DOI:
10.1039/C2PY20646C
(Paper)
Polym. Chem., 2013,
4, 124-132
Poly(glycidyl methacrylate): a highly versatile polymeric building block for post-polymerization modifications†
Received
13th August 2012
, Accepted 10th September 2012
First published on 12th September 2012
Abstract
The treatment of poly(glycidyl methacrylate) with nucleophilic agents yielded new linear homo/co-polymers. The materials obtained have different physicochemical properties depending upon the nucleophiles utilized and thus, by this way, water soluble polymers are easily accessible. This approach is also very convenient for the synthesis of amphiphilic block copolymers. Additional chemical modifications can further transform the obtained polymers after the post-polymerization treatment. Starting from RAFT pre-synthesized/pre-functionalized polymers, the described procedures allow us to obtain a wide variety of chemically diverse materials characterized by controlled structure and specific functions.
Introduction
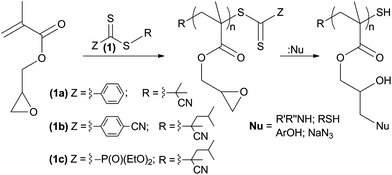
The adoption of controlled radical polymerization techniques has contributed to the evolution of material sciences into a new era of nanotechnologies.1 Among the many reported techniques, Atom Transfer Radical Polymerization (ATRP)2 and Reversible Addition–Fragmentation chain Transfer polymerization (RAFT)3 have risen to the forefront for an ever increasing number of scientists. Indeed, these extremely versatile techniques allow for the precise control of molecular weight, structure and end group functionalization for polymers obtained from a wide variety of monomers. RAFT polymerization, besides being a very robust system, also delivers metal free materials suitable for biomedical applications4 that can readily exploit the intrinsic thiol RAFT functional group.5,6 Using this technique, it is nowadays possible to obtain very sophisticated functionalized molecular architectures7 such as hetero-telechelic, comb and star (co)polymers8,9 and/or structures that are capable of spatial self-organization10,11 into micelles or polymersomes.11 Nevertheless, a limitation of the RAFT process is that polymerization conditions must be optimized for each monomer, since the choice of solvent, RAFT agent and experimental procedures is of crucial importance for gaining the desired control. The use of monomers carrying extra functional groups, that are inert towards the polymerization process, represents an elegant way to overcome this complication.12 These secondary reactive groups allow for further chemical transformation once the polymer is obtained. Thus, in principle, utilizing a single reactive polymer it is possible to obtain a variety of different materials characterized by the properties introduced by the modifying reagent. Notably, these reactions must be very efficient, selective, quantitative, and possibly able to achieve an orthogonal reactivity. The most commonly used monomers for this purpose belong to the class of active esters, and examples of succinyl ester,13,14 pentafluorophenyl ester15 and p-nitrophenyl ester16 derivatized monomers have been used for the preparation of reactive polymers that can undergo subsequent chemical transformation upon reaction with amine substrates. Thiol reactive (co)polymers bearing the 2-pyridyl disulfide pendent moiety have been reported,14 with subsequent chemical modification of these kinds of materials achieved by thiol exchange reactions. For this post-polymerization approach, other reactive monomers like 2-isopropenyl-2-oxazoline,17 which is susceptible to ring-opening reactions in the presence of carboxylic acids and thiols, or monomers which carry an azalactone function,18 which is reactive towards primary amines or alkyl alcohols, have also been used. These types of processes are very promising and can be useful for the systematic study of termini functionalized polymers, allowing for the correlation of chemical properties with differing chain pendent moieties within a consistent length series. This enables a high throughput combinatorial approach19 that can accelerate research progress to satisfy the needs of nanotechnology disciplines. On the other hand, the reactive monomers required for this purpose are either not commercially available or very expensive. Furthermore, some post-polymerization processes can also suffer from undesirable side reactions.20 Glycidyl methacrylate, having both vinyl and epoxy functions, is a reactive monomer which meets the requirements for post-polymerization modification. It is also a low-cost reagent because it is widely used for the industrial production of epoxy functional methacrylic resins to be employed as coatings and adhesives.
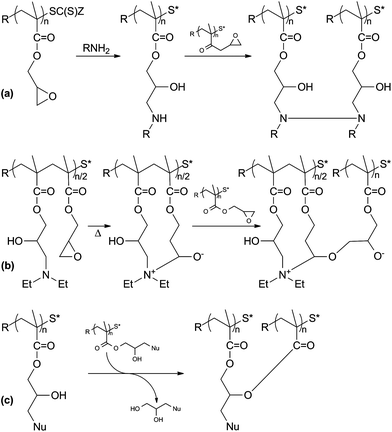
Herein we report the post-polymerization treatments of RAFT synthesized poly(glycidyl methacrylate) (PGMA) that allowed us to obtain a series of polymers with varied properties. This process relies on the well known nucleophilic ring-opening reaction of the epoxy group. This reaction can become difficult to control when applied to polymers (PGMA) because of the high reactivity of the oxirane ring and the derived hydroxyl group which may lead to chain-branching. Furthermore, the long reaction time required for the full conversion can increase the formation of micro/macro gels. Other authors have reported the treatment of PGMA with primary amines,21–23 and with secondary amines.23–25 With primary amines this can lead to branched polymers since a more nucleophilic secondary amine is obtained upon the ring-opening reaction (Scheme 1(a)). However, these reports did not investigate or show evidence of whether the polymers obtained maintained linearity during the process. The treatment we propose is performed in DMSO, requires a short time and makes use of such nucleophilic reagents as secondary amines, thiols, aromatic alcohols and the azide anion (Scheme 2). The resulting polymers maintain linearity and gain the properties conferred by the individual nucleophilic agent used. Thus, it is possible to achieve functional polymers by choosing appropriate nucleophilic agents. Moreover, in a subsequent post-polymerization process, the derivatized polymer can be further modified taking advantage of the hydroxyl group formed in the epoxide ring-opening reaction.
