Facile synthesis of gradient copolymers via semi-batch copper(0)-mediated living radical copolymerization at ambient temperature
Yin-Ning
Zhou
ab and
Zheng-Hong
Luo
*ab
aDepartment of Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, P. R. China. E-mail: luozh@sjtu.edu.cn
bDepartment of Chemical and Biochemical Engineering, Xiamen University, Xiamen 361005, P. R. China. E-mail: luozh@xmu.edu.cn
First published on 29th August 2012
Abstract
In this work, we report an example of the facile synthesis of methyl methacrylate/tert-butyl acrylate (MMA/tBA) gradient copolymers (poly(MMA-grad-tBA) using the Cu(0) and conventional ATRP ligands as catalysts in DMF solvent at 25 °C. Semi-batch copper(0)-mediated living radical copolymerization technique (Cu(0)-mediated LRP) was used for achieving the chain gradient microstructure of the resulting copolymers. We also compared copolymerizations with two different ATRP ligands at ambient temperature allowing control over the molecular weight and polydispersity with a quarter of catalyst concentration versus a conventional ATRP in dipolar protic solvent (i.e. DMF), while the reaction temperature up to 80 °C in a non-polar medium (i.e. toluene) in order to reach the above polymerization efficiency. The addition of a small amount of reducing agent (i.e. hydrazine hydrate) into the reaction system allows the reaction proceeding in the oxygen tolerant system without losing control and decreasing total conversion such as using the reagents without deoxygenating.
Introduction
Gradient copolymer, in which the instantaneous composition of copolymer changes continuously from one end of the chain to the other, constitutes a relatively new class of polymer with a molecular structure that differs from those of random and block copolymers.1,2 As a novel type of chain microstructure, synthesis of gradient copolymers and evaluation of their properties have received increasing interest recently.3–8 On the other hand, both theoretical and experimental investigations demonstrated that the gradient composition change of the gradient copolymers can result in less intrachain and interchain repulsion compared to block copolymers, leading to unique behaviors, especially unique thermal properties in bulk gradient copolymers.2,9–13 Arising from their unique structures and properties, the behavior of gradient copolymers is useful for some featured applications including designing biosensors, membranes, and substrates for separation of biomolecules,14 compatibilizers in polymer blends15 and damping materials,16etc.The recent advent of controlled/“living” radical polymerization (CLRP) techniques makes the control of chain structure no longer a formidable task.17 Currently, the most used CLRP involves nitroxide mediated polymerization (NMP),18 atom transfer radical polymerization (ATRP),19,20 reversible addition fragmentation chain transfer polymerization (RAFT),21 and single-electron transfer and single-electron transfer degenerative chain transfer living radical polymerization (SET-LRP and SET-DTLRP).22,23 More recently, more attention has been paid to the reductions in the amount of copper required for ATRP through the techniques of activators (re)generated by electron transfer (A(R)GET), initiators for continuous activator regeneration (ICAR).24,25 In addition, although the mechanism of SET-LRP is still under debate in the literature,22,23,26–32 the SET-LRP with Cu(0) as catalyst has shown some distinct advantages over other CLRPs since its emergence in 2006,23 including low temperature polymerization conditions, a small amount of catalyst, an ultrafast polymerization rate and obtaining high molecular weight polymers with narrow molecular weight distribution. Putting the undefined mechanism aside, the direct use of Cu(0) as the catalyst is attractive since Cu(0) is cheaper and easier to handle than Cu(I) and Cu(II) complexes. Moreover, the SET-LRPs of acrylate monomers can be easily performed in these dipolar protic and aprotic solvents (i.e. dimethyl sulfoxide (DMSO), N,N-dimethylformamide (DMF)) at ambient temperatures,33–39 while Perrier et al. reported the Cu(0)-mediated LRP of methyl methacrylate (MMA) in toluene at 30 °C with well-controlled behavior based on the use of an active ligand.40 In addition, Perrier et al.'s recent report implied that the Cu(0)-mediated polymerization of methacrylate (MA) may be an intricate control process and the early loss of control in polymerization can be prevented by delaying the addition of monomer or addition of a small amount of 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO).41 Percec et al. reported a detailed kinetic investigation and structural analysis of the polymer products prepared by the Cu(0)-mediated LRP in solvents mediating different degrees of disproportionation.26 Additionally, Percec et al.'s previous work,42 which is an important inspiration significance and reference value to today's research, showed that the addition of reducing agents such as phenol and phenolates as well as hydrazine or ascorbic acid reduces the amount of Cu(II) species. The potential benefit of these additives was believed to be a regeneration of active Cu(I) species from Cu(II) species via a redox process.42–44
