DOI:
10.1039/C2PY20559A
(Paper)
Polym. Chem., 2013,
4, 113-123
Orthogonal self-assembly of stimuli-responsive supramolecular polymers using one-step prepared heterotelechelic building blocks†
Received
24th July 2012
, Accepted 18th August 2012
First published on 21st August 2012
Abstract
The one-step preparation of heterodifunctional telechelic polymers containing 2,2′:6′,2′′-terpyridine (tpy) and 2-ureido-4[1H]-pyrimidinone (UPy) end-groups, as orthogonal supramolecular moieties, is reported. The utilization of an appropriately functionalized alkoxyamine, as an initiator for the nitroxide-mediated radical polymerization (NMP), directly constitutes the end-groups of the resultant polymers. The targeted alkoxyamines are based on the nitroxide structure of 2,2,5-trimethyl-3-(1-phenylethoxy)-4-phenyl-3-azahexane (TIPNO) and were obtained via stepwise functionalization of a heterodifunctional alkoxyamine skeleton. Controlled radical polymerization of styrenics using the alkoxyamine tpy–TIPNO–UPy, as an initiator, is demonstrated to generate well-defined telechelic polymers in one step. These telechelics represent promising building blocks for supramolecular architectures via self-assembly processes, yielding linear chain-extended polymers of high molar masses. Due to the orthogonality of the metal ion complexation and hydrogen bonding, the system can be addressed selectively by external stimuli. Besides for various applications, e.g. as self-healing materials, the strategy is highly attractive for tailoring the material's properties of supramolecular polymers, since the nature and the length of the polymer chain between the terminal supramolecular motifs can be controlled in a facile way.
Introduction
The preparation of well-defined adaptive macromolecular architectures is one of the main challenges in today's polymer science. Nowadays, the fast progress in living and controlled polymerization techniques provides straightforward access to advanced macromolecules, for instance, (multi)block copolymers, star-shaped (co)polymers as well as graft and comb-like (co)polymers.1–3 Although excellent control over the molecular structure is generally assured, these systems suffer from poor control over the structures of higher order and adaptivity when compared to natural macromolecules. In particular, the spontaneous assembly/disassembly of natural systems driven by the surrounding environmental conditions has to be named in this context. Nature utilizes a combination of tailor-made macromolecular structures and non-covalent interactions to create responsive systems that adapt to changes in the environment. Thereby, solvophobic and ionic interactions as well as hydrogen bonding and metal-to-ligand coordination serve as non-covalent, i.e. supramolecular, interactions. Inspired by these natural systems, polymer chemists have investigated the usage of non-covalent interactions for the design of supramolecular polymers featuring responsive behavior, i.e. their properties can be changed by varying the environmental conditions.4–14 If reversibility of the change in properties is guaranteed, the material can be switched between a self-assembled and a disassembled state by simply applying an external stimuli: e.g. a change in temperature, pH value, redox potential and concentration. Any change between the two different states will also be accomplished by changes in the polymer’s macroscopic properties, thus enabling the application in property changing, e.g. for self-healing materials.15 From the range of non-covalent interactions, in particular metal-to-ligand coordination and hydrogen bonding can offer both complementarity and directionality – essential features for the formation of complex supramolecular polymers, including block and graft copolymers.8,16–18
The combination of different non-covalent interactions in one supramacromolecular ensemble provides the opportunity to selectively address one of them via an external stimulus, if they are orthogonal to each other, thus expanding the range of responsiveness of the system. The concept of orthogonal supramolecular interactions in the field of polymer science has been evaluated in several review articles.18–23 As a key example, Weck and co-workers were able to synthesize supramolecular ABC triblock copolymers utilizing orthogonal binding units, namely a hydrogen bonding unit (Hamilton wedge) and pyridine, which can coordinate to a Pd pincer.24 Recently, the authors showed the versatility of the combination of metal–ligand interactions with hydrogen bonding units: a multiresponsive reversible polymer network was synthesized using the same binding units.25
Moreover, we have shown that the incorporation of a terpyridine (tpy)26–28 and a 2-ureido-4H-pyrimidone (UPy) moiety, as a metal-binding site and quadruple hydrogen bonding entity (Scheme 1), into polymers gives rise to the formation of chain-extended supramolecular polymers in the presence of divalent transition metal ions.29–32 In these materials, the polymeric building blocks were held together by bis-terpyridine complexes and UPy dimers in an alternating fashion. The initial synthetic strategies towards the heterotelechelic building blocks were based on a monofunctionalized initiator29,30 with a postmodification to attach the second moiety. More recently, we reported a heterodifunctional alkoxyamine initiator29,30 for the nitroxide-mediated radical polymerization (NMP) bearing already both end-groups of the resultant polymer. This latter approach provides a significantly higher versatility since the chain length and composition between the supramolecular binding sites can be varied without compromising the end-group fidelity and simple purification by precipitation, which is less straightforward for the postmodification procedure.
