DOI:
10.1039/C2PY20573D
(Paper)
Polym. Chem., 2013,
4, 85-94
Supramolecular amphiphilic multiarm hyperbranched copolymer: synthesis, self-assembly and drug delivery applications†
Received
27th July 2012
, Accepted 25th August 2012
First published on 28th August 2012
Abstract
Novel supramolecular amphiphilic multiarm hyperbranched copolymers were successfully constructed through the molecular recognition of nucleobases. First, adenine-terminated H40-star-poly(ε-caprolactone)-adenine (H40-star-PCL-A) and uracil-terminated poly(ethylene glycol) (PEG-U) were successfully prepared. Due to the molecular recognition between A and U moieties, supramolecular multiarm hyperbranched copolymers were obtained by simply mixing the hydrophobic H40-star-PCL-A core and hydrophilic PEG-U shell. They not only had similar properties to conventional covalent-linked multiarm hyperbranched copolymers, but also possessed a dynamic and tunable nature. These supramolecular hyperbranched copolymers were found to self-assemble into pH-responsive micelles with low critical micelle concentration (CMC) because of non-covalent connection and hyperbranched architecture. The size of the self-assembled micelles could be easily tailored by changing the ratio of hydrophobic H40-star-PCL-A core and hydrophilic PEG-U arm. Moreover, encapsulation and controlled drug release were demonstrated with the chemotherapeutic drug doxorubicin (DOX). These supramolecular hyperbranched copolymer systems represent an evolution over conventional stimuli-responsive covalent-bonded hyperbranched copolymer systems and display a significant reduction in the viability of HeLa cells upon triggered release of DOX from the supramolecular micelles.
Introduction
Dendritic polymers including dendrimers and hyperbranched polymers have received great interest in both academic and industrial fields due to their peculiar topological structure and interesting physical/chemical properties.1–5 They possess a highly branched and globular void-containing topological structure and a large number of terminal functional groups for further conjugation of drug or targeting/imaging ligands, which provides a unique advantage in encapsulating small molecules.6 These features are very attractive for biomedical treatment, especially for drug delivery applications. In particular, multiarm hyperbranched copolymers can form unimolecular micelles or self-assemble into multimolecular micelles with a very low CMC. Therefore, they are widely regarded as promising biomaterials for constructing drug delivery systems.7–9 To date, many double hydrophilic or amphiphilic multiarm hyperbranched copolymers have been prepared as drug carriers.10–15
Although the multiarm hyperbranched copolymers have demonstrated great potential in delivering drugs, it is still of great importance to develop delivery systems with better drug-release properties. One of the most promising strategies is to design hyperbranched polymers that respond to specific stimulation, such as redox reaction, pH variation and temperature change.16–22 Among these activating signals, pH triggers belong to the most extensively studied stimuli due to the mildly acidic pH values encountered in tumor sites and inflammatory tissues, as well as in intracellular compartments, such as endosomes and lysosomes of cells.23,24 Thus, some multiarm hyperbranched copolymers with acid-labile covalent bonds, such as imine and hydrazone, have been designed and applied in the drug delivery field.19,25,26 However, these multiarm hyperbranched polymers based on acidic-sensitive covalent bonds exhibit sustained release of drugs over tens of hours. Therefore, it is still a challenge to exploit novel multiarm hyperbranched copolymers with improved responsive ability to acidic stimuli.
Supramolecular chemistry offers a new strategy for designing dynamic multiarm hyperbranched copolymers.27–31 In particular, complementary hydrogen bonding from nucleobases, which are moderately strong, highly directional and sensitive to acidic pH, have experienced increasing interest in recent years.32–36 In the present work, we constructed supramolecular multiarm hyperbranched copolymer H40-star-PCL-A:U-PEG based on the molecular recognition of nucleobases. This supramolecular approach offers versatility and simplicity in the preparation of hyperbranched copolymers compared to conventional covalent techniques for polymer synthesis. These supramolecular hyperbranched polymers not only have similar properties to conventional covalent-linked multiarm hyperbranched copolymers, but also possess a dynamic and tunable nature. The use of complementary hydrogen bonding offers a pH-responsive nature for hyperbranched copolymers. These supramolecular hyperbranched copolymers could self-assemble into micelles with a low CMC in aqueous solution. The micelle sizes were easily tailored by simply adjusting the ratio of hydrophilic/hydrophobic segments. To our knowledge, this is the first attempt to construct supramolecular multiarm hyperbranched copolymers based on molecular recognition of nucleobases. Thanks to the perfect combination of hyperbranched architecture and supramolecular dynamic nature, supramolecular hyperbranched copolymer H40-star-PCL-A:U-PEG may be a promising carrier for drug delivery.
Experimental
Materials
Dendritic aliphatic polyester, Boltorn® H40 (H40) (64 hydroxyl groups per molecule theoretically and Mn of 2833) was obtained from Perstorp Polyols AB, Sweden and purified by fractional precipitation according to the literature procedure.20 ε-Caprolactone (ε-CL, 99%, Alfa Aesar) was dried over calcium hydride (CaH2) for 24 h and then purified by distillation under reduced pressure prior to use. Tin(II) octoate (Sn(Oct)2), uracil (U, 99%), adenine (A, 99%) and 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) were all purchased from Sigma and used as received. Ethyl acrylate (EA) was first washed with 2 mol L−1 NaOH solution, dried and distilled over CaH2 under reduced pressure. 1-Ethyl-3-(3-dimethyllaminopropyl)carbodiimide hydrochloride (EDC) and 4-dimethylamino-pyridine (DMAP) were purchased from Aladdin. N,N-Dimethylformamide (DMF) was dried over CaH2 for 48 h and then distilled before use. Toluene was dried by refluxing with the fresh sodium-benzophenone complex under nitrogen gas and distilled just before use. Monomethoxy poly(ethylene glycol) (PEG) with Mn = 2000 g mol−1 was purchased from Fluka and dried by azeotropic distillation in the presence of dry toluene. Doxorubicin hydrochloride (DOX·HCl) was purchased from Beijing Huafeng United Technology Corporation and used as received. Clear polystyrene tissue culture-treated 12-well and 96-well plates were obtained from Corning Costar. All other reagents and solvents were purchased from the domestic suppliers and used as received unless mentioned.
