Facile synthesis of soluble nonlinear polymers with glycogen-like structures and functional properties from “simple” acrylic monomers†
Rongrong
Hu
ab,
Jacky W. Y.
Lam
ab,
Yong
Yu
ab,
Herman H. Y.
Sung
a,
Ian D.
Williams
a,
Matthew M. F.
Yuen
c and
Ben Zhong
Tang
*abd
aDepartment of Chemistry, Institute of Molecular Functional Materials, Institute for Advanced Study, State Key Laboratory of Molecular Neuroscience and Division of Biomedical Engineering, The Hong Kong University of Science & Technology (HKUST), Clear Water Bay, Kowloon, Hong Kong, China. E-mail: tangbenz@ust.hk
bHKUST Fok Ying Tung Research Institute, Nansha, Guangzhou, China
cDepartment of Mechanical Engineering, The Hong Kong University of Science & Technology, Clear Water Bay, Kowloon, Hong Kong, China
dState Key Laboratory of Luminescent Materials and Devices, Institute of Polymer Optoelectronic Materials and Devices, South China University of Technology, Guangzhou 510640, China
First published on 24th August 2012
Abstract
A facile approach for the synthesis of hyperbranched polyacrylates is developed. Tetraphenylethene-containing di- and tetra-acrylates are synthesized by esterification of bis(4-hydroxylphenyl)diphenylethene and tetrakis(4-hydroxylphenyl)ethene with acryloyl chloride and polymerized using azobisisobutyronitrile in refluxing THF, furnishing high molecular weight (Mw up to 87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100) polyacrylates with glycogen-like structures and closed-loops in nearly quantitative yields (isolation yield up to 99%). All the polymers are soluble in common organic solvents and are thermally stable with degradation temperatures up to 357 °C under nitrogen. Whereas the monomers and polymers are nonemissive or faintly emissive in solutions, they become strong emitters in the aggregated state, demonstrating a phenomenon of aggregation-induced or enhanced emission. The emission of the monomer and polymer spots on the TLC plates can be turned “off” and “on” continuously and reversibly by wetting and dewetting processes by vapors of organic solvents. The polymers are photosensitive and UV irradiation of their films through copper masks crosslinks the exposed parts, generating fluorescent negative photoresist patterns. The polymers possess good optical transparency and show high refractive indices (RI = 1.6825–1.5653) in a wide wavelength region (400–1700 nm) with low chromatic dispersion. The emission of the polymer nanoaggregates can be quenched efficiently by picric acid with large quenching constants up to 2.07 × 105 M−1, suggesting that they are sensitive fluorescent sensors for explosive detection. The fluorescent polymers become weakly emissive when hydrolyzed under appropriate conditions, implicative of their utility in controlled drug delivery.
100) polyacrylates with glycogen-like structures and closed-loops in nearly quantitative yields (isolation yield up to 99%). All the polymers are soluble in common organic solvents and are thermally stable with degradation temperatures up to 357 °C under nitrogen. Whereas the monomers and polymers are nonemissive or faintly emissive in solutions, they become strong emitters in the aggregated state, demonstrating a phenomenon of aggregation-induced or enhanced emission. The emission of the monomer and polymer spots on the TLC plates can be turned “off” and “on” continuously and reversibly by wetting and dewetting processes by vapors of organic solvents. The polymers are photosensitive and UV irradiation of their films through copper masks crosslinks the exposed parts, generating fluorescent negative photoresist patterns. The polymers possess good optical transparency and show high refractive indices (RI = 1.6825–1.5653) in a wide wavelength region (400–1700 nm) with low chromatic dispersion. The emission of the polymer nanoaggregates can be quenched efficiently by picric acid with large quenching constants up to 2.07 × 105 M−1, suggesting that they are sensitive fluorescent sensors for explosive detection. The fluorescent polymers become weakly emissive when hydrolyzed under appropriate conditions, implicative of their utility in controlled drug delivery.
Introduction
Development of polymeric materials with advanced properties is of great academic value and technological implication. Thanks to the enthusiastic efforts by scientists, a wealth of functional linear polymers has been prepared and their structure–property relationship has been well evaluated.1 Studies show that the property of a polymer depends largely on its topology,2 which triggers much research interest in creating polymers with architectures and topologies of higher dimension.3 Among them, materials with three-dimensional structures such as dendrimers and hyperbranched polymers4 have received particular interest because of the expectation that their numerous terminal functional groups and molecular cavities as well as intrinsic globular structures will impart them with unusual properties.5 Dendrimers with perfect shapes, however, are difficult to synthesize and those with high generation numbers are generally prepared via multistep reactions and purification.6 In contrast, hyperbranched polymers can be readily prepared by one-pot, single-step procedures and are thus, in most cases, regarded as more rapidly prepared and economical substitutes for dendrimers without sacrificing their fascinating and unique features such as low viscosity, good solubility, high functionality, etc.7Various approaches have been proposed for the construction of hyperbranched polymers since the first report in a patent in 1987.8 From the aspect of monomers, hyperbranched polymers are typically fabricated from ABx, A2 + B3 or A2-type monomers.9 According to the reaction protocol, polycondensation, polyaddition, chain polymerization and condensative chain polymerization are widely adopted for polymer synthesis,10 through which a broad range of hyperbranched polyphenylenes, poly(ester amide)s, polyimides, polyesters, polyethers and polycarbosiloxanes are synthesized.11
Although polymers synthesized from acetylene monomers show unique properties such as conductivity and optical nonlinearity,12 vinyl polymer is still the main stream in polymer research. The synthesis of hyperbranched vinyl polymer, however, is also challenging. In 1995, Fréchet and co-workers developed a novel approach to hyperbranched vinyl polymers coined as self-condensing vinyl polymerization (SCVP). This protocol utilizes vinyl monomers with pendant groups that can be transformed into initiating species in the presence of external stimuli.13 Polymerization can occur at the olefin double bond and/or the initiating site, resulting in the generation of branched or hyperbranched polymers. Although SCVP is a versatile method for hyperbranched polymer synthesis, it possesses some limitations. For example, the molecular weight distributions of the obtained polymers are generally broad. The metal catalyst used in the polymerization is toxic and shows little tolerance to monomers with functional groups, which limits the variety of polymers prepared. Despite a new SCVP approach being developed recently,14 it involves the complicated syntheses of tailored vinyl monomers with specialized functional groups. Transition-metal catalysts, strong base, or other toxic catalytic species are used for the polymerization, making large-scale production a daunting task.
Hyperbranched vinyl polymers can be achieved also by traditional radical polymerization.15 In one typical approach, the polymers are constructed in the presence of divinyl monomer such as 1,4-divinylbenzene or ethylene glycol diacrylate (EGDMA) (Chart 1). The radical polymerization of vinyl monomers produces high molecular weight polymer chains, which undergo further propagation with divinyl monomer to furnish a branched polymer. However, to avoid gelation at low conversion, the amount of divinyl monomer added should be well controlled and small (<3 mol%).16 In 2000, Sherrington and co-workers reported a one-pot versatile synthesis of high molecular weight branched vinyl polymers by copolymerization of a vinyl molecule and a divinyl monomer in the presence of a controlled amount of chain transfer agent (typically a thiol) for inhibition of crosslinking reaction and hence gelation.17 Though the strategy is described as a new “Strathclyde” route, it is still rather tedious. It would be very rewarding if methods can be developed that can generate soluble branched polymers from solely divinyl monomers as they will simplify and shorten the fabrication cost as well as reduce the production cost. Unfortunately, only a few successful examples are available in the literature.
 | ||
| Chart 1 Typical examples of divinyl monomers. | ||
In this work, we aim to investigate the feasibility of synthesizing hyperbranched vinyl polymers from divinyl or tetravinyl monomers by traditional free radical polymerization in the absence of transition-metal catalyst and strong bases under mild reaction conditions. We replaced the ethyl chain in EGDMA by tetraphenylethene (TPE), an archetypal luminogen with aggregation-induced emission (AIE) characteristic,18,19 and generated TPE-containing diacrylates and tetra-acrylate with different substitution patterns. The A4- or A8-type monomers are polymerized in the presence of azobisisobutyronitrile (AIBN), furnishing soluble hyperbranched polymers with high molecular weights in high yields. All the polymers inherit the AIE feature of TPE. They are photosensitive and can generate negative fluorescent photoresist patterns by UV irradiation. They exhibit high refractive indices with low chromatic dispersion. The emission of their nanoparticles can be quenched efficiently by picric acid, enabling them to work as sensitive fluorescent chemosensors for explosive detection. The polymers undergo hydrolysis reaction under appropriate conditions, making them promising candidates for drug delivery.
