, allowing direct calculation of the fraction of VBC repeating units in the polymer. For all three cases, a significantly higher fraction of acid chloride-containing monomers was incorporated in the resulting copolymer than the feed. This is consistent with the low reactivity ratio of styrene when copolymerized with the acid chloride-containing monomers reported in the literature.40 In cases of AC and MAC, reactivity ratios of both styrene and the acid chloride-containing monomer are small (rS = 0.10 and rAC = 0.03 for styrene and AC polymerized in benzene at 67 °C initiated by AIBN,24rS = 0.13 and rMAC = 0.075 for styrene and MAC polymerized in 1,2-dichloroethane41), suggesting a strong alternating character in each copolymer sequence. In the case of VBC, the reactivity ratio of VBC is much larger than that of styrene (rS = 0.17 and rVBC = 1.80 for styrene and VBC polymerized in bulk at 60 °C36) suggesting a composition drift from the VBC-rich sequence to the styrene-rich sequence as the copolymerization proceeds.
 | ||
| Fig. 1 1H NMR spectra of methyl ester analogues of P(S-co-AC) (a), P(S-co-MAC) (b), and P(S-co-VBC) (c) synthesized by RAFT copolymerization with 5 mol% of acid chloride-containing monomers in the feed. Grey areas denote signals corresponding to the –COOCH3 protons in the copolymer incorporated by the reaction of acid chloride with methanol. Other signals not originated from the polymer are noticed on the figure (* denotes residual styrene). | ||
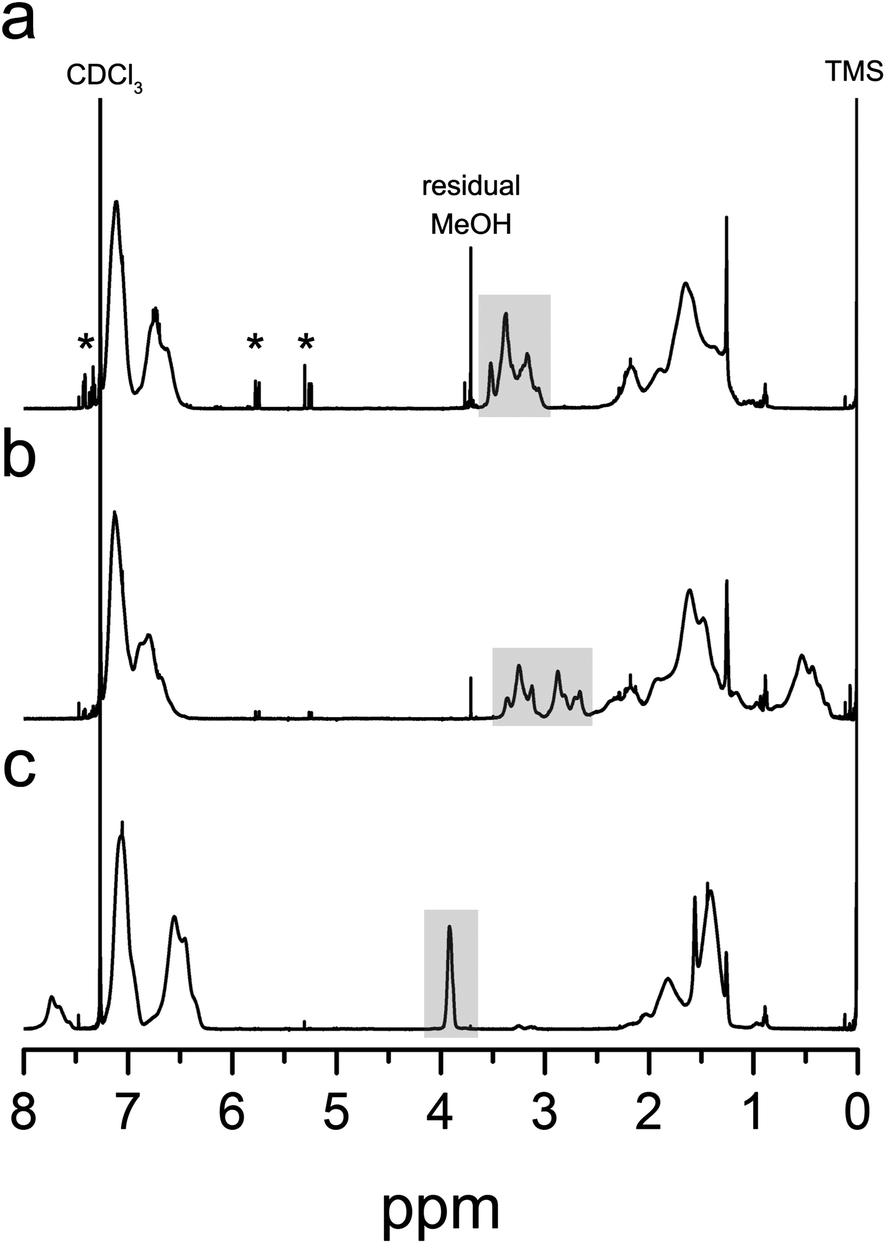
Fig. 2 shows SEC traces of the methyl ester analogues of P(S-co-AC), P(S-co-MAC), and P(S-co-VBC) with the feed ratio of 5% and 10% of acid chloride-containing monomers, respectively. While dispersity (Đ) of the polymers increased as the amount of the acid chloride-containing monomer was increased in the feed, it was possible