Characterization
All nuclear magnetic resonance (NMR) spectra were recorded on a Bruker AVANCE III 400 spectrometer with deuterated chloroform (CDCl3), dimethyl sulfoxide-d6 (DMSO-d6) and 1,1,2,2-tetrachloroethane-d2 as solvents. The number-average molecular weight (Mn), weight-average molecular weight (Mw) and polydispersity index (PDI) were determined by gel permeation chromatography/multi-angle laser light scattering (GPC-MALLS). The gel permeation chromatography system consisted of a Waters degasser, a Waters 515 HPLC pump, a 717 automatic sample injector, a Wyatt Optilab DSP differential refractometer detector and a Wyatt miniDAWN multi-angle laser light scattering detector. Three chromatographic columns (styragel HR3, HR4, and HR5) were used in series. Tetrahydrofuran (THF) was used as the mobile phase at a flow rate of 1 mL min−1 at 30 °C. The refractive index increment dn/dc was determined with a Wyatt Optilab DSP differential refractometer at 690 nm. Data analysis was performed with Astra software (Wyatt Technology). Fourier transform infrared (FTIR) spectra were recorded on a Paragon 1000 instrument by the KBr sample holder method. Dynamic light scattering (DLS) measurements were performed with a Malvern Zetasizer Nano S apparatus equipped with a 4.0 mW laser operating at λ = 633 nm. All samples were measured at a scattering angle of 173°. The data were the mean of three tests. Transmission electron microscopy (TEM) studies were performed with a JEOL JEM-100CX-II instrument at a voltage of 200 kV. Samples were prepared by drop-casting micelle solutions onto carbon-coated copper grids and then air-drying at room temperature before measurement.
Synthesis of ethyl 3-(9-adeninyl)propionate
Ethyl 3-(9-adeninyl)propionate was prepared according to the literature.37 A mixture containing adenine (8 g, 59 mmol), metal sodium (60 mg, 26 mmol) and ethyl acrylate (19.2 mL, 176 mmol) in anhydrous ethanol (200 mL) was refluxed for 2 h. Then, the solvent was removed by filtration and the precipitate was recrystallized from ethanol to give ethyl 3-(9-adeninyl)propionate. Yield: 77%. 1H NMR (400 MHz, CDCl3, 298 K) δ ppm: 8.35 (s, 1H), 7.90 (s, 1H), 5.85 (s, 2H), 4.48–4.51 (t, 2H), 4.10–4.15 (q, 2H), 2.90–2.94 (t, 2H), 1.19–1.23 (t, 3H).
Synthesis of 3-(9-adeninyl)propionic acid
3-(9-Adeninyl)propionic acid was prepared according to the literature.38 The ethyl 3-(9-adeninyl)propionate (4.7 g, 20 mmol) in 150 mL of 3 M HCl was refluxed for 5 h. The reaction solution was cooled to room temperature and followed by neutralization of the hydrochloride salt of the product by sodium carbonate solution. The resulting precipitates were collected and dried under vacuum to a constant weight. Yield: 70%. 1H NMR (400 MHz, DMSO-d6, 298 K) δ ppm: 8.12 (s, 1H), 8.07 (s, 1H), 7.19 (s, 2H), 4.30–4.33 (t, 2H), 2.83–2.86 (t, 2H).
Synthesis of H40-star-poly(ε-caprolactone) (H40-star-PCL)
H40-star-PCL was prepared by the ring-opening polymerization (ROP) of ε-CL using the purified H40 as a macroinitiator and Sn(Oct)2 as a catalyst. In a typical polymerization procedure, H40 (0.40 g, 3.5 mmol hydroxyl groups) was added into a 50 mL flask and dried under vacuum at 100 °C for 1 h. Then, ε-CL (10.3 g, 90 mmol) was charged into the flask and heated to 120 °C under an argon atmosphere. After H40 was completely dissolved in ε-CL, Sn(Oct)2 (36.6 mg, 0.090 mmol) was introduced to start the polymerization. The reaction mixture was stirred for 24 h at 120 °C. After cooling to room temperature, the crude product was dissolved in dichloromethane (CH2Cl2), precipitated into cold methanol, filtrated and dried under vacuum to afford a white powder (8.6 g, yield: 80.4%). Mn,GPC = 8.5 × 104 g mol−1, Mw/Mn = 1.2. 1H NMR (400 MHz, CDCl3, 298 K) δ ppm: 3.9–4.2 (polycaprolactone, –COOCH2–), 3.6 (polycaprolactone, –CH2CH2–OH), 2.2–2.4 (polycaprolactone, –COCH2CH2–), 1.5–1.8 (polycaprolactone, –COCH2CH2–, –COOCH2CH2–), 1.2–1.4 (polycaprolactone, –COCH2CH2CH2–).
Synthesis of H40-star-PCL-A
H40-star-PCL-A was prepared by coupling 3-(9-adeninyl)propionic acid with H40-star-PCL. In a typical procedure, H40-star-PCL-A (1.0 g, 0.75 mmol OH), 3-(9-adeninyl)propionic acid (0.23 g, 1.10 mmol), EDC (0.42 g, 2.20 mmol) and DMAP (0.136 g, 1.10 mmol) were dissolved in DMF (60 mL) and stirred at 45 °C for 24 h. DMF was evaporated under reduced pressure and the residue was precipitated into cold methanol three times. The product was dried under vacuum to a constant weight. Yield: 64.5%. Mn,GPC = 9.1 × 104 g mol−1, Mw/Mn = 1.1. 1H NMR (400 MHz, CDCl3, 298 K) δ ppm: 8.3 (1H, –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHN–), 8.0 (1H, NCHN–), 4.5 (2H, –NCH2CH2–), 3.9–4.2 (polycaprolactone, –COOCH2–), 2.9 (2H, –NCH2CH2–), 2.2–2.4 (polycaprolactone, –COCH2CH2–), 1.5–1.8 (polycaprolactone, –COCH2CH2–, –COOCH2CH2–), 1.2–1.4 (polycaprolactone, –COCH2CH2CH2–).
CHN–), 8.0 (1H, NCHN–), 4.5 (2H, –NCH2CH2–), 3.9–4.2 (polycaprolactone, –COOCH2–), 2.9 (2H, –NCH2CH2–), 2.2–2.4 (polycaprolactone, –COCH2CH2–), 1.5–1.8 (polycaprolactone, –COCH2CH2–, –COOCH2CH2–), 1.2–1.4 (polycaprolactone, –COCH2CH2CH2–).
Synthesis of PEG-U
Uracil-terminated PEG, PEG-U, was synthesized via a two-step synthesis. First, PEG–sulfanilic acid ester was obtained by the reaction of PEG with excess p-toluenesulfonyl chloride (TsCl) in the presence of sodium hydroxide (NaOH). Then, PEG-sulfanilic acid ester (1.60 g, 0.8 mmol), uracil (0.18 g, 1.6 mmol) and potassium carbonate (K2CO3) (0.22 g, 1.6 mmol) were dissolved in 30 mL DMF in a round-bottomed flask and reacted at 80 °C for 8 h. The resultant mixture was poured into water and extracted with CH2Cl2. After drying over anhydrous magnesium sulfate (MgSO4), most of the solvent was removed by evaporation. The residue was precipitated into cold diethyl ether and dried under vacuum to a constant weight. 1H NMR (400 MHz, CDCl3, 298 K) δ ppm: 8.4 (1H, –CONHCO–), 7.4 (1H, –NCHCH–), 5.6 (1H, –CHCH–CO–), 3.9 (2H, –CH2CH2N–), 3.5–3.7 (PEG, 2H per repeating unit, –OCH2CH2–, 2H per repeating unit, –OCH2CH2–), 3.4 (3H, CH3OCH2–). 13C NMR (100 MHz, CDCl3, 298 K) δ: 47.0 (–CH2CH2N–), 58.0 (CH3O–), 67.8 (–CH2CH2N–), 69.7 (–CH2CH2O–), 71.2 (CH3OCH2–), 100.2 (–CHCHCO–), 146.3 (–CHCHCO–), 150.9 (–NCONH–), 163.8 (–CHCONH–).