Results and discussion
Monomer and polymer syntheses
To develop a facile route for the preparation of functional hyperbranched polymers, we designed TPE-containing diacrylates (1 and 2) and tetra-acrylate (3) and elaborated a two-step reaction route for their synthesis (Scheme 1). Briefly, cross McMurry coupling reaction of benzophenone (4) with 4,4′-dihydroxybenzophenone (5) catalyzed by TiCl4–Zn in THF gave a mixture of TPE and dihydroxyl- and tetrahydroxyl tetraphenylethene (6 and 7). Since 6 and 7 readily underwent oxidation reaction in air, they were treated with acryloyl chloride once isolated to furnish the desirable products 1 and 3. Similarly, McMurry coupling reaction of 4-hydroxybenzophenone (8) generated intermediate 9, whose reaction with acryloyl chloride produced diacrylate 2. Unlike 1, 2 was isolated as a mixture of E and Z isomers with a molar ratio of ∼1:1. The isomers cannot be separated by column chromatography due to their similar physical properties. Previous studies showed that the Z- and E-isomers had almost identical orbital distributions and hence exhibited similar fluorescence properties.20 Thus, the existence of both isomers in 2 would not alter significantly the overall properties of the material. The structures of 1 and E-2 were analyzed by single crystal X-ray diffraction. Their ORTEP drawings are given in Fig. 1 and the associated crystal data are provided in Tables S1 and S2.†
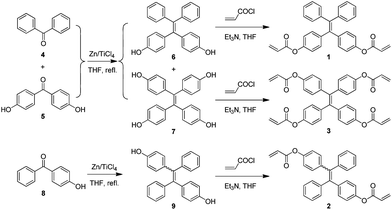 | ||
| Scheme 1 Synthesis of tetraphenylethene-containing acrylates 1–3. | ||
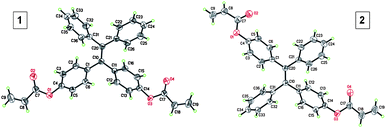 | ||
| Fig. 1 ORTEP drawings of crystals of 1 and E–2. | ||
After the success in preparing the monomers, we then converted them into polymers by a traditional free radical polymerization technique. Since 1–3 are mainly constructed from phenyl rings with 4 or 8 active sites, theoretically, insoluble micro or nanogels are anticipated to be formed in the absence of chain transfer agent.
To search for optimal conditions for the polymerization, we first studied the effect of monomer concentration on the polymerization of 1 catalyzed by AIBN in refluxing THF at a constant initiator/monomer ratio. As shown in Table 1, insoluble gel was formed quantitatively after 1 h when the polymerization was carried out at a relatively high monomer concentration of 300 mM. Decreasing the monomer concentration to 100 mM helps suppress the undesirable crosslinking reaction and hence the solubility of the resulting polymer: a soluble polymeric product with a high molecular weight of 87100 was, delightfully, isolated in a 97% yield after 10 h. Lowering further the monomer concentration has a negative effect on both the isolated yield and molecular weight though the obtained polymers still possess good solubility in common organic solvents. At a low monomer concentration of 10 or 5 mM, only oligomers are formed after 10 h.
| No. | [M]0 (mM)b | [I]0 (mM)c | Time (h) | Yield (%) | M w d | M w/Mnd |
|---|---|---|---|---|---|---|
| a Carried out under nitrogen in reflux THF using AIBN as an initiator at a constant [I]0/[M]0 value of 0.6. b Monomer concentration. c Initiator concentration. d Estimated by GPC in THF on the basis of a linear polystyrene calibration. | ||||||
| 1 | 300 | 180 | 1 | 99 | Gel | |
| 2 | 100 | 60 | 10 | 97 | 87100 |
6.1 |
| 3 | 50 | 30 | 10 | 91 | 11300 |
2.3 |
| 4 | 10 | 6 | 10 | 80 | 3400 | 1.3 |
| 5 | 5 | 3 | 10 | 55 | 2700 | 1.1 |
The influence of initiator concentration was then examined at an optimal monomer concentration of 100 mM (Table 2). The initiator concentration does not affect the polymerization yield much but exerts a strong effect on the molecular weight of the polymer. At a concentration window of 30–80 mM, the obtained polymers are soluble and possess high molecular weights (Mw = 50900–78100). When the initiator concentration was further lowered to 12 mM or below, insoluble gel started to form though the soluble portion showed a very high Mw value up to 214800.
| No. | [I]0 (mM)b | Yield (%) | M w c | M w/Mnc |
|---|---|---|---|---|
| a Carried out under nitrogen in reflux THF at a monomer concentration of 100 mM using AIBN as an initiator for 10 h. b Initiator concentration. c Estimated by GPC in THF on the basis of a linear polystyrene calibration. d THF-soluble fraction. | ||||
| 1 | 80 | 90 | 50900 |
4.5 |
| 2 | 60 | 97 | 87100 |
6.1 |
| 3 | 30 | 97 | 64100 |
5.3 |
| 4 | 12 | 97 | 214800d |
11.7 |
| 5 | 6 | 94 | 161500d |
10.0 |
Table 3 shows the time course of the polymerization under optimal monomer and initiator concentrations. As expected, the polymerization results are improved at longer reaction time, with the yield and molecular weight of the polymer isolated at 10 h being 1.7 and 6.6-fold higher than those achieved at 0.5 h.
| No. | Time (h) | Yield (%) | M w b | M w/Mnb |
|---|---|---|---|---|
| a Carried out under nitrogen in reflux THF at monomer and initiator concentrations of 100 and 60 mM, respectively, using AIBN as an initiator. b Estimated by GPC in THF on the basis of a linear polystyrene calibration. | ||||
| 1 | 0.5 | 57 | 13100 |
2.6 |
| 2 | 1 | 84 | 18200 |
3.0 |
| 3 | 2 | 80 | 25200 |
3.2 |
| 4 | 10 | 97 | 87100 |
6.1 |
The above systematic studies enable us to polymerize other monomers under optimal polymerization conditions. Similar to 1, the polymerization of 2 proceeds smoothly, giving soluble P2 with an Mw value of 19600 in a nearly quantitative yield (Table 4, no. 2). Since monomer 3 contains four acrylate functional groups, it is expected that it will readily undergo crosslinking reaction to give insoluble gel. Indeed, the polymerization carried out at [M]0 = 50 mM for 1 h yields only a partially soluble polymer. Luckily, soluble polymer was finally isolated by further lowering the monomer concentration to 25 mM.
| No. | Monomer | [M]0 (mM)b | [I]0 (mM)c | Time (h) | Yield (%) | M w d | M w/Mnd |
|---|---|---|---|---|---|---|---|
| a Carried out under nitrogen in reflux THF using AIBN as an initiator. b Monomer concentration. c Initiator concentration. d Estimated by GPC in THF on the basis of a linear polystyrene calibration. e Data taken from Table 1, no. 2. f THF-soluble fraction. | |||||||
| 1e | 1 | 100 | 60 | 10 | 97 | 87100 |
6.1 |
| 2 | 2 | 100 | 60 | 10 | 99 | 19600 |
4.3 |
| 3 | 3 | 50 | 30 | 1 | 99 | 22000f |
5.9 |
| 4 | 3 | 25 | 15 | 10 | 99 | 33400 |
6.1 |
Structure characterization
All the intermediates and polymers were soluble, allowing us to characterize their structures by “wet” spectroscopic methods with satisfactory analysis data (see the ESI† for details). The IR spectra of P1 and its monomer 1 are given in Fig. S1† as an example. The olefin C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–H stretching vibrations of 1 occur at 1634, 1403 and 3052 cm−1, which disappear completely in the spectrum of P1. Instead, new peaks associated with the saturated C–H stretching vibrations emerge after the polymerization. The transformation of the olefin double bond of 1 into an aliphatic single bond of P1 shifts the carbonyl absorption peak at 1740 cm−1, which is now observed at 1756 cm−1. Similar phenomena are observed in the IR spectra of P2 and P3 (Fig. S2 and S3†) due to the complete consumption of the acrylate functional groups in the monomers by the polymerization reaction.