to obtain a relatively well-defined acid chloride-containing copolymer by copolymerization with VBC.42 With 5 mol% of VBC in the feed, it was possible to obtain P(S-co-VBC) with a dispersity of 1.26. When more VBC was added into the feed, a high molar mass shoulder was more pronounced.
 | ||
| Fig. 2 SEC traces of P(S-co-AC) (a), P(S-co-MAC) (b), and P(S-co-VBC) (c). Solid and dashed lines denote polymers synthesized from the feed containing 5% and 10% of acid chloride-containing monomers, respectively. | ||
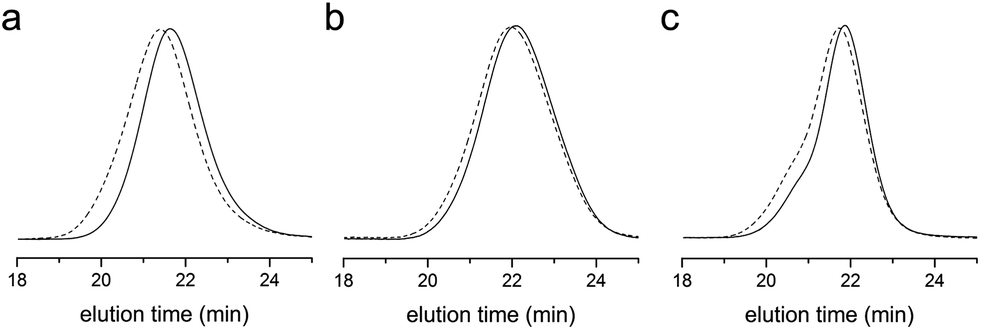
RAFT copolymerization kinetics of styrene with 5% of VBC in the feed was investigated more in detail at 60 °C. Starting from a polymerization mixture of [S]:[VBC]:[CTA] = 570:30:1, we monitored conversion of styrene and VBC over time by analyzing the crude by 1H NMR spectroscopy (Fig. 3a). As expected from the reactivity ratios, VBC was consumed much faster than styrene but copolymerization took place effectively without formation of homoPVBC. Using the total monomer concentration (M) calculated from the conversion data, the first order kinetic plot (ln([M]0/[M]) versus time) was presented and showed a linear dependence consistent with the RAFT mechanism (Fig. 3b). SEC traces of the methyl ester analogues of the polymers formed by precipitation in methanol showed steady growth of molar masses and reduction of Đ over time supporting successful control over copolymerization by RAFT (Fig. 3c). Fig. 3d summarizes evolution of number-average molar masses (Mn) and Đ of the methyl ester analogues determined by SEC based on linear polystyrene standards. Analysis of the composition of the polymers by 1H NMR spectroscopy indicated that the fraction of VBC in the copolymer decreased as polymerization proceeded, from 21% after 8 h to 18% after 24 h of polymerization, due to the composition drift.