Fabrication of H40-star-PCL-A:U-PEG micelles
Briefly, H40-star-PCL-A with different molar PEG-U (the ratio of adenine and uracil was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:0.8, 1:0.6, 1:0.4) was dissolved in THF at room temperature. After being fully dissolved, the polymer solution was added dropwise into deionized water under stirring with a magnetic bar. Thereafter, the mixture was dialyzed against deionized water using a dialysis tube (Mw cutoff: 3500 Da) for 24 h, during which the water was renewed every 4 h.
:1, 1:0.8, 1:0.6, 1:0.4) was dissolved in THF at room temperature. After being fully dissolved, the polymer solution was added dropwise into deionized water under stirring with a magnetic bar. Thereafter, the mixture was dialyzed against deionized water using a dialysis tube (Mw cutoff: 3500 Da) for 24 h, during which the water was renewed every 4 h.
Preparation of DOX-loaded micelles
Typically, 5 mg H40-star-PCL-A and 7 mg PEG-U were dissolved in 2 mL of THF at room temperature, followed by adding a predetermined amount of DOX·HCl and an equal molar amount of triethylamine (TEA) and stirred for 6 h. Then, the mixture was slowly added into 5 mL of phosphate buffered saline (PBS, 50 mM, pH 7.4) and stirred for another 2 h. Subsequently, the solution was dialyzed against deionized water for 18 h (Mw cutoff: 3500 Da) at room temperature and the deionized water was exchanged every 4 h. The contents inside the dialysis tube were filtered and lyophilized. The amount of DOX was determined with fluorescence (QM/TM/IM Steady-State & Time-Resolved Fluorescence Spectrofluorometer) measurement (excitation at 480 nm). To determine the total loading of the drug, DOX-loaded micelles were dissolved in DMSO and analyzed with fluorescence spectroscopy, wherein calibration curve was obtained with DOX–DMSO solutions with different DOX concentrations.
Drug loading content (DLC) and drug loading efficiency (DLE) were calculated according to the following formulae:
| DLC (wt%) = (weight of loaded |
| DLE (%) = (weight of loaded |
| /weight of drug in feed) × 100% |
In vitro release measurements
A total of 6 mL of DOX-loaded micelles was transferred to a dialysis bag with a molecular weight cutoff of 2000 Da. It was immersed in 50 mL of phosphate buffer (pH 7.4) or acetate buffer (pH 5.0) solutions with gentle shaking (100 rpm) at 37 °C in a laboratory shaker. At predetermined time intervals, 2.0 mL buffer solution outside the dialysis bag was extracted and it was replaced by an equal volume of fresh media to keep the sink condition. The amount of released DOX was analyzed with fluorescence measurement (QM/TM/IM Steady-State & Time-Resolved Fluorescence Spectrofluorometer, excitation at 480 nm). Each experiment was done in triple and the results were the average data.
Cell culture
NIH/3T3 normal cells (a mouse embryonic fibroblast cell line) and HeLa cancer cells (a human uterine cervix carcinoma cell line) were cultured in DMEM (Dulbecco's Modified Eagle's Medium) supplied with 10% FBS (fetal bovine serum) and antibiotics (50 units per mL penicillin and 50 units per mL streptomycin) at 37 °C in a humidified atmosphere containing 5% CO2.
Cytotoxicity measurements of micelles
The relative cytotoxicity of H40-star-PCL-A:U-PEG micelles was estimated by a MTT viability assay against NIH/3T3 cells. In the MTT assay, NIH/3T3 cells were seeded into 96-well plates with a density of 1 × 104 cells per well in 200 μL of medium. After 24 h of incubation, the culture medium was removed and replaced with 200 μL of a medium containing serial dilutions of H40-star-PCL-A:U-PEG micelles. The cells were grown for another 24 h. Then, 20 μL of 5 mg mL−1 MTT assays stock solution in PBS was added to each well. After incubating the cells for 4 h, the medium containing unreacted dye was carefully removed. The obtained blue formazan crystals were dissolved in 200 μL per well DMSO and the absorbance was measured in a BioTek® SynergyH4 at a wavelength of 490 nm.
Intracellular drug release
The experiments of intracellular drug release were performed on flow cytometry and confocal laser scanning microscopy (CLSM).
Flow cytometry.
HeLa cells were seeded in six-well plates at 5 × 105 cells per well in 1 mL complete DMEM and cultured for 24 h. Then, the DOX-loaded micelles dissolved in DMEM culture medium at a final DOX concentration of 9 μg mL−1 were added to different wells and the cells were incubated at 37 °C for 5, 15, 30, 60 and 180 min. Thereafter, the culture medium was removed and cells were washed with PBS twice and treated with trypsin. Subsequently, 2 mL of PBS was added to each culture well and the solutions were centrifuged for 5 min (1000 rpm). After the supernates were removed, the cells were resuspended in 0.5 mL of PBS. Data for 1.0 × 104 gated events were collected and analysis was performed by means of a BD FACSCalibur flow cytometer and CELLQuest software.
CLSM.
HeLa cells were seeded in six-well plates at 2 × 105 cells per well in 1 mL complete DMEM and cultured for 24 h, followed by removing the culture medium and adding DOX-loaded micelles (1 mL DMEM medium) at a final DOX concentration of 9 μg mL−1. The cells were incubated at 37 °C for predetermined intervals. Then, the culture medium was removed and cells were washed with PBS three times. Subsequently, the cells were fixed with 4% paraformaldehyde for 30 min at room temperature and the slides were rinsed with PBS three times. Finally, the slides were mounted and observed with a LSM510 META.
In vitro cytotoxicity evaluation
The cytotoxicity of DOX-loaded micelles and free DOX against HeLa cells was evaluated in vitro by MTT assay. HeLa cells were seeded into 96-well plates at an initial density of 6 × 103 cells per well in 200 μL of medium. After incubating for 24 h, the culture medium was replaced with a fresh one and the cells were treated with DOX-loaded micellar solution at a predetermined concentration. The cells were grown in a humidified environment with 5% CO2 at 37 °C for another 48 h. After the incubation, the culture medium was removed and washed with PBS twice. Then 200 μL of DMEM and 20 μL of 5 mg mL−1 MTT assays stock solution in PBS were added to each well. After incubating the cells for 4 h, the medium containing unreacted dye was carefully removed. The obtained blue formazan crystals were dissolved in 200 μL per well DMSO and the absorbance was measured in a BioTek® SynergyH4 at a wavelength of 490 nm.