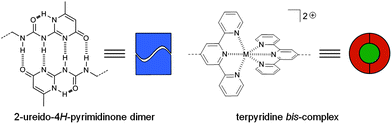 |
| | Scheme 1 Schematic representation of the chemical structure of the 2-ureido-4H-pyrimidinone quadruple hydrogen bonding array (left) and the terpyridine metal ion complexes (right). | |
In a continuation of this previous work,30 the ability of the heterodifunctional initiator tpy–TIPNO–UPy (TIPNO: 2,2,5-trimethyl-4-phenyl-3-azahexane-3-nitroxide), bearing both UPy and tpy supramolecular binding sites, in NMP is further explored in the current work. The orthogonality of the two non-covalent interactions within the supramolecular polymer is also evaluated by addressing the selective responsivity to external stimuli. Functionalized TIPNO derivatives were initially reported by Hawker and co-workers as universal initiators in NMP featuring a high end-group fidelity and a high tolerance towards a broad range of functional groups.33 Lohmeijer and Schubert expanded the scope of functional TIPNOs to the field of terpyridine chemistry by using tpy–TIPNO, as an initiator, for the synthesis of terpyridine-functionalized block copolymers via NMP.34
Results and discussion
Synthesis and characterization of the TIPNO-based initiator
The synthesis of the heterodifunctional initiator tpy–TIPNO–UPy (5) via stepwise functionalization of the nitroxide building block 1 has already been published elsewhere.30 As outlined in Scheme 2, the terpyridine-containing initiator 3, as a key building block, was obtained by a nucleophilic substitution reaction of 1 with 2,2′:6′,2′′-terpyridin-4′(1′H)-one35 and subsequent cleavage of the tetrahydropyran (THP) protecting group of the intermediate 2. The heterodifunctional alkoxyamine system 5 was then prepared in good yields from 3 by isocyanate-coupling with the UPy derivative 4. Due to the presence of two stereogenic centers within the central TIPNO unit, 5 was obtained as a mixture of two diastereomers. Consequently, two main sets of signals (in an almost 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) could be observed in the 1H and (partially) also the 13C NMR spectra.30 Full unambiguous assignment of the spectra was achieved by 2D NMR experiments. It should be noted that additional signals were observed in the 1H NMR spectra arising from the presence of the analogous meta-substituted isomer of 5 since commercial 4-chloromethylstyrene was used for the synthesis of 1, which consists of a mixture of para- and meta-isomers in a ca. 9:1 ratio. The presence of this second isomer is not expected to interfere or moderate the polymerization behavior of this alkoxyamine initiator. The MALDI-TOF mass spectrometric analysis of 5 showed the expected [M + H]+ adduct at m/z = 910.64; also the experimental isotopic pattern was in good agreement with the one for calculated m/z = 910.49.30
:1 ratio) could be observed in the 1H and (partially) also the 13C NMR spectra.30 Full unambiguous assignment of the spectra was achieved by 2D NMR experiments. It should be noted that additional signals were observed in the 1H NMR spectra arising from the presence of the analogous meta-substituted isomer of 5 since commercial 4-chloromethylstyrene was used for the synthesis of 1, which consists of a mixture of para- and meta-isomers in a ca. 9:1 ratio. The presence of this second isomer is not expected to interfere or moderate the polymerization behavior of this alkoxyamine initiator. The MALDI-TOF mass spectrometric analysis of 5 showed the expected [M + H]+ adduct at m/z = 910.64; also the experimental isotopic pattern was in good agreement with the one for calculated m/z = 910.49.30
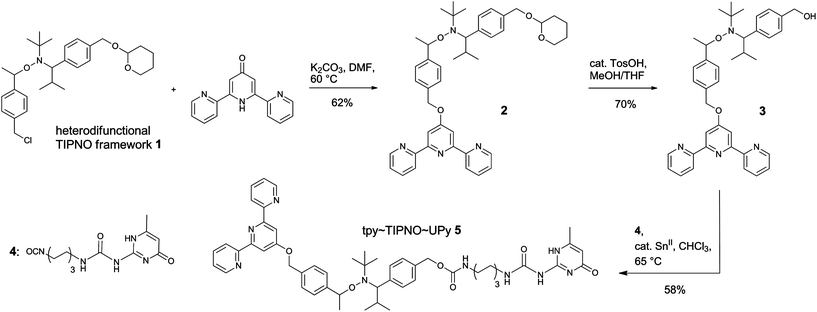 |
| | Scheme 2 Schematic representation of the synthesis of the hetero- and homodifunctional initiators via sequential functionalization of the heterodifunctional alkoxyamine 1. | |
Synthesis of the polymers.
For the successful utilization of alkoxyamine 5 as an initiator for NMP to obtain telechelic polymers some criteria have to be met: in particular, the polymerization has to be highly controlled leading to a high end-group fidelity, i.e. the functional groups at both chain ends must not be lost in the course of the polymerization due to extensive chain transfer or termination reactions. To evaluate the control over the polymerization, the NMP of styrene initiated by alkoxyamine 5 was investigated (Fig. 1a). Polymerizations with different monomer-to-initiator (M/I) ratios and different polymerization times were performed; the results are summarized in Table 1. With increasing polymerization time, the SEC traces of tpy–PSm–UPy (6c–g) were shifted to shorter elution times corresponding to an increase of the molar mass; all polymers revealed symmetrical curves with a minor tailing to lower molar masses, indicating good control over the polymerization (Fig. 1b). However, low polydispersity index (PDI) values do not provide unambiguous evidence for a controlled polymerization and, thus, a kinetic study was also performed. For this purpose, a stock solution with the telechelic initiator and styrene (solvent: anisole) was divided over different reaction vessels; after the removal of oxygen, the vessels were heated in an oil bath and the reactions were stopped after different polymerization times. The resulting tpy–PS–UPy (6c–g) Mn values (PDI < 1.2) were in good agreement with the theoretical values (Table 1). However, the first order kinetic plot of ln([M0]/[M]) as a function of time revealed a non-linear time dependency, indicating a variation in radical concentration during the polymerization (Fig. 2). The obtained data could be satisfactorily fitted to a time dependency of t2/3, which evidences the persistent radical effect (PRE):36,37 the radical initiation rate of another species, e.g. the monomer itself, is much lower than the dissociation rate of the alkoxyamine.36 This is pronounced, since the autoinitiation of the monomer was tremendously reduced due to the dilution with an inert solvent and provided the excess of persistent radicals within the non-steady-state polymerization. Under these conditions, the rate of self-termination of the propagating radical is much lower than the cross-reaction to the dormant species. The decomposition rate of the nitroxide is negligible, since this would hinder the certain build-up of the persistent radical with respect to the t2/3-dependency. Finally, the controlled character of the polymerization is evident from the linear increase of the number average molar mass plotted against the conversion (Fig. 3).
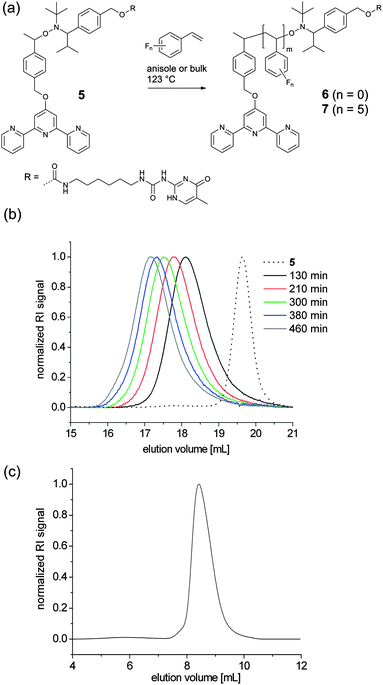 |
| | Fig. 1 Schematic representation of the polymerization of styrenic monomers using 5 as an initiator (a) and SEC traces of polystyrenes 6c–g with increasing polymerization time (b) and of poly(pentafluorostyrene) 7 (c). Eluent: N,N-dimethyl acetamide for polystyrene and chloroform for poly(pentafluorostyrene). | |
Table 1 Details for nitroxide-mediated polymerizations using alkoxyamine 5 as the initiator
| |
Time [min] |
Monomer |
M/I |
Conv. [%] (GC) |
M
n [g mol−1] theor.a |
M
n [g mol−1] (SEC)b,c |
PDI (SEC) |
|
M
n (theor.) represents the targeted Mn with respect to the conversion (GC).
