Reversible maleimide–thiol adducts yield glutathione-sensitive poly(ethylene glycol)–heparin hydrogels†
Aaron D.
Baldwin
a and
Kristi L.
Kiick
*ab
aDepartment of Materials Science and Engineering, University of Delaware, 201 DuPont Hall, Newark, DE 19716, USA. E-mail: kiick@udel.edu
bDelaware Biotechnology Institute, 15 Innovation Way, Newark, DE 19716, USA
First published on 13th September 2012
Abstract
We have recently reported that retro Michael-type addition reactions can be employed for producing labile chemical linkages with tunable sensitivity to physiologically relevant reducing potentials. We reasoned that such strategies would also be useful in the design of glutathione-sensitive hydrogels for a variety of targeted delivery and tissue engineering applications. In this report, we describe hydrogels in which maleimide-functionalized low molecular weight heparin (LMWH) is crosslinked with various thiol-functionalized poly(ethylene glycol) (PEG) multi-arm star polymers. Judicious selection of the chemical identity of the thiol permits tuning of degradation via previously unstudied, but versatile chemical methods. Thiol pKa and hydrophobicity affected both the gelation and degradation of these hydrogels. Maleimide–thiol crosslinking reactions and retro Michael-type addition reactions were verified with 1H NMR during the crosslinking and degradation of hydrogels. PEGs esterified with phenylthiol derivatives, specifically 4-mercaptophenylpropionic acid or 2,2-dimethyl-3-(4-mercaptophenyl)propionic acid, induced sensitivity to glutathione as shown by a decrease in hydrogel degradation time of 4-fold and 5-fold respectively, measured via spectrophotometric quantification of LMWH. The degradation proceeded through the retro Michael-type addition of the succinimide thioether linkage, with apparent pseudo-first order reaction constants derived from oscillatory rheology experiments of 0.039 ± 0.006 h−1 and 0.031 ± 0.003 h−1. The pseudo-first order retro reaction constants were approximately an order of magnitude slower than the degradation rate constants for hydrogels crosslinked via disulfide linkages, indicating the potential use of these Michael-type addition products for reduction-mediated release and/or degradation, with increased blood stability and prolonged drug delivery timescales compared to disulfide moieties.
Introduction
Hydrogels have been widely adopted as tools for the study of many diverse types of biological phenomena and in applications including tissue engineering,1–3 biological sensor and microarrays,4,5 protein and polymer purification6,7 and drug delivery.8–11 They are composed mostly of water and maintain their self-supporting and elastic nature by a network of hydrophilic polymers that are chemically, physically, and/or ionically crosslinked, leading to a material that swells in the presence of water and that can yield mechanical and/or chemical properties similar to those of biological tissues.12 Polymers of synthetic and natural origin – including poly(ethylene glycol) (PEG), poly(vinyl alchol), poly(N-isopropylacrylamide), gelatin, collagen, alginate, and hyaluronic acid – have been exploited for the above applications, with the application dictating the requisite properties of the chosen hydrogel.1,12We have shown that the fast reaction kinetics and specificity of reactions between maleimides and thiols are useful for the in situ crosslinking of thiolated polyethylene glycol with maleimide-functionalized heparins.13–15 Most recently, we have reported the utility of PEG–low molecular weight heparin (LMWH) hydrogels for injectable anticoagulant therapies.16 Heparin, a highly sulfated and variable glycosaminoglycan, is a widely employed anticoagulant in subcutaneous and intravenous therapies,17 although currently not in a long-term hydrogel delivery format. The anionic character of heparin mediates binding to numerous proteins which in turn mediates many cell fate processes including cell proliferation, differentiation and control of chemokine signaling.18,19 Indeed, many types of heparins have been investigated for antimetastatic properties given heparin's role in tumor metastasis;20 heparin has also been used as a drug in reduction-sensitive delivery vehicles.21,22 Accordingly, there have been many investigations of heparin-containing polymeric hydrogels, based on PEG, hyaluronic acid or other polymeric matrices, as drug delivery platforms and tissue engineering scaffolds.13,23–28 The ability to control degradation in these and related polymeric materials thus has potential for programmed temporal control of degradation for in vitro cellular studies29–32 and future clinical applications.
In hydrogels developed for drug delivery, particularly for targeted release of chemotherapeutic drugs, a variety of strategies based on labile covalent linkages have been used to afford environmentally triggered release of therapeutic agents. Many of these have focused on the use of matrix metalloproteinase (MMP)-sensitive crosslinks, or on hydrolytically labile bonds between the network and tethered therapeutics.33–36 Many strategies have used environmentally triggered degradation of hydrogels to direct bulk degradation and delivery of entrapped therapeutic molecules; in addition to the use of degradable synthetic polymer hydrogels, some additional examples include enzymatic cleavage of gelatin-37,38 or chitosan-based networks,37–40 or through reduction of disulfide-based crosslinks.21,22,26,41–44
Reduction-sensitive bonds have also been widely used in bioconjugates employed for biomedical applications, particularly for intracellular-triggered gene and drug delivery.45–47 In all cases, the degradation or release relies on the reduction of disulfide bonds by glutathione (GSH), a reducing agent found naturally in circulation and in cellular compartments.48 Typically, the extracellular concentration of GSH is relatively low (ca. 1–20 μM in plasma) and relatively high in cells (ca. 0.5–10 mM), providing a level of stability for conjugates and hydrogels outside the cell and aiding in rapid degradation of disulfides intracellularly.49,50 Accordingly, disulfides have been utilized, mainly with aims in tumor therapies, in many drug conjugates and delivery matrices, including disulfide-stabilized micelles for delivery of anticancer drugs,51–56 disulfide-stabilized vesicles,57 disulfide crosslinked nanoparticles,58 carbon nanotubes with disulfide-tethered drugs59 and targeting molecules with disulfide-linked taxoid anticancer agents.60
We have recently shown that succinimide–thioether linkages, formed via the Michael-type addition of aromatic thiols to maleimides, are sensitive to reducing potential and can be cleaved by exogenous glutathione. The cleavage rate of the arylthioether–succinimide adducts was significantly lower than that of the analogous cleavage of disulfide linkages (10–100× reduction in rate) and more rapid than that reported for the cleavage of select cysteine–maleimide adducts.61,62 Arylthioether–succinimide linkages can thus be used as GSH-sensitive crosslinks with expanded timescales of reduction compared to disulfides and other common adducts. In the work reported here, we investigate the degradation of reduction-sensitive PEG–LMWH hydrogels crosslinked by maleimide–thiol chemistries. The chemical crosslinking and degradation of the gels was monitored via1H NMR, oscillatory rheology, and spectrophotometric studies. The gelation kinetics and degradation sensitivity to reducing agents was investigated to illustrate the utility of the retro Michael-type addition for temporal control of degradation.