Results and discussion
Synthesis and characterization of H40-star-PCL-A and PEG-U
As a kind of commercially available hyperbranched polyester, H40 is widely used as the core of amphiphilic multiarm copolymers for drug delivery.19,39,40 Biodegradable PCL is a U. S. Food and Drug Administration approved biomedical polymer and has been increasingly investigated for pharmacological and biomedical applications. Therefore, H40 and PCL were separately selected as the hydrophobic core and inner shell of the supramolecular amphiphilic multiarm copolymer. On the other hand, PEG is used as the outer hydrophilic shell in amphiphilic copolymers because of its hydrophilicity, biocompatibility, non-cytotoxicity and lack of immunogenicity.19
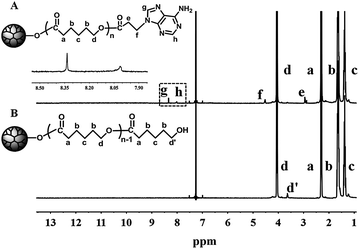
The synthesis of the H40-star-PCL-A was carried out according to Scheme 1. H40 was processed and characterized as in our previous report.41 The purified H40 had a degree of branching (DB) of about 0.38, degree of polymerization (DP) near 61, number-average molecular weight (Mn) of 7300 g mol−1 and terminal hydroxyl group number close to 64. Multiarm hyperbranched copolymer H40-star-PCL was prepared via the bulk ROP of ε-CL using the purified H40 as the macroinitiator and Sn(Oct)2 as the catalyst at 120 °C for 24 h. The structure of H40-star-PCL is confirmed by the 1H NMR in Fig. 1. 13C NMR spectra of H40 and H40-star-PCL are shown in Fig. S1.† It has been reported that the chemical shift of quaternary carbon in H40 is sensitive to the degree of substitution.42 The signal of the quaternary carbon appears at 50.6 ppm if both hydroxyl groups are not substituted, at 48.8 ppm if one hydroxyl group is substituted and at 46.8 ppm if both hydroxyl groups are substituted. The 13C NMR spectrum of H40-star-PCL is shown in Fig. S1A.† The signals at 50.6 and 48.8 ppm disappear, which confirms that all of the hydroxyl groups of H40 initiate the ROP of ε-CL to produce H40-star-PCL. Therefore, it is reasonable to assume that H40-star-PCL possesses about 64 PCL arms.40,43 The degree of polymerization (DP) of PCL arm within H40-star-PCL is 20, which is calculated from the integral ratio of the characteristic peaks at 3.6 ppm (methylene protons adjacent to the terminal hydroxyl group) and 2.3 ppm (methylene protons adjacent to carbonyl groups).
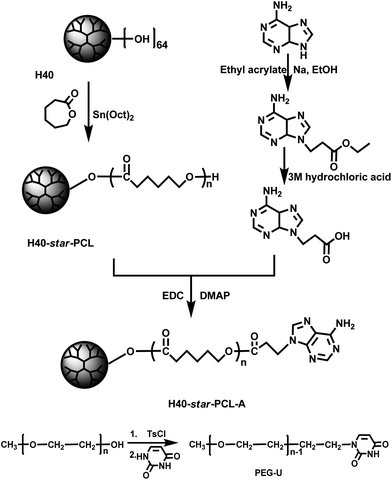 |
| | Scheme 1 The detailed synthetic route of H40-star-PCL-A and PEG-U. | |
In the next step, H40-star-PCL-A was synthesized by esterification reaction of the terminal hydroxyl groups of H40-star-PCL and 3-(9-adeninyl)propionic acid at 45 °C in the presence of EDC and DMAP. The adenine derivate, 3-(9-adeninyl)propionic acid, was prepared according to previous reports37,38 and well characterized by NMR measurements (see the data in the experimental section). The primary amino groups in the adenine derivate are reasonably deactivated due to the resonance effect originated by two conjugated rings, so there is no need to protect the amino groups in the esterification reaction. The 1H NMR spectrum of H40-star-PCL-A is shown in Fig. 1. After esterification reaction, the methylene protons adjacent to terminal hydroxyl groups of H40-star-PCL at 3.6 ppm almost disappear in the spectrum of H40-star-PCL-A. Four new signals appear in the spectrum of H40-star-PCL-A at 8.3, 8.0, 4.5 and 2.9 ppm, which can be attributed to the protons on the adenine and the protons of methylene connected with adenine, respectively. According to the 1H NMR results, the esterification efficiency of H40-star-PCL-A is calculated to be 72% based on the integration ratio of the methylene signal on the PCL polymer backbone (–CH2OCO–, 3.9–4.2 ppm) to the signal of methylene connected with adenine (–NCH2CH2–, 4.5 ppm).
The molecular weights and their distributions of all intermediates and H40-star-PCL-A were measured by GPC and the data are summarized in Table 1. The representative GPC profiles of H40-star-PCL and H40-star-PCL-A are shown in Fig. S2.† The GPC analysis of them reveals a relatively unimodal and symmetric elution peak. A clear shift toward higher molecular weight is observed by comparing the GPC curves of the H40-star-PCL-A and H40-star-PCL. This indicates that the corresponding esterification reaction is successful.
Table 1 Characterization data of the intermediates and the resulting polymers
| Polymers |
M
n (g mol−1; × 104) |
M
w(g mol−1; ×104) |
M
w/Mn |
|
From Perstorp data sheet.
|
| H40 |
0.3a |
0.5a |
1.8a |
| H40-star-PCL |
8.5 |
10.5 |
1.2 |
| H40-star-PCL-A |
9.1 |
10.2 |
1.1 |
Uracil-terminated PEG was synthesized in a two-step reaction. In the first step, PEG–sulfanilic acid ester was prepared by the coupling reaction of PEG and TsCl. Then PEG-U was obtained by the grafting reaction of PEG-sulfanilic acid ester and excess of uracil.
The hydrogen bonding interactions of supramolecular multiarm hyperbranched copolymer H40-star-PCL-A:U-PEG
The proposed molecular structure of supramolecular multiarm hyperbranched copolymer H40-star-PCL-A:U-PEG is illustrated in Scheme 2. The formation of complementary multiple hydrogen bonds between H40-star-PCL-A and PEG-U was analyzed by variable-temperature 1H NMR spectroscopy for the blend of two polymers with an equivalent mole of adenine to uracil units in 1,1,2,2-tetrachloroethane-d2. The temperature dependence of the NH proton chemical shift of the H40-star-PCL-A and PEG-U complexes at 2.0 wt% in 1,1,2,2-tetrachloroethane-d2 is shown in Fig. 2. The chemical shift of the NH resonance moves upfield systematically from 8.3 to 7.9 ppm when the temperature is raised from 30 to 100 °C. The gradual decrease in the NH resonance with the increase of temperature can be attributed to the dissociation of the complementary hydrogen bonds.44 However, as soon as the sample is cooled from 100 to 30 °C, the NH resonance returns to its original position at 8.3 ppm. All of these data suggest that complementary hydrogen bonds generate between H40-star-PCL-A and PEG-U, and the supramolecular multiarm hyperbranched copolymers are thermo-reversible in 1,1,2,2-tetrachloroethane-d2.