 | ||
| Fig. 3 RAFT copolymerization kinetics of styrene with VBC ([S]:[VBC]:[CTA] = 570:30:1) at 60 °C. (a) Time–conversion plots for each monomer. (b) Time–ln([M]0/[M]) plot for the total monomer concentration [M]. (c) SEC traces over time. (d) Conversion–Mn and Đ plot. | ||
Taking advantage of the RAFT polymerization, reactive block polymers consisting of the acid chloride functionality in one of the blocks were also readily synthesized. As an example, RAFT copolymerization of styrene and VBC in the presence of polylactide possessing the CTA moiety at the terminus (PLA-CTA) was conducted in 1,4-dioxane with AIBN at 60 °C. Following the previous protocol, the reactive polymer was obtained by precipitation in hexanes and the methyl ester analogue for the characterization was obtained by precipitation in methanol and stirring overnight. Fig. 4a shows a representative 1H NMR spectrum of the methyl ester analogue. Successful formation of the acid chloride-containing block polymer was evident by appearance of signals corresponding to PLA (5.3–5.1 and 1.7–1.5 ppm), P(S-co-VBC), and –COOCH3 protons as a result of esterification with methanol. SEC traces of the methyl ester analogues of PLA-b-P(S-co-VBC) shifted to higher molar mass compared to PLA-CTA, supporting growth of P(S-co-VBC) from the end of PLA-CTA. The traces were narrow and unimodal indicating well-defined PLA-b-P(S-co-VBC)s with different molar masses (i.e., different weight fractions of PLA) were successfully synthesized by RAFT polymerization (Fig. 4b). Table 2 summarizes the characterization data of the block polymers.
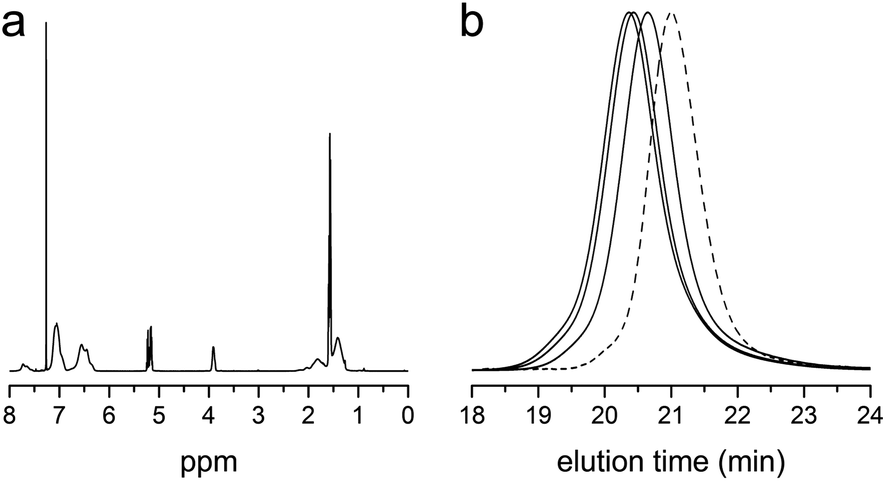 | ||
| Fig. 4 (a) 1H NMR spectrum of the methyl ester analogue of PLA-b-P(S-co-VBC) (entry 3 in Table 2). (b) SEC traces of the methyl ester analogues of PLA-b-P(S-co-VBC) (solid lines). From right to left, the traces correspond to entry 1–3 in Table 2. The trace of PLA-CTA is also shown as a reference (dashed line). | ||
| Entry | Nucleophile | Conversiona (%) | M n,SEC (kg mol−1) | Đ |
|---|---|---|---|---|
| a Determined by 1H NMR spectroscopy. b Calculated based on linear polystyrene standards. c Reaction by precipitating in the excess of the nucleophile and stirring overnight. d Reaction with 1.2 eq. of the nucleophile in the presence of triethylamine. | ||||
| 1c |
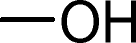
|
94 | 4.1 | 1.45 |
| 2d |
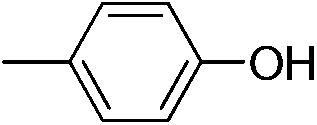
|
92 | 7.4 | 1.50 |
| 3d |
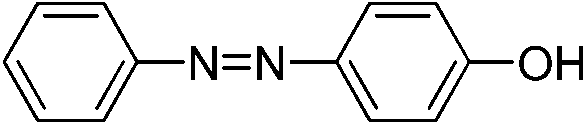
|
95 | 7.1 | 1.48 |
| 4d |
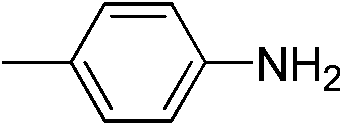
|
100 | 6.8 | 1.47 |
| 5d |
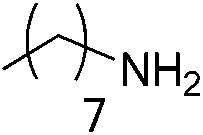
|
100 | 6.1 | 1.22 |