Experimental section
Materials
Four-arm, hydroxyl-functionalized PEG (Mn 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) was purchased from JenKem Technology USA Inc. (Allen, TX, USA). Four-arm, thiol-functionalized PEG (PEG–SH, Mn 10000 g mol−1) was purchased from Creative PEG Works (Winston Salem, NC, USA). Mercaptoisobutyric acid (MIB) was purchased from TCI America (Portland, OR, USA). 4-mercaptophenylpropionic acid (MPP) and 2,2-dimethyl-3-(4-mercaptophenyl)propionic acid (DMMPP) were purchased from Toronto Research Chemicals (North York, Ontario, Canada). Nitrous acid depolymerized low molecular weight heparin (LMWH) was purchased from Celsus (Cincinnati, OH, USA). N-(2-Aminoethyl)maleimide, trifluoroacetate salt (AEM), 1-hydroxybenzotriazole hydrate (HOBT), 2-(N-morpholino)ethanesulfonic acid (MES), 3-mercaptopropionic acid (MP), p-toluenesulfonic acid monohydrate (PTSA), and light mineral oil were purchased from Sigma-Aldrich (St. Louis, MO, USA). N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl), and all other reagents and materials were purchased from Fisher Scientific unless noted (Pittsburgh, PA, USA). Proton nuclear magnetic resonance (1H NMR) spectra were acquired under standard quantitative conditions at ambient temperature on a Bruker DRX-400 NMR spectrometer (Billerica, MA). The spectra of all purified compounds were recorded in deuterated chloroform or deuterium oxide.
000 g mol−1) was purchased from JenKem Technology USA Inc. (Allen, TX, USA). Four-arm, thiol-functionalized PEG (PEG–SH, Mn 10000 g mol−1) was purchased from Creative PEG Works (Winston Salem, NC, USA). Mercaptoisobutyric acid (MIB) was purchased from TCI America (Portland, OR, USA). 4-mercaptophenylpropionic acid (MPP) and 2,2-dimethyl-3-(4-mercaptophenyl)propionic acid (DMMPP) were purchased from Toronto Research Chemicals (North York, Ontario, Canada). Nitrous acid depolymerized low molecular weight heparin (LMWH) was purchased from Celsus (Cincinnati, OH, USA). N-(2-Aminoethyl)maleimide, trifluoroacetate salt (AEM), 1-hydroxybenzotriazole hydrate (HOBT), 2-(N-morpholino)ethanesulfonic acid (MES), 3-mercaptopropionic acid (MP), p-toluenesulfonic acid monohydrate (PTSA), and light mineral oil were purchased from Sigma-Aldrich (St. Louis, MO, USA). N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl), and all other reagents and materials were purchased from Fisher Scientific unless noted (Pittsburgh, PA, USA). Proton nuclear magnetic resonance (1H NMR) spectra were acquired under standard quantitative conditions at ambient temperature on a Bruker DRX-400 NMR spectrometer (Billerica, MA). The spectra of all purified compounds were recorded in deuterated chloroform or deuterium oxide.
Synthesis of PEG–thiol
The synthesis of thiolated four-arm PEG was performed as previously reported.13 In short, PEG (1 meq.), mercaptoacid (40 meq. MP, MIB, MPP or DMMPP) and PTSA (0.4 meq.) were dissolved in toluene. Under a flow of nitrogen the reaction was refluxed with stirring for 48 h (Scheme 1). Water was collected by using a Dean Stark trap. Toluene was removed under reduced pressure and the polymer was precipitated 3 times in cold ether. The polymer was reduced by dissolving 1 meq. polymer in methanol with DTT (1 meq.) and triethylamine (1 meq.) under nitrogen for 5 hours. The finished reaction was acidified with trifluoroacetic acid (1.1 meq.), and the polymer was precipitated in ether and rinsed with 2-propanol then hexane. Functionality was determined via1H NMR spectroscopy and was ∼4 (>95%) for all derivatives (Fig. S1–S4†). PEG–MP (Fig. S1†) 1H NMR (CDCl3): δ = 4.28 (8H, t), 3.90–3.35 (900H, bs), 2.84–2.63 (16H, m), 1.69 (4H, t). PEG–MPP (Fig. S2†) 1H NMR (CDCl3): δ = 7.24–7.18 (8H, d), 7.11–7.06 (8H, d), 4.22 (8H, t), 3.90–3.35 (900H, bs), 2.94 (8H, t). PEG–MIB (Fig. S3†) 1H NMR (CDCl3): δ = 4.28 (8H, m), 3.90–3.35 (900H, bs), 2.84–2.60 (12H, m), 1.57 (4H, t), 1.31–1.23 (12H, d). PEG–DMMPP (Fig. S4†) 1H NMR (CDCl3): δ = 7.22–7.15 (8H, d), 7.05–6.98 (8H, d), 4.21 (8H, t), 3.90–3.35 (900H, bs), 2.81 (8H, s), 1.18 (24H, s). All products were stored under argon or vacuum at room temperature to maintain the reduced thiol during storage.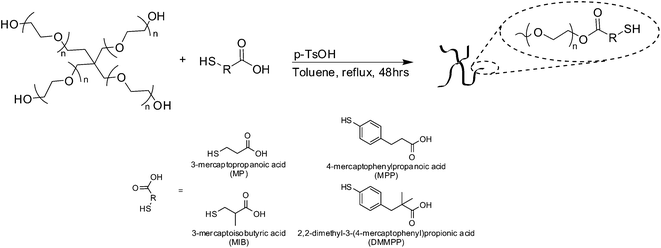 | ||
| Scheme 1 Mercaptoacid esterification of PEG. | ||
Synthesis of maleimide-functionalized LMWH (Mal–LMWH)
The molecular weight of LMWH was characterized by size exclusion chromatography (SEC) using previously described methods.63 The SEC system comprised a Waters 515 HPLC pump (Milford, MA, USA), two Waters Ultrahydrogel (7.8 × 300 mm) columns in series, a Waters 2414 refractive index detector, a Waters 2996 photodiode array detector and a Precision Detectors light scattering unit (Bellingham, MA, USA). The number average molecular weight was determined to be 8300 g mol−1.The synthesis of maleimide-functionalized heparin was performed as previously described with slight modification of reactant quantities to control the extent of modification.13 Briefly, 500 mg LMWH (0.06 mmol) was dissolved with 103 mg HOBT (0.67 mmol), 103 mg AEM (0.67 mmol) and 103 mg EDC·HCl (0.54 mmol) dissolved in 50 ml of 0.1 M MES pH 6.0 (Scheme 2). The reaction proceeded overnight at room temperature with stirring. The product was purified by dialysis (MWCO 1000) against 4 l of 1M NaCl solution and then subsequently against de-ionized water each with 4 volumes exchanges over 24 h. The freeze-dried sample was characterized via1H NMR indicating a degree of functionalization of 2.6. 1H NMR (400 MHz, D2O): δ = 6.83 (2H, s), 5.60–5.05 (29H, heparin anomeric proton, bs) (Fig. S5†).