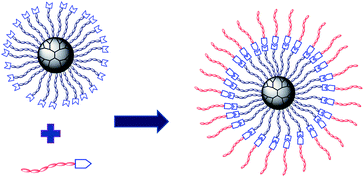 |
| | Scheme 2 The schematic structure of supramolecular multiarm hyperbranched copolymer H40-star-PCL-A:U-PEG. | |
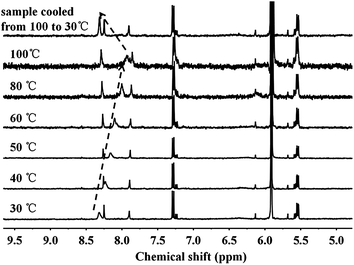 |
| | Fig. 2
1H NMR NH chemical shift of the H40-star-PCL-A and PEG-U complexes (the molar ratio of adenine and uracil was 1:1) as a function of temperature in 1,1,2,2-tetrachloroethane-d2. The sample was allowed to equilibrate for 5 min at each temperature. | |
The molecular recognition of nucleobases between H40-star-PCL-A and PEG-U was also analyzed by variable-temperature FTIR. The partial IR spectra of H40-star-PCL-A and PEG-U complexes with equivalent molar PCL-A and PEG-U in the bulk state are shown in Fig. 3. With the temperature increasing from 25 to 180 °C, the band in the range of 1750–1710 cm−1 ascribed to the CO stretching vibration shifts to higher frequency from 1724 to 1735. When the sample is cooled from 180 to 25 °C, the CO stretching peak returns to its original position at 1724 cm−1. According to previous reports, the CO stretching band shifts to lower frequencies upon hydrogen bond formation.45 The results of the FTIR spectra suggest the existence of the hydrogen bonds between H40-star-PCL-A and PEG-U and the complementary hydrogen bonds dissociate with increasing temperature. It is concluded that H40-star-PCL-A and PEG-U are linked through complementary hydrogen bonds to form a supramolecular multiarm hyperbranched copolymer with thermo-reversibility.
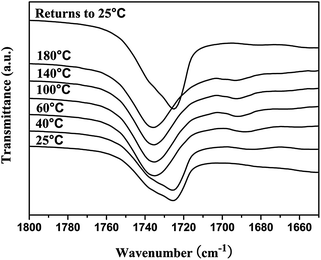 |
| | Fig. 3 Variable temperature FTIR spectra in the 1800–1650 cm−1 region of H40-star-PCL-A and PEG-U complexes (the molar ratio of adenine and uracil was 1:1). | |
Micellization of H40-star-PCL-A:U-PEG and pH-triggered destabilization
The multiarm hyperbranched copolymer H40-star-PCL-A and linear PEG-U could form a supramolecular amphiphilic star block copolymer via molecular recognition. Due to its amphiphilicity and globular architecture, the supramolecular hyperbranched copolymer can form stable micelles in an aqueous solution.13 The hydrophobic H40-star-PCL-A segment serves as the core, while the hydrophilic PEG-U forms a shell to maintain a hydration barrier to provide a stable interface between the hydrophobic core and the external medium. Through tuning the ratios of hydrophobic and hydrophilic segments, a series of micelles with different sizes were obtained by simply mixing the H40-star-PCL-A with PEG-U. As shown in Fig. 4A–D, the diameter of supramolecular copolymer micelles increases from 10 nm to 200 nm as the ratio of the hydrophilic segments decreases. In comparison to conventional chemical synthesis, the supramolecular preparation of amphiphilic multiarm hyperbranched copolymer provides a facile approach to adjust the ratio of hydrophobic and hydrophilic blocks using noncovalent interactions. Therefore, the micelle size of the supramolecular copolymer can be easily tailored and hence the drug loading and release profile of the resulting micelles can be tuned, which is significant in drug delivery systems.
 |
| | Fig. 4 TEM microscope images of self-assembled H40-star-PCL-A:U-PEG micelles with different ratios of hydrophobic and hydrophilic segments. The ratio of adenine and uracil was: (A) 1:1; (B) 1:0.8; (C) 1:0.6; (D) 1:0.4. | |
The size of the micelles is an important parameter that influences the cellular internalization process, as well as in vivo performance. It has been well reported that small particle sizes (<200 nm) can lower the level of reticuloendothelial system (RES) uptake, minimize renal excretion and strengthen accumulation of micelle-encapsulated drugs within tumors via enhanced permeability and retention (EPR) effect.19 Furthermore, the outer hydrophilic PEG shell can protect the micelles from attacks in the biological media, thereby increasing the stability of micelles, as well as prolonging their circulation time in vivo. Therefore, supramolecular multiarm hyperbranched copolymer H40-star-PCL-A:U-PEG with high PEG content (the molar ratio of adenine and uracil was 1:1) was selected as a drug carrier. The CMC of supramolecular hyperbranched copolymer H40-star-PCL-A:U-PEG was investigated by UV/Vis spectra using DPH as a hydrophobic probe. As shown in Fig. 5, this supramolecular copolymer has a relatively low CMC (0.003 mg mL−1), indicating the rather high stability of H40-star-PCL-A:U-PEG micelles.7,9 The characteristic size and morphology of H40-star-PCL-A:U-PEG micelles were determined by DLS and TEM. As shown in Fig. 6A, the size distribution histogram of the micelles in aqueous solution gives a monomodal distribution with a hydrodynamic diameter of 37 nm and a PDI of 0.18 (a relatively narrow polydispersity). The morphology of the supramolecular multiarm hyperbranched copolymer micelles visualized by TEM is shown in Fig. 6C. The supramolecular copolymer self-assembles into approximate spherical micelles in aqueous solution and has a diameter of 18 nm. The diameter of the micelles observed by TEM is smaller than its diameter determined by DLS. The diameter of the micelles determined by DLS reflects the hydrodynamic diameter of the micelles, whereas the diameter visualized by TEM gives that of dried micelles. Therefore, compared with that of TEM, the size increase of the micelles determined by DLS can be attributed to the hydration of the shell portion of the micelles.19 These results indicate that the size of H40-star-PCL-A:U-PEG micelles is suitable for intracellular drug delivery. It should be noted that the resulting H40-star-PCL-A:U-PEG micelles are stable in aqueous media over several weeks and display no notable changes in visual appearance and size/size distribution during this period.