We also explored postpolymerization modification of P(S-co-VBC) with various nucleophiles as summarized in Table 3. To construct a library of functional polymers with identical average chain length and distribution, P(S-co-VBC) synthesized from a single batch (estimated as Mn,SEC = 4.1 kg mol−1 and Đ = 1.45 by analyzing the methyl ester analogue (entry 1)) was used for all reactions (entries 2–5). Typically, the reaction was conducted in dry dichloromethane with 1.2 eq. of the nucleophile relative to acid chloride groups and 2.4 eq. of triethylamine at rt for 24 h and the modified polymer was obtained by precipitation in methanol. 1H NMR spectra of the resulting polymers showed appearance of signals corresponding to the functional groups of the nucleophiles in addition to those from P(S-co-VBC), indicating successful incorporation of functional groups by the reaction (see ESI, Fig. S1–S5†). No peak corresponding to –COOCH3 protons of the methyl ester was observed for the entries 2–5 even though the polymers were precipitated in methanol at the end of reaction, suggesting every reactive acid chloride moiety was consumed by the reaction. Conversion was calculated by comparing the integral of VBC protons and that of the protons corresponding to the functional groups. For every case conversion was >92% indicating postpolymerization modification was highly efficient. SEC analysis of the functional polymers showed slight increase in the molar masses relative to the methyl ester analogue of P(S-co-VBC), which was reasonable considering the molecular weight of the nucleophiles (Fig. S6†). The molar mass distribution of the polymers was not significantly changed by the reaction reflected by similar values of Đ except for the entry 5, where Đ was reduced by 0.2. The origin of the Đ reduction is not clear at this moment.43
Conclusion
RAFT copolymerization of styrene with three acid chloride-containing monomers was investigated as a means to synthesize well-defined reactive polymers possessing the acid chloride functionality. P(S-co-VBC) produced by copolymerization of styrene with VBC showed relatively narrow molar mass distributions compared to P(S-co-AC) and P(S-co-MAC). Kinetics of the copolymerization at the early stage was consistent with the RAFT polymerization mechanism. The RAFT copolymerization scheme was further utilized for the synthesis of reactive block polymers with the acid chloride functionality on one of the blocks, which was demonstrated by copolymerization of styrene and VBC in the presence of PLA-CTA. Successful formation of various functional polymers derived from P(S-co-VBC) by postpolymerization modification suggests promise of this methodology.Experimental details
Materials
Unless specifically noted, all chemicals were purchased from Sigma-Aldrich (St. Louis, MO). Octylamine (99%), p-cresol (99%), p-toluidine (99%), 4-phenylazophenol (98%), triethylamine (≥99%), methyl acrylate (99%), and 1,4-dioxane (anhydrous, 99.8%) were used without further purification. Acryloyl chloride (97.0%) and methacryloyl chloride (97%) were purified by vacuum distillation. Styrene (≥99%) was passed through a basic alumina column prior to use. Azobisisobutyronitrile (AIBN) (98%) was recrystallized from methanol and dried under reduced pressure