In general, introducing a comonomer in CLRP offers the possibility of preparing tailor-made copolymers with specific desired characteristics. However, in a batch process, composition drift is very common due to differences in the reactivity ratios of the monomers. To synthesize forced gradient copolymers, a semi-batch operation is commonly used, which is a method without limitation of reactivity of co-monomers. It was experimentally and theoretically demonstrated that, these copolymers with uniform composition or linear gradient composition can be successfully designed and controlled.45–48 In this contribution, we reported the facilely controlled synthesis of MMA/tBA gradient copolymers mediated by copper powder at ambient temperature in a polar solvent (i.e. DMF), using the commercially available ligand N,N,N′,N′′,N′′-pentamethyl diethylene triamine (PMDETA) via semi-batch Cu(0)-mediated LRP (see Scheme 1). The process of reaction was also investigated in non-polar solvent (toluene), using the same ligand (PMDETA). In particular, all the reagents used were not deoxygenated, which was instead of the addition of a small amount of reducing agent hydrazine hydrate to the reaction mixture.
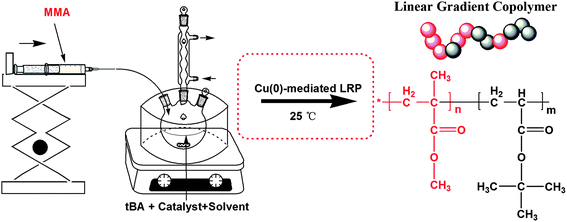 | ||
| Scheme 1 Experimental apparatus for the semibatch Cu(0)-mediated LRP. | ||
Experimental section
Materials
t-Butyl acrylate (tBA, 99%, Sinopharm Chemical Reagent Co., Ltd (SCRC)) and methyl methacrylate (MMA, 99%, SCRC) were rinsed with 5 wt% aqueous NaOH solution to remove inhibitor, dried with MgSO4 over night and distilled before use. Ethyl 2-bromoisobutyrate (Eib-Br, 98%) was obtained from A Better Choice for Research Chemicals GmbH & Co. KG. (ABCR). 4,4′-Dinonyl-2,2′-bipyridyl (diNbpy, Nanjing Chemzam Pharmtech, 99%) was recrystallized three times from ethanol. 1,1,4,7,7-Pentamethyldiethylenetriamine (PMDETA, Aldrich, 98%). Copper powder (75 μm, 99%, Sigma-Aldrich). Hydrazine monohydrate (Alfa Aesar, 98+%) dimethyl sulfoxide (DMSO, 99.5%, SCRC) and N,N-dimethylformamide (DMF, 99.5%, SCRC). All other reagents and solvents were obtained from SCRC and used without further purification.Measurements
The copolymer compositions were determined by nuclear magnetic resonance (1H NMR) spectroscopy (Bruker AV400 MHz) in CDCl3 and tetramethylsilane (TMS) as an internal standard. To obtain the relative amounts of the co-monomers incorporated in polymer chains were estimated from the areas under assigned peaks of the spectra. The copolymer composition was determined by comparing the integrated intensities of these resonance signals as above mention. The molecular weight (Mn) and molecular weight distribution (Mw/Mn, PDI) of the polymer was determined at 40 °C by gel permeation chromatography (GPC) equipped with a waters 1515 isocratic HPLC pump, three Styragel columns (Waters HT4, HT5E, and HT6) and a waters 2414 refractive index detector (set at 30 °C), using THF as the eluent at the flow rate of 1.0 mL min−1. A series of poly(methyl methacrylate) (PMMA) narrow standards were used to generate a universal calibration curve.Typical procedures for the synthesis of gradient copolymer poly(tBA-grad-MMA) via semi-batch Cu(0)-mediated LRP technique
Solvent (DMF, 4.5 ml), catalyst (copper powder, 4.77 mg, 0.075 mmol), and hydrazine hydrate (3.6 μl, 0.075 mmol) were first added to a 25 ml dried round-bottom flask, and then the mixture was stirred for 5 min in a water bath with a thermostat at 25 ± 0.1 °C. After that, tBA (4.35 ml, 30 mmol), ligand (PMDETA, 31.35 μl, 0.075 mmol) and initiator (Eib-Br, 43.89 μl, 0.3 mmol) were added. In addition, MMA (3.21 ml, 30 mmol) was transferred into an airtight syringe and assembled to a syringe pump. Synchronously, the continuous addition of MMA to the flask was started at an optimized rate (0.642 ml h−1), which corresponds to targeted monomer conversion (“Test Reaction” method49). Samples were taken of about 0.5–0.6 mL every 1 h. After 5 h, the MMA addition was complete. The reaction was stopped after 5.5 h by exposing the reaction mixture to the air. All of the above polymer solutions were diluted with CHCl3 and passed over an alumina column to remove the catalyst. The solvent was distilled off under vacuum using a rotary evaporator at 25 °C, and then the polymer was precipitated twice in the mixed solvent of methanol and water (v/v, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The purified product was dried under vacuum until constant weight was reached.