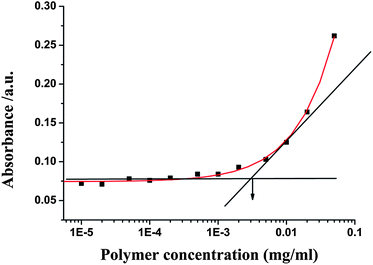 |
| | Fig. 5 The relationship of the absorbance and the concentration of H40-star-PCL-A:U-PEG solutions (λ = 313 nm, 25 °C). | |
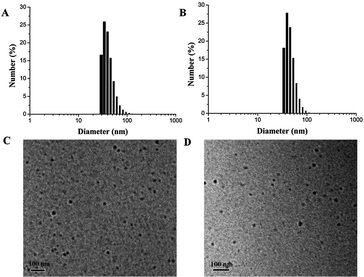 |
| | Fig. 6 DLS plots: (A) H40-star-PCL-A:U-PEG micelles; (B) DOX-loaded micelles. TEM microscope images: (C) H40-star-PCL-A:U-PEG micelles; (D) DOX-loaded micelles. | |
The hydrogen bonding interactions between hydrophobic H40-star-PCL-A core and hydrophilic PEG-U shell made micelles unstable at acidic pH. To evaluate the responsive ability, the copolymer micelles were treated with pH 5.0 acetate buffer (50 mM) and the particle sizes were followed by DLS measurements at different time intervals. Fig. 7A shows that H40-star-PCL-A:U-PEG micelles aggregate rapidly at pH 5.0. The size of the copolymer micelles increases from 37 nm to about 100 nm in 2 h and reaches over 400 nm after 6 h. The photographs in Fig. 7B show that the micelle solution becomes turbid at low pH value after 12 h, while still keeps clear under physiological pH conditions (pH 7.4). The driving force for the change of micelle size at low pH value is attributed to the protonation of the nucleobase nitrogen atoms, which leads to the shedding of the hydrophilic PEG shell from the micelles and the aggregation of the hydrophobic PCL core.20
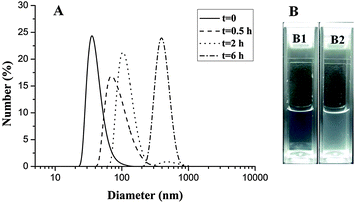 |
| | Fig. 7 Characterization of H40-star-PCL-A:U-PEG micelles: (A) size change of pH-sensitive micelles over time at pH 5.0 at 25 °C monitored by DLS; (B) photographs of unimolecular micelles over 12 h (B1) pH = 7.4, (B2) pH = 5.0. | |
In vitro cytotoxicity of H40-star-PCL-A:U-PEG micelles
The cytotoxicity of H40-star-PCL-A:U-PEG micelles was evaluated by MTT assay using a mouse embryonic fibroblast NIH/3T3 cell line. The MTT assay is based on the ability of a mitochondrial dehydrogenation enzyme in viable cells to cleave the tetrazolium rings of the pale-yellow MTT and form formazan crystals with a dark-blue color. Therefore, the number of surviving cells is directly proportional to the level of formed formazan.41 Fig. S3† shows the cell viability after 24 h incubation with the H40-star-PCL-A:U-PEG micelles at different concentrations. The results demonstrate that no obvious cytotoxicity against NIH/3T3 cells is observed even when the concentration of copolymer micelles is up to 500 μg mL−1. Apparently, these H40-star-PCL-A:U-PEG micelles have low cytotoxicity to NIH/3T3 cells.
Drug loading and pH-triggered drug release
It is well known that DOX, one of the most potent anticancer drugs, is widely used to treat different types of solid malignant tumors through interacting with DNA by interaction and inhibition of macromolecular biosynthesis.46,47 To investigate the potential of supramolecular multiarm copolymer micelles based on molecular recognition of adenine and uracil nucleobases, DOX was selected to evaluate the drug loading and release properties. Supramolecular multiarm copolymer H40-star-PCL-A:U-PEG possessed an H40 core, an inner hydrophobic PCL segment and a hydrophilic PEG outer corona. The hydrophobic DOX was encapsulated into the hydrophobic inner cores of the micelles. When the feed ratio of copolymer to DOX was 10:1, the final DLC and encapsulation efficiency of H40-star-PCL-A:U-PEG micelles were 2.8% and 28%, respectively. After drug loading, the size of micelles shows a slightly increased average size from 18 nm to 20 nm, measured by TEM (Fig. 6C and D).
The in vitro release behavior of DOX-loaded micelles was investigated under simulated physiological conditions (PBS, pH 7.4) and in an acidic environment (acetate buffer, pH 5.0) at 37 °C to assess the feasibility of using H40-star-PCL-A:U-PEG micelles as an anticancer drug delivery carrier. The drug release profiles of DOX are shown in Fig. 8. The release rate of DOX from the copolymer micelles at pH 5.0 is much faster than that at pH 7.4, which indicates that pH of the medium has a strong effect on the DOX release from the H40-star-PCL-A:U-PEG micelles. At pH 7.4, the release rate of DOX is relatively low. At pH 5.0, the DOX release rate is much faster with approximately 80% of the drug released within 5 h. The fast release of DOX from the DOX-loaded micelles in an acidic environment is likely due to the protonation of the amino group of nucleobase and shedding of the micelle shell in acidic conditions. The accelerated drug release at lower pH is of particular interest in achieving tumor-targeted DOX delivery with micelles as the pH value at the solid tumor is reported to be one order of magnitude lower than that of normal physiological conditions.48 Therefore, a sufficiently high concentration of DOX can be achieved within a reasonably short period of time once the micelles are taken up by the tumor cells via endocytosis, thereby greatly enhancing tumor-directed therapeutic efficacy.
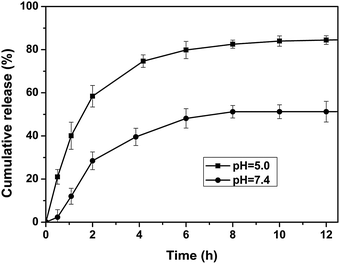 |
| | Fig. 8 Release profiles of DOX from DOX-loaded H40-star-PCL-A:U-PEG micelles at different pH values (7.4 and 5.0) at 37 °C. | |
Cell internalization
The cellular uptake of DOX-loaded H40-star-PCL-A:U-PEG micelles was evaluated to determine the effect of micelles on DOX delivery at the cellular level. Flow cytometry analysis was performed to test the cellular uptake using a human cervical carcinoma HeLa cell line. DOX-loaded micelles with a DOX concentration of 9 μg mL−1 were added to culture medium and the HeLa cells were further cultured at 37 °C for the predetermined time intervals. HeLa cells without DOX-loaded micelles were set as a control. As displayed in Fig. 9, the histogram of cells shifts to the direction of high fluorescence intensity with increasing time. The relative geometrical mean fluorescence intensities of DOX-loaded micelle pretreated cells increase from 35- to 150-fold of non-pretreated cells with the incubation time increasing from 5 min to 3 h. The result demonstrates effective internalization of DOX-loaded H40-star-PCL-A:U-PEG micelles and faster intracellular release of DOX inside HeLa cells.