C and C–H stretching vibrations of 1 occur at 1634, 1403 and 3052 cm−1, which disappear completely in the spectrum of P1. Instead, new peaks associated with the saturated C–H stretching vibrations emerge after the polymerization. The transformation of the olefin double bond of 1 into an aliphatic single bond of P1 shifts the carbonyl absorption peak at 1740 cm−1, which is now observed at 1756 cm−1. Similar phenomena are observed in the IR spectra of P2 and P3 (Fig. S2 and S3†) due to the complete consumption of the acrylate functional groups in the monomers by the polymerization reaction.
The NMR spectroscopy offers more detailed information on the structures of the polymers. The 1H NMR spectra of 1 and P1 in dichloromethane-d2 are given in Fig. 2 for discussion. The spectrum of P1 shows no olefin proton resonances of 1 at δ 6.52, 6.25 and 5.96. New peaks assigned to aliphatic proton absorption, however, appear at δ 2.60 and 1.98. The spectrum of P1 shows aromatic proton resonances at chemical shifts similar to those of 1, suggesting that the TPE core remains intact after polymerization.
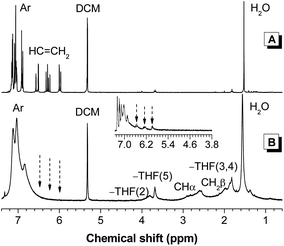 | ||
| Fig. 2 1H NMR spectra of (A) monomer 1 and (B) polymer P1 (sample taken from Table 1, no. 2) in dichloromethane-d2. Inset: 1H NMR spectrum of chloroform-d solution of P1 obtained from polymerization in toluene at δ 7.3–3.8. | ||
Interestingly, three new signals are detected at ∼δ 3.81, 3.68 and 1.82. These peaks are still observed even after repeated polymer purification, suggesting that they originate from the polymer structure. For peak assignment, we checked whether polymers prepared under different conditions also exhibit such a phenomenon. We changed the solvent from THF to toluene and conducted the polymerization under similar conditions as stated in Table 1, no. 2. After 1 h, the reaction mixture was added into hexane and the precipitates were collected and dried to a constant weight. We analyzed the obtained polymer by 1H NMR spectroscopy and found that the spectrum displays no peak at ∼δ 3.8 (Fig. 2 inset). Instead, the spectrum gives weak signals owing to the proton resonances of the unreacted double bond at δ 6.55, 6.28 and 5.98. Clearly, polymers with different structures are obtained at different solvents. Considering that the protons of THF resonate at δ 3.76 and 1.85, it is possible that the propagated polymer chains are terminated by THF molecules or in other words, P1 contains THF moieties as chain ends.
To demonstrate such possibility and rule out the effect of the TPE unit, phenyl acrylate was synthesized and utilized as a model compound. The polymerization was carried out in THF under nitrogen with a monomer concentration of 0.1 M and an initiator concentration of 0.03 M for 24 h. No precipitate was formed when the reaction mixture was added into hexane, suggesting that the product is oligomeric in nature. Similar to P1, the 1H NMR spectrum of the oligo(phenyl acrylate) exhibits no olefin proton resonances at δ 6.62, 6.32 and 6.02 (Fig. S4†). Besides the resonance peaks of aliphatic protons at δ < 3.0, the spectrum shows THF signals at δ 3.95 and 3.87. Evidently, the THF molecules take part in the polymerization and work as chain transfer agents. This modulates the chain propagation and thus gives rise to polymers with good processability. Similar observations are found in the 1H NMR spectra of P2 and P3 (Fig. S5 and S6†). Compared with its monomer, the 13C NMR spectrum of P3 shows new aliphatic carbon resonance peaks at ∼δ 68.38, 32.04, 31.42 and 26.49, which is in good agreement with the 1H NMR result (Fig. S7†).
From the above investigation, the polymerization of 1–3 may follow the routes as depicted in Scheme 2. The initial step is the formation of 2-cyanoprop-2-yl radical (R˙) by thermal dissociation of AIBN. Sequential addition of monomer to R˙ generates a propagated polymer chain 10 with multiple free radicals at its ends. Chain transfer reaction of 10 with THF yields a dead polymer molecule and meanwhile gives 2-tetrahydrofuranyl radical (THF˙) that reacts with a monomer to initiate the growth of a new chain. The propagated polymer chain carrying the THF moiety can be terminated in three pathways. The first route is the chain transfer reaction with THF to form 13. It may also undergo radical coupling with THF˙ to form 14 or self-coupling reaction to form 15. No matter by which pathway the termination occurs, it will furnish tetrahydrofuranyl-ended polymer chains that give rise to the absorption peaks at ∼δ 3.81, 3.68 and 1.82 in the 1H NMR spectrum.
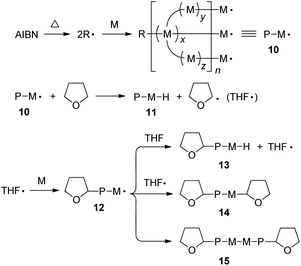 | ||
| Scheme 2 Schematic presentation of some key steps in the radical polymerization of a multi-functional acrylic monomer. Abbreviation: AIBN = azobisisobutyronitrile (initiator), R˙ = 2-cyanoprop-2-yl radical (initiator fragment), M = monomer (unit), M˙ = monomer radical, x, y, z = length of chain branch, n = degree of polymerization, P = polymer chain, and THF˙ = 2-tetrahydrofuranyl radical. | ||
Through the polymerization mechanism, we proposed a nonlinear polymer structure with hyperbranched glycogen-like topology and closed-loops for P1 (Chart 2). Glycogen contains many branches of glucose molecules and acts as a storage of energy. The unique feature of such structure is that the branches can be cut off simultaneously for large and quick energy generation. The two olefin double bonds of 1 offer four active sites for chain transfer reaction with THF and combination with THF˙ and polymer radical 12. If three sites are terminated, two possible structures I and II are formed, which serve as terminal units in P1. On the other hand, if only two sites are available for further reaction, four possible structures III–VI will be produced that function as linear units in the polymer. If only one site is terminated, two possible structures VII and VIII will be created that contribute to the branching of the polymer. It is also feasible for all the active sites to be occupied by polymer chains to form a hyperbranched structure. The irregular connection of these building units will result in a nonlinear structure similar to glycogen but with a certain number of closed loops. P2 is expected to possess a structure similar to P1, while P3 will have an even more complicated structure as monomer 3 possesses eight active sites for various skeleton constructions. Nevertheless, the present work demonstrates a strategy for ready access to synthetic polymers with structures similar to those of biomacromolecules such as glycogen, which is rarely reported.