:1). The purified product was dried under vacuum until constant weight was reached.
Results and discussion
It is well-known that the reactivation of the oxidized metal catalyst, such as Cu2O, is often performed with hydrogen in the laboratory.50 On the other hand, it has been reported that Cu2O on Cu(0) surface can be reduced effectively by hydrazine.51 Furthermore, the Cu(0) powder can be synthesized by the in situ reduction of Cu(II) salts with hydrazine.52,53 Percec et al. reported that a successful SET-LRP can also be carried out using Cu2O as catalyst, nevertheless, the process is less reactive.23In order to assure the stringent anaerobic experimental conditions (deoxygenated reagents) and eliminate the time consuming deoxygenating cycles before the semi-batch Cu(0)-mediated LRP process for the synthesis of gradient copolymers, the reducing agent hydrazine hydrate was added into the reactive flask, which therefore resulted in an activation of the copper powder by reducing Cu2O to Cu(0), and then the fresh Cu(0) acts an effective scavenger for oxygen in reagents. Thus, we believe that the benefit of hydrazine hydrate addition in Cu(0)-mediated LRP clearly stands on its own. Herein, an equimolar (with respect to Cu(0)) amount of hydrazine hydrate was employed for producing the gradient copolymers with linear gradient composition.
For the sake of monitoring the sequence distribution of monomers in products, during the synthesis of gradient copolymers, reaction aliquots were taken from reactor to verify the change in cumulative composition (Fcum) of feeding monomer (M2) as a function of chain length. It should be pointed out that the instantaneous composition (Finst) of the copolymer chains is not experimentally measurable. Fcum is the one that is measured by 1H NMR. And then the Finst of feeding monomer was in turn calculated according to the following equation: Finst = [Conv.total,i × Fcum,M2,i − Conv.total,i−1 × Fcum,M2,i−1]/[Conv.total,i − Conv.total,i−1], where Conv.total is the total conversion of both monomers (tBA and MMA).54–56 Note that the Finst curve of feeding monomer as a function of polymerization degree virtually shows a true gradient profile along a chain in contrast to that of Fcum (Fig. 1). The linear relationship shows that an ideal gradient composition is formed clearly and gradually. The compositions of gradient copolymers are summarized in Table 1.