overnight. Vinylbenzoyl chloride (VBC) was either purchased from Aldlab and purified by vacuum distillation, or synthesized from 4-vinylbenzoic acid (Sigma-Aldrich, 97%) using thionyl chloride44 or oxalyl chloride.11c The use of oxalyl chloride produced VBC with higher yield. All bulk solvents were purchased from Avantor (Hazelwood, MO) and used as received unless otherwise specified. Dichloromethane (HPLC grade) was purified on a MBraun solvent purification system. S-1-Dodecyl-S′-(R,R′-dimethyl-R′′-acetic acid) trithiocarbonate (CTA) was prepared according to the literature.39 Hydroxyl-terminated polylactide (PLA-OH) and CTA-endcapped PLA (PLA-CTA) were prepared by following procedures reported elsewhere.6,45Characterization
1H nuclear magnetic resonance (NMR) spectroscopy was conducted using a Varian Inova 500 MHz instrument spectrometer at 20 °C. Deuterated chloroform was used as a solvent and the residual proton solvent signal was chosen as an internal reference at 7.26 ppm. Size exclusion chromatography (SEC) was performed in chloroform at 35 °C on a Hewlett-Packard 1100 series liquid chromatograph (Palo Alto, CA) equipped with three PLgel 5 μm Mixed-C columns in series with the molecular weight range of 400–400000 g mol−1. A Hewlett-Packard 1047A refractive index detector was employed. The molecular weights and dispersity (Đ) of the polymers were calculated relative to linear polystyrene standards from Varian, Inc. (Palo Alto, CA).
RAFT copolymerization of styrene with acid chloride-containing monomers
Copolymerization of styrene with VBC (4.8 mol% feed) is exemplified. CTA (7.5 mg, 21 μmol) and AIBN (0.3 mg, 2.0 μmol (as a 1.3 wt% benzene solution)) were dissolved in a mixture of styrene (1.19 g, 11.4 mmol), VBC (0.096 g, 0.58 mmol), and 1,4-dioxane (0.2 mL) and the solution was transferred into an ampoule. After three cycles of freeze–pump–thaw, the ampoule was flame sealed under vacuum and placed in the oil bath preset at 60 °C. After heating for 24 h, the polymerization mixture was cooled to rt and the ampoule was opened. The mixture was diluted with dichloromethane and precipitated in hexanes. A part of the mixture was precipitated in methanol and stirred overnight at rt to generate a methyl ester analogue of P(S-co-VBC) for analytical purposes. P(S-co-VBC) and its methyl ester analogue were obtained by filtration and dried under vacuum overnight at rt.Postpolymerization modification of P(S-co-VBC)
To a dichloromethane solution of the nucleophile (1.2 eq. relative to the number of acid chloride groups) and triethylamine (2.4 eq.), 0.1 g of P(S-co-VBC) in dichloromethane was added. The total amount of dichloromethane was 2.4 mL. The reaction mixture was stirred for 24 h at rt, and the functionalized polymer was collected by precipitation in methanol and filtration, and then dried under vacuum overnight at 40 °C.RAFT copolymerization of styrene with VBC in the presence of PLA-CTA