After precipitation of the polymer into methanol.
A linear polystyrene calibration with DMA as eluent was used.
Bulk polymerization lacks a reference signal for GC measurements.
A linear polystyrene calibration with chloroform as eluent was used.
|
|
6a
|
400 |
St |
35 |
50 |
2700 |
6800 |
1.07 |
|
6b
|
360 |
St |
200 |
35 |
8000 |
11400 |
1.16 |
|
6c
|
130 |
St |
200 |
21 |
5700 |
5900 |
1.18 |
|
6d
|
210 |
St |
200 |
29 |
7400 |
7600 |
1.18 |
|
6e
|
300 |
St |
200 |
35 |
8700 |
9000 |
1.17 |
|
6f
|
380 |
St |
200 |
39 |
9800 |
10400 |
1.18 |
|
6g
|
460 |
St |
200 |
44 |
10800 |
11500 |
1.19 |
|
7
|
300 |
PFS |
200 |
—d |
—d |
10500e |
1.11 |
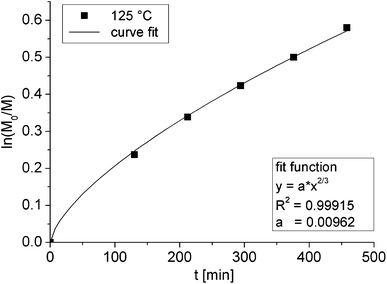 |
| | Fig. 2 Kinetic plot for the polymerization of styrene 6c–g as an initiator with the semilogarithmic monomer conversion plotted against the polymerization time. | |
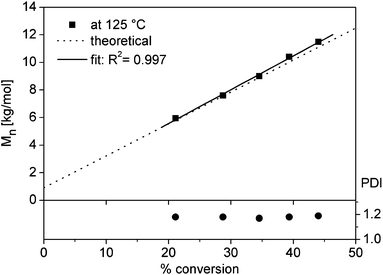 |
| | Fig. 3 Kinetic plot for the polymerization of styrene 6c–g with the molar mass plotted against the conversion showing a linear increase of the number average molar mass with the conversion. | |
Similarly, pentafluorostyrene (PFS) was utilized as a monomer in the NMP, yielding tpy–PPFS–UPy (7) (Fig. 1a, 1c and Table 1). As for the non-fluorinated styrene monomer good control over the polymerization, in bulk, was observed after 5 h with a Mn value of 10500 g mol−1 and a PDI value of 1.13. Since PPFS 7 was not soluble in DMA, a linear polystyrene calibration with chloroform as an eluent was used. Pentafluorostyrene was polymerized to show that the use of this initiator is not limited to styrene. An additional value relies on the potential of a click-like side-chain modification via the para-fluoro position.38–40
NMR spectroscopic analysis.
Polystyrenes 6 and 7 were investigated by 1H NMR spectroscopy in order to confirm the incorporation of the telechelic functional groups into the polymeric material. The 1H NMR spectra of 6 and 7 revealed signals for both end-groups where the characteristic terpyridine signals (7.2 to 8.8 ppm) as well as the hydrogen bonding signals for the UPy unit (10 to 13 ppm) were clearly present (Fig. S1and S2†). According to 1H NMR spectroscopy, the alkoxyamine initiator 5 as well as the polymers appeared exclusively as the dimeric species (5)2, (6)2, and (7)2, respectively, in non-polar solvents, i.e. dichloromethane and chloroform. Temperature-dependent NMR experiments (in d4-ortho-dichlorobenzene) revealed that these dimers were stable up to at least 75 °C. According to diffusion-ordered-spectroscopy (DOSY) measurements, a second signal at δ = 1.7 ppm, with almost the same diffusion behavior as for dichloromethane, was present in (5)2 (Fig. 4, left). Moreover, an exchange between one of the hydrogen bonding protons and this signal could be observed. Most likely this second signal can thus be ascribed to residual water: apparently, the proton-exchange process hindered the removal of water from the product by drying. In addition, the 2D DOSY spectrum confirmed that all signals (apart from the solvents) belonged to the same compound due to the same range of the relative diffusion coefficient, thereby proving that both UPy and tpy are attached to the TIPNO.
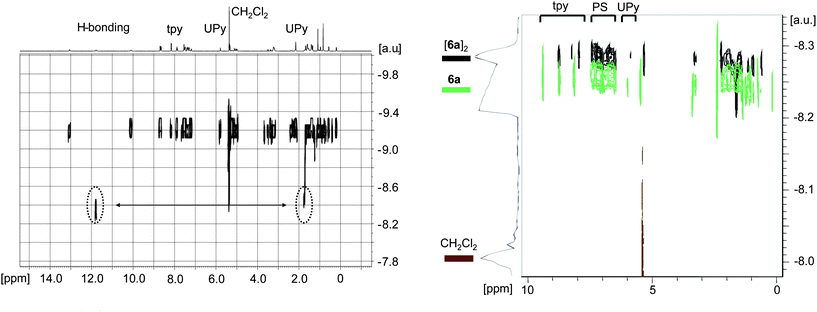 |
| | Fig. 4 Left: 2D DOSY spectrum of (5)2 revealing the exchange of water with a hydrogen bonding proton of the UPy moiety (400 MHz, CD2Cl2, 298 K). Right: comparison of the 2D DOSY spectra for 6a (green, CD2Cl2 and TFA) and (6a)2 (black, CD2Cl2) demonstrating the same significant change of the diffusion coefficients for signals of tpy, UPy and polystyrene (for both spectra: 400 MHz, 298 K). | |
In this vein, the 2D DOSY spectrum of the resultant telechelic polystyrene (62) also shows the same relative diffusion coefficient for the signals of both end-groups and the polymer backbone proving that UPy and tpy are attached to the polystyrene (Fig. 4, right). The addition of trifluoroacetic acid (TFA), as a strong competitor for hydrogen bonding, led to significant changes in the NMR spectrum.