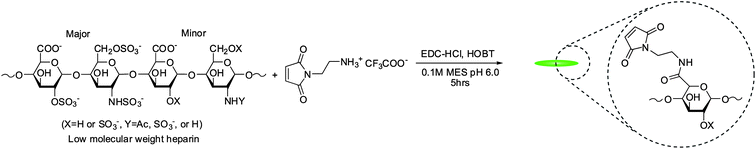 | ||
| Scheme 2 Synthesis of maleimide-functionalized LMWH. | ||
Hydrogel formation
Hydrogel formation was accomplished by mixing separate solutions of the functionalized PEG and LMWH, prepared individually in phosphate buffered saline (PBS, 10 mM phosphate and 150 mM NaCl). Hydrogels prepared with alkyl thiol-functionalized PEGs (MP and MIB) were gelled at a pH of 7.0 and an initial temperature of 25 °C, while hydrogels prepared with phenylthiol-functionalized PEG were gelled at a pH 6.6 and an initial mixing temperature of 4 °C to slow the gelation and permit mixing before gelation occurred. Reducing the pH and temperature allowed sufficient time to briefly vortex mix and load samples before gelation occurred. Self-supporting hydrogels were formed upon mixing the two solutions. For simplicity of analysis, 5 wt% gels were used in all experiments. Control gels for the oscillatory rheology degradation experiments were crosslinked with disulfide bonds and were formed as previously described.64 In short, 5.3 wt% PEG–SH was dissolved in 10 mM glycine buffer containing 0.01× (molar ratio to PEG–SH) iron lactate and cysteine.1H NMR of hydrogels
The Michael-type addition of the thiol to the maleimide for gelation (Scheme 3), as well as the hydrolysis or retro-Michael-type additions for degradation (Scheme 4), was verified using 1H NMR of hydrogels formed in deuterated buffers. Gelation experiments were conducted by loading 60 μl of thiol-functionalized PEG, maleimide-functionalized LMWH, or mixtures of both into a single opening 1.5–1.8 × 90 mm borosilicate glass capillary. The filled capillary was placed in a standard NMR tube containing 700 μl of the deuterated buffer. Measurements (32 scans, room temperature) were taken under standard acquisition parameters for both MP and MPP functionalized PEG at 10, 30, 60 minutes, and 2 and 8 h. Degradation experiments to verify reduction sensitivity were completed on MPP-functionalized PEG–LMWH hydrogels. A 60 μl hydrogel was formed in the bottom of a standard NMR tube (below the receiver coils of the NMR); the liberation of small molecules into the buffer above the hydrogel was monitored in the NMR experiment. The degradation experiments were conducted on gels that were swelled at 37 °C (using deuterated buffers containing 10 mM glutathione at pH 7.4), with measurements taken at every 24 h. To increase the sensitivity of the spectral runs, an increased acquisition time of 8.2 s was used.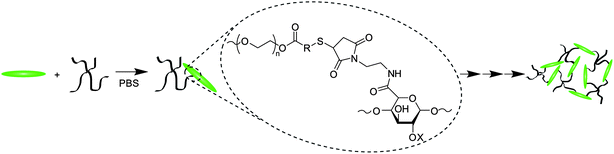 | ||
| Scheme 3 Hydrogel formation using Mal–LMWH and PEG–thiols. | ||
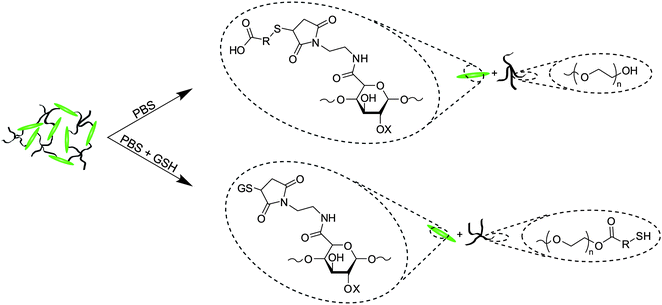 | ||
| Scheme 4 Degradation mechanisms for ester and succinimide thioether groups. Mixtures of both hydrolysis and retro-Michael-type addition products are possible for these hydrogels. | ||
Hydrogel formation monitored by oscillatory rheology
Samples were gelled in situ by co-injection of the separately dissolved functionalized polymers onto the rheometer Peltier plate. An AR-2000 rheometer (TA Instruments, New Castle, DE) was used to measure oscillatory shear properties at 37 °C with maximum shearing amplitude of 1.0%. A 20 mm, 1°56′′ cone plate geometry with a 33 μm truncation requiring 40 μl of solution was used in all experiments. Time sweep studies were performed at a constant 6 rad s−1 while frequency sweeps were conducted over a logarithmic scale from 0.1 rad s−1 to 100 rad s−1. The rheometer Peltier plate was chilled to 25 °C for alkyl thiol-functionalized PEG-containing gels, and 4 °C for phenylthiol-functionalized PEG-containing gels, before co-injection of polymers onto the plate. Time sweep data collection, and a 3-minute temperature ramp to 37 °C, was initiated once the polymers were injected onto the stage. Light mineral oil was applied to the perimeter of the sample to prevent evaporation over the course of the experiment.Hydrogel degradation monitored by oscillatory rheology
The reduction in modulus as a function of degradation was monitored by oscillatory rheology by swelling the perimeter of the hydrogel with standard or reducing buffers. Hydrogels were formed as described above with slight modification to the procedure. Before adding the solutions to the Peltier plate, a polyvinylchloride (PVC) ring with a height of 7 mm, inside diameter of 35 mm and an outside diameter of 42 mm was pre-coated with a thin film of silicone vacuum grease on the bottom and positioned above the geometry by a ring stand and clamp. After the crosslinked hydrogel had reached equilibrium the rheometer bearing was locked to prevent disruption of the hydrogel while the mineral oil was washed away three times with hexane. The PVC tube was then firmly pressed onto the Peltier plate creating a water-tight seal with the vacuum grease (Fig. S6†). The volume between the gel and cylinder walls was filled with 2 ml of buffer (50 mM phosphate, 150 mM NaCl, 10 mM or 10 μM GSH at pH 7.4) and topped with mineral oil to prevent evaporation. A buffer concentration of 50 mM (versus 10 mM as described above) with was used to maintain a constant pH throughout the experiment. Data points were collected every 5 minutes over 5 days.Hydrogel degradation monitored by heparin release
40 μl hydrogels were formed in a 0.5 ml tuberculin syringe with the tip removed using the same mixing method described above. The resulting hydrogels had a diameter of 3.5 mm and a length of 4 mm. The truncated syringe was sealed with Parafilm® to minimize evaporation during gelation. Cast gels were incubated at 37 °C for the length of time, indicated by oscillatory rheology experiments, required for the storage modulus to reach a plateau. Gels were then ejected by advancing the syringe plunger. Released gels were incubated in a bath of 4 ml buffer at 37 °C. Highly reducing conditions in vivo were mimicked by using 10 mM phosphate buffer with 150 mM sodium chloride and 10 mM GSH at pH 7.4. Standard reducing conditions were mimicked using lower concentrations of GSH (10 μM). 1 ml samples were taken and replenished with fresh buffer at time points of 0, 1, 6, 12, and 24 h initially, then every 12 h for 2 days, every 24 h for 4 additional days, every 48 h for 4 additional days, every 72 h for 18 additional days, and every 96 h for the remaining 23 days.The concentration of heparin in solution was measured using established toluidine blue measurements with slight modification.65 Briefly, 100 μl of collected sample were mixed with 800 μl toluidine blue solution (0.005% toluidine blue in 50 mM HCl) and 100 μl dichloromethane in 1.5 ml centrifuge tubes. The solutions were mixed and incubated overnight at room temperature followed by centrifugation at 20000 × g for 20 minutes. The absorbance of the aqueous phase was measured using an Agilent 8453 spectrophotometer (Santa Clara, CA, USA) in a Hellma 3 mm quartz cuvette (Plainview, NY, USA). Absorbance maxima at 590 and 632 nm were recorded. Calibration curves were constructed using functionalized LMWH. No difference in calibration was noted when stock solution of functionalized LMWH was reacted with thiolated PEGs or small molecule thiols.