 |
| | Scheme 1 Possible chain-branching mechanisms. By using primary amines (a). Anionic ring-opening crosslinking (b). Transesterification crosslinking (c). | |
Through this methodology polymers can be accessed with solubilities that can be varied from hydrophilic to hydrophobic and therefore also amphiphilic block copolymers can be easily prepared. These kinds of polymers are usually obtained with the sequential synthesis of the two polymeric blocks.26 This technique requires the use of solvents capable of solubilizing both the hydrophilic and the hydrophobic monomers/blocks which, as a consequence, limits the choice of the polymerizable species. Click reactions7,27 have also been successfully used, but this requires the particular functionalization groups to be present at the end of the polymers. The recently reported switchable RAFT agents28 have introduced a new and innovative system for the synthesis of block copolymers by controlled polymerization of more activated and less activated monomers. Here, access to amphiphilic chains can be achieved through the choice of appropriated monomers. More conveniently, with our procedure, amphiphilic block copolymers are obtained in a novel, very simple and flexible way. This greatly increases the variety of functional hydrophilic blocks obtainable.
Results and discussion
PGMA synthesis
The controlled polymerization of PGMA by the RAFT technique has been already reported for homopolymers29 and for copolymers.30 We synthesized PGMA using the RAFT agents 2-cyanoprop-2-yl dithiobenzoate (1a) and 2-cyano-4-methylpent-2-yl 4-cyanodithiobenzoate (1b) (Scheme 2). Compound 1b, which we have found to be very efficient in controlling the polymerization of methyl methacrylate even at low conversion,31 yielded polymers with very narrow molar mass distribution (Table 1). This better control is given by the simultaneous action of the electron-withdrawing Z group and of the 2-cyano-4-methylpent-2-yl moiety which is an excellent leaving group; as a consequence, the RAFT initiation stage is shortened. For these reasons, and having previously synthesized similar phosphorylated chain transfer agents,32 the new RAFT agent 2-cyano-4-methylpent-2-yl diethoxyphosphoryldithioformate (1c) was synthesized and successfully utilized with GMA. The choice of RAFT agents with electron withdrawing Z groups was further prompted by the higher reactivity of the dithiocarbonyl moiety towards aminolysis. In fact this reaction, followed by monitoring the disappearance of the characteristic red/purple color of the dithioester, was found to be faster for RAFT groups carrying the 4-cyanophenyl and the diethoxyphosphoryldithioformate moieties compared to the phenyl moiety as Z substituent. Overall, the polymers were obtained with good control in the range between 1700 and 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 (Table 1). In particular compound 1b yielded PGMA characterized by a low and a narrow distribution of the molecular weight. With this RAFT agent, the polymers' molar mass was conveniently determined by means of 1H NMR experiments integrating the signals of the aromatic protons of the 4-cyanodithiobenzoate terminal group (see spectra in the ESI†).
000 g mol−1 (Table 1). In particular compound 1b yielded PGMA characterized by a low and a narrow distribution of the molecular weight. With this RAFT agent, the polymers' molar mass was conveniently determined by means of 1H NMR experiments integrating the signals of the aromatic protons of the 4-cyanodithiobenzoate terminal group (see spectra in the ESI†).
Table 1 GMA polymerization conditions and results
| Entry |
RAFT (mg) |
GMA (mL) |
AIBN (mg) |
Time (min) |
Conversion (%) |
GPC
M
n
|
NMR
M
n
|
PDI |
|
Gravimetric.
By NMR.
|
| 1 |
1a (19.5) |
7.5 |
0.7 |
240 |
31a |
30b |
18600 |
— |
1.24 |
| 300 |
36a |
35b |
22000 |
— |
1.23 |
| 2 |
1b (105.8) |
7.5 |
1.06 |
210 |
3.9a |
4.2b |
1700 |
3200 |
1.20 |
| 270 |
4.7a |
6.2b |
3200 |
3500 |
1.13 |
| 330 |
8.5a |
9.6b |
3800 |
4900 |
1.11 |
| 390 |
10.2a |
11b |
4600 |
6000 |
1.12 |
| 3 |
1c (89) |
7.5 |
1.07 |
240 |
45a |
40b |
11000 |
— |
1.14 |
| 330 |
55a |
54b |
14500 |
— |
1.14 |
Ring-opening reaction
Treatment with amines.