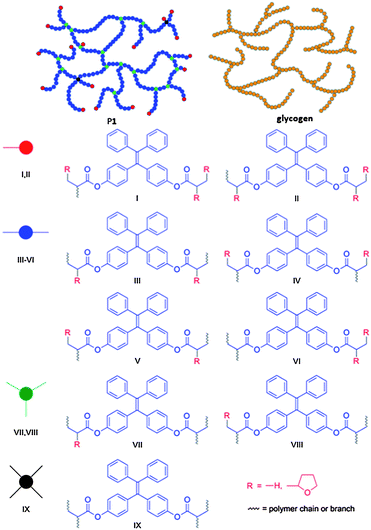
Solubility and thermal stability
Despite their high molecular weights with numerous chain interconnected points, P1–P3 possess good solubility in common organic solvents. They dissolve readily in toluene, dichloromethane, chloroform and THF but are insoluble in water and methanol. They show good film-forming ability and can form tough solid films by spin-coating or a solution-casting process. The thermal stability of the polymers was investigated by thermogravimetric analysis. As shown in Fig. S8,† P1–P3 show a 5% loss of their weight at 317–357 °C under nitrogen, revealing their strong resistance to thermolysis at high temperature.Photophysical properties
The absorption spectra of 1–3 and P1–P3 in diluted THF solutions (10 μM) are shown in Fig. S9.† Both 1 and P1 exhibit an absorption peak at 314 nm owing to the electronic transition of the TPE unit. The substitution pattern affects little the ground-state electronic transition: the UV spectra of 2 and P2 are practically similar to those of 1 and P1. Surprisingly, 3 and P3 do not show any peaks at wavelengths longer than 300 nm, presumably due to the severe steric hindrance between the substituents, which results in a more twisted structure and hence a lower conjugation.Since TPE is a typical AIE luminogen, monomers 1–3 and their polymers P1–P3 are thus expected to be AIE-active. The direct impression on the AIE nature of 1 and P1 in THF solutions and THF–water mixtures under UV irradiation is shown in Fig. 3. The emission from 1 is observable only in THF–water mixtures with a water content of >90 vol%, while P1 starts to emit at a lower water content of 50 vol%.
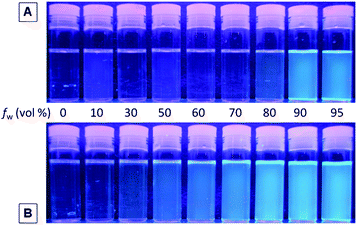 | ||
| Fig. 3 Photographs of (A) monomer 1 and (B) polymer P1 in THF–water mixtures with different water fractions (fw) taken under UV illumination from a hand-held UV lamp. | ||
In addition to visual observation, we examined the emission behaviors of 1 and P1 in THF and THF–water mixtures by a photoluminescence (PL) spectrometer. The PL spectrum of 1 in THF is basically a flat line parallel to the abscissa (Fig. S10†). However, when a large amount of water (≥90 vol%) was added to the THF solution, an emission peak emerged at 468 nm, demonstrating a typical AIE phenomenon. Its polymer P1, on the other hand, behaves differently. Despite being weak, the polymer is somewhat fluorescent in THF solution (Fig. 4). Even when a small amount of water was added to the THF solution, obvious fluorescence enhancement was observed. The emission becomes stronger progressively with increasing the amount of water in the solvent mixture. At 95 vol% water content, the PL intensity is 33-fold higher than that of pure THF solution, while the emission maximum is still located at 460 nm. The change in the relative PL intensity (I/I0) with the solvent composition of THF–water mixtures of 1 and P1 is given in Fig. 4B. While a smooth emission enhancement with the water content was observed in P1, the PL intensity of solution 1 rises abruptly after threshold water content. We have proposed that restriction of intramolecular rotation (RIR) is the main cause for the AIE phenomenon. Being a low molecular weight compound, the molecules of 1 are free to rotate in pure THF solution as well as THF–water mixtures with low water contents. In contrast, the polymer chains in P1 are knitted together by covalent bonds, which partially activate the RIR process and thus make the polymer somewhat emissive in pure THF solution. Moreover, due to its higher hydrophobicity or comparatively low solubility in aqueous mixture, aggregates are more readily formed in THF–water mixtures with low water fractions, thus making the PL process of the polymer more sensitive to the change in its surrounding solvent medium.
![(A) PL spectra of polymer P1 in THF–water mixtures with different water contents (fw). (B) Dependence of emission intensities on the solvent compositions of the THF–water mixtures of 1 and P1. [1] = [P1] = 10 μM; excitation wavelength (nm): 313 (1) and 314 (P1).](/image/article/2013/PY/c2py20485a/c2py20485a-f4.gif) | ||
| Fig. 4 (A) PL spectra of polymer P1 in THF–water mixtures with different water contents (fw). (B) Dependence of emission intensities on the solvent compositions of the THF–water mixtures of 1 and P1. [1] = [P1] = 10 μM; excitation wavelength (nm): 313 (1) and 314 (P1). | ||
Monomer 2 and polymer P2 show similar fluorescence behaviors. As shown from the photographs given in Fig. S11,†2 is nonemissive in THF but emits blue light when more than 80% of water was added to its THF solution. In contrast, a noticeable emission was observed in P2 in the THF–water mixture with a much lower water content of 50%. PL analysis shows that 2 becomes emissive in a 70% aqueous mixture (Fig. S12†) and the PL intensity reaches its maximum at 95% water content, being 71-fold higher than that of pure THF solution. Similar to P1, P2 shows faint emission in pure THF solution, whose intensity is enhanced with gradual addition of water and has attained its maximum value at 90% water content (Fig. S13†). Surprisingly, the THF solutions and THF–water mixtures of 3 and P3 emit weakly at ∼400 nm. The exact reason is still unclear at present but may be related to their highly twisted structure and low conjugation, thus resulting in inefficient emission at the shorter wavelength.
It is noteworthy that both aqueous mixtures of P1 and P2 are macroscopically homogeneous and visually transparent without precipitates even at a high water content of 95%, suggesting that the aggregates are of nano-dimension. To prove this, we analyzed the aqueous mixtures of P1 and P2 using a Zeta-potential analyzer and the data are summarized in Table S3.† No signals were detected in both polymers in THF solutions as well as in solvent mixtures with 10% water content, suggesting that the polymer chains are well-dispersed without forming any aggregates. In 30% aqueous mixture, particles with sizes of several hundred nanometers are formed. Further increment of the water fraction leads to the formation of “smaller” aggregates, revealing that the particles shrink in the presence of poor solvent.
Analysis by scanning electron microscopy shows the formation of nanoparticles upon evaporation of solution and aqueous suspension of P2 (Fig. S14†). The particles formed from THF solution have various sizes ranging from 50 to 160 nm, while those from the THF–water mixture have a much narrower size distribution (50–80 nm). Since THF is volatile, the fast evaporation of the solvent molecules may aggregate the polymer chains quickly and randomly, which gives nanoparticles of varied sizes. The polymer aggregates in the THF–water mixture, on the other hand, are formed by dropwise addition of water into the THF solution under vigorous stirring and thus are inherently more uniform. Moreover, the comparative lower volatility of the aqueous mixture also enables the dissolved polymer chains to cluster slowly to form more uniform nanoparticles. Similar spherical nanoparticles with different sizes are also formed from the THF solutions of P1 and P3 (Fig. S15†).