As expected, the quantitative disappearance of the signals arising from hydrogen bonding as well as a high-field shift of the terpyridine signals, due to protonation, could be observed in the 1H NMR spectrum (Fig. S3†). It was concluded that predominantly the monomeric form of 6 was present under these conditions. Subsequent DOSY measurements revealed a significant shift for the diffusion coefficients of all signals of the tpy and UPy moieties as well as the polystyrene with respect to the diffusion coefficient in dichloromethane (Fig. 4, right). Thus, the change in molar mass related to the monomeric and dimeric forms of 6 can be probed by the relative diffusion coefficients. It can be concluded within the sensitivity limits of NMR spectroscopy that both end-groups are attached to the polymer chain and the presence of auto-polymerized non-functionalized polystyrene or monofunctional tpy–PS could be excluded, as they would reveal a different diffusion coefficient range for the terpyridine and polystyrene signals.
Mass spectrometric analysis.
The polymers were also investigated by mass spectrometry. The MALDI-TOF MS of 6 revealed three distributions that could all be correlated to polystyrenes (i.e. in all cases m/z-differences of 104 amu between two signals, Fig. 5). No distribution could be directly related to the heterodifunctional telechelic polymer 6, thus fragmentation by abstraction of the nitroxide residue has to be assumed (the proposed fragmentation products are shown in Fig. 5). Nitroxide-functionalized polymers are well-known to be prone to fragmentation during the MALDI process;41 however, the characteristic fragmentation due to loss of the tert-butyl group of the TIPNO moiety could not be observed. The observed fragmentations for series B and C could also be characteristic products of termination reactions during the polymerization. On this note, it is not fully clear if these fragments were formed during the MALDI process or during the polymerization—the latter possibility can only be neglected within the limits of 1H NMR spectroscopy and SEC. The proposed mechanism for the chain-end degradation for 6a during the MALDI process according to Schulte et al. is shown in Fig. 6.42 The distribution at higher molar mass up to m/z = 10000 could be assigned to the recombination product of two polystyrene chains (B). Taking the slight tailing of the SEC traces of 6a to higher molar masses into account, a small degree of recombination during the NMP appears to be plausible, which, however, will not strongly interfere with the formation of supramolecular polymer.
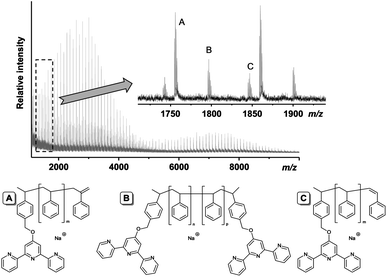 |
| | Fig. 5 MALDI-TOF mass spectrum of polystyrene 6a. The inset shows the three observed modes of fragmentation (A–C). The proposed corresponding structures are shown on the bottom. | |
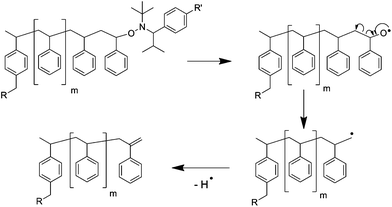 |
| | Fig. 6 Schematic representation of the proposed mechanism for the chain-end degradation of the polymer (formation of series C). | |
Chain extension via NMP.
The control and end-group fidelity of the telechelic polymers was further confirmed by using them as macroinitiators for a chain-extension experiment. More specifically, the heterodifunctional polystyrene 6c was applied as a macroinitiator for chain extension with styrene. The polymerization was conducted in anisole, as a solvent, in order to allow a correlation to the NMP performance of the heterodifunctional alkoxyamine initiator 5. As shown in Fig. 7, a complete shift of the Mn value from 6000 g mol−1 to 14600 g mol−1 was observed while retaining the narrow polydispersity index (PDI < 1.2), demonstrating the controlled character of the NMP initiated by 6c and, thus, proving that almost all of the polymer chains of 6c still bear the UPy-functionalized nitroxide as an end-group. Thus, the telechelic polymers represent suitable building blocks for the preparation of block copolymers containing two orthogonal supramolecular end-groups via CRP.
Chain extension via metal ion complexation
The combination of hydrogen bonding and metal-to-ligand coordination was applied by Hofmeier et al. for the preparation of supramolecular chain-extended poly(ε-caprolactone) of high molar masses.20,29 In particular, the divalent cations of the first row transition metals are suited for this purpose, since the formation of octahedral bis-complexes with terpyridine ligands is highly preferred.43 However, the low kinetic stability of some bis-terpyridine complexes (according to the Irving–Williams series) on the one hand and the paramagnetic nature of some metal ions on the other hand limit the range of potential candidates with respect to the preparation and characterization of these materials. Chiper et al. showed that metallo-supramolecular polymers based on [Ni(tpy)2]2+ linkages are stable enough to be investigated by size exclusion chromatography.44,45 However, NMR spectroscopy cannot be applied for such polymers due to the paramagnetic nature of the NiII ions. In this respect, FeII or ZnII ions might be used; however, in particular the latter ions lead to rather weak metal complexes. In the present study, these three metal ions were utilized for the preparation of supramolecular chain-extended polymers {[(6)M(6)](PF6)2}n (Scheme 3, M = ZnII, FeII or NiII). The metallo-polymerization reactions were performed in a chloroform–methanol mixture at a 2:1 stoichiometry of macroligand 6d or 6e and the corresponding metal MII ions. In the case of FeII ions, an equimolar amount of stannous chloride was added to the reaction mixture to prevent oxidation of FeII to the paramagnetic FeIII. Subsequent to the anion exchange with PF6−, the polymers {[(6e)M(6e)](PF6)2}n were characterized by SEC or 1H NMR spectroscopy (when applicable). The expected significant increase of