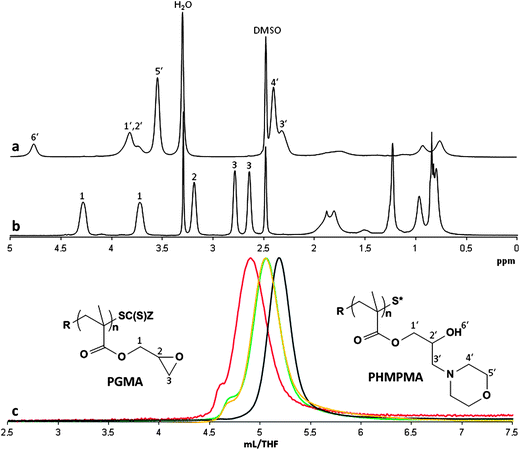
The post-polymerization treatments were carried out in DMSO at 80 °C with an excess of the nucleophilic reagent (2- to 10-fold). A variety of different cyclic or open-chain secondary amines were chosen due to their reactivity towards oxiranes. Primary amines were not considered in order to avoid the formation of branched polymers/gels. Other authors have reported the thermic treatment of PGMA with primary amines in acetonitrile24 or THF/DMF21 in order to obtain branched polymers used as nucleic acid transporters. Here, the ring-opening processes described have been carried out respectively for 16 hours24 or 36 hours.21 The lengthy reaction times led us to believe that the optimal conditions were not obtained. With our procedure, using DMSO as the solvent, a complete conversion of the epoxide rings is reached in 30 minutes to two hours for all of the amines investigated (Table 2), with the exception of the sterically encumbered benzimidazole, which required 16 hours for an exhaustive reaction. The ring-opening reaction proceeded quantitatively and the GPC analysis showed that branching reactions did not occur, with the only exception of diethylamine which displayed a multimodal molar mass distribution with a PDI of 1.36. The modification of the GPC trace of the starting PGMA from monomodal to multimodal, following the treatment with diethylamine, is a clear sign of the partial formation of branched polymeric material characterized by higher molecular weight. This branching process is concurrent with the nucleophilic oxirane ring-opening reaction, and a possible explanation of it can be given by the anionic polymerization occurring between the unreacted epoxy side groups (Scheme 1(b)). This ring-opening crosslinking would be self-initiated by the tertiary amines originated from the reaction of diethylamine with the epoxy groups. A similar, but extensive branching process has been previously reported by Jiang et al.,33 which consists of the thermic treatment of poly(2-diethylaminoethyl methacrylate)-b-poly(glycidyl methacrylate) block copolymer at 80 °C for 9 hours. Another cause of branching can be possibly due to inter-chain transesterification reactions between the hydroxy groups (derived from the oxirane ring-opening) and the side ester groups of the methacrylic backbone (Scheme 1(c)). This phenomenon might become more relevant when a long reaction time is required to achieve an exhaustive ring-opening reaction. The use of DMSO proved fundamental to increase the rate of this reaction and, at the same time, to avoid gelification. Moreover, strong hydrogen bond interactions occurring between the hydroxyl groups and the solvent may prevent the transesterification side reaction. To better understand this phenomenon we tried the post-polymerization treatment of PGMA with morpholine at 80 °C using N-methylpyrrolidone as a solvent. The ring-opening reaction was slightly slower as it needed two hours to completely convert the epoxy groups. Nevertheless, the polymer obtained was linear and, very similarly to the process in DMSO, the GPC trace (Fig. 1) did not show any high molecular weight branched material.
Table 2 Treatment of PGMA with different nucleophiles in DMSO at 80 °C and GPC analysisa
| Nucleophile |
Time/h |
Mn/g mol−1 |
PDI |
|
Calibration using polystyrene standards.
In NMR tube in DMSO-d6.
With 2-fold 2-mercaptoethanol.
With 2-fold phenol at 120 °C.
Details provided in the experimental section. GPC analysis run in:
THF at 22 °C;
DMF at 70 °C;
pyridine at 70 °C.
After further derivatization with phenyl isocyanate.