Results and discussion
Gelation kinetics and storage modulus
Oscillatory rheology was used to determine the gelation kinetics and final modulus for all four types of LMWH and PEG hydrogels (PEG–MP, –MIB, –MPP, –DMMPP). The mixing of the solutions of functional PEG and LMWH resulted in the rapid in situ formation of an elastic hydrogel on the rheometer stage. Results for PEG–MPP, which was the most rapid hydrogelator, are shown in the oscillatory rheology time-sweep data presented in Fig. 1; the PEG–MPP hydrogel was formed within seconds of mixing the dissolved components (<6 s). Although the mixed components were added to the rheometer Peltier plate before an increase of viscosity was observed, the first data point recorded at 6 seconds indicated that the storage modulus (G′) had increased to 3.8 ± 2.1 Pa, greater than the loss modulus (G′). The reduction in G′′ with time most likely indicates the initial presence of kinetically trapped, unreacted functionalities between covalent crosslinks that eventually react to form elastic crosslinks as time progresses.66 This occurrence was observed only for the phenylthiol PEG-containing hydrogels and was absent in alkylthiol PEG-containing gels (Fig. S7†), possibly due to fewer kinetically entrapped network defects.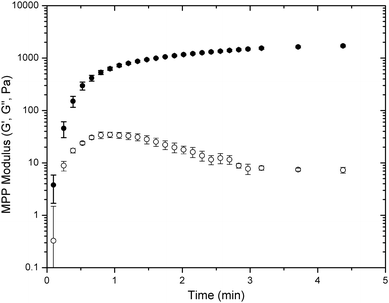 | ||
| Fig. 1 Oscillatory rheology time-sweep data for the gelation of PEG–MPP-containing PEG–LMWH hydrogels. G′ values are indicated by closed symbols, and G′′ values by open symbols. Error bars represent the standard deviation of measurements of 3 separate hydrogels. | ||
Variations in the rate of gelation were also assessed from the rheology experiments; representative data for comparison to Fig. 1 are presented in Fig. S7.† Gelation occurred within 15 s, 50 s and 90 s for PEG–DMMPP, PEG–MP, and PEG–MIB hydrogels, respectively (Fig. S7†). The rate of gelation of the hydrogels was related to the Michael donor reactivity of the thiols; the phenylthiol derivatives, with lower pKa values relative to the alkyl thiols (6.6 (ref. 67) vs. 10.3 (ref. 68)), thus had faster reaction rates.69 The same dependence on Michael donor reactivity was observed for the final equilibration of the storage modulus of the hydrogels, with gels achieving 90% of their final modulus in 9 minutes for PEG–MPP, 20 minutes for PEG–DMMPP, 60 minutes for PEG–MP and 70 minutes for PEG–MIB. Regardless of the identity of the mercaptoacid-functionalized PEG, the resulting hydrogels exhibited stable elastic moduli over all frequencies measured (0.1 rad s−1 to 100 rad s−1, Fig. S7†) with the ratio of G′′/G′ or tan(δ) being less than 0.01 for all hydrogels over all frequencies.
The ultimate storage modulus measured in the rheology experiments was used to compare the consistency of the elastic storage modulus between the various gels; data are shown in Fig. 2. The measured equilibrium storage moduli for the various gels were all approximately 2.1 kPa (p = 0.79), regardless of the identity of the mercaptoacid-functionalized PEG and the corresponding differences in gelation kinetics. The apparently low elastic moduli values, compared to the predicted modulus for these hydrogels from classical rubber elasticity theory (19.1 kPa (ESI†)),70 is likely due to the formation of non-elastically active chains, such as cycles or loops, arising from the crosslinking of these materials under relatively dilute conditions.71,72 An increase in hydrogel concentration (e.g., to 10–15 wt%) increased the storage modulus of the gels to near that of the theoretically predicted value (data not shown). However, a significant decrease in gelation time was observed, and obtaining uniform samples with the more rapidly gelling materials (PEG–MPP and PEG–DMMPP) was difficult. A maximum hydrogel concentration of 5 wt% was thus used to maintain sample uniformity and modulus across all PEG–LMWH hydrogels. Furthermore, the statistically similar final moduli exhibited by these networks suggest that the mechanism of gelation is conserved, over the timescales of network formation, and that other reactions, such as disulfide bond formation or ring opening of the maleimide, are minimized. Therefore, direct comparisons of the degradation kinetics of the gels are possible.
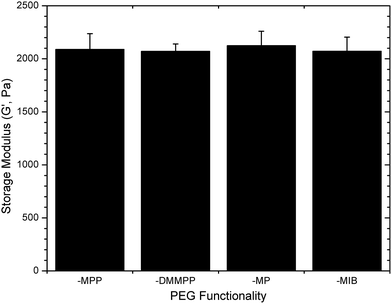 | ||
| Fig. 2 Final storage moduli measured for various PEG–LMWH hydrogels. The identity of the thiol derivative does not impact the final modulus (ANOVA; p = 0.79). | ||
Gelation
Auto-oxidation of thiols is well known to occur in solution,73 and could possibly occur prior to or during hydrogel formation. Hydrogelation (Scheme 3) was thus monitored via1H NMR to confirm the extent to which any disulfide bond formation occurred during gelation. The spectra in Fig. S8† show complete disappearance of the maleimide protons (6.82 and 6.8 ppm) as well as the protons from the mercaptoacid moieties (centered at 2.7 ppm for MP, and 2.6, 2.8, 7.0 and 7.2 ppm for MPP) without the appearance of new peaks for disulfide bond or ring-opened maleimides. Rates of disulfide bond formation under these conditions were also determined via standard Ellman's assay (Fig. S9†).74 These data illustrate that the rate of disulfide formation occurred the fastest for phenylthiol-functionalized PEG, with an oxidation half-life of 13.5 h, and the slowest for alkyl thiol-functionalized PEG with an oxidation half-life of 4.7 days. The rate of disulfide bond formation was significantly slower than the rate of maleimide–thiol reactions under these conditions, as expected.Degradation monitored by 1H NMR
The degradation of the hydrogels was monitored via1H NMR to verify the mechanism of degradation under reducing conditions. As we previously reported for small-molecule model compounds, the succinimide thioether products of the addition of aromatic thiols to maleimides are susceptible to thiol exchange in the presence of exogenous thiols; indeed, 4-mercaptophenylacetic acid–maleimide adducts were shown to cleave up to 85% after 70 h.61 In the experiments here, the degradation of PEG–MPP-containing PEG–LMWH hydrogels was assessed via1H NMR. Hydrogels were formed on the bottom of standard NMR tubes such that the positioning of the hydrogel was below the receiver coils of the instrument. Deuterated PBS lacking GSH was used for the gelation; the hydrogels were then swelled in deuterated PBS containing 10 mM GSH and the liberated compounds detected in solution.Fig. 3 shows the results of these experiments; the spectra showing the progression from the initial hydrogel to soluble macromers are shown (traces A–D) with a magnified view showing the position of the aromatic protons (6.7 ppm to 7.6 ppm), which were sensitive to the chemical substituents of the thiol and thus provided a facile metric for confirming the mechanism of degradation. Trace A shows the spectrum of the initial PEG–MPP hydrogel (from NMR experiments of gelation), with clear broadening of the aromatic protons owing to the conjugation of the thiol with the maleimide. Trace D shows unreacted and soluble PEG–MPP for comparison. The amount of soluble PEG–MPP in the solution above the degrading hydrogel was not significant until the hydrogel was no longer visibly distinguishable from the solution; trace B shows the clear appearance of the protons from the free PEG–MPP at day 3. At this point there is a mixture of PEG–MPP conjugated to LMWH, as indicated by the presence of both the broad peaks centered at 7.2 and 7.4 ppm and the PEG–MPP aromatic protons centered at 6.9 and 7.2 ppm. The slight shift in the position of the aromatic protons from the positions in the gelation studies above (i.e. 6.9 versus 7.0 ppm) is most likely due to slight differences in the solution pH values between the experiments. Trace C displays the NMR spectrum at day 4; at this timepoint most of the succinimide thioether linkages have degraded, yielding mainly the soluble PEG–MPP. Hydrogels incubated in the absence of GSH did not show the appearance of the peaks from the soluble PEG–MPP (data not shown).