Synthesis of PLA-b-P(S-co-VBC) shown in entry 1 of Table 2 is given here as an exemplary case. A solution of PLA-CTA (Mn,NMR = 20 kg mol−1, Mn,SEC = 33 kg mol−1, Đ = 1.11, 1.40 g, 70 μmol) and AIBN (2.3 mg, 14 μmol (as a 1.1 wt% benzene solution)) in a mixture of styrene (13.81 g, 130 mmol), VBC (1.17 g, 7 mmol), and 1,4-dioxane (2.8 mL) was prepared and placed in a Schlenk flask. The mixture was degassed by three cycles of freeze–pump–thaw and then placed in the oil bath preset at 60 °C. After heating for 19 h, the polymerization mixture was cooled to rt and the flask was opened. The mixture was diluted with dichloromethane and precipitated in hexanes. A part of the mixture was precipitated in methanol and stirred overnight at rt to generate a methyl ester analogue of PLA-b-P(S-co-VBC) for analytical purposes. PLA-b-P(S-co-VBC) and its methyl ester analogue were obtained by filtration and dried under vacuum overnight at rt.Using the above procedure, PLA-b-P(S-co-VBC)s shown in entries 2 and 3 were synthesized from the reaction mixtures of [PLA-CTA]:[styrene]:[VBC] = 1:1900:96 and heating for 19 h and 21 h, respectively.
Acknowledgements
We thank Dow Chemical Company for financial support. Partial support for this work was also provided by the National Science Foundation (DMR-1006370).References
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS PubMed.
- (a) M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS PubMed; (b) K. A. Günay, P. Theato and H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1–28 CrossRef.
- (a) J. B. Beck, K. L. Killops, T. Kang, K. Sivanandan, A. Bayles, M. E. Mackay, K. L. Wooley and C. J. Hawker, Macromolecules, 2009, 42, 5629–5635 CrossRef CAS PubMed; (b) A. van der Ende, T. Croce, S. Hamilton, V. Sathiyakumar and E. Harth, Soft Matter, 2009, 5, 1417–1425 RSC.
- (a) H. Huang, T. Kowalewski, E. E. Remsen, R. Gertzmann and K. L. Wooley, J. Am. Chem. Soc., 1997, 119, 11653–11659 CrossRef CAS; (b) R. K. O'Reilly, M. J. Joralemon, C. J. Hawker and K. L. Wooley, Chem.–Eur. J., 2006, 12, 6776–6786 CrossRef CAS PubMed; (c) Y. Li, I. Akiba, S. Harrisson and K. L. Wooley, Adv. Funct. Mater., 2008, 18, 551–559 CrossRef CAS.
- (a) U. Jeong, D. Y. Ryu, J. K. Kim, D. H. Kim, X. Wu and T. P. Russell, Macromolecules, 2003, 36, 10126–10129 CrossRef CAS; (b) N. Zhou, F. S. Bates and T. P. Lodge, Nano Lett., 2006, 6, 2354–2357 CrossRef CAS PubMed; (c) A. Okumura, Y. Nishikawa and T. Hashimoto, Polymer, 2006, 47, 7805–7812 CrossRef CAS PubMed.
- M. Seo, M. A. Amendt and M. A. Hillmyer, Macromolecules, 2011, 44, 9310–9318 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed.
- (a) E. Pedone, X. Li, N. Koseva, O. Alpar and S. Brocchini, J. Mater. Chem., 2003, 13, 2825–2837 RSC; (b) Z. Hu, Y. Liu, C. Hong and C. Pan, J. Appl. Polym. Sci., 2005, 98, 189–194 CrossRef CAS; (c) S. Ghosh, S. Basu and S. Thayumanavan, Macromolecules, 2006, 39, 5595–5597 CrossRef CAS; (d) S. Y. Wong and D. Putnam, Bioconjugate Chem., 2007, 18, 970–982 CrossRef CAS PubMed.