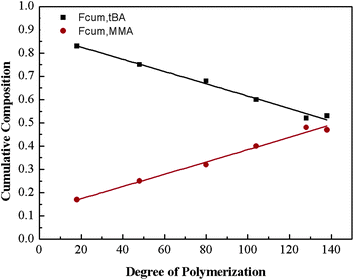 | ||
| Fig. 1 Copolymer cumulative composition in gradient copolymer as a function of the degree of polymerization. | ||
| Entry | MMA (ml) | Adding time (h) | Conv.tBA (%) | Conv.MMA (%) | F cum,MMA | Gradient composition |
|---|---|---|---|---|---|---|
| 1 | 1.284 | 2 | 69.31 | 37.32 | 0.35 | Poor |
| 2 | 3.210 | 5 | 72.05 | 58.95 | 0.45 | Good |
| 3 | 3.210 | 5 | 72.30 | 65.51 | 0.47 | Excellent |
| 4 | 3.210 | 5 | 70.39 | 43.14 | 0.38 | General |
| 5 | 3.210 | 5 | 81.97 | 56.97 | 0.41 | General |
| 6 | 1.926 | 3 | 68.16 | 38.34 | 0.36 | Poor |
| 7 | 1.284 | 2 | 84.89 | 43.73 | 0.34 | Poor |
| 8 | 3.210 | 5 | 83.95 | 90.94 | 0.52 | None (random) |
| 9 | 3.210 | 5 | 92.70 | 85.57 | 0.48 | Excellent |
The overall ratio of incorporated monomer in the resulting copolymer poly(tBA-grad-MMA) and the monomer conversion were determined using the 1H NMR spectra of pure copolymer and polymerization solution respectively at an interval time. The changing characteristic signals for PtBA and PMMA in Fig. 2–4 show that the MMA units are gradually incorporated into the polymer chains. Firstly, the overall ratio of incorporated monomer in the resulting copolymer was determined using 1H NMR (Fig. 2, taking entry 3 for example) measurement by comparing the peak area ratio of characteristic signals for PtBA (1.350–1.550 ppm 9H, –OC(CH3)3) and PMMA (3.500–3.750 ppm, 3H, –OCH3). The equations for calculation are as follows:
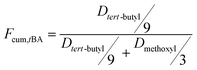
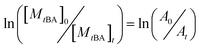
where, At = Dpropenyl,t/Dtert-butyl,t, Dpropenyl,t and Dtert-butyl,t are peak areas of the double-bond and tert-butyl in the 1H NMR spectra at different times, respectively. A0 = Dpropenyl,0/Dtert-butyl,0 is the normalized peak areas of double-bond in 1H NMR spectra, here Dtert-butyl,t = Dtert-butyl,0. The conversion of MMA is also obtained from above equations based on the same standard.
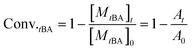
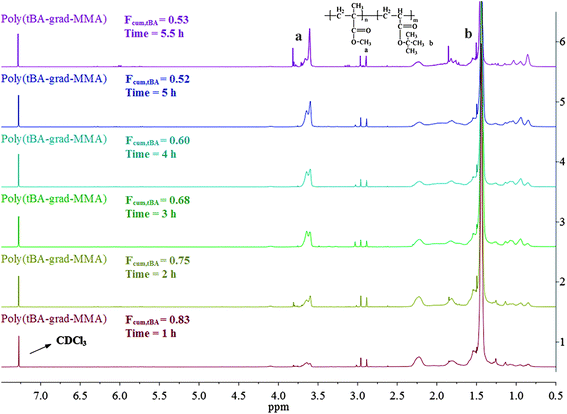 | ||
| Fig. 2 1H NMR spectrum of the tBA/MMA gradient copolymers at different reaction time. | ||
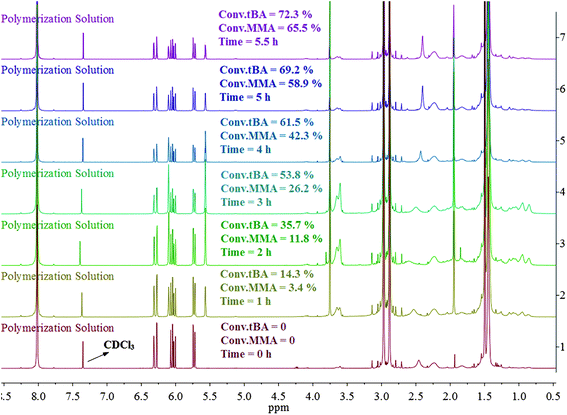 | ||
| Fig. 3 1H NMR spectrum of the polymerization solutions at different reaction time. | ||
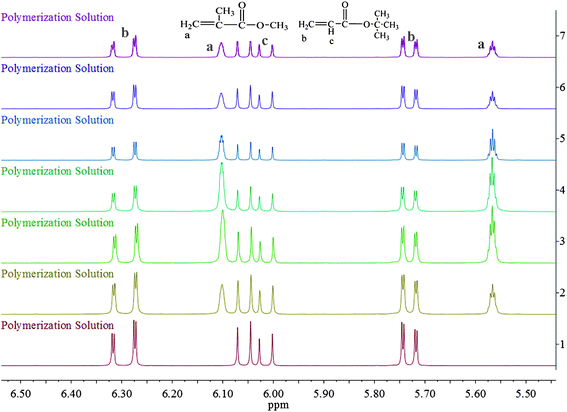 | ||
| Fig. 4 Partial enlarged details for 1H NMR spectrum of the polymerization solutions at different reaction time from 5.50 to 6.50 ppm. | ||
Fig. 5 describes the number-average molecular weight (Mn,GPC) and molecular weight distribution (Mw/Mn) as function of the overall monomer conversion. The relatively high value of Mw/Mn, which declines gradually with the increase of the conversion indicates the slight broadening of the polydispersity and also exhibits a slight tailing towards low molecular weight, which possible due to the poor initiation efficiency caused by recombination. Furthermore, the copolymerization kinetics data were also recorded (see Fig. 6 and 7). The kinetic plots for the copolymerization in DMF initiated with Eib-Br in the presence of Cu(0)/PMDETA at 25 °C (Table 2, entry 3) can be observed in Fig. 6. The approximate linearity of the kinetic plot with an inducing period of about 30 min may results from Cu2O on the surface of Cu(0) powder, which indicates that a successful Cu(0)-LRP in the presence of limited amount of air with hydrazine additive requires the complete consumption of oxygen and rapid regeneration of Cu(0) by reduction of the resulting Cu2O