|
| Morpholine |
0.5b |
14400f |
1.18 |
| Piperidine |
0.5b |
27700g |
1.04 |
| 4-Hydroxypiperidine |
0.5b |
38000h |
1.09 |
| 3-Hydroxypiperidine |
0.5b |
38500h |
1.12 |
| 1-Methylpiperazine |
0.5b |
26800h |
1.04 |
| Diethylamine |
1.5b |
41900g |
1.36 |
|
N-Methylethanolamine |
2 |
32000g |
1.04 |
| Diethanolamine |
1b |
25000f,i |
1.23 |
| Benzimidazole |
16b |
45600h,i |
1.35 |
| 2-Mercaptoethanol |
2c |
32600h |
1.17 |
| Phenol |
16d |
22500f |
1.03 |
| Morpholine/NaN3 |
2/2e |
14000f |
1.15 |
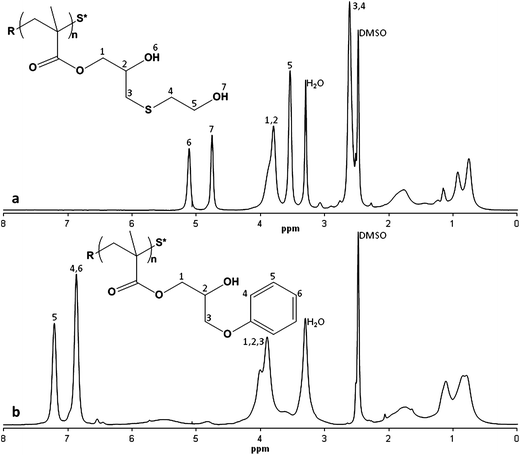
As an example, the treatment of PGMA with a 10-fold amount of morpholine in DMSO at 80 °C for 30 minutes yielded the linear poly(2-hydroxy-3-morpholinopropyl methacrylate) homopolymer (PHMPMA) which was fully converted, as evidenced by NMR and GPC analysis (Fig. 1). The small shoulder present in the PHMPMA GPC trace corresponds to double the molecular weight of the main peak, which we attribute to the formation of a disulfide bridge between two thiol ended chains. In fact, upon aminolysis of the dithioester end group, the derived thiol is partially oxidized to disulfane. This dimerization can be minimized by reacting the formed thiol with phenyl maleimide or methyl maleimide before the ring-opening reaction. In order to avoid Michael addition onto the maleimide substrate, the amine has to be added in two stages: initially the amount needed for the aminolysis then, after the completion of the thiol addition, the amount needed for the ring-opening reaction. The coupling reaction is evidenced by the aromatic protons of the added phenyl maleimide (see NMR spectra in the ESI Fig. S6†). Notably, this thiol-ene addition can also be used for introducing specific functionalities at one end of the polymer.6 For this purpose we also obtained PHMPMA functionalized with fluorescein by reacting the thiol ended polymer with N-(5-fluoresceinyl) maleimide. The modification of PGMA by means of secondary amines leads to functional polymeric materials that are also pH sensitive, and the range of pH sensitivity can be varied by selecting different amines as nucleophilic agents.
Treatment with other nucleophiles.
Thiols and aromatic alcohols were also considered for the post-polymerization processing of PGMA. In particular, we used 2-mercaptoethanol and phenol. In these cases, triethylamine is required as a reaction activator. Thus, two equivalents of 2-mercaptoethanol and 0.5 equivalents of Et3N in DMSO at 80 °C for 2 hours yielded the linear homopolymer derived from total conversion of the starting PGMA. Instead, using the same reactant ratio, phenol required 16 hours at 120 °C for a total conversion (Fig. 2). By using mercaptanes or aromatic alcohols it is then possible to obtain pH insensitive hydrophilic or lipophilic polymers. To the best of our knowledge, the treatment of PGMA with thiol-functionalized compounds or with phenol is unprecedented.
Random copolymers.
With the above procedures, random copolymers may be obtained using different nucleophilic agents in the same post-polymerization reaction, or in subsequent additions. As an example, we have synthesized poly(2-hydroxy-3-morpholino propyl methacrylate)-co-poly(2-hydroxy-3-azidopropylmethacrylate) (PHMPMA-co-PHAPMA) by initially treating PGMA with morpholine, and then by adding sodium azide before the total conversion of the epoxide rings (Scheme 3). The resulting azido-functionalized copolymer displayed the characteristic asymmetric stretching frequency of the azido N3 group, observed via FT-IR spectroscopy at 2104 cm−1 (see spectra in the ESI†). These azide groups can be reacted with alkynyl-functionalized species via the Huisgen 1,3-dipolar cycloaddition click reaction.
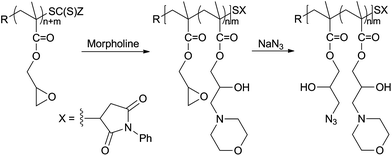 |
| | Scheme 3 Synthesis of PHMPMA-co-PHAPMA. | |
A different approach for random copolymers containing azido groups (PMMA-co-PHAPMA) starting from pre-synthesized PMMA-co-PGMA followed by the treatment with sodium azide have been reported by Tsarevsky et al.34
Amphiphilic block-copolymers.
The physicochemical properties of the materials obtained after treatment of PGMA can be tuned by choosing the appropriate nucleophilic reagent. Hence, it is possible to obtain a series of polymers with different grades of hydrophilicity. For example, the morpholine derivatized PHMPMA displays a hybrid behaviour, being soluble either in water or in organic solvents such as THF and chloroform. This property can be very conveniently exploited to obtain block copolymers with amphiphilic characteristic. In this light, PGMA macro-RAFT was chain-extended with polystyrene (PS), poly(methyl methacrylate) (PMMA) and poly(butyl acrylate) (PBA) (Table 3). The subsequent treatment with morpholine readily yielded the block copolymers PHMPMA-b-PS, PHMPMA-b-PMMA and PHMPMA-b-PBA which, when dispersed in water, proved capable of spatial self-organization into micelles. Dynamic Light Scattering (DLS) measurements showed these particles to have a hydrodynamic diameter in the range of 30–60 nm. They also proved to possess loading abilities by dispersing in water a highly hydrophobic substance like the fluorescent dye Nile Red (Fig. 3). Ongoing experiments presently study this feature in order to administer lipophilic substances such as fluorescent dyes and/or therapeutic agents to cultured cells. This can satisfy the recently introduced theranostic concept.30
Table 3 PGMA copolymerization conditions and results
| Entry |
PGMA (g) |
Monomer (mL) |
Initiator (mg) |
Temp. (°C) |
Time (min) |
Conversion (%) |
Mn |
PDI |
| 1 |
1.13 |
MMA (7) |
AIBN (0.44) |
70 |
300 |
27.7 |
47300 |
1.10 |
| 2 |
0.8 |
Styrene (6) |
VAZO88 (1.3) |
90 |
270 |
7.7 |
39000 |
1.25 |
| 3 |
0.58 |
BA (4.7) |
AIBN (0.44) |
70 |
120 |
15 |
47000 |
1.16 |
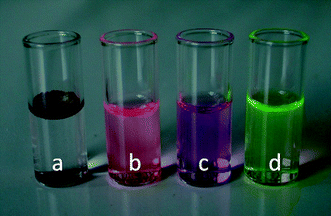 |
| | Fig. 3 Nile Red: dispersed in pure water (a), dissolved in THF (b), dispersed in water by means of PMMA-b-PHMPMA micelles (c). Aqueous solution of PHMPMA homopolymer covalently bound to fluorescein (d). | |
Further treatments.