- A. Desai, N. Atkinson, F. Rivera Jr, W. Devonport, I. Rees, S. E. Branz and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1033–1044 CrossRef CAS.
- (a) C. M. Schilli, A. H. E. Müller, E. Rizzardo, S. H. Thang and Y. K. Chong, ACS Symp. Ser., 2003, 854, 603–618 CrossRef CAS PubMed; (b) J. J. Vosloo, M. P. Tonge, C. M. Fellows, F. D'Agosto, R. D. Sanderson and R. G. Gilbert, Macromolecules, 2004, 37, 2371–2382 CrossRef CAS; (c) A. Favier, F. D'Agosto, M.-T. Charreyre and C. Pichot, Polymer, 2004, 45, 7821–7830 CrossRef CAS PubMed; (d) J. Yanjarappa, K. V. Gujraty, A. Joshi, A. Saraph and R. S. Kane, Biomacromolecules, 2006, 7, 1665–1670 CrossRef PubMed.
- (a) M. Eberhardt, R. Mruk, R. Zentel and P. Théato, Eur. Polym. J., 2005, 41, 1569–1575 CrossRef CAS PubMed; (b) M. Eberhardt and P. Théato, Macromol. Rapid Commun., 2005, 26, 1488–1493 CrossRef CAS; (c) K. Nilles and P. Théato, Eur. Polym. J., 2007, 43, 2901–2912 CrossRef CAS PubMed.
- C. S. Marvel and C. L. Levesque, J. Am. Chem. Soc., 1939, 61, 3244–3246 CrossRef CAS.
- M. Vrancken and G. Smets, J. Polym. Sci., 1954, 14, 521–534 CrossRef CAS.
- V. R. C. Schulz, P. Elzer and W. Kern, Makromol. Chem., 1960, 42, 189–196 CrossRef.
- A. Z. El-Sonbati and M. A. Diab, J. Therm. Anal., 1988, 34, 769–776 CrossRef CAS.
- M. A. Diab, Acta Polym., 1990, 41, 351–354 CrossRef CAS.
- Y. S. Yang, G. R. Qi, J. W. Qian and S. L. Yang, J. Appl. Polym. Sci., 1998, 68, 665–670 CrossRef CAS.
- L. Wu, X. Tuo, H. Cheng, Z. Chen and X. Wang, Macromolecules, 2001, 34, 8005–8013 CrossRef CAS.
- S. Rondou, G. Smets and M. C. de Wilde-Delvaux, J. Polym. Sci., 1957, 24, 261–274 CrossRef CAS.
- P. E. Blatz, J. Polym. Sci., 1962, 58, 755–769 CrossRef CAS.
- C. Chovino and P. Gramain, Macromol. Chem. Phys., 1996, 197, 1411–1418 CrossRef CAS.
- P. Strohriegl, Makromol. Chem., 1993, 194, 363–387 CrossRef CAS . This paper reviews the (co)polymerization of acryloyl chloride and methacryloyl chloride as well as utilization of the reactive polymers for postpolymerization reactions.
- G. Smets, A. Poot and G. L. Duncan, J. Polym. Sci., 1961, 54, 65–81 CrossRef CAS.
- G. Smets and E. Dysseleer, Makromol. Chem., 1966, 91, 160–174 CrossRef CAS.
- A. C. Constantinescu, S. Dumitriu and C. I. Simionescu, Acta Polym., 1983, 34, 60–61 CrossRef CAS.
- Y. A. Aggour, Eur. Polym. J., 2000, 36, 2231–2234 CrossRef CAS.
- T. Otsu and T. Tazima, Kobunshi Ronbunshu, 1974, 31, 181–185 CrossRef CAS.
- I. Gitsov, J. Appl. Polym. Sci., 1990, 41, 2705–2710 CrossRef CAS.
- V. R. C. Schulz, P. Elzer and W. Kern, Makromol. Chem., 1960, 42, 197–204 CrossRef.