during the early stage of the polymerization in this system.57 In addition, from Fig. 7, the plots show excellent linearity, which proves an excellent living polymerization. Fig. 7 also demonstrates that a good agreement between experimental and theoretical molecular weight is obtained at the interval time of reaction and the polymer molecular weight distribution index is in the range of 1.78–1.39. We propose the cause of the poor control observed during the early stages of the reactions is that Cu(I)X/L species do not instantaneously disproportionate, thus resulting in a high radical concentration.44 The relatively broader distribution at high conversion may stem from the highly reactive Cu(0) surface and high concentration catalyst (in the following discussion) which facilitates a rapid initiation with an increasing radical concentration which favors irreversible termination; meanwhile, the reduction of Cu(II) by an excess of hydrazine leads to the decrease of deactivator concentration.38,42 As a whole, all the above data showed that the gradient copolymers were synthesized successfully via the semi-batch Cu(0)-mediated LRP.
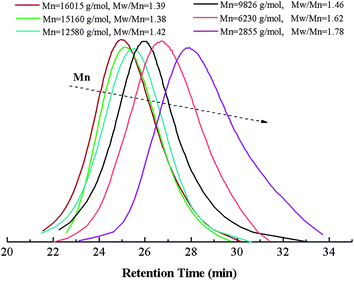 | ||
| Fig. 5 Number-average molecular weight (Mn,GPC) and molecular weight distribution (Mw/Mn) as function of the overall monomers conversion at different reaction time. | ||
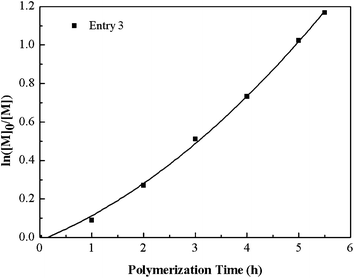 | ||
| Fig. 6 Kinetic plot for the semi-batch copolymerization of tBA and MMA using Cu(0) and PMDETA as catalyst system in DMF. (Table 2, entry 3). | ||
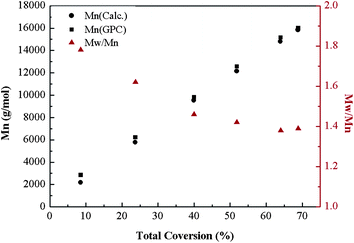 | ||
| Fig. 7 Dependence of poly(tBA-grad-MMA) gradient copolymer molecular weight (Mn) and molecular weight distribution (Mw/Mn) on the total conversion for the copolymerization in DMF (Table 2, entry 3). | ||
A large quantity of catalyst used in ATRP, which is difficult to remove and results in the catalyst contaminated polymeric materials, is the main limitation for the industrialization of ATRP. In this work, a series of experiments were performed with different amounts of Cu(0) powder in DMF with VtBA/Vsol. = 1/1 and [MMA]0/[tBA]0/[Eib-Br]0 = 100/100/1 at 25 °C. From the results summarized in Table 2 (entries 1–4) and Fig. 8, it is found that the total conversion increases first and then decreases with the increase of the amount of the Cu(0) powder, while the value of Mw/Mn raises from 1.38 to 1.48. In practice, the reaction solution became exceedingly viscous just after 2 h and the reactants experienced diffusion limitation, which results in the decline of the total conversion at the highest concentration of catalyst ([Cu(0)]0/[initiator]0 = 1/1, entry 1); accordingly, it causes the poor gradient composition. The reason may be that the increase of the amount of Cu(0) causes a higher concentration of Cu(II)/N-ligand produced by the disproportionation of in situ Cu(I) species, leading to a faster polymerization.20 However, the higher radical concentration leads to more pronounced termination and the formation of the increasing levels of persistent radicals (XMtn+1/L),58 which reduces the radical concentration and self-regulates the system. These phenomena imply that the lower concentration of catalyst gives rise to the improvement in control of molecular weight (MW) and polydispersity, but there exists a maximum value of total conversion. Therefore, a suitable catalyst concentration should be chosen to provide a balance between polymerization rate and controllability.