the molar mass due to the metallo-polymerization of 6 and 7 could not be observed by SEC since the applied polar and protic conditions suppress the formation of UPy dimers. Consequently, only dimers of the type [Ni(6)2](PF6)2 could be detected – as pointed out above, the kinetic stability of NiIIbis-terpyridine complexes allows the SEC measurements, while for all other metal ions investigated, dissociation occurs.44,45 The SEC trace of [Ni(6d)2](PF6)2 was shifted to a higher molar mass compared to the one obtained for the precursor polymer 6 under the same conditions (Fig. S4†; eluent: DMF containing 0.005 M NH4PF6). A shoulder at higher molar masses was observed that cannot be explained but has also been reported for other NiII-based systems in the literature.45 Thus, this shoulder is most likely not caused by any impurity, but rather originates from different solvation, interactions with the column or higher aggregates. The SEC traces recorded with a photodiode array (PDA) detector confirmed the complete shift to lower retention times, i.e. higher molar masses (Fig. 8). The corresponding UV-vis absorption spectrum proved the formation of the NiIIbis-terpyridine complex by the appearance of the characteristic ligand-centered (LC) bands at 315 and 329 nm.45
2}n and {[(7)M(7)](PF6)2}n by metal-to-ligand coordination (SnCl2 was added to the reaction mixture in order to prevent oxidation of the FeII ion).](/image/article/2013/PY/c2py20559a/c2py20559a-s3.gif) |
| | Scheme 3 Schematic representation of the synthesis of supramolecular chain-extended polymers {[(6)M(6)](PF6)2}n and {[(7)M(7)](PF6)2}n by metal-to-ligand coordination (SnCl2 was added to the reaction mixture in order to prevent oxidation of the FeII ion). | |
The formation of the MIIbis-terpyridine complexes of 6b and 7 was also monitored by UV-vis titration experiments with FeII ions (Fig. 9). Since methanol was used as a solvent, the formation of hydrogen bonds between the UPy moieties and, thereby, of the chain-extended polymers does not occur. The evolution of the LC and the metal-to-ligand charge-transfer (MLCT) absorptions at ca. 320 and ca. 560 nm, respectively, were plotted as a function of added FeII ions. The point-of-equivalence indicates the consumption of free tpy sites by complexation at a 2:1 ratio of macroligand and FeII ions (i.e. formation of [Fe(tpy)2]2+ complexes). From this equivalence point the number average molar mass of the polymers can also be calculated (Table 2). For both 6b and 7, similar Mn values as determined by 1H NMR spectroscopy were obtained. Apparently, the SEC measurements underestimated the Mn value for 7 due to the unavailability of a suitable calibration.
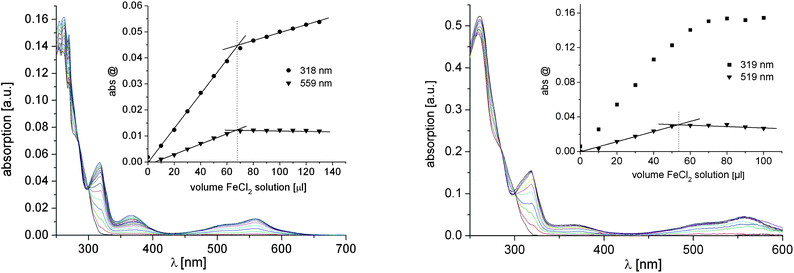 |
| | Fig. 9 UV-vis titration curves for the complexation of 6b (left) and 7 (right) with FeII ions (solvent: methanol). The insets show the evolution of the LC and MLCT absorption bands as a function of added FeII ions (the left figure was created from data published in ref. 30). | |
| Polymer |
M
n [g mol−1] |
| SECa |
NMRb |
UV-visc |
|
A linear polystyrene calibration was used.
Determined by comparison of the integrals of the tpy and polystyrene signals.
Calculated from the point-of-equivalence in the UV-vis titration experiments.
|
|
6b
|
11400 |
13700 |
9000 |
|
7
|
10500 |
21500 |
18500 |
Due to the diamagnetic character of the FeII and ZnII ions, the respective metallopolymers {[(6e)M(6e)](PF6)2}n could also be investigated by NMR spectroscopy. In the 1H NMR spectra, the signals of the terpyridine protons were shifted upon complexation, whereas the signals assigned to hydrogen bonding remained unchanged (Fig. 10 and S5†). Thus, the formation of a supramolecular polymer based on two different non-covalent interactions in CD2Cl2 was demonstrated. The 2D DOSY measurement further confirmed the presence of the two types of supramolecular bonds within the chain-extended polymer: the same relative diffusion coefficients were observed for all tpy, polystyrene and UPy signals of {[(6e)Fe(6e)](PF6)2}n (Fig. 11, left).
2}n (bottom). For both spectra: 400 MHz, CD2Cl2, 298 K.](/image/article/2013/PY/c2py20559a/c2py20559a-f10.gif) |
| | Fig. 10 Zoom into the low-field region of the 1H NMR spectra of (6e)2 (top) and {[(6e)Fe(6e)](PF6)2}n (bottom). For both spectra: 400 MHz, CD2Cl2, 298 K. | |
2}n (400 MHz, CD2Cl2, 298 K) demonstrating the same diffusion coefficients for signals of tpy complex, UPy and polystyrene. Right: 2D DOSY spectrum of [(6e)Fe(6e)](PF6)2 (400 MHz, CD2Cl2, 298 K) after addition of trifluoroacetic acid demonstrates the cleavage of the hydrogen bonding units (diffusion coefficient of dichloromethane was taken as a reference).](/image/article/2013/PY/c2py20559a/c2py20559a-f11.gif) |
| | Fig. 11 Left: 2D DOSY spectrum of {[(6e)Fe(6e)](PF6)2}n (400 MHz, CD2Cl2, 298 K) demonstrating the same diffusion coefficients for signals of tpy complex, UPy and polystyrene. Right: 2D DOSY spectrum of [(6e)Fe(6e)](PF6)2 (400 MHz, CD2Cl2, 298 K) after addition of trifluoroacetic acid demonstrates the cleavage of the hydrogen bonding units (diffusion coefficient of dichloromethane was taken as a reference). | |
In order to study the orthogonal responsiveness of the supramolecular entities, TFA was added to the chain-extended FeII polymer. As expected, the quadruple hydrogen bonding of the UPy-dimer was disrupted without affecting the more pH robust metal–ligand binding (Fig. S6†). The disappearance of the signals arising from hydrogen bonding in the 1H NMR spectrum in concert with a significant change in the diffusion behavior confirmed the selective responsiveness (Fig. 11). The comparison of the 2D DOSYs of the polymer {[(6e)Fe(6e)](PF6)2}n and the dimer [Fe(6e)2](PF6)2 clearly shows the influence of the pH value on the molecular structure.