- C. M. Paleos, G. Margomenou-Leonidopoulou, S. E. Filippakis and A. Malliaris, J. Polym. Sci., Polym. Chem. Ed., 1982, 20, 2267–2275 CrossRef CAS.
- N. Ogata, K. Sanui, M. Watanabe and I. Yahagi, J. Polym. Sci., Polym. Lett. Ed., 1985, 23, 349–352 CrossRef CAS.
- I. Yahagi, M. Watanabe, K. Sanui and N. Ogata, J. Polym. Sci., Part A: Polym. Chem., 1987, 25, 727–730 CrossRef CAS.
- Y. H. Park, K. W. Kim and W. H. Jo, Polym. Adv. Technol., 2002, 13, 670–677 CrossRef CAS.
- H. Wang, Y. He, X. Tuo and X. Wang, Macromolecules, 2004, 37, 135–146 CrossRef CAS.
- E.-R. Kenawy, M. El-Newehy, F. Abdel-Hay and R. M. Ottenbrite, Biomacromolecules, 2007, 8, 196–201 CrossRef CAS PubMed.
- Y. Iwakura, K. Uno, N. Nakabayashi and T. Kojima, Bull. Chem. Soc. Jpn., 1968, 41, 186–191 CrossRef CAS.
- (a) G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669–692 CrossRef CAS; (b) G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS; (c) G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 1402–1472 CrossRef; (d) M. J. Monteiro, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3189–3204 CrossRef CAS; (e) L. Barner, T. P. Davis, M. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2007, 28, 539–559 CrossRef CAS.
- C. Barner-Kowollik and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5715–5723 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- Compared to free radical copolymerization that has been used to obtain the literature values, the RAFT process seems to allow more incorporation of the monomer with a larger reactivity ratio but does not significantly alter the general tendency for comonomer incorporation. See A. Feldermann, A. A. Toy, H. Phan, M. H. Stenzel, T. P. Davis and C. Barner-Kowollik, Polymer, 2004, 45, 3997–4007 CrossRef CAS PubMed.
- M. Ueda and T. Kumakura, J. Polym. Sci., Part A: Polym. Chem., 1982, 20, 2829–2838 CrossRef CAS . The authors took the values from U. Kh. Usmanov, Sh. Nadzhimutdinov and A. S. Turaev, Dokl. Akad. Nauk SSSR, 1980, 252, 663 Search PubMed.
- This also indicates that anhydride formation by reaction of the acid chloride moiety and the carboxylic acid in the chain transfer agent did not occur to any significant extent as this would lead to formation of branched polymers via the self-condensing vinyl polymerization mechanism and broadening of SEC traces for all three acid chloride-containing monomers.
- Octylamine might also induce aminolysis of the trithiocarbonate moiety and generate a free thiol, which might affect Đ of the resulting polymer. Thus a reference experiment was conducted in which methyl acrylate (10 eq. relative to the number of CTA) was added at the end of the reaction and stirred for another 18 h to endcap thiols via the Michael addition. While the 1H NMR spectrum of the resulting polymer suggested ca. 18% of the trithiocarbonate moiety was aminolyzed and endcapped with methyl acrylate to form a thioether linkage, its SEC trace and the Đ value were virtually identical to the ones obtained from the original experiment.
- A. Hirao, Y. Ishino and S. Nakahama, Macromolecules, 1988, 21, 561–565 CrossRef CAS.
- J. Rzayev and M. A. Hillmyer, J. Am. Chem. Soc., 2005, 127, 13373–13379 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR and SEC data of functional polymers obtained by postpolymerization modification of P(S-co-VBC). See DOI: 10.1039/c3py00867c |
| ‡ Current address: Graduate School of Nanoscience and Technology, KAIST, Daejeon 305-701, Korea. |
| This journal is © The Royal Society of Chemistry 2014 |