Chemical sensor
There is an urgent call for the development of new chemosensors that can sensitively detect volatile organic compounds of potential health hazards. As shown in Fig. 5, the light emission from the spot of 1 on a TLC plate (panel A) was turned off when the plate was put into a Petri-dish saturated with dichloromethane vapor (panel B). The emission was recovered again when the solvent was evaporated. The solvent vapor may have condensed and formed a thin liquid layer on the surface of the TLC plate in the saturated vapor environment, which dissolves the adsorbed luminogen molecule and consequently quenches the light emission. After solvent evaporation, the molecules aggregate and hence emit again (panel C). This luminescent sensoric “on/off” switching process is completely reversible and reliably repeatable many times as the involved process is a nondestructive physical cycle of dissolution-aggregation. The same effect can also be achieved by other low boiling point organic solvents such as acetone and THF. A similar phenomenon was also found in P1. Instead of complete quenching, weak emission was still observed in the fumed plate (Fig. 5E). Again, such process is reversible and can be repeated many times without fatigue. | ||
| Fig. 5 Photographs of spots of (A–C) monomer 1 and (D–F) polymer P1 deposited on TLC plates (A and D) before and (B and E) after exposure to vapor of dichloromethane for 1 min. The photographs in (C and F) were taken after the organic vapor in (B and E) had been evacuated. The fluorescent images were taken under UV illumination from a hand-held UV lamp. | ||
Photo-cross-linking and photopatterning
As stated before, all the polymers can form uniform, tough thin films by spin-coating of their solutions. When the prepared films are exposed to UV light through copper photomasks, the irradiated parts are crosslinked while the unexposed areas are rendered soluble. After development in 1,2-dichloroethane, negative photoresist patterns with sharp edges are developed, which emit under UV irradiation (Fig. 6). Interestingly, monomers 1–3 are also film-forming and photosensitive and can generate similar fluorescent patterns using the same process, which is truly impressive (Fig. S16†). | ||
| Fig. 6 Negative-tone photoresist patterns generated by photo-crosslinking of thin films of (A) P1 (sample from Table 1, no. 2), (B) P2 (Table 4, no. 2) and (C) P3 (Table 4, no. 4) through copper masks. The photographs were taken under UV illumination from a hand-held UV lamp. Square length: 100 μm. | ||
The photo-crosslinking reaction of 1–3 can be explained easily by the UV-induced polymerization of the acrylate functional units. The underlying mechanism for the polymers, however, remains unknown since they possess neither photo-crosslinkable triple or double bonds at their chains or ends21 nor phenyl benzoate functionality that can undergo photo Fries rearrangement to form a photosensitive benzophenone unit.22 Thus, the vinyl core of the TPE unit should be, in most cases, responsible for the photo-crosslinking of the polymers. To prove this, we prepared compound 16 by replacement of the TPE unit in 1 with a 9,9-diphenylfluorene core (Scheme 3). The single crystal structure of the model compound resembles that of 1 (Table S4†). It was polymerized under the conditions stated in Table 1, no. 2. The resulting polymer, similar to P1, possesses a glycogen-like structure and shows good film-forming ability. It, however, undergoes no crosslinking reaction in the presence of UV light. Through such investigation, we proposed the mechanism for the photo-crosslinking reaction using P1 as an example. As depicted in Scheme 4, two polymer radicals (B and C) were initially formed from polymer A. Radical B was then reacted with the vinyl core of the TPE unit of polymer A to generate a new radical D. Combination of polymer radical D with radical B or C completed the crosslinking reaction and produced polymer E or F, respectively.
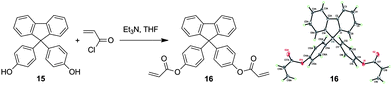 | ||
| Scheme 3 Synthesis of acrylic monomer 16 and its single crystal structure. | ||
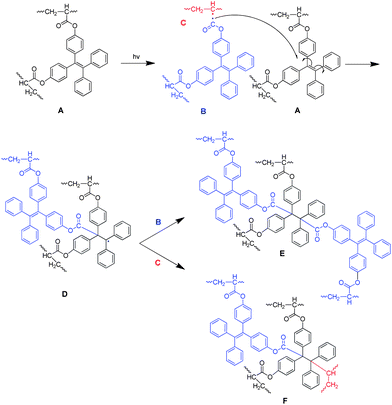 | ||
| Scheme 4 Proposed mechanism for the photocrosslinking of P1. | ||
Optical transparency and light refractivity
Polyesters are promising candidates for photonic applications because of their high optical clarity. Polyesters such as poly(methyl methacrylate) (PMMA), poly(ethylene teraphthalate) and polycarbonate (PC) are well known and widely used “organic glasses”. Since P1–P3 are polyesters, they are thus expected to show high optical clarity. The light transmission spectra of P1–P3 are given in Fig. S17.† All the polymers absorb little light in the visible spectral region and allow all the light with wavelengths longer than 400 nm to transmit through, suggesting that they possess excellent optical transparency.The polymers are comprised of polarizable aromatic rings and ester groups and may possess high refractive indices (RI). As can be seen from Fig. 7, P1 displays high RI values (n = 1.6825–1.5653) in a wide wavelength region (400–1700 nm). Similarly, P2 and P3 exhibit also high RI values (n = 1.6756–1.5871 for P2 and 1.6283–1.5800 for P3), which are much higher than those of the commercially important optical plastics (e.g. n = ∼1.49 for PMMA). The RI values of P1–P3 at 632.8 nm are also measured using a Gaertner ellipsometer and summarized in Table 5.
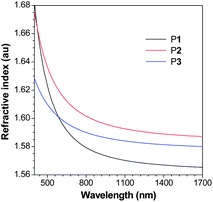 | ||
| Fig. 7 Wavelength dependence of refractive index of thin films of P1 (sample from Table 1, no. 2), P2 (sample from Table 4, no. 2) and P3 (sample from Table 4, no. 4). | ||
| polymer | n 632.8 | ν D | ν D′ | D | D′ |
|---|---|---|---|---|---|
| a All data taken from Fig. 7, except the refractive index (n) at 632.8 nm, which was measured by a Gaertner ellipsometer. Abbreviation: νD = Abbé number, νD′ = modified Abbé number and D(′) = chromatic dispersion. | |||||
| P1 | 1.608 | 15.80 | 118.23 | 0.0633 | 0.00846 |
| P2 | 1.605 | 21.51 | 111.25 | 0.0465 | 0.00899 |
| P3 | 1.587 | 38.26 | 153.13 | 0.0261 | 0.00653 |
For a material to be useful for practical applications, its chromatic dispersion should be small. The Abbé number (νD) of a material is a measure of the variation or dispersion in its RI value with wavelength and is defined as: νD = (nD − 1)/(nF − nC), where nD, nF and nC are the RI values at wavelengths of Fraunhofer D, F and C spectral lines of 589.2, 486.1 and 656.3 nm, respectively. A modified Abbé number (νD′) has also been proposed to evaluate the application potential of an optical material, using its RI values at the non-absorbing wavelengths of 1064, 1319 and 1550 nm.23 The modified Abbé number is defined as: νD′ = (n1319 − 1)/(n1064 − n1550), where n1319, n1064 and n1550 are the RI values at 1319, 1064 and 1550 nm, respectively. The chromatic dispersion (D or D′) is the constringency of the Abbé number (νD or νD′) and is defined as: D(′) = 1/νD(′). The νD′ value of P1 is 118.23, from which a D′ value of 8.46 × 10−3 is derived. P2 exhibits similar νD′ and D′ values. On the other hand, P3 shows a much lower chromatic dispersion. The D value of P3 is 26.1 × 10−3, which is comparable to those of PC (D = 29.7 × 10−3) and PMMA (D = 17.5 × 10−3).24
Explosive detection
2,4,6-Trinitrotoluene (TNT) and 2,4-dinitrotoluene (DNT) are important chemical species to detect in munition remediation sites25 and mine fields.26 As they are also warfare explosives, their detection has great value to homeland security27 and antiterrorism implications.28 We have recently developed fluorescent chemosensors for the efficient detection of ultra-trace explosive analytes by using nanoaggregates of AIE polymeric materials.29 Such an approach can avoid the emission quenching problem caused by intrinsic autoaggregation of polymer chains and analyte-induced aggregation, thus generating less “false” signals and resulting in a higher signal-to-noise ratio and sensitivity. The fluorescence annihilation is proved by a static quenching mechanism, in which the electron-rich polymer strands bind to the electron-deficient analytes and are in the non-emissive or dark state.30 To investigate whether the present polymers can work as efficient fluorescent chemosensors for explosive detection, nanoaggregates of P1 in the THF–water mixture (1:9 v/v) were used as a probe, while picric acid (PA) was used as a model explosive analyte instead of TNT and DNT due to its commercial availability. Although P1–P3 are not fully conjugated, their glycogen-like structures with closed-loops may help trap the analyte molecules and thus facilitate the PL quenching process.