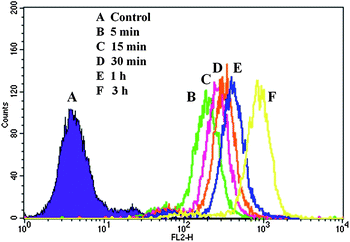 |
| | Fig. 9 Flow cytometry histogram profiles of HeLa cells that were incubated with DOX-loaded H40-star-PCL-A:U-PEG micelles for different time intervals. | |
Moreover, the cellular uptake and intracellular drug release of DOX-loaded H40-star-PCL-A:U-PEG micelles were further studied by CLSM. HeLa cells were cultured at 37 °C for 15 min, 1 h, and 6 h, respectively after DOX-loaded micelles were added to culture medium with a DOX concentration of 9 μg mL−1. As a control, HeLa cells were also incubated with free DOX. Then, the nucleus was stained with Hoechst 33342. All of the pretreated cells were observed under CLSM. We took advantage of the red autofluorescence from DOX and blue fluorescence from Hochest33342 to study the intracellular localization of drug within the cells. As shown in Fig. 10, the DOX fluorescence in all cells pretreated by DOX-loaded micelles for 15 min and 1 h is accumulated preferentially in the perinuclear region instead of the nucleus. Furthermore, the strong red fluorescence of DOX is presented in the cell nucleus when the cells are cultured in the DOX-loaded H40-star-PCL-A:U-PEG micelles for 6 h. However, the DOX red fluorescence is mostly localized in the cell nucleus when cells are pretreated with free DOX for 15 min (Fig. 10D). These results show that DOX-loaded H40-star-PCL-A:U-PEG micelles may be transported by the endocytosis mechanism and free DOX is taken up by simple passive diffusion through the cell membrane.41 Also, it suggests that the DOX molecules from acid-sensitive micelles are released rapidly in the endocytic compartment and finally reach the nucleus. Therefore, the H40-star-PCL-A:U-PEG micelles can be internalized efficiently, which is in agreement with the flow cytometric analysis. These properties of H40-star-PCL-A:U-PEG are very important for its use as an ideal drug carrier.
 |
| | Fig. 10 CLSM images of HeLa cells incubated with DOX-loaded H40-star-PCL-A:U-PEG micelles for (A) 15 min, (B) 1 h and (C) 6 h, and (D) free DOX for 15 min. Cell nuclei were stained with Hoechst 33342. | |
In vitro cytotoxicity of drug-loaded micelles
The proliferation inhibition of drug-loaded micelles against tumor cells is an important consideration for cancer therapy. Thus, the in vitro cytotoxicity of DOX-loaded H40-star-PCL-A:U-PEG micelles compared with that of free DOX was studied using a MTT assay against HeLa cells. It should be noted that blank micelles of H40-star-PCL-A:U-PEG did not show significant cytotoxicity (Fig. S3†). The HeLa cells were treated with DOX-loaded micelles at different doses from 0.05 to 2 μg mL−1. The HeLa cell proliferation results are shown in Fig. 11. It is found that DOX-loaded hyperbranched copolymer micelles exhibit improved inhibition to HeLa cell proliferation after 48 h culture in comparison with free-form DOX. The IC50 values, at which 50% cells proliferation was inhibited, were ca. 0.8 and 0.7 μg mL−1 for free DOX and DOX-loaded H40-star-PCL-A:U-PEG micelles, respectively. It has been well reported that DOX-loaded micelles are less potent in cell proliferation inhibition than free DOX at the same dose.49,50 This may be attributed to the time-consuming DOX-release from micelles and delayed nuclear uptake. In the present work, the enhanced inhibition to HeLa cell proliferation of DOX-loaded micelles indicates that the destabilization of H40-star-PCL-A:U-PEG micelles by intracellular acidic environment accelerates the DOX release, and the cell proliferation inhibition is thus enhanced because of the faster intracellular DOX release and the improved cellular uptake, as shown by the in vitro DOX release and internalization studies by CLSM.
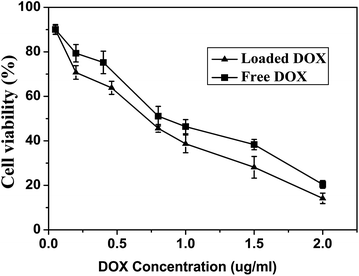 |
| | Fig. 11 The viability of HeLa cells after being cultured for 48 h with DOX-loaded H40-star-PCL-A:U-PEG micelles and free DOX was used as the control. | |
Conclusion
A novel type of supramolecular multiarm hyperbranched copolymer H40-star-PCL-A:U-PEG with the molecular recognition of nucleobases was synthesized. These supramolecular hyperbranched copolymers could self-assemble into pH-sensitive micelles with a low CMC in aqueous solution. Different size micelles were obtained by simply adjusting the ratio of hydrophilic/hydrophobic segments. These micelles were found to release DOX inside the cells rapidly in an acidic environment to yield significantly enhanced drug efficacy. In vitro study demonstrated that the supramolecular hyperbranched copolymer was nontoxic. Supramolecular hyperbranched copolymers exhibited a dynamic nature over conventional covalent-bonded hyperbranched polymers, since they could potentially combine the advantages of multiarm hyperbranched copolymer and dynamic supramolecular properties together. They are expected to be attractive new carriers for drug delivery or other medical applications.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (20974062, 30700175) and National Basic Research Program 2009CB930400, Shanghai Leading Academic Discipline Project (no. B202) and China National Funds for Distinguished Young Scientists (21025417).
Notes and references
- A. W. Bosman, H. M. Janssen and E. W. Meijer, Chem. Rev., 1997, 97, 1681–1712 CrossRef.
- J. M. J. Fréchet, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4782–4787 CrossRef.
- C. Gao and D. Y. Yan, Prog. Polym. Sci., 2004, 29, 183–275 CrossRef CAS.
- B. I. Voit and A. Lederer, Chem. Rev., 2009, 109, 5924–5973 CrossRef CAS.
- A. Carlmark, C. Hawker, A. Hult and M. Malkoch, Chem. Soc. Rev., 2009, 38, 352–362 RSC.
- Y. F. Zhou, W. Huang, J. Y. Liu, X. Y. Zhu and D. Y. Yan, Adv. Mater., 2010, 22, 4567–4590 CrossRef CAS.
- H. Tian, C. Deng, H. Lin, J. Sun, M. Deng, X. Chen and X. Jing, Biomaterials, 2005, 26, 4209–4217 CrossRef CAS.
- I. Gitsov and J. M. J. Fréchet, J. Am. Chem. Soc., 1996, 118, 3785–3786 CrossRef CAS.
- B. Lele and J. C. Leroux, Polymer, 2002, 43, 5595–5606 CrossRef CAS.
- C. Ternat, L. Ouali, H. Sommer, W. Fieber, M. I. Velazco, C. J. G. Plummer, G. Kreutzer, H.-A. Klok, J. E. Månson and A. Herrmann, Macromolecules, 2008, 41, 7079–7089 CrossRef CAS.
- J. Y. Liu, Y. Pang, W. Huang, X. Y. Zhu, Y. F. Zhou and D. Y. Yan, Biomaterials, 2010, 31, 1334–1341 CrossRef CAS.
- L.-A. Tziveleka, C. Kontoyianni, Z. Sideratou, D. Tsiourvas and C. M. Paleos, Macromol. Biosci., 2006, 6, 161–169 CrossRef CAS.