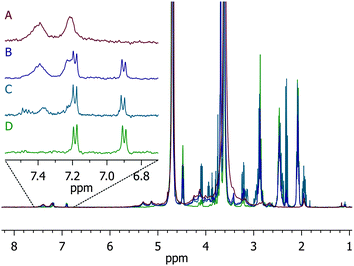 | ||
| Fig. 3 1H NMR of degradation of PEG–MPP-containing PEG–LMWH hydrogels. Trace A is the starting hydrogel, B when hydrogel is no longer visibly apparent, C is a later timepoint showing almost complete regeneration of the thiol, and D is the starting four-arm PEG–MPP for reference. | ||
Degradation monitored by oscillatory rheology
Oscillatory rheology experiments provided a means to assess both the kinetics of degradation of the hydrogels as well as the changes in mechanical properties that occur with degradation. For comparison, the extended degradation timescales of the maleimide–thiol crosslinked hydrogels were compared with PEG–SH hydrogels crosslinked by disulfide linkages. The control disulfide hydrogel was composed of 5.3 wt% four-arm thiolated PEG with a storage modulus of 3 kPa. The formation of the control hydrogel required approximately 24 h even when using the Fenton-type catalyst as previously described.64 Degradation of the gels was monitored in the presence of GSH-containing buffer via direct measurement of the modulus using oscillatory rheology (Fig. 4). The hydrogels were constricted between the rheometer geometry and Peltier plate, whereby the swelling of the hydrogel was prevented axially. Radial swelling was negligible; therefore, the reduction in storage moduli was directly related to the scission of active crosslinks absent of swelling effects. Furthermore, a bath volume of 2 ml (50-fold greater than the volume of the hydrogel) provided an adequate sink so that pH and reductant concentration were unaffected during the experiment (data not shown).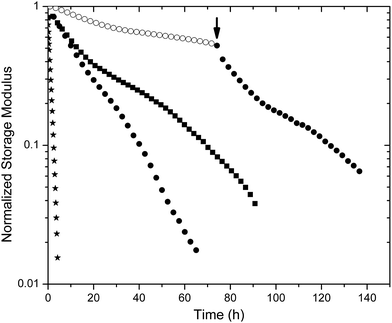 | ||
| Fig. 4 Comparison of storage moduli for select degrading hydrogels: PEG–SH hydrogel (★) LMWH–PEG–MPP (●) and –DMMPP (■) under high reducing conditions (10 mM GSH) and LMWH–PEG–MPP (○) under standard reducing conditions (10 μM GSH). At 72 h (arrow) the buffer of the standard reducing condition hydrogel was exchanged for high reducing buffer, showing an increase in rate of degradation. | ||
Fig. 4 shows the reduction of a normalized storage modulus versus time for the hydrogels that are most sensitive to reductant (PEG–SH, PEG–MPP and PEG–DMMPP). The PEG–SH hydrogels rapidly degraded when exposed to high concentrations of reductant, with a near-zero modulus observed in 4 hours. PEG–MPP- and PEG–DMMPP-containing hydrogels show much lower susceptibility to the GSH, consistent with the fact that the rate constants for the retro Michael-type addition are an order of magnitude lower than those for glutathione–disulfide exchange.61 The sensitivity to GSH is maintained over 72 hours, indicated by the increased rate of degradation observed upon exchange of the surrounding buffer with 10 mM GSH at this timepoint (indicated by the arrow in Fig. 4). The fact that this degradation rate is slightly slower than that for when the hydrogels are initially immersed in 10 mM GSH buffer is consistent with the ring opening of the succinimide, which renders the gels less sensitive to GSH.61
The degradation profiles were evaluated via standard rubber elasticity theory assuming that the rate of scission of any network active chain correlates with a reduction in modulus; a slight modification in the value of f was made to account for the less defined maleimide functionality of the LMWH (ESI, Fig. S10†). The complicated kinetics of the retro Michael-type addition produced an overall non-first order degradation curve, as evident by the non-linear curve displayed on the log-linear scale shown in Fig. 4. As degradation proceeds, the amount of available succinimide thioethers diminishes, resulting in an apparent reduction in the degradation rate. This trend is then followed by an accelerated degradation as the hydrogel enters the depercolation regime. Thus, the pseudo-first order kinetics of the retro reaction can only be approximated under the initial timescales when a majority of the succinimide rings are intact and susceptible to thiol exchange via the retro Michael-type addition. Given that the half-life of succinimide rings for similar small molecules was reported to be on the order of 200 h,61 the first 15 hours were fit to exponential decay equations to minimize the impact of the ring opening on the estimates of the retro-Michael addition rate constants (Fig. S11†).
The determined rate constants were 0.039 ± 0.006 h−1 for PEG–MPP- and 0.031 ± 0.003 h−1 for PEG–DMMPP-containing PEG–LMWH hydrogels (ESI†). Given the linearity of the degradation curve for the PEG–SH gels, these rate constants were estimated by fitting the entire curve, yielding pseudo-first order constants of 0.81 ± 0.1 h−1. These pseudo-first order rates are consistent with typical values (5 to 0.9 h−1) reported for disulfide cleavage,75 and also with those previously derived for succinimide thioethers comprising phenylthiol substituents (0.0371 h−1).61 The agreement of the rate constants determined from the degradation experiments with those previously reported for small-molecule model studies suggests that the diffusion of glutathione to the center of the 20 mm rheometer geometry (calculated to require 10 hours (based on an approximated diffusion coefficient for glutathione of 6 × 10−6 cm2 s−1)76) does not introduce significant non-uniformity in the degradation to complicate our analysis. Notably, complete degradation of the PEG–SH hydrogel occurs within 4 hours, suggesting that diffusion of glutathione may be accelerated by the degradation of the hydrogel network as well as an increase in molecular motion provided by the oscillatory straining of the hydrogel.