As shown in Fig. 8, the PL of nanoaggregates of P1 in a 90% aqueous mixture is progressively weakened when an increasing amount of PA is added. The PL quenching can be discernible at a PA concentration less than 1 ppm. No signal was detected at a PA concentration of 0.67 mM. The Stern–Volmer plot of relative emission intensity (I0/I) versus PA concentration gives an upward-bending curve instead of a straight line, revealing that the emission quenching becomes more efficient with increasing the quencher concentration. Large quenching constants up to 207000 M−1 are deduced from the plot, which are much higher than those of polysiloles and polygermoles (6710–11000 M−1) reported previously.31
![(A) PL spectra of nanoaggregates of P1 in a THF–water mixture (1 : 9 v/v) containing different amounts of picric acid (PA). (B) Stern–Volmer plot of PL quenching of P1 by PA. I0 = PL intensity in the absence of PA (i.e. [PA] = 0 mM).](/image/article/2013/PY/c2py20485a/c2py20485a-f8.gif) | ||
| Fig. 8 (A) PL spectra of nanoaggregates of P1 in a THF–water mixture (1:9 v/v) containing different amounts of picric acid (PA). (B) Stern–Volmer plot of PL quenching of P1 by PA. I0 = PL intensity in the absence of PA (i.e. [PA] = 0 mM). | ||
Polymer hydrolysis
Being polyacrylates, P1–P3 may hydrolyze under appropriate conditions, causing the polymer skeleton to collapse abruptly. This releases dihydroxyl- or tetrahydroxyl-substituted TPE that can undergo free rotation in solution with little constraint. Moreover, these compounds are oxidized readily in air to form nonemissive quinone derivatives.32 All these attributes have quenched the light emission of the polymers, making monitoring of their hydrolysis process possible.The change in the PL spectrum of P1 in a 90% aqueous mixture with time upon addition of KOH aqueous solution is shown in Fig. 9. The emission becomes weaker progressively with the hydrolysis time. At 170 min, the PL intensity drops 30-fold. Since TPE derivatives are biocompatible,33 the present polymers are promising materials for biodegradable drug delivery that serve both as drug carrier and fluorescent probe. The drug can be loaded in the solution state. Water is then added to aggregate and shrink the polymer chains, which turns their emission on and traps the drug molecules inside their network. UV irradiation of the nanoparticles will further crosslink the polymers and lock the drug molecules. When the drug-embedded nanoparticles enter the cell with basic environment, they will undergo hydrolysis. This collapses their structure and releases the drug, whose rate can be followed by monitoring the PL change during the process. We are now working on the exploration of the drug delivery application of these luminescent polyacrylates, details of which will be presented in due course.
![(A) Change in the PL spectrum of nanoaggregates of P1 in a THF–water mixture (1 : 9 v/v) with time upon addition of a small amount of KOH solution. (B) Plot of emission intensity versus hydrolysis time. [P1] = 10 μM; [KOH] = 13.4 M; excitation wavelength = 314 nm.](/image/article/2013/PY/c2py20485a/c2py20485a-f9.gif) | ||
| Fig. 9 (A) Change in the PL spectrum of nanoaggregates of P1 in a THF–water mixture (1:9 v/v) with time upon addition of a small amount of KOH solution. (B) Plot of emission intensity versus hydrolysis time. [P1] = 10 μM; [KOH] = 13.4 M; excitation wavelength = 314 nm. | ||
Conclusions
In this work, a facile approach for the synthesis of hyperbranched polyacrylates is developed. Polymerization of TPE-containing di- and tetra-acrylates was initiated by AIBN in reflux THF, generating soluble, high molecular weight polymers with glycogen-like structures and closed loops in nearly quantitative yields.All the polymers are thermally stable, losing little of their weight when heated to above 300 °C. Both the monomers and polymers are nonemissive or weakly fluorescent in solutions but become strong emitters when aggregated in poor solvents, demonstrating a phenomenon of aggregation-induced or enhanced emission. Zeta-potential analysis reveals that the polymer nanoparticles shrink upon aggregation. The emission of the monomer and polymer spots on the TLC plates can be turned “off” and “on” by wetting and dewetting processes by vapors of organic solvents. The polymers are photo-responsive and their thin films can be readily crosslinked by UV irradiation to furnish fluorescent negative photoresist patterns with good resolution. Their thin films are optically transparent and exhibit high refractive indices as well as low chromatic aberration. The emission of the polymer nanoaggregates can be quenched efficiently by picric acid with large quenching constants, suggesting that they are promising fluorescent chemosensors for explosive detection. The fluorescent polymers can be hydrolyzed readily under appropriate conditions, implicative of their utility in the controlled drug delivery. These unique features make the polymers promising fluorescent materials for high-tech applications.
Experimental section
Materials
THF was distilled under normal pressure from sodium benzophenone ketyl under nitrogen immediately prior to use. Benzophenone (4), 4,4′-dihydroxybenzophenone (5), 4-hydroxybenzophenone (8), zinc powder, titanium(IV) chloride (TiCl4), triethylamine, acryloyl chloride and azobisisobutyronitrile (AIBN) were used as received without further purification.Instrumentation
1H and 13C NMR spectra were measured on a Bruker ARX 300 or 400 NMR spectrometer using chloroform-d or dichloromethane-d2 as a solvent and tetramethylsilane (TMS; δ = 0 ppm) as an internal standard. IR spectra were recorded on a Perkin-Elmer 16 PC FT-IR spectrophotometer. HR-MS spectra were recorded on a GCT premier CAB048 mass spectrometer. Single crystal X-ray diffraction intensity data were collected at 100 K on a Bruker-Nonices Smart Apex CCD diffractometer with graphite monochromated Mo Kα radiation. Processing of the intensity data was carried out using the SAINT and SADABS routines, and the structure and refinement were conducted using the SHELTL suite of X-ray programs (version 6.10). UV-vis absorption and light transmission spectra were measured on a Milton Roy Spectronic 3000 array spectrophotometer. PL spectra were recorded on a Perkin-Elmer LS 55 spectrofluorometer. Thermogravimetric analysis (TGA) was carried out under nitrogen on a Perkin-Elmer TGA 7 analyzer at a heating rate of 10 °C min−1. Images of scanning electron microscopy were taken on a JSM-6700F electron microscope. Particle sizes of aggregates in THF–water mixtures were measured on a Brookhaven Instruments Corporation 90 Plus/B1-MAS Zetaplus Zeta Potential Analyzer. Relative number- (Mn) and weight-average (Mw) molecular weights and polydispersity indices (Mw/Mn) of the polymers were estimated by a Waters Associates gel permeation chromatography (GPC) system equipped with RI and UV detectors. THF was used as eluent at a flow rate of 1.0 mL. A set of monodisperse linear polystyrenes covering a molecular weight range of 103 to 107 was used as standards for molecular weight calibration. RI values were measured on a Gaertner L116C ellipsometric thin film thickness measurement system using a 1 mW He–Ne laser beam (λ = 632.8 nm) as the light source or determined on a J. A. Woollam variable angle ellipsometry system with a wavelength tunability from 300 to 1700 nm. To fit the acquired Ψ and Δ curves with the data obtained from the 3-layer optical model consisting of crystalline silicon substrate, 2 nm SiO2 layer and a uniform polymer film, the Levenberg–Marquardt regression algorithm was employed. The Cauchy dispersion law was applied to describe the polymer layer from visible to IR spectral region.Preparation of aggregates
Stock THF solutions of the monomers and polymers with a concentration of 1 × 10−4 M were prepared. 1 mL of these stock solutions was transferred to 10 mL volumetric flasks. After adding an appropriate amount of THF, water was added dropwise under vigorous stirring to furnish 1 × 10−5 M solutions in THF–water mixtures with specific water fractions (0–90 vol%). THF–water mixtures (1 × 10−5 M) with 95 vol% water fractions were prepared from THF solution (1 × 10−3 M) in the same manner. Absorption, emission and particle size of the resulting solutions or aggregates were measured immediately after the sample preparation.Photopatterning
Photo-cross-linking reactions of the polymer films were conducted in air at room temperature using 365 nm light obtained from a Spectroline ENF-280C/F UV lamp at a distance of 5 cm from the light source. The incident light intensity was ∼18.5 mW cm−2. The films were prepared by spin-coating the polymer solution (10% w/w in 1,2-dichloroethane) at 1200 rpm for 1 min on a silicon wafer. The photoresist patterns were generated by UV irradiation of the polymer films through copper photomasks for 20 min followed by development in 1,2-dichloroethane. The photos were taken on an optical microscope (Nikon ECLIPSE 80i) under a UV light source.Explosive detection and polymer hydrolysis
A stock solution of picric acid with a concentration of 2 mg mL−1 was prepared by dissolving an appropriate amount of PA in THF. PL titration was carried out by adding aliquots of PA solution in nanoaggregates of P1 in a THF–water mixture (1:9 v/v).