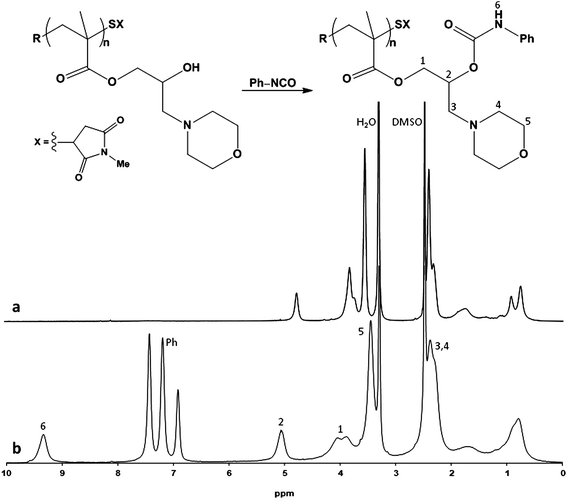
Another interesting feature of the polymers obtained after the post-polymerization treatment is the presence of hydroxyl groups originating in the ring-opening reaction of the pendent epoxides. Their reactivity can be further exploited for additional post-polymerization modifications. Thus, treating PHMPMA with phenyl isocyanate we obtained polymeric chains completely substituted with the phenyl carbamate group. The NMR spectrum (Fig. 4) clearly shows the aromatic protons at 6.8, 7.2, 7.5 ppm, the shift of the proton adjacent to the carbamate function from 3.8 to 5.1 ppm (this makes distinguishable the two diastereotopic protons at 4 ppm) and the disappearance of the hydroxyl proton at 4.8 ppm while the carbamoyl proton is then visible at 9.3 ppm. GPC analysis in THF at 22 °C displayed number average molecular weights (Mn) of 20000 g mol−1 and a PDI of 1.20 (Fig. 1c). This reaction was also exploited to further derivatize those polymers that proved not easily analyzable by GPC for solubility reason or because of their stable intra-/inter-chain hydrogen bonds. Therefore, PGMA treated with diethanolamine and benzimidazole was further reacted with phenyl isocyanate. The resulting polymers were analyzed by GPC (Table 2). The reaction with isocyanates is very convenient in terms of reactivity and efficiency, but also due to the wide commercial availability of substituted isocyanates. Other classes of compounds such as acyl halides, phosphoryl halides and sulfonyl halides can also be reacted with the hydroxyl group, which significantly enhances the variety of differing post-polymerization functional moieties that can be introduced on a pre-synthesized/pre-functionalized PGMA.
Conclusions
PGMA, which had been previously synthesized under RAFT conditions, was reacted with different classes of nucleophilic agents through a fast and efficient process that yielded new linear homopolymers having the side moieties derivatized with secondary amines, thiols and aromatic alcohols. A random copolymer derivatized with morpholine containing azide groups was also synthesized. Using this methodology it is not only possible to obtain a library of different polymers starting from a pre-synthesized/end-functionalized single one, but also to have access to materials not otherwise obtainable. The described procedures afford functionalized polymers showing different physicochemical properties, e.g. solubilities ranging from hydrophilic to hydrophobic. This propensity provides a new way to easily obtain amphiphilic block copolymers which are capable of self-organization into micelles. Furthermore, the polymeric chains derivatized with hydroxyl/ether-carrying nucleophiles, being highly hydrated, can have anti-nonspecific protein adsorption properties (stealth properties), and so are not recognizable as alien to the immune system. This makes them very good candidates as therapeutic and/or diagnostic agent carriers.
In an additional treatment, the pendent hydroxy groups derived from the treatment of PGMA can be further functionalized, as indicated by the isolation of the phenyl carbamate substituted polymer when reacting the morpholine derivatized polymer with phenyl isocyanate.
All in all, we wish to emphasize that the described methodology is capable of combining the useful qualities of the RAFT technique together with the versatility of PGMA post-polymerization treatments. Thus, while chain length, structure and end functionalization are controlled by the very well known RAFT process, the chemical composition of the polymeric material can be tuned by appropriately selecting the reactive agents for the post-polymerization modifications. The two systems together enormously increase the possibility of obtaining new functional polymers with predefined structure and with monomeric units that were never reported before. The obtainable materials can be widely useful in many emerging fields, ranging from bio-nanotechnology (e.g. as drug, nucleic acid and diagnostic agent carriers) to the sustainable energy area (e.g. as charge and proton carriers for solar and fuel cells).