LMWH release
The increased stability of the succinimide–thioether bond, relative to the disulfide bond, provides opportunities to utilize these chemistries for a more sustained release of drugs under the high reducing loads present in intracellular compartments and tumor microenvironments.61 The release of LMWH from the hydrogels was thus investigated; the amount of LMWH released from the hydrogels was quantified using metachromatic dye measurements, with resulting data shown in Fig. 5. The figure shows the release of LMWH, for all hydrogels, under standard reducing conditions in blood circulation (10 μm GSH, open symbols) compared to that under the high reductant load commensurate with intracellular compartments and tumor microenvironments (10 mM GSH, solid symbols).49,50,77 All materials showed some burst release of LMWH within the first day (∼23%), likely due to the large polydispersity of LMWH and potentially poor efficiency of crosslinking. The average functionalization of the LMWH is approximately 2.6; therefore, a small amount of low molecular weight species (roughly 3% from GPC data) are without maleimide functionality and free to diffuse from the hydrogel. Hydrogels were also formed in dilute solutions (5 wt%), as discussed above, and exhibited low crosslinking efficiencies; consequently, the formation of loops and intramolecular crosslinks enable free or partially reacted but non-crosslinked LMWH to be released within the first day.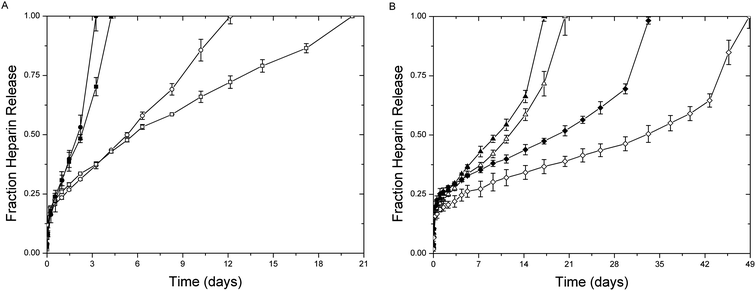 | ||
| Fig. 5 Release of LMWH from (A) PEG–MPP (●) PEG–DMMPP (■) and (B) PEG–MP (▲) and PEG–MIB (◆) containing hydrogels under standard reducing conditions (10 μM GSH, open) and high reductant load (10 mM GSH, closed). | ||
The hydrogels containing the GSH-sensitive succinimide thioether linkages (PEG–MPP and PEG–DMMPP) release 100% LMWH over 12 and 20 days under standard reducing conditions, while they rapidly degrade under the high reducing conditions, completely releasing LMWH within 3 and 4 days (Fig. 5A). Comparatively, the release profiles for the other hydrogels (PEG–MP and PEG–MIB) show significantly less sensitivity to reducing conditions, requiring 20 and 49 days for PEG–MP and PEG–MIB under standard reducing conditions and 17 and 33 days under high reductant load. This slight increase in LMWH release rate for the PEG–MP and PEG–MIB gels was not expected, given the lack of retro Michael-type reactions observed in our previous studies even when glutathione was present in 100× excess over the succinimide thioether substituent.61 The sensitivity of the gels to a 1:1 stoichiometry of GSH in this hydrogel experimental format may suggest that GSH transesterification is promoted by the relatively high concentration of thiolate (0.1 mM) over hydroxyl groups (0.3 μM) in the gel.78,79
The acceleration in LMWH release for the PEG–MP and PEG–MIB gels (Fig. 5B) at the later timepoints in the experiments may be due to a depercolation transition in the hydrogels as degradation proceeds.32,80–83 The absence of this defined point in the PEG–MPP and PEG–DMMPP gels (Fig. 5A), particularly under standard reducing conditions, suggests that some process resulting in a reduction in degradation rate competes with this depercolation. This is likely due to the ring opening of the succinimide, which eliminates their capacity for the retro Michael addition and thiol exchange, as we have previously demonstrated.61
These GSH-sensitive maleimide–thiol adduct hydrogels have useful properties that recommend their use in multiple applications. First, the rapid reaction kinetics of the maleimide–thiol offers opportunities to employ these gels in vivo, with in situ formation of hydrogels at subcutaneous injection sites.16 The ability to control the degradation of these matrices, and corresponding delivery of drugs, should be useful in tailoring degradation in drug delivery applications. Devices such as disulfide-linked micelles for anticancer-related drug delivery utilize the disulfides to permit triggered release in reducing environments.51–55,58 Thus, employing maleimide–arylthiol adducts may increase blood stability and prolong the timescale of drug delivery.
As briefly mentioned in the introduction, heparins have been investigated for their antimetastatic properties via oral delivery,84,85 nanoparticle,86 amphiphilic polymers28 cationic polymer/heparin pairs21,87 and nanogel22 systems, and there is evidence that subcutaneously administered heparin88 or orally administered heparin89 can cross barriers and enter cells. Given that it has been shown that cancerous tissues contain heightened concentrations of glutathione compared with non-pathological tissues,77,90–92 injecting or implanting these PEG–LMWH hydrogels in the vicinity of tumor tissues should increase the rate of LMWH heparin release as in similar studies of nanogels crosslinked via disulfide bonds.21,22,26,41–44 In particular, Bae et al. described a system in which thiolated heparin was complexed with poly(ethylene glycol) in organic solvents, forming nanogels which released heparin in a reducing environment and inhibited the proliferation of mouse melanoma cells.22 It has also been shown that tumor tissues exhibit slightly lower pH values than normal tissues with ranges of experimental values varying from 6.8–7.2.93 This reduction in pH should not substantially increase the rate of ester hydrolysis in the PEG–LMWH hydrogels reported here, as the ester hydrolysis is relatively slow compared to the retro reaction kinetics; indeed, since the stability of the succinimide ring to ring-opening should be slightly increased at lower pH values,13,61,94,95 degradation by the retro reaction in the tumor microenvironment may be further favored over that by ester hydrolysis. Though not tested here, it is likely that the GSH-sensitive PEG–LMWH hydrogels may also exhibit similar antimetastatic properties if injected near the site of a tumor, and opportunities to produce various nanogel formulations of these hydrogels are underway.
Conclusions
Hydrogels containing GSH-sensitive succinimide–thioether crosslinks were successfully synthesized from maleimide-functionalized LMWH and thiolated, four-arm star PEGs. Four different PEGs – functionalized with 3-mercaptopropionic acid, mercaptoisobutyric acid, 4-mercaptophenylpropionic acid, or 2,2-dimethyl-3-(4-mercaptophenyl)propionic acid – were used to determine the impact of crosslink chemical identity on GSH-sensitive LMWH release and hydrogel degradation. 1H NMR characterization validated the absence of disulfide bond formation during crosslinking, and also confirmed that the GSH-sensitivity of the PEG–MPP and PEG–DMMPP hydrogels resulted from retro Michael-addition-mediated cleavage of the succinimide–thioether linkage. The kinetics of degradation and rate of LMWH release, under both standard and reducing solutions, were dictated by the identity of the mercaptoacid-functionalized PEG. Oscillatory rheology experiments confirmed the stability of the succinimide–thioether-containing hydrogels compared to disulfide-crosslinked hydrogels, with the former exhibiting 10-fold slower rates of degradation. These PEG–LMWH hydrogels establish a new application for widely used thiol–maleimide adducts in imparting glutathione-sensitivity to crosslinked hydrogels. These strategies should be widely applicable in tissue engineering platforms, micro- or nanogel technologies, and in the addition of glutathione-sensitive linkages in micelles, vesicles, and tethered drugs.Acknowledgements
This work was supported in part by the Nemours Foundation, the National Science Foundation (DGE-0221651) and the National Institutes of Health (5-P20-RR016472-10). The contents of the manuscript are the sole responsibility of the authors and do not necessarily reflect the official views of the National Institutes of Health. The authors would like to thank Dr S. Bai for assistance with water suppression 1H NMR techniques.References
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1880 CrossRef CAS.
- A. C. Jen, M. C. Wake and A. G. Mikos, Biotechnol. Bioeng., 1996, 50, 357–364 CrossRef CAS.
- A. H. Zisch, M. P. Lutolf and J. A. Hubbell, Cardiovasc. Pathol., 2003, 12, 295–310 CrossRef CAS.
- T. Miyata, T. Uragami and K. Nakamae, Adv. Drug Delivery Rev., 2002, 54, 79–98 CrossRef CAS.