The fluorescence from nanoaggregates of P1 in a 90% aqueous mixture (10 μM) was recorded continuously from 0 to 170 min after addition of 1 drop of 13.4 M KOH aqueous solution.
Acknowledgements
The work reported in this paper was partially supported by the National Science Foundation of China (20974028), the RPC and SRFI Grants of HKUST (RPC10SC13, RPC11SC09, and SRFI11SC03PG), the Research Grants Council of Hong Kong (604711, 603509, HKUST2/CRF/10, and N_HKUST620/11), the Innovation and Technology Commission (ITCPP/17-9), the University Grants Committee of Hong Kong (AoE/P-03/08), and Guangdong Innovative Research Team Program.Notes and references
- P. C. Hiemenz and T. P. Lodge, Polymer Chemistry, CRC Press, Boca Raton, FL, 2007 Search PubMed.
- (a) C. J. Hawker and J. M. J. Fréchet, ACS Symp. Ser., 1996, 624, 132 CrossRef CAS; (b) Z. B. Guan, J. Am. Chem. Soc., 2002, 124, 5616 CrossRef CAS.
- (a) S. M. Grayson and J. M. J. Fréchet, Chem. Rev., 2001, 101, 3819 CrossRef CAS; (b) D. A. Tomalia, A. M. Naylor and W. A. Goddard, Angew. Chem., 1990, 102, 119 CrossRef CAS; (c) S. Hecht and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2001, 40, 74 CrossRef CAS; (d) Y. H. Kim, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1685 CrossRef CAS.
- (a) J. W. Chen and Y. Cao, Macromol. Rapid Commun., 2007, 28, 1714 CrossRef CAS; (b) B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2679 CrossRef CAS; (c) D. A. Tomalia, Prog. Polym. Sci., 2005, 30, 294 CrossRef CAS; (d) C. R. Yates and W. Hayes, Eur. Polym. J., 2004, 40, 1257 CrossRef CAS; (e) C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183 CrossRef CAS; (f) S. Hecht, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1047 CrossRef CAS; (g) Y. H. Kim, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1685 CrossRef CAS; (h) D. Y. Sogah. US Pat., 4906713, 1990.
- (a) A. Hult, M. Johansson and E. Malmstrom, Adv. Polym. Sci., 1999, 143, 1 CrossRef CAS; (b) Y. H. Kim, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1685 CrossRef CAS; (c) D. A. Tomalia and J. M. J. Fréchet, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2719 CrossRef CAS.
- (a) G. R. Newkome, C. N. Moorefield and F. Vçgtle, Dendrons and Dendrimers: Concepts, Synthesis and Applications, Wiley-VCH, Weinheim, 2001 Search PubMed; (b) R. M. Crooks, M. Q. Zhao, L. Sun, W. Chechik and L. K. Yeung, Acc. Chem. Res., 2001, 34, 181 CrossRef CAS; (c) M. Ballauff and C. N. Likos, Angew. Chem., Int. Ed., 2004, 43, 2998 CrossRef CAS; (d) A. M. Caminade and J. P. Majoral, Acc. Chem. Res., 2004, 37, 341 CrossRef CAS; (e) D. Méry and D. Astruc, Coord. Chem. Rev., 2006, 250, 1965 CrossRef; (f) J. L. Mynar, T. Yamamoto, A. Kosaka, T. Fukushima, N. Ishii and T. Aida, J. Am. Chem. Soc., 2008, 130, 1530 CrossRef CAS; (g) W. S. Li and T. Aida, Chem. Rev., 2009, 109, 6047 CrossRef CAS; (h) B. M. Rosen, C. J. Wilson, D. A. Wilson, M. Peterca, M. R. Imam and V. Percec, Chem. Rev., 2009, 109, 6275 CrossRef CAS.
- (a) Y. H. Kim, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1685 CrossRef CAS; (b) B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2505 CrossRef CAS; (c) M. Jikei and M. Kakimoto, Prog. Polym. Sci., 2001, 26, 1233 CrossRef CAS; (d) P. Froehling, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3110 CrossRef CAS; (e) M. Jikei and M. Kakimoto, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1293 CrossRef CAS; (f) C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183 CrossRef CAS; (g) B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2679 CrossRef CAS; (h) D. Yan, C. Gao and H. Frey, Hyperbranched Polymers: Synthesis, Properties, and Applications, John Wiley & Sons, Inc., New Jersey, 2011 Search PubMed.
- Y. H. Kim, US Pat., 4857630, 1987.
- P. J. Flory, J. Am. Chem. Soc., 1953, 74, 2718 CrossRef.
- I. Mita, R. F. T. Stepto and U. W. Suter, Pure Appl. Chem., 1994, 66, 2483 CrossRef CAS.
- (a) Y. H. Kim and O. W. Webster, J. Am. Chem. Soc., 1990, 112, 4592 CrossRef CAS; (b) X. R. Li, J. Zhan and Y. S. Li, Macromolecules, 2004, 37, 758 CrossRef; (c) X. R. Li, Y. S. Li, Y. J. Tong, L. Shi and X. Liu, Macromolecules, 2003, 36, 5537 CrossRef CAS; (d) S. H. Mansour, N. N. Rozik, K. Dirnberger and N. E. Ikladious, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3278 CrossRef CAS; (e) T. Biela and I. Polanczyk, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4214 CrossRef CAS; (f) L. Ye, Z. G. Feng, Y. M. Zhao, F. Wu, S. Chen and G. Q. Wang, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4214 CrossRef; (g) J. Kong, X. D. Fan, Q. F. Si, G. B. Zhang, S. J. Wang and X. Wang, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3930 CrossRef CAS; (h) J. F. Miravet and J. M. J. Fréchet, Macromolecules, 1998, 31, 3461 CrossRef CAS.
- (a) J. Liu, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2009, 109, 5799 CrossRef CAS; (b) J. W. Y. Lam and B. Z. Tang, Acc. Chem. Res., 2005, 38, 745 CrossRef CAS.
- J. M. J. Fréchet, M. Henmi, I. Gitsov, S. Aoshima, M. R. Leduc and R. B. Grubbs, Science, 1995, 269, 1080 Search PubMed.
- (a) D. M. Knauss and H. A. Al-Muallem, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4289 CrossRef CAS; (b) C. J. Hawker, J. M. J. Fréchet, R. B. Grubbs and J. Dao, J. Am. Chem. Soc., 1995, 117, 10763 CrossRef CAS; (c) C. Cheng, K. L. Wooley and E. Khoshdel, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4754 CrossRef CAS; (d) Z. Wang, J. He, Y. Tao, L. Yang, H. Jiang and Y. Yang, Macromolecules, 2003, 36, 7446 CrossRef CAS; (e) P. F. W. Simon, W. Radke and A. H. E. Müller, Macromol. Rapid Commun., 1997, 18, 865 CrossRef CAS; (f) Y. Y. Xu, C. Gao, H. Kong, D. Y. Yan, P. Luo, W. W. Li and Y. Y. Mai, Macromolecules, 2004, 37, 6264 CrossRef CAS; (g) S. G. Gaynor, S. Edelman and K. Matyjaszewski, Macromolecules, 1996, 29, 1079 CrossRef CAS.