Conclusion
The one-step preparation of heterotelechelic polymers via NMP using an alkoxyamine as inhibitor bearing the desired heterotelechelic end-groups of the resultant polymer was accomplished. The polymerization of styrene was proven to be a controlled process, while the bulky end-groups, namely tpy and UPy, did not disturb the performance of the initiator. Therefore, the polymeric spacer between the supramolecular binding sites can be easily varied in length by NMP. This approach has a wider scope by means of construction flexibility compared to a postmodification approach for telechelic polymers, as the chain length and composition between the supramolecular binding sites can be varied in less reaction steps and purification can be simply done by precipitation. Therefore, the strategy of functional initiators represents a powerful method for investigating the influence of the polymer spacer between functional groups within supramolecular architectures.
The self-assembly of the prepared building blocks towards linear supramolecular polymers with alternating metal ion complex and hydrogen bonding linkage was investigated in apolar solvents with complexes of zinc(II), iron(II) and nickel(II) metal ions. The selective responsiveness of the system against pH value as an external stimulus was proven where the hydrogen bonding linkage could be switched off while the metal ion complex remained intact.
The presented strategy of functionalized initiators provides a promising strategy for the preparation of various tailor-made supramolecular architectures using polymeric building blocks, with respect to future progress in the field of stimuli-responsive and self-healing materials.
Experimental section
Methods and instrumentation
All chemicals were received from Aldrich, Fluka and Acros; solvents were purchased from Biosolve. Unless otherwise stated, the chemicals and solvents were used without further purifications. DMF was dried over molecular sieves (pore size of 4 Å). THF and toluene were dried using a PureSolv-EN™ solvent purification system. Column chromatography was carried out on silica gel 60 and standardized aluminium oxide 90 (Merck). Styrene was freshly purified prior to use by filtration over neutral aluminium oxide 90 (Merck). 1D (1H, 13C) and 2D (1H–1H-COSY) nuclear magnetic resonance (NMR) spectra were recorded on a Varian Mercury 400 MHz spectrometer at 298 K, 348 K or 368 K. Chemical shifts are reported in parts per million (ppm) downfield from tetramethylsilane (TMS) and were calibrated to the residual solvent peaks. 2D DOSY experiments were conducted on a Bruker 400 MHz spectrometer at 298 K. Deuterated solvents for NMR spectroscopy were obtained from Cambridge Isotope Laboratories. Two different set-ups for size-exclusion chromatography (SEC) were used: a Shimadzu system equipped with a SCL-10A system controller, a LC-10AD pump, a RID-10A refractive index (RI) detector, an UV-vis absorption detector (250 and 290 nm) and a PSS SDV column utilizing (i) N,N-dimethyl acetamide with 2.1 g L−1 LiCl or (ii) chloroform/iso-propanol/triethylamine (94/2/4), as eluent. Linear polystyrene was used as the calibration standard. Additionally, SEC was measured on a Waters SEC system consisting of an isocratic pump, solvent degasser, column oven, photodiode array (PDA) detector, RI detector and a Styragel HT-4 SEC column with a precolumn installed. The eluent was DMF containing 0.005 M NH4PF6 at a flow speed of 0.5 mL min−1. The column temperature was 323 K. Gas chromatographic (GC) measurements were performed on an Interscience Trace GC with a Trace Column RTX-5 connected to a PAL autosampler. UV-vis absorption spectra were recorded in diluted chloroform solutions on a Perkin-Elmer Lambda-45 UV-vis spectrophotometer at room temperature (1 cm cuvettes). Elemental analyses were carried out on a EuroVector EuroEA300 elemental analyzer. GC mass spectrometry was conducted on a Shimadzu GC17A connected to a mass spectrometer. Infrared spectra were recorded on a Bruker IFS55 FT-IR spectrometer. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) was performed on a Voyager-DE™ PRO Biospectrometry™ Workstation (Applied Biosystems) time-of-flight mass spectrometer using linear and reflector mode for operation. Dithranol or 2-[(2E)-3-(4-tert-butylphenyl)-2-methylprop-2-enylidene]malononitrile (DCTB) were used as matrices and NaI as an additive. The spectra were recorded in the positive ion mode and ionization was performed with a 337 nm pulsed nitrogen laser. The MALDI-TOF mass spectra of polymers were also measured on a Bruker Ultraflex™ III TOF/TOF spectrometer.
The following compounds were prepared according to literature procedures: 2,2′:6′,2′′-terpyridin-4′(1′H)-one,46N-(tert-butyl)-O-(1-(4-(chloromethyl)phenyl)ethyl)-N-(2-methyl-1-(4-(((tetrahydro-2H-pyran-2-yl)oxy)methyl)phenyl)-propyl)hy-droxylamine (1), O-(1-(4-((2,2′:6′,2′′-terpyridin-4′-yloxy)-methyl)phenyl)ethyl)-N-(tert-butyl)-N-(2-methyl-1-(4-(((tetra-hydro-2H-pyran-2-yl)-oxy)methyl)phenyl)propyl)-hydroxylamine (2),30 (4-(1-((1-(4-((2,2′:6′,2′′-terpyridin-4′-yloxy)methyl)-phenyl)ethoxy)(tert-butyl)amino)-2-methylpropyl)phenyl)metha-nol (3),30 1-(6-isocyanato-hexyl)-3-(6-methyl-4-oxo-1,4-dihydro-pyrimidin-2-yl)urea (4)30 and 4-(1-((1-(4-((2,2′:6′,2′′-terpyridin-4′-yloxy)methyl)phenyl)ethoxy)(tert-butyl)amino)-2-methyl-propyl)benzyl (6-(3-(6-methyl-4-oxo-1,4-dihydropyrimidin-2-yl)ureido)hexyl)carbamate (tpy–TIPNO–UPy, 5).30
Styrene monomers were freshly purified by filtration over Al2O3 prior to use in order to remove the inhibitor. The freshly purified styrene was added to a clear anisole solution of the TIPNO-based initiator in a polymerization vessel. After applying three freeze–pump–thaw-cycles to remove the oxygen, the vessel was purged with argon and immersed in an oil bath at 123 °C for a certain period of time. To remove residual monomer the reaction mixture was precipitated from CHCl3 into cold methanol. The monomer conversion was determined by GC measurements (before precipitation), the molar masses (i.e. Mn and Mw) and the polydispersity index (PDI) values were determined by SEC, whereas 1H NMR spectroscopy was applied for the determination of the end-group functionality (by integration and comparison of the corresponding end-group signals) and Mn values (by integration of the polymer backbone to the terpyridine signals).
tpy–PS50–UPy (6c).