- H. J. van der Linden, S. Herber, W. Olthuis and P. Bergveld, Analyst, 2003, 128, 325–331 RSC.
- J. Kim and K. Park, Bioseparation, 1998, 7, 177–184 CrossRef CAS.
- I. Y. Galaev, I. Galaev and B. Mattiasson, Smart Polymers for Bioseparation and Bioprocessing, Taylor & Francis, 2002 Search PubMed.
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS.
- N. A. Peppas, P. Bures, W. Leobandung and H. Ichikawa, Eur. J. Pharm. Biopharm., 2000, 50, 27–46 CrossRef CAS.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2002, 54, 3–12 CrossRef CAS.
- T. Nie, A. Baldwin, N. Yamaguchi and K. L. Kiick, J. Controlled Release, 2007, 122, 287–296 CrossRef CAS.
- T. Nie, R. E. Akins Jr and K. L. Kiick, Acta Biomater., 2009, 5, 865–875 CrossRef CAS.
- K. G. Robinson, T. Nie, A. D. Baldwin, E. C. Yang, K. L. Kiick and R. E. Akins, J. Biomed. Mater. Res., Part A, 2012, 100, 1356–1367 CrossRef.
- A. D. Baldwin, K. G. Robinson, J. L. Militar, C. D. Derby, K. L. Kiick and R. E. Akins Jr, J. Biomed. Mater. Res., Part A, 2012, 100A, 2106–2118 CrossRef CAS.
- J. Hirsh, T. E. Warkentin, S. G. Shaughnessy, S. S. Anand, J. L. Halperin, R. Raschke, C. Granger, E. M. Ohman and J. E. Dalen, Chest, 2001, 119, 64S–94S CrossRef CAS.
- R. Lever and C. P. Page, Nat. Rev. Drug Discovery, 2002, 1, 140–148 CrossRef CAS.
- P. W. Park, O. Reizes and M. Bernfield, J. Biol. Chem., 2000, 275, 29923–29926 CrossRef CAS.
- L. Borsig, Semin. Thromb. Hemostasis, 2007, 33, 540–546 CrossRef CAS.
- K. Bae, C. Moon, Y. Lee and T. Park, Pharm. Res., 2009, 26, 93–100 CrossRef CAS.
- K. H. Bae, H. Mok and T. G. Park, Biomaterials, 2008, 29, 3376–3383 CrossRef CAS.
- B. L. Seal and A. Panitch, Biomacromolecules, 2003, 4, 1572–1582 CrossRef CAS.
- S. Cai, Y. Liu, X. Z. Shu and G. D. Prestwich, Biomaterials, 2005, 26, 6054–6067 CrossRef CAS.
- N. Yamaguchi and K. L. Kiick, Biomacromolecules, 2005, 6, 1921–1930 CrossRef CAS.
- H. Lee, H. Mok, S. Lee, Y.-K. Oh and T. G. Park, J. Controlled Release, 2007, 119, 245–252 CrossRef CAS.
- L. W. Hosack, M. A. Firpo, J. A. Scott, G. D. Prestwich and R. A. Peattie, Biomaterials, 2008, 29, 2336–2347 CrossRef CAS.
- K. Ono, M. Ishihara, K. Ishikawa, Y. Ozeki, H. Deguchi, M. Sato, H. Hashimoto, Y. Saito, H. Yura, A. Kurita and T. Maehara, Br. J. Cancer, 2002, 86, 1803–1812 CrossRef CAS.
- S. J. Bryant and K. S. Anseth, J. Biomed. Mater. Res., 2002, 59, 63–72 CrossRef CAS.
- K. H. Bouhadir, K. Y. Lee, E. Alsberg, K. L. Damm, K. W. Anderson and D. J. Mooney, Biotechnol. Prog., 2001, 17, 945–950 CrossRef CAS.
- D. S. W. Benoit, A. R. Durney and K. S. Anseth, Tissue Eng., 2006, 12, 1663–1673 CrossRef CAS.
- K. S. Anseth, A. T. Metters, S. J. Bryant, P. J. Martens, J. H. Elisseeff and C. N. Bowman, J. Controlled Release, 2002, 78, 199–209 CrossRef CAS.
- J. R. Tauro and R. A. Gemeinhart, Bioconjugate Chem., 2005, 16, 1133–1139 CrossRef CAS.
- J. R. Tauro, B.-S. Lee, S. S. Lateef and R. A. Gemeinhart, Peptides, 2008, 29, 1965–1973 CrossRef CAS.
- H. Li, Y. Liu, X. Z. Shu, S. D. Gray and G. D. Prestwich, Biomacromolecules, 2004, 5, 895–902 CrossRef CAS.
- M. Ehrbar, S. C. Rizzi, R. G. Schoenmakers, B. S. Miguel, J. A. Hubbell, F. E. Weber and M. P. Lutolf, Biomacromolecules, 2007, 8, 3000–3007 CrossRef CAS.
- M. Konishi, Y. Tabata, M. Kariya, H. Hosseinkhani, A. Suzuki, K. Fukuhara, M. Mandai, K. Takakura and S. Fujii, J. Controlled Release, 2005, 103, 7–19 CrossRef CAS.
- M. Konishi, Y. Tabata, M. Kariya, A. Suzuki, M. Mandai, K. Nanbu, K. Takakura and S. Fujii, J. Controlled Release, 2003, 92, 301–313 CrossRef CAS.
- E. Ruel-Gariépy, M. Shive, A. Bichara, M. Berrada, D. Le Garrec, A. Chenite and J.-C. Leroux, Eur. J. Pharm. Biopharm., 2004, 57, 53–63 CrossRef.
- S. Seo, H. Han, K. Noh, T. Kim and S. Son, Clin. Exp. Metastasis, 2009, 26, 179–187 CrossRef CAS.
- E. Kohli, H.-Y. Han, A. D. Zeman and S. V. Vinogradov, J. Controlled Release, 2007, 121, 19–27 CrossRef CAS.
- H. Koo, G.-w. Jin, H. Kang, Y. Lee, H. Y. Nam, H.-s. Jang and J.-S. Park, Int. J. Pharm., 2009, 374, 58–65 CrossRef CAS.
- J. K. Oh, D. J. Siegwart, H.-i. Lee, G. Sherwood, L. Peteanu, J. O. Hollinger, K. Kataoka and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 5939–5945 CrossRef CAS.
- K. H. Sun, Y. S. Sohn and B. Jeong, Biomacromolecules, 2006, 7, 2871–2877 CrossRef CAS.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581–593 CrossRef CAS.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS.
- F. Meng, W. E. Hennink and Z. Zhong, Biomaterials, 2009, 30, 2180–2198 CrossRef CAS.
- G. Saito, J. A. Swanson and K.-D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199–215 CrossRef CAS.
- G. Wu, Y.-Z. Fang, S. Yang, J. R. Lupton and N. D. Turner, J. Nutr., 2004, 134, 489–492 CAS.
- D. P. Jones, J. L. Carlson, P. S. Samiec, P. Sternberg Jr, V. C. Mody Jr, R. L. Reed and L. A. S. Brown, Clin. Chim. Acta, 1998, 275, 175–184 CrossRef CAS.
- A. N. Koo, H. J. Lee, S. E. Kim, J. H. Chang, C. Park, C. Kim, J. H. Park and S. C. Lee, Chem. Commun., 2008, 6570–6572 RSC.
- J.-H. Ryu, R. Roy, J. Ventura and S. Thayumanavan, Langmuir, 2010, 26, 7086–7092 CrossRef CAS.