- (a) S. Graham, P. A. G. Cormack and D. C. Sherrington, Macromolecules, 2005, 38, 86 CrossRef CAS; (b) F. Isaure, P. A. G. Cormack, S. Graham, D. C. Sherrington, S. P. Armes and V. Buetuen, Chem. Commun., 2004, 1138 RSC; (c) G. Saunders, P. A. G. Cormack, S. Graham and D. C. Sherrington, Macromolecules, 2005, 38, 6418 CrossRef CAS; (d) F. Isaure, P. A. G. Cormack and D. C. Sherrington, J. Mater. Chem., 2003, 13, 2701 RSC; (e) R. M. England and S. Rimmer, Polym. Chem., 2010, 1, 1533 RSC.
- (a) N. Ide and T. Fukuda, Macromolecules, 1997, 30, 4268 CrossRef CAS; (b) N. Ide and T. Fukuda, Macromolecules, 1999, 32, 95 CrossRef CAS; (c) I. Bannister, N. C. Billingham, S. P. Armes, S. P. Rannard and P. Findlay, Macromolecules, 2006, 39, 7483 CrossRef CAS; (d) Y. Lin, X. Liu, X. Li, J. Zhan and Y. Li, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 26 CrossRef CAS; (e) A. T. Slark, D. C. Sherrington, A. Titterton and I. K. Martin, J. Mater. Chem., 2003, 13, 2711 RSC.
- N. O. Brien, A. McKee, D. C. Sherrington, A. T. Slark and A. Titterton, Polymer, 2000, 41, 6027 CrossRef.
- (a) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC; (b) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332 RSC; (c) J. W. Chen, B. Xu, X. Y. Ouyang, B. Z. Tang and Y. Cao, J. Phys. Chem. A, 2004, 108, 7522 CrossRef CAS; (d) H. Tong, Y. Dong, Y. Hong, M. Häussler, J. W. Y. Lam, H. H. Y. Sung, X. Yu, J. Sun, I. D. Williams, H. S. Kwok and B. Z. Tang, J. Phys. Chem. C, 2007, 111, 2287 CrossRef CAS.
- J. D. Luo, Z. L. Xie, J. W. Y. Lam, L. Cheng, H. Y. Chen, C. F. Qiu, H. S. Kwok, X. W. Zhan, Y. Q. Liu, D. B. Zhu and B. Z. Tang, Chem. Commun., 2001, 1741 Search PubMed.
- (a) R. Hu, J. L. Maldonado, M. Rodriguez, C. Deng, C. K. W. Jim, J. W. Y. Lam, M. M. F. Yuen, G. R. Ortiz and B. Z. Tang, J. Mater. Chem., 2012, 22, 232 RSC; (b) Y. Liu, S. Chen, J. W. Y. Lam, P. Lu, R. T. K. Kwok, F. Mahtab, H. S. Kwok and B. Z. Tang, Chem. Mater., 2011, 23, 2536 CrossRef CAS.
- (a) R. Hu, J. W. Y. Lam, J. Liu, H. H. Y. Sung, I. D. Williams, Z. Yue, K. S. Wong, M. M. F. Yuen and B. Z. Tang, Polym. Chem., 2012, 3, 1481 RSC; (b) M. Häussler, R. Zheng, J. W. Y. Lam, H. Tong, H. Dong and B. Z. Tang, J. Phys. Chem. B, 2004, 108, 10645 CrossRef.
- (a) C. K. W. Jim, A. J. Qin, J. W. Y. Lam, M. Häussler, J. Liu, M. M. F. Yuen, J. K. Kim, K. M. Ng and B. Z. Tang, Macromolecules, 2009, 42, 4099 CrossRef CAS; (b) A. Qin, J. W. Y. Lam, H. Dong, W. Lu, C. K. W. Jim, Y. Q. Dong, M. Häussler, H. H. Y. Sung, I. D. Williams, G. K. L. Wong and B. Z. Tang, Macromolecules, 2007, 40, 4879 CrossRef CAS; (c) H. Dong, R. Zheng, J. W. Y. Lam, M. Häussler and B. Z. Tang, Macromolecules, 2005, 38, 6382 CrossRef CAS.
- (a) C. J. Yang and S. A. Jenekhe, Chem. Mater., 1995, 7, 1276 CrossRef CAS; (b) C. J. Yang and S. A. Jenekhe, Chem. Mater., 1994, 6, 196 CrossRef CAS.
- (a) J. C. Seferis, J. Brandrup and E. H. Immergut, Polymer Handbook, Wiley, New York, 3rd edn, 1989, p. 451 Search PubMed; (b) N. J. Mills and J. I. Kroschwitz, Concise Encyclopedia of Polymer Science and Engineering, Wiley, New York, 1990, p. 683 Search PubMed.
- J. I. Steinfeld and J. Wormhoudt, Annu. Rev. Phys. Chem., 1998, 49, 203 CrossRef CAS.
- A. M. Rouhi, Chem. Eng. News, 1997, 75, 14 Search PubMed.
- (a) A. Fainberg, Science, 1992, 255, 1531 CAS; (b) J. C. Sanchez, A. G. Dipasquale, A. L. Rheingold and W. C. Trogler, Chem. Mater., 2007, 19, 6459 CrossRef CAS; (c) H. Sohn, M. J. Sailor, D. Magde and W. C. Trogler, J. Am. Chem. Soc., 2003, 125, 3821 CrossRef CAS; (d) D. Zhao and T. M. Swager, Macromolecules, 2005, 38, 9377 CrossRef CAS; (e) J. C. Sanchez and W. C. Trogler, J. Mater. Chem., 2008, 18, 3134 Search PubMed; (f) T. Naddo, Y. Che, W. Zhang, K. Balakrishnan, X. Yang, M. Yen, J. Zhao, J. S. Moore and L. Zhang, J. Am. Chem. Soc., 2007, 129, 6978 CrossRef CAS; (g) Y. Long, H. Chen, Y. Yang, H. Wang, Y. Yang, N. Li, K. Li, J. Pei and F. Liu, Macromolecules, 2009, 42, 6501 CrossRef CAS.
- (a) D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537 CrossRef CAS; (b) K. J. Albert, N. S. Lewis, C. L. Schauer, G. A. Sotzing, S. E. Stitzel, T. P. Vaid and D. R. Walt, Chem. Rev., 2000, 100, 2595 CrossRef CAS.
- P. Lu, J. W. Y. Lam, J. Liu, C. K. W. Jim, W. Yuan, N. Xie, Y. Zhong, Q. Hu, K. S. Wong, K. K. L. Cheuk and B. Z. Tang, Macromol. Rapid Commun., 2010, 31, 834 CrossRef CAS.
- J. Liu, Y. Zhong, P. Lu, Y. Hong, J. W. Y. Lam, F. Mahtab, Y. Yu, K. S. Wong and B. Z. Tang, Polym. Chem., 2010, 1, 426 RSC.
- H. Sohn, M. J. Sailor, D. Magde and W. C. Trogler, J. Am. Chem. Soc., 2003, 125, 3821 CrossRef CAS.
- (a) P. Deya, M. Dopico and A. G. Raso, Tetrahedron, 1987, 43, 3523 CrossRef CAS; (b) D. H. Barton, J. P. Finet and M. Thomas, Tetrahedron, 1988, 44, 6397 CrossRef CAS.
- (a) M. Faisal, Y. Hong, J. Liu, Y. Yu, J. W. Y. Lam, A. Qin, P. Lu and B. Z. Tang, Chem.–Eur. J., 2010, 16, 4266 CrossRef CAS; (b) Y. Hong, C. Feng, Y. Yu, J. Liu, J. W. Y. Lam, K. Q. Luo and B. Z. Tang, Anal. Chem., 2010, 82, 7035 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section for general information, synthesis and detailed characterization data, crystal data for 1, E-2 and 16, IR, NMR and UV spectra of 1–3 and P1−P3, TGA thermograms of P1–P3, PL spectra of 1, 2 and P2, photographs of 2 and P2, particle sizes of P1 and P2, SEM images of nanoparticles of P1–P3, photoresist patterns generated from thin films of 1–3 and light transmission spectra of THF solutions of P1–P3. CCDC 885935–885937. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2py20485a |
| This journal is © The Royal Society of Chemistry 2013 |