- G. Kreutzer, C. Ternat, T. Q. Nguyen, C. J. G. Plummer, J.-A. E. Månson, V. Castelletto, I. W. Hamley, F. Sun, S. S. Sheiko, A. Herrmann, L. Ouali, H. Sommer, W. Fieber, M. I. Velazco and H.-A. Klok, Macromolecules, 2006, 39, 4507–4516 CrossRef CAS.
- M. R. Radowski, A. Shukla, H. Berlepsch, C. Böttcher, G. Pickaert, H. Rehage and R. Haag, Angew. Chem., Int. Ed., 2007, 46, 1265–1269 CrossRef CAS.
- W. Du, Z. Xu, A. M. Nyström, K. Zhang, J. R. Leonard and K. L. Wooley, Bioconjugate Chem., 2008, 19, 2492–2498 CrossRef CAS.
- Y. Haba, A. Harada, T. Takagishi and K. Kono, J. Am. Chem. Soc., 2004, 126, 12760–12761 CrossRef CAS.
- Z. F. Jia, H. Chen, X. Y. Zhu and D. Y. Yan, J. Am. Chem. Soc., 2006, 128, 8144–8145 CrossRef CAS.
- M. Calderón, M. A. Quadir, S. K. Sharma and R. Haag, Adv. Mater., 2010, 22, 190–218 CrossRef.
- M. Prabaharan, J. J. Grailer, S. Pilla, D. A. Steeber and S. Q. Gong, Biomaterials, 2009, 30, 5757–5766 CrossRef CAS.
- J. Y. Liu, Y. Pang, W. Huang, X. H. Huang, L. L. Meng, X. Y. Zhu, Y. F. Zhou and D. Y. Yan, Biomacromolecules, 2011, 12, 1567–1577 CrossRef CAS.
- S. Luo, X. Hu, C. Ling, X. Liu, S. Chen and M. Han, Polym. Int., 2011, 60, 717–724 CrossRef CAS.
- H. Tai, W. Wang, C. Alexander, K. Shakesheff and S. Howdle, J. Controlled Release, 2008, 132, e48–e50 CrossRef CAS.
- J. Z. Du, X. J. Du, C. Q. Mao and J. Wang, J. Am. Chem. Soc., 2011, 133, 17560–17563 CrossRef CAS.
- J. Jung, I. H. Lee, E. Lee, J. Park and S. Jon, Biomacromolecules, 2007, 8, 3401–3407 CrossRef CAS.
- S. J. Xu, Y. Luo and R. Haag, Macromol. Biosci., 2007, 7, 968–974 CrossRef CAS.
- X. Y. Sun, Y. F. Zhou and D. Y. Yan, Sci. China, Ser. B: Chem., 2009, 52, 1703–1710 CrossRef CAS.
- J. Wang and M. Jiang, J. Am. Chem. Soc., 2006, 128, 3703–3708 CrossRef CAS.
- G. Zhou and I. Harruna, Macromolecules, 2005, 38, 4114–4123 CrossRef CAS.
- O. A. Scherman, G. B. W. L. Ligthart, H. Ohkawa, R. P. Sijbesma and E. W. Meijer, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11850–11855 CrossRef CAS.
- S. Liu, G. Zhang and M. Jiang, Polymer, 1999, 40, 5449–5453 CrossRef CAS.
- M. N. Higley, J. M. Pollino, E. Hollembeak and M. Weck, Chem.–Eur. J., 2005, 11, 2946–2953 CrossRef CAS.
- J. Lutz, A. Thünemann and K. Rurack, Macromolecules, 2005, 38, 8124–8126 CrossRef CAS.
- R. Deans, F. Ilhan and V. Rotello, Macromolecules, 1999, 32, 4956–4960 CrossRef CAS.
- S. Sntjens, R. Sijbesma, M. van Genderen and E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 7487–7493 CrossRef.
- M. Sotiropoulou, G. Bokias and G. Staikos, Macromolecules, 2003, 36, 1349–1354 CrossRef CAS.
- M. J. Krische and J.-M. Lehn, Struct. Bonding, 2000, 96, 3–29 CrossRef CAS.
- M. Weisser, J. Käshammer, B. Menges, J. Matsumoto, F. Nakamura, K. Ijiro, M. Shimomura and S. Mittler, J. Am. Chem. Soc., 2000, 122, 87–95 CrossRef CAS.
- J. C. Kim, J. Jung, Y. Rho, M. Kim, W. Kwon, H. Kim, I. J. Kim, J. R. Kim and M. Ree, Biomacromolecules, 2011, 12, 2822–2833 CrossRef CAS.
- M. Prabaharan, J. J. Grailer, S. Pilla, D. A. Steeber and S. Q. Gong, Biomaterials, 2009, 30, 3009–3019 CrossRef CAS.
- X. J. Li, Y. F. Qian, T. Liu, X. L Hu, G. Y. Zhang, Y. Z. You and S. Y. Liu, Biomaterials, 2011, 32, 6595–6605 CrossRef CAS.
- J. Y. Liu, W. Huang, Y. Pang, X. Y. Zhu, Y. F. Zhou and D. Y. Yan, Langmuir, 2010, 26, 10585–10592 CrossRef CAS.
- S. Chen, X. Z. Zhang, S. X. Cheng, R. X. Zhuo and Z. W. Gu, Biomacromolecules, 2008, 9, 2578–2585 CrossRef CAS.
- M. Ornatska, S. Peleshanko, K. L. Genson, B. Rybak, K. N. Bergman and V. V. Tsukruk, J. Am. Chem. Soc., 2004, 126, 9675–9684 CrossRef CAS.
- A. S. Karikari, B. D. Mather and E. Timothy, Biomacromolecules, 2007, 8, 302–308 CrossRef CAS.
- A. Dong, T. Wan, S. Feng and D. Sun, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 2642–2650 CrossRef CAS.
- M. L. Adams, A. Lavasanifar and G. S. Kwon, J. Pharm. Sci., 2003, 92, 1343–1355 CrossRef CAS.
- D. A. Gewirtz, Biochem. Pharmacol., 1999, 57, 727–741 CrossRef CAS.
- K. Kataoka, T. Matsumoto, M. Yokoyama, T. Okano, Y. Sakurai, S. Fukushima, K. Okamoto and G. S. Kwon, J. Controlled Release, 2000, 64, 143–153 CrossRef CAS.
- X. T. Shuai, H. Ai, N. Nasongkla, S. Kim and J. M. Gao, J. Controlled Release, 2004, 98, 415–426 CrossRef CAS.
- K. S. Soppimath, L. H. Liu, W. Y. Seow, S. Q. Liu, R. Powell, P. Chan and Y. Y. Yang, Adv. Funct. Mater., 2007, 17, 355–362 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 13C NMR spectra recorded for H40-star-PCL and purified H40, GPC-MALLS curves of H40-star-PCL and H40-star-PCL-A, cell viability of NIH/3T3 cells against H40-star-PCL-A:U-PEG micelles. See DOI: 10.1039/c2py20573d |
| ‡ These authors are joint first authors. |
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.