| Entry | [MMA]0/[tBA]0/[Eib-Br]0/[Cu(0)]0/[N2H4·H2O]/[Ligand] | Solvent | Time (h) | Conv. (%) | M n b (g mol−1) (theo) | M n (g mol−1) (GPC) | M w/Mn (GPC) |
|---|---|---|---|---|---|---|---|
| a Temperature = 25 °C, ligand = PMDETA, VtBA/Vsol. = 1/1. b M n (theo) = ([tBA]0/[Initiator]0) × conv.tBA × 128.17 + ([MMA]0/[Initiator]0) × conv.MMA × 100.12. c Ligand = diNbpy. d V tBA/Vsol. = 2/1. e Temperature = 80 °C. | |||||||
| 1 | 100/100/1/1/1/1 | DMF | 2.5 | 53.3 | 12620 |
12957 |
1.48 |
| 2 | 100/100/1/0.5/0.5/0.5 | DMF | 5.5 | 65.5 | 15136 |
15269 |
1.45 |
| 3 | 100/100/1/0.25/0.25/0.25 | DMF | 5.5 | 68.9 | 15825 |
16015 |
1.39 |
| 4 | 100/100/1/0.1/0.1/0.1 | DMF | 5.5 | 56.8 | 13341 |
13767 |
1.38 |
| 5c | 100/100/1/0.25/0.25/0.25 | DMF | 5.5 | 69.5 | 16210 |
16409 |
1.89 |
| 6d | 100/100/1/0.25/0.25/0.25 | DMF | 3.5 | 53.2 | 12575 |
13681 |
1.66 |
| 7 | 100/100/1/0.25/0.25/0.25 | DMSO | 2.5 | 64.3 | 15258 |
16617 |
2.08 |
| 8 | 100/100/1/0.25/0.25/0.25 | Toluene | 20.0 | 87.4 | 19865 |
20308 |
1.58 |
| 9e | 100/100/1/0.25/0.25/0.25 | Toluene | 5.5 | 89.1 | 20449 |
21059 |
1.29 |
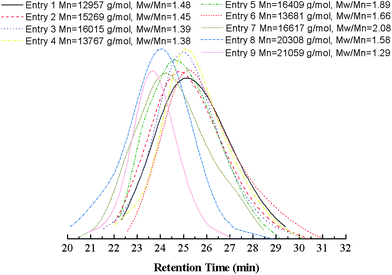 | ||
| Fig. 8 Number-average molecular weight (Mn,GPC) and molecular weight distribution (Mw/Mn) at different entries (corresponding to Table 2). | ||
From Tables 1 and 2 (entries 1–4), these results demonstrate that this polymerization proceeded by the Cu(0)-mediated LRP at a relatively lower concentration of catalyst ([Cu(0)]0/[initiator]0 = 1/4, entry 3) features the better control of MW and its distribution with a “living”/controlled manner, which leads to form the good gradient composition in polymer chains.