Experimental section
Materials
Glycidyl methacrylate (GMA) and butyl acrylate (BA) (Sigma-Aldrich) were filtered through alumina. Styrene and methyl methacrylate (MMA) were flash distilled. 2,2′-Azobis(2-methylpropionitrile) (AIBN) (Sigma-Aldrich) was crystallized from methanol. 1,1′-Azobis(cyclohexanecarbonitrile) (VAZO88), morpholine, piperidine, 4-hydroxypiperidine, 3-hydroxypiperidine, 1-methylpiperazine, diethylamine, N-methylethanolamine, diethanolamine, imidazole, benzimidazole, 2-mercaptoethanol, phenol, N-(5-fluoresceinyl) maleimide, Nile Red, DMSO, N-methylpyrrolidone, toluene and ethyl ether were purchased from Sigma-Aldrich and used as received. 2,2′-Azobis(2,4-dimethylpentanenitrile) (VAZO52) from Du Pont was used as received. The dialysis tube (cutoff 12000–14000 Dalton) was purchased from Medicell International Ltd.
Instrumentation
1H-NMR spectra were recorded at 400 MHz on a Varian Mercury 400. The polymer's molar mass and its distribution were determined using a GPC instrument (MSI Concept PU III with a refractive index detector Shodex RI-71) equipped with PLgel 5 μm MIXED-D, 300 × 7.5 mm column calibrated with polystyrene standards. DLS measurements were taken with a Malvern Instruments – Zetasizer 3000 HAS. Infrared spectra were recorded on a FT/IR PerkinElmer BX spectrophotometer.
Procedures
Synthesis of 2-cyano-4-methylpent-2-yl diethoxyphosphoryl-dithioformate (1c).
The RAFT agent 1c was synthesized by following the previously reported procedure32 with the use of 2,2′-azobis(2,4-dimethylpentanenitrile) as the azo compound. Purification with flash chromatography using ethyl ether/DCM 95/5 yielded 85% of the title product. 1H NMR (400 MHz, CDCl3), δ: [1.03 (d, J = 6.6 Hz, 3H); 1.07 (d, J = 6.6 Hz, 3H) (CH(CH3)2]; 1.36 (t, J = 7 Hz, 6H, OCH2CH3); [1.81 (dd, 1J = 13.9 Hz, 2J = 5.2 Hz, 1H); 2.1 (dd, 1J = 13.9 Hz, 2J = 5.9 Hz, 1H) (CH2CHMe2)]; 1.87 (s, 3H, C(CH3)CN); 2.08 (m, 1H, CH2CHMe2); 4.24 (m, 4H, OCH2CH3) (see spectra in the ESI†).
PGMA synthesis.
Glycidyl methacrylate, the RAFT agent and AIBN were placed in a 10 mL volumetric flask, toluene was added to fill the flask. Aliquots were taken and placed in ampoules which were degassed with four freeze–thaw cycles. The ampoules were placed in a thermostatic bath set at 70 °C (see details in Table 1). The polymerization was stopped by cooling the ampoule. A sample was taken for the conversion determination by NMR. The polymer was precipitated in ethyl ether. The NMR conversion was calculated by comparing the integral signals of GMA vinyl hydrogens at 5.53 and 6.08 ppm with those at 2.42–2.96, 3.16, 3.61–4.10 and 4.12–4.65 ppm of the glycidyl moiety that are overlapped or partially overlapped in their monomeric/polymeric form. The number average molar mass of PGMA polymerized with 2-cyano-4-methylpent-2-yl 4-cyanodithiobenzoate (Table 1, entry 2) was also estimated by NMR by comparing the integral signals at 7.66 and 7.91 ppm of the 4-cyanodithiobenzoate end aromatic hydrogens with those at 2.46–2.97, 3.23, 3.81 and 4.30 ppm of the polymeric glycidyl moiety (see spectra in the ESI†).
Synthesis of PGMA block copolymers.
PGMA macroRAFT (22000 g mol−1, Table 1; entry 1), the monomer and the initiator were placed in a 10 mL volumetric flask, that was topped up with acetonitrile (see details in Table 3). The solution was placed in an ampoule which was degassed with four freeze–thaw cycles. The ampoule was placed in a thermostatic bath. The polymerization was stopped by cooling the ampoule. A sample was taken for the conversion determination by NMR. The volatiles were removed in a rotary evaporator under reduced pressure, the residue was dissolved in chloroform and it was precipitated in ethyl ether.
Treatment of PGMA with morpholine.
PGMA (Mn 11000 g mol−1; 100 mg) was dissolved in DMSO (2.5 mL), the solution was degassed by bubbling nitrogen for 15 minutes, and then morpholine (1.6 μL, 2 eq. relative to the RAFT function) was added. After 30 minutes phenyl maleimide (3.2 mg, 2 eq. relative to the RAFT function) was added and the solution was allowed to react for 2 hours. Further morpholine (0.62 mL, 10 eq. relative to the repeating units) was added before heating at 80 °C for two hours. The volatiles were removed in a rotary evaporator at 1 mbar, the residue was dissolved in chloroform and it was precipitated in cyclohexane. After filtration PHMPMA was recovered as a white solid. The same procedure was used with N-methylpyrrolidone as the solvent and it is also a general way for the treatment with amines.