1H NMR (400 MHz, CD2Cl2): δ = 0.05–2.71 (set of multiplets), 3.11–3.38 (m, 4H), 3.85–4.37 (m, 1H), 4.97–5.22 (m, 2H), 5.26–5.34 (m, 3H), 5.83 (s, 1H), 6.25–7.54 (m, 200H), 7.91 (dt, J3 = 7.8 Hz, J4 = 1.6 Hz, 2H), 8.15–8.24 (m, 2H), 8.64–8.76 (m, 4H), 10.14 (s, 1H), 11.88 (s, 1H), 13.12 (s, 1H) ppm. SEC (CHCl3, RI): Mn = 6800 g mol−1, PDI = 1.07.
tpy–PPFS100–UPy (7).
1H NMR (400 MHz, CD2Cl2): δ = 0.05–2.96 (set of multiplets), 3.13–3.32 (m, 4H), 4.7–4.9 (m, 1H), 5.01–5.16 (m, 2H), 5.18–5.32 (m, 3H), 5.83 (s, 1H), 6.97–7.47 (m, 10H), 7.91 (dt, J3 = 7.8 Hz, J4 = 1.6 Hz, 2H), 8.11–8.18 (m, 2H), 8.63–8.76 (m, 4H), 10.14 (s, 1H), 11.86 (s, 1H), 13.13 (s, 1H) ppm. SEC (CHCl3, RI): Mn = 10500 g mol−1, PDI = 1.13.
General procedure for the chain-extension polymerization
The macroinitiator tpy–PS50–UPy (6, 70 mg, 0.012 mmol) was dissolved in anisole (200 μL) and freshly purified styrene. After following the general polymerization procedure (vide supra) the reaction was stopped after 6 h.
tpy–PS212–UPy (8).
The chain-extension polymerization was conducted with a monomer-to-initiator ratio of M/I = 145:1. 1H NMR (400 MHz, CD2Cl2): δ = 0.05–2.71 (set of multiplets), 3.11–3.38 (m, 4H), 3.85–4.37 (m, 1H), 4.97–5.22 (m, 2H), 5.26–5.34 (m, 3H), 5.83 (s, 1H), 6.25–7.54 (m, 850H), 7.91 (dt, J3 = 7.8 Hz, J4 = 1.6 Hz, 2H), 8.15–8.24 (m, 2H), 8.64–8.76 (m, 4H), 10.14 (s, 1H), 11.88 (s, 1H), 13.12 (s, 1H) ppm. SEC (RI): Mn = 14600 g mol−1, PDI = 1.18.
UV-vis titration experiments
The macroligand (6b or 7, 0.852 mg) was dissolved in chloroform (20 mL) and FeCl2 (0.913 mg, 0.0072 mmol) was dissolved in methanol (10 mL). The methanol solution was subsequently added to the polymer solution in steps of 10 μL while the formation of FeIIbis-complex 16 was monitored by UV-vis spectroscopy after each addition. The point-of-equivalence and the Mn value of the macroligand were obtained by plotting the UV-vis absorption intensity at the LC and the MLCT as a function of added FeII ions.
Metallo-polymerization of the macroligands
To a solution of macroligand 6 or 7 (0.003 mmol) in chloroform (200 μL) was added a solution of MCl2 (MII = NiII, FeII or ZnII; 0.002 mmol) in methanol (100 μL). For FeII complexation SnCl2 was used stoichiometric to avoid partial oxidation. The resultant clear solution was stirred under an argon atmosphere at 40 °C for 24 h. The chain-extended polymer was precipitated by adding an excess of NH4PF6 (ca. 10 eq.).
{[(6d)Ni(6d)](PF6)2}n.
The material was analyzed without further purification by SEC coupled with a PDA detector to prove the formation of the NiIIbis-terpyridine complex; SEC (PDA/RI).
{[(6e)Fe(6e)](PF6)2}n.
1H NMR (400 MHz, CD2Cl2): δ = 0.39–2.58 (set of multiplets), 3.13–3.37 (m, 4H), 4.05–4.25 (m, 1H), 4.97–5.29 (m, 3H), 5.59–5.66 (m, 2H), 5.79–5.92 (s, 1H), 6.28–7.50 (m, 500 H), 7.51–7.67 (m, 2H), 7.80–7.93 (m, 2H), 8.29–8.40 (m, 2H), 8.41–8.50 (m, 2H), 10.16 (s, 1H), 11.88 (s, 1H), 13.12 (s, 1H) ppm.
{[(6e)Zn(6e)](PF6)2}n.
1H NMR (400 MHz, CD2Cl2): δ = 0.38–2.62 (set of multiplets), 3.12–3.38 (m, 4H), 4.04–4.31 (m, 1H), 4.99–5.21 (m, 3H), 5.52–5.62 (m, 2H), 5.82 (s, 1H), 6.28–7.63 (m, 760H), 7.73–7.8 (m, 2H), 8.06–8.25 (m, 4H), 8.38–8.48 (m, 2H), 10.13 (s, 1H), 11.86 (s, 1H), 13.11 (s, 1H) ppm.
Acknowledgements
The authors kindly acknowledge financial support from the Dutch Polymer Institute (DPI, technology area HTE), the Thüringer Ministerium für Bildung, Wissenschaft und Kultur (Grant no. B514-09049: PhotoMic) and the Deutsche Forschungsgemeinschaft (DFG, SPP 1568). Moreover, the authors thank Grit Festag and Anja Baumgaertel for their help with SEC and MALDI-TOF MS measurements, respectively.
Notes and references
- N. Hadjichristidis, M. Pitsikalis, S. Pispas and H. Iatrou, Chem. Rev., 2001, 101, 3747–3792 CrossRef CAS.
- K. Matyjaszewski, Prog. Polym. Sci., 2005, 30, 858–875 CrossRef CAS.
- C. J. Hawker, Adv. Polym. Sci., 1999, 147, 113–160 CrossRef CAS.
-
A. Winter, M. D. Hager and U. S. Schubert, Supramolecular Polymers, in Polymer Science: A Comprehensive Reference, ed. M. Möller and K. Matyjaszewski, Elsevier Ltd., Oxford, 2012, vol. 5 Search PubMed.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS.
- C. K. Ober and G. Wegner, Adv. Mater., 1997, 9, 17–31 CrossRef CAS.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS.