Two different ligands widely used in ATRP were chose to investigate the effect of ligand on the Cu(0)-mediated copolymerization. The ligand 4,4′-dinonyl-2,2′-bipyridine (diNbpy) has been used extensively in ATRP to control synthesis of gradient copolymers.48,49,59 PMDETA is a commercially available ligand with intermediate activity,37,38,40 and also it was verified by Perrier et al. that PMDETA as a SET-LRP ligand was superior to diNbpy and hexamethylene tris(2-aminoethyl)amine (Me6TREN) in toluene.60 The comparison experiments were carried out with 0.25 equimolar of Cu(0) relative to initiator in DMF at 25 °C (Table 2, entries 3 and 5), which is inspired by Perrier et al.'s contribution.60 Although the overall conversion for above two systems at the end of copolymerization remains the nearly same, using diNbpy, the polymerization can give broader polydispersity (Fig. 8). Uncontrolled free radical polymerization during the early stages of the reaction as a result of insufficient concentration of Cu(II) deactivator can be regarded as rational explanation for this consequence. Therefore, the general gradient composition is introduced in this reaction system.
There are many achievements about the effect of solvents on the process of Cu(0)-mediated “living”/controlled radical polymerization.26,27,41,61–64 In this work, the attempts launched in DMF, DMSO, toluene in combination with PMDETA were applied to analyze the influence of solvents on the polymerization and controllability. The results are shown in Tables 1 and 2, entries 3, 6–9, and Fig. 8. First, the proportion of solvent in reaction mixture was reduced from VtBA/Vsol. = 1/1 to VtBA/Vsol. = 2/1. During the polymerization, we found that the reaction solution became exceedingly viscous just after 3 h. Comparing with the results at entry 3, one can find that the total conversion declines, polydispersity becomes slightly broader at entry 6 and the gradient composition of resulting copolymers is also poor, which may be the same as the explanation for the increase of catalyst concentration that causes the similar consequence mentioned above. Second, a widely used disproportionating solvent, DMSO was chosen as the solvent instead of DMF. The extraordinary viscous reaction appeared just after 2 h. From the results summarized in Table 1, entry 7, the polymerization rate is significantly elevated comparison with DMF as solvent under the same reaction conditions (entry 3), and the conversion of tBA monomer soars to 84.9% at the end of reaction, while it of MMA just arrives 43.7%. The value of Fcum,MMA (0.34) indicates the poor gradient composition. In addition, the value of Mw/Mn is larger than the result of entry 3 at the comparative total conversion. The highly polymerization rate obtained by SET-LRP has been attributed to the self-regulated generation of Cu(0) activator and Cu(II)X2/L deactivator from the disproportionation of Cu(I)X/L produced in situ during activation in DMSO.26 Finally, we performed the polymerization in toluene with the same experimental conditions (entry 8). It can be expected that the polymerization rate is extremely low at 25 °C. The reaction time was prolonged to 20 h after the commoner was added. Therefore, the composition of resulting copolymer should be random. It seems not to be the same catalyst activation mechanism with SET-LRP reported by some research groups.26,41 They proposed that Cu(0) is oxidized to Cu(I) while activating the initiator, which triggers uncontrolled polymerization. Cu(I) then reacts with the alkyl halide initiator to generate active species and Cu(II), leading to the establishment of the equilibrium between Cu(I) and (II). However, interestingly, when the reaction temperature was raised up to 80 °C, the polymerization rate was noticeably accelerated and the polydispersity index was narrowed to 1.29, which is lower than that in the other experiments carried out in this work. The values of Fcum,MMA(0.48) and conversion of both comonomers (85.6% and 92.7% for MMA and tBA respectively) manifest the good gradient composition. The reason is not clear now, more intensive investigations should be needed.
Conclusion
In conclusion, the semi-batch Cu(0)-mediated LRP technique using Cu(0) and PMDETA as a catalyst system in DMF in the presence of hydrazine hydrate at 25 °C was used to produce linear gradient copolymers. For this method, we observed that the addition of an equimolar (with respect to ligand) amount of hydrazine hydrate to the reaction mixture allowed the reaction to proceed in the reagents without deoxygenating. Besides, the results regarding the effect of the reaction conditions on the copolymerization demonstrated that the suitable catalyst concentration was required to balance the polymerization rate with its controllability. The commercially available ligand PMDETA was regarded as a promising one, with moderate polydispersity and relatively high conversion. Also, the different kinds of solvents had great impact on the copolymerization; using DMF, the polymerization gave a more appropriate polymerization rate for controlling synthesis of gradient copolymer than DMSO and toluene at 25 °C, but the copolymerization performed in toluene at 80 °C featuring low polydipersity and high yield had excellent prospects. Accordingly, we thought that the above method was a facile and efficient one for the synthesis of gradient copolymer poly(tBA-grad-MMA).Acknowledgements
The authors thank the National Natural Science Foundation of China (no. 21076171, 21276213).References and notes
- K. Matyjaszewski, M. J. Ziegler, S. V. Arehart, D. Greszta and T. Pakula, J. Phys. Org. Chem., 2000, 13, 775–786 CrossRef CAS.