Treatment of PGMA with morpholine and N-(5-fluoresceinyl) maleimide.
PGMA (Mn 11000 g mol−1; 100 mg) was dissolved in DMSO (2.5 mL), the solution was degassed by bubbling nitrogen for 15 minutes, and then morpholine (0.8 μL, 1 eq. relative to the RAFT function) was added. After 30 minutes N-(5-fluoresceinyl) maleimide (3.9 mg, 1 eq. relative to the RAFT function) was added and the solution was allowed to react for 2 hours. Further morpholine (0.62 mL, 10 eq. relative to the repeating units) was added before heating at 80 °C for two hours. The volatiles were removed in a rotary evaporator at 1 mbar, the residue was dissolved in deionized water and purified by osmosis against 10 mM NaHCO3 followed by deionized water. The purified aqueous solution was freeze-dried obtaining a yellow solid.
Synthesis of PHMPMA-co-PHAPMA.
PGMA (Mn 11000 g mol−1; 100 mg) was dissolved in DMSO (2.5 mL), the solution was degassed by bubbling nitrogen for 15 minutes, then morpholine (1.6 μL, 2 eq. relative to the RAFT function) was added. After 30 minutes phenyl maleimide (3.2 mg, 2 eq. relative to the RAFT function) was added and the solution was left to react for 2 hours. Further morpholine (123 μL, 2 eq. relative to the repeating units) was added before heating at 80 °C for two hours. Then glacial acetic acid (4 μL) was added followed by NaN3 (4.6 mg). After heating for a further two hours at 80 °C, the volatiles were removed in a rotary evaporator at 1 mbar, the residue was dissolved in chloroform and it was precipitated in cyclohexane. Unreacted sodium azide was eliminated by dialysis. After lyophilization PHMPMA-co-PHAPMA was recovered as a white solid.
Treatment of PGMA with 2-mercaptoethanol.
PGMA (Mn 11000 g mol−1; 100 mg) was dissolved in DMSO (2.5 mL) then triethylamine (50 μL, 0.5 eq. relative to the repeating units), 2-mercaptoethanol (100 μL, 2 eq. relative to the repeating units) and phenyl maleimide (3.2 mg, 2 eq. relative to the RAFT function) were added. After heating for two hours at 80 °C, the volatiles were removed in a rotary evaporator at 1 mbar, the residue was dissolved in methanol and it was precipitated in ethyl ether.
Treatment of PGMA with phenol.
PGMA (Mn 11000 g mol−1; 100 mg) was dissolved in DMSO (2.5 mL) then triethylamine (50 μL, 0.5 eq. relative to the repeating units), phenol (124 μL, 2 eq. relative to the repeating units) and methyl maleimide (2 mg, 2 eq. relative to the RAFT function) were added. After heating for 16 hours at 120 °C, the volatiles were removed in a rotary evaporator at 1 mbar, the residue was dissolved in methanol and it was precipitated in ethyl ether.
Treatment of PHMPMA with phenyl isocyanate.
PHMPMA (50 mg) was dissolved in anhydrous THF (2 mL) then phenyl isocyanate (50 μL, 2 eq. relative to the repeating units) was added before heating at 60 °C for two hours. The reaction was quenched with methanol and the volatiles were removed in a rotary evaporator. The residue was dissolved in chloroform and it was precipitated in ethyl ether. A white solid was recovered.
Derivatization of polymers with phenyl isocyanate for GPC analysis.
The polymer (20 mg) was dissolved in anhydrous DMSO (1.5 mL) and phenyl isocyanate (4 eq.) was added. The solution was heated at 120 °C for 2 hours, the volatiles were removed in a rotary evaporator at 1 mbar. The residue was triturated with THF (5 mL) in which 1–2 drops of triethylamine had been added. The filtrate was used for the GPC analysis.
Micelles formation.
The block copolymer (20 mg) [and Nile Red (1.2 mg) when loading with it] was dissolved in THF (2.5 mL) and deionized water (7.5 mL) was added dropwise under gentle stirring. The suspension was placed in a dialysis tube which was left in deionized water in order to eliminate the THF. The aqueous suspension was used for DLS measurements.
Notes and references
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- K. Matyjaszewski and N. V. Tsarevsky, Nat. Chem., 2009, 1, 276–288 CrossRef CAS.
-
(a) G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS;
(b) G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669–692 CrossRef CAS;
(c) G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Q. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS.
-
(a) C. Boyer, J. Liu, V. Bulmus and T. P. Davis, Aust. J. Chem., 2009, 62, 830–847 CrossRef CAS;
(b) C. Boyer, V. Bulmus and T. P. Davis, Macromol. Rapid Commun., 2009, 30, 493–497 CrossRef CAS.
- P. J. Roth, C. Boyer, A. B. Lowe and T. P. Davis, Macromol. Rapid Commun., 2011, 32, 1123–1143 CrossRef CAS.
- A. Gregory and M. H. Stenzel, Prog. Polym. Sci., 2012, 37, 38–105 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- L. Barner, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2007, 28, 539–559 CrossRef CAS.