- H. Sun, B. Guo, X. Li, R. Cheng, F. Meng, H. Liu and Z. Zhong, Biomacromolecules, 2010, 11, 848–854 CrossRef CAS.
- L.-Y. Tang, Y.-C. Wang, Y. Li, J.-Z. Du and J. Wang, Bioconjugate Chem., 2009, 20, 1095–1099 CrossRef CAS.
- H.-Y. Wen, H.-Q. Dong, W.-j. Xie, Y.-Y. Li, K. Wang, G. M. Pauletti and D.-L. Shi, Chem. Commun., 2011, 47, 3550–3552 RSC.
- B. Khorsand Sourkohi, R. Schmidt and J. K. Oh, Macromol. Rapid Commun., 2011, 32, 1652–1657 CrossRef CAS.
- S. Cerritelli, D. Velluto and J. A. Hubbell, Biomacromolecules, 2007, 8, 1966–1972 CrossRef CAS.
- N. Abdullah Al, H. Lee, Y. S. Lee, K. D. Lee and S. Y. Park, Macromol. Biosci., 2011, 11, 1264–1271 CrossRef.
- J. Chen, S. Chen, X. Zhao, L. V. Kuznetsova, S. S. Wong and I. Ojima, J. Am. Chem. Soc., 2008, 130, 16778–16785 CrossRef CAS.
- I. Ojima, Acc. Chem. Res., 2007, 41, 108–119 CrossRef.
- A. D. Baldwin and K. L. Kiick, Bioconjugate Chem., 2011, 22, 1946–1953 CrossRef CAS.
- B.-Q. Shen, K. Xu, L. Liu, H. Raab, S. Bhakta, M. Kenrick, K. L. Parsons-Reponte, J. Tien, S.-F. Yu, E. Mai, D. Li, J. Tibbitts, J. Baudys, O. M. Saad, S. J. Scales, P. J. McDonald, P. E. Hass, C. Eigenbrot, T. Nguyen, W. A. Solis, R. N. Fuji, K. M. Flagella, D. Patel, S. D. Spencer, L. A. Khawli, A. Ebens, W. L. Wong, R. Vandlen, S. Kaur, M. X. Sliwkowski, R. H. Scheller, P. Polakis and J. R. Junutula, Nat. Biotechnol., 2012, 30, 184–189 CrossRef CAS.
- S. Bertini, A. Bisio, G. Torri, D. Bensi and M. Terbojevich, Biomacromolecules, 2004, 6, 168–173 CrossRef.
- A. Goessl, N. Tirelli and J. A. Hubbell, J. Biomater. Sci., Polym. Ed., 2004, 15, 895–904 CrossRef CAS.
- F. C. Macintosh, Biochem. J., 1941, 35, 776–782 CAS.
- M. P. Lutolf and J. A. Hubbell, Biomacromolecules, 2003, 4, 713–722 CrossRef CAS.
- T. V. DeCollo and W. J. Lees, J. Org. Chem., 2001, 66, 4244–4249 CrossRef CAS.
- J. P. Danehy and C. J. Noel, J. Am. Chem. Soc., 1960, 82, 2511–2515 CrossRef CAS.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487–531 CrossRef CAS.
- P. C. Hiemenz and T. Lodge, Polymer Chemistry, CRC Press, Boca Raton, Fla, 2007 Search PubMed.
- J. E. Elliott, M. Macdonald, J. Nie and C. N. Bowman, Polymer, 2004, 45, 1503–1510 CrossRef CAS.
- A. Metters and J. Hubbell, Biomacromolecules, 2004, 6, 290–301 CrossRef.
- G. A. Bagiyan, I. K. Koroleva, N. V. Soroka and A. V. Ufimtsev, Russ. Chem. Bull., 2003, 52, 1135–1141 CrossRef CAS.
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef CAS.
- S. P. Trimble, D. Marquardt and D. C. Anderson, Bioconjugate Chem., 1997, 8, 416–423 CrossRef CAS.
- B. Amsden, Macromolecules, 1998, 31, 8382–8395 CrossRef CAS.
- P. Kuppusamy, H. Li, G. Ilangovan, A. J. Cardounel, J. L. Zweier, K. Yamada, M. C. Krishna and J. B. Mitchell, Cancer Res., 2002, 62, 307–312 CAS.
- S. G. Tajc, B. S. Tolbert, R. Basavappa and B. L. Miller, J. Am. Chem. Soc., 2004, 126, 10508–10509 CrossRef CAS.
- D. E. Ponde, V. H. Deshpande, V. J. Bulbule and A. Sudalai, J. Org. Chem., 1998, 63, 1058–1063 CrossRef.
- A. T. Metters, K. S. Anseth and C. N. Bowman, Polymer, 2000, 41, 3993–4004 CrossRef CAS.
- A. T. Metters, C. N. Bowman and K. S. Anseth, J. Phys. Chem. B, 2000, 104, 7043–7049 CrossRef CAS.
- S. K. Reddy, K. S. Anseth and C. N. Bowman, Polymer, 2005, 46, 4212–4222 CrossRef CAS.
- A. E. Rydholm, S. K. Reddy, K. S. Anseth and C. N. Bowman, Biomacromolecules, 2006, 7, 2827–2836 CrossRef CAS.
- D. Y. Lee, K. Park, S. K. Kim, R.-W. Park, I. C. Kwon, S. Y. Kim and Y. Byun, Clin. Cancer Res., 2008, 14, 2841–2849 CrossRef CAS.
- K. Park, G. Y. Lee, Y.-S. Kim, M. Yu, R.-W. Park, I.-S. Kim, S. Y. Kim and Y. Byun, J. Controlled Release, 2006, 114, 300–306 CrossRef CAS.
- K. Lee, H. Lee, K. H. Bae and T. G. Park, Biomaterials, 2010, 31, 6530–6536 CrossRef CAS.
- D. Berry, D. M. Lynn, R. Sasisekharan and R. Langer, Chem. Biol., 2004, 11, 487–498 CrossRef CAS.
- L. Bara, E. Billaud, G. Gramond, A. Kher and M. Samama, Thromb. Res., 1985, 39, 631–636 CrossRef CAS.
- L. M. Hiebert, Clin. Lab., 2002, 48, 111 CAS.
- E. Ferruzzi, R. Franceschini, G. Cazzolato, C. Geroni, C. Fowst, U. Pastorino, N. Tradati, J. Tursi, R. Dittadi and M. Gion, Eur. J. Cancer, 2003, 39, 1019–1029 CrossRef CAS.
- D. Y.-K. Wong, Y.-L. Hsiao, C.-K. Poon, P.-C. Kwan, S.-Y. Chao, S.-T. Chou and C.-S. Yang, Cancer Lett., 1994, 81, 111–116 CrossRef CAS.
- T. C. Chang, M. J. W. Chang and S. Hsueh, Gynecol. Obstet. Invest., 1993, 36, 52–55 CrossRef CAS.
- L. E. Gerweck and K. Seetharaman, Cancer Res., 1996, 56, 1194–1198 CAS.
- M. N. Khan and A. A. Khan, J. Org. Chem., 1975, 40, 1793–1794 CrossRef CAS.
- S. Matsui and H. Aida, J. Chem. Soc., Perkin Trans. 2, 1978, 1277–1280 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2py20576a |
| This journal is © The Royal Society of Chemistry 2013 |