- U. Beginn, Colloid Polym. Sci., 2008, 286, 1465–1474 CAS.
- K. Karaky, L. Billon, C. Pouchan and J. Desbrières, Macromolecules, 2007, 40, 458–464 CrossRef CAS.
- N. Cherifi, A. Issoulie, A. Khoukh, A. Benaboura, M. Save, C. Derail and L. Billon, Polym. Chem., 2011, 2, 1769–1777 RSC.
- W. Yuan, M. M. Mok, J. Kim, C. L. H. Wong, C. M. Dettmer, S. T. Nguyen, J. M. Torkelson and K. R. Shull, Langmuir, 2010, 26, 3261–3267 CrossRef CAS.
- S. Harrisson, F. Ercole and B. Wuir, Polym. Chem., 2010, 1, 326–332 RSC.
- Y. Zhao, Y. W. Luo, B. G. Li and S. P. Zhu, Langmuir, 2011, 27, 11306–11315 CrossRef CAS.
- Y. Milonaki, E. Kaditi, S. Pispas and C. Demetzos, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1226–1237 CrossRef CAS.
- M. D. Lefebvre, M. O. de la Cruz and K. R. Shull, Macromolecules, 2004, 37, 1118–1123 CrossRef CAS.
- M. D. Lefebvre, C. M. Dettmer, R. L. McSwain, C. Xu, J. R. Davila, R. J. Composto, S. T. Nguyen and K. R. Shull, Macromolecules, 2005, 38, 10494–10502 CrossRef CAS.
- R. Wang, W. H. Li, Y. W. Luo, B. G. Li, A. C. Shi and S. P. Zhu, Macromolecules, 2009, 42, 2275–2286 CrossRef CAS.
- C. L. H. Wong, J. Kim and J. M. Torkelson, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 2842–2849 CrossRef CAS.
- M. M. Mok, C. J. Ellison and J. M. Torkelson, Macromolecules, 2011, 44, 6220–6226 CrossRef CAS.
- M. Y. Zaremski, D. I. Kalugin and V. B. Golubev, Polym. Sci., 2009, 51, 103–122 Search PubMed.
- J. Kim, M. K. Gray, H. Y. Zhou, S. T. Nguyen and J. M. Torkelson, Macromolecules, 2005, 38, 1037–1040 CrossRef CAS.
- M. M. Mok, J. Kim and J. M. Torkelson, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 48–58 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamto, Chem. Rev., 2001, 101, 3689–3746 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffrey, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069–5119 CrossRef CAS.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- D. Konkolewicz, A. J. D. Magenau, S. E. Averick, A. Simakova, H. He and K. Matyjaszewski, Macromolecules, 2012, 45, 4461–4468 CrossRef CAS.
- N. H. Nguyen, M. E. Levere, J. Kulis, M. J. Monteiro and V. Percec, Macromolecules, 2012, 45, 4606–4622 CrossRef CAS.
- N. H. Nguyen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4227–4240 CAS.
- K. Matyjaszewski, S. Coca, S. G. Gaynor, M. L. Wei and B. E. Woodworth, Macromolecules, 1997, 30, 7348–7350 CrossRef CAS.
- K. Matyjaszewski, N. V. Tsarevsky, W. A. Braunecker, H. Dong, J. Huang, W. Jakubowski, Y. Kwak, R. Nicolay, W. Tang and J. A. Yoon, Macromolecules, 2007, 40, 7795–7806 CrossRef CAS.
- C. Y. Lin, M. L. Coote, A. Gennaro and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 12762–12774 CrossRef CAS.
- Y. Zhang, Y. Wang, C.-H. Peng, M. Zhong, W. Zhu, D. Konkolewicz and K. Matyjaszewski, Macromolecules, 2012, 45, 78–86 CrossRef