- M. H. Stenzel, Chem. Commun., 2008, 3486–3503 RSC.
- A. E. Smith, X. Xu and C. L. McCormick, Prog. Polym. Sci., 2010, 35, 45–93 CrossRef CAS.
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS.
-
(a) A. Favier, F. D'Agosto, M.-T. Charreyre and C. Pichot, Polymer, 2004, 45, 7821–7830 CrossRef CAS;
(b) K. A. Aamer and G. N. Tew, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5618–5625 CrossRef CAS.
- S. Ghosh, S. Basu and S. Thayumanavan, Macromolecules, 2006, 39, 5595–5597 CrossRef CAS.
-
(a) K. Nilles and P. Théato, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3683–3692 CrossRef CAS;
(b) M. Eberhardt, R. Mruk, R. Zentel and P. Théato, Eur. Polym. J., 2005, 41, 1569–1575 CrossRef CAS;
(c) K. Nilles and P. Théato, Eur. Polym. J., 2007, 43, 2901–2912 CrossRef CAS;
(d) M. Eberhardt and P. Théato, Macromol. Rapid Commun., 2005, 26, 1488–1493 CrossRef CAS.
-
(a) Y. Liu, L. Wang and C. Pan, Macromolecules, 1999, 32, 8301–8305 CrossRef CAS;
(b) Y.-C. Hu, Y. Liu and C.-Y. Pan, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4862–4872 CrossRef CAS;
(c) R. C. Li, J. Hwang and H. D. Maynard, Chem. Commun., 2007, 3631–3633 RSC.
- C. Weber, T. Neuwirth, K. Kempe, B. Ozkahraman, E. Tamahkar, H. Mert, C. R. Becer and U. S. Schubert, Macromolecules, 2012, 45, 20–27 CrossRef CAS.
- M. E. Buck and D. M. Lynn, Polym. Chem., 2012, 3, 66–80 RSC.
- D. J. Siegwart, K. A. Whitehead, L. Nuhn, G. Sahay, H. Cheng, S. Jiang, M. Ma, A. Lytton-Jean, A. Vegas, P. Fenton, C. G. Levins, K. T. Love, H. Lee, C. Cortez, S. P. Collins, Y. F. Li, J. Jang, W. Querbes, C. Zurenko, T. Novobrantseva, R. Langer and D. G. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 12996–13001 CrossRef CAS.
- S. R. A. Devenish, J. B. Hill, J. W. Blunt, J. C. Morris and M. H. G. Munro, Tetrahedron Lett., 2006, 47, 2875–2878 CrossRef CAS.
- F. J. Xu, M. Y. Chai, W. B. Li, Y. Ping, G. P. Tang, W. T. Yang, J. Ma and F. S. Liu, Biomacromolecules, 2010, 11, 1437–1442 CrossRef CAS.
- F. J. Xu, Y. Zhu, M. Y. Chai and F. S. Liu, Acta Biomater., 2011, 7, 3131–3140 CrossRef CAS.
- I. O. Strube, L. Nothdurft, Z. Abisheva and G. Schmidt-Naake, Macromol. Chem. Phys., 2012, 213, 1274–1284 CrossRef.
- H. Gao, X. Lu, Y. Ma, Y. Yang, J. Li, G. Wu, Y. Wang, Y. Fan and J. Ma, Soft Matter, 2011, 7, 9239–9247 RSC.
- H. Gao, M. Elsabahy, E. V. Giger, D. Li, R. E. Prud'homme and J. C. Leroux, Biomacromolecules, 2010, 11, 889–895 CrossRef CAS.
- A. W. York, S. E. Kirkland and C. L. McCormick, Adv. Drug Delivery Rev., 2008, 60, 1018–1036 CrossRef CAS.
-
(a) A. J. Inglis, S. Sinnwell, M. H. Stenzel and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2009, 48, 2411–2414 CrossRef CAS;
(b) C. F. Hansell, P. Espeel, M. M. Stamenovic, I. A. Barker, A. P. Dove, F. E. Du Prez and R. K. O'Reilly, J. Am. Chem. Soc., 2011, 133, 13828–13831 CrossRef CAS.
- M. Benaglia, J. Chiefari, Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, J. Am. Chem. Soc., 2009, 131, 6914–6915 CrossRef CAS.
- J. Zhu, D. Zhou, X. L. Zhu and G. J. Chen, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2558–2565 CrossRef CAS.
- C. S. Gudipati, M. B. H. Tan, H. Hussain, Y. Liu, C. He and T. P. Davis, Macromol. Rapid Commun., 2008, 29, 1902–1907 CrossRef CAS.
- M. Benaglia, E. Rizzardo, A. Alberti and M. Guerra, Macromolecules, 2005, 38, 3129–3140 CrossRef CAS.
- A. Alberti, M. Benaglia, M. Laus and K. Sparnacci, J. Org. Chem., 2002, 67, 7911–7914 CrossRef CAS.
- P. Jiang, Y. Shi, P. Liu and Y. Cai, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2947–2958 CrossRef CAS.
- N. V. Tsarevsky, S. A. Bencherif and K. Matyjaszewski, Macromolecules, 2007, 40, 4439–4445 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.