- W. H. Binder and R. Zirbs, Adv. Polym. Sci., 2007, 207, 7–78 CrossRef.
- U. S. Schubert and C. Eschbaumer, Angew. Chem., Int. Ed., 2002, 41, 2892–2926 CrossRef CAS.
- D. G. Kurth and M. Higuchi, Soft Matter, 2006, 2, 915–927 RSC.
- J. D. Fox, Macromolecules, 2009, 42, 6823–6835 CrossRef CAS.
- J.-F. Gohy, B. G. G. Lohmeijer and U. S. Schubert, Chem.–Eur. J., 2003, 9, 3472–3479 CrossRef CAS.
- J.-F. Gohy, B. G. G. Lohmeijer, A. Alexeev, X.-S. Wang, I. Manners, M. A. Winnik and U. S. Schubert, Chem.–Eur. J., 2004, 10, 4315–4323 CrossRef CAS.
- C. D. Eisenbach and U. S. Schubert, Macromolecules, 1993, 26, 7372–7374 CrossRef CAS.
- M. D. Hager, P. Greil, C. Leyens, S. van der Zwaag and U. S. Schubert, Adv. Mater., 2010, 22, 5424–5430 CrossRef CAS.
- R. Shunmugam, G. J. Gabriel, K. A. Aamer and G. N. Tew, Macromol. Rapid Commun., 2010, 31, 784–793 CrossRef CAS.
- B. Happ, A. Winter, M. D. Hager and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 2222–2255 RSC.
- R. Hoogenboom, D. Fournier and U. S. Schubert, Chem. Commun., 2008, 155–162 RSC.
- W. Gerhardt, M. Črne and M. Weck, Chem.–Eur. J., 2004, 10, 6212–6221 CrossRef CAS.
- H. Hofmeier and U. S. Schubert, Chem. Commun., 2005, 2423–2432 RSC.
- J. M. Pollino and M. Weck, Chem. Soc. Rev., 2005, 34, 193–207 RSC.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS.
- S.-K. Yang, A. V. Ambade and M. Weck, Chem. Soc. Rev., 2012, 40, 129–137 RSC.
- A. V. Ambade, S.-K. Yang and M. Weck, Angew. Chem., Int. Ed., 2009, 48, 2894–2898 CrossRef CAS.
- K. P. Nair, V. Breedveld and M. Weck, Macromolecules, 2011, 44, 3346–3357 CrossRef CAS.
- U. S. Schubert, C. Eschbaumer, P. Andres, H. Hofmeier, C. H. Weidl, E. Herdtweck, E. Dulkeith, A. Morteani, N. E. Hecker and J. Feldmann, Synth. Met., 2001, 121, 1249–1252 CrossRef CAS.
- U. S. Schubert, O. Hien and C. Eschbaumer, Macromol. Rapid Commun., 2000, 21, 1156–1161 CrossRef CAS.
- U. S. Schubert, C. Eschbaumer and G. Hochwimmer, Synthesis, 1999, 5, 779–782 CrossRef.
- H. Hofmeier, R. Hoogenboom, M. E. L. Wouters and U. S. Schubert, J. Am. Chem. Soc., 2005, 127, 2913–2921 CrossRef CAS.
- U. Mansfeld, M. D. Hager, R. Hoogenboom, C. Ott, A. Winter and U. S. Schubert, Chem. Commun., 2009, 3386–3388 RSC.
- S. Schmatloch, A. M. J. van den Berg, A. S. Alexeev, H. Hofmeier and U. S. Schubert, Macromolecules, 2003, 36, 9943–9949 CrossRef CAS.
- H. Hofmeier, S. Schmatloch, D. Wouters and U. S. Schubert, Macromol. Chem. Phys., 2003, 204, 2197–2203 CrossRef CAS.
- D. Benoit, V. Chaplinski, R. Braslau and C. J. Hawker, J. Am. Chem. Soc., 2001, 121, 3904–3920 CrossRef.
- B. G. G. Lohmeijer and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4016–4027 CrossRef CAS.
- M. Heller and U. S. Schubert, J. Org. Chem., 2002, 67, 8269–8272 CrossRef CAS.
- H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS.
- T. Fukuda, T. Terauchi, A. Goto, K. Ohno, Y. Tsuji, T. Miyamoto, S. Kobatake and B. Yamada, Macromolecules, 1996, 29, 6393–6398 CrossRef CAS.
- C. Ott, R. Hoogenboom and U. S. Schubert, Chem. Commun., 2008, 3516–3518 RSC.
- C. R. Becer, K. Babiuch, D. Pilz, S. Hornig, T. Heinze, M. Gottschaldt and U. S. Schubert, Macromolecules, 2009, 42, 2387–2394 CrossRef CAS.
- C. Ott, C. Ulbricht, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2012, 33, 556–561 CrossRef CAS.
- W. Dempwolf, S. Flakus and G. Schmidt-Naake, Macromol. Symp., 2007, 259, 416–420 CrossRef CAS.
- T. Schulte, K. O. Siegenthaler, H. Luftmann, M. Letzel and A. Studer, Macromolecules, 2005, 38, 6833–6840 CrossRef CAS.
-
U. S. Schubert, A. Winter and G. R. Newkome, Terpyridine-based Materials, Wiley-VCH, Weinheim, 2011 Search PubMed.
- M. Chiper, M. A. R. Meier, D. Wouters, S. Hoeppener, C.-A. Fustin, J.-F. Gohy and U. S. Schubert, Macromolecules, 2008, 41, 2771–2777 CrossRef CAS.
- M. Chiper, M. A. R. Meier, J. M. Kranenburg and U. S. Schubert, Macromol. Chem. Phys., 2007, 208, 679–689 CrossRef CAS.
- E. C. Constable and M. D. Ward, J. Chem. Soc., Dalton Trans., 1990, 1405–1409 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2py20559a |
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here. 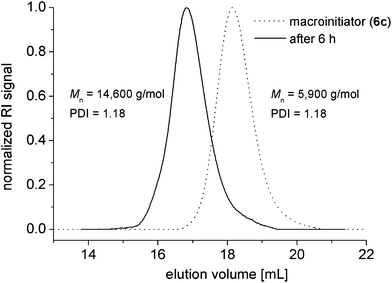
2 recorded with a PDA detector (eluent: DMF containing 0.005 M NH4PF6).](/image/article/2013/PY/c2py20559a/c2py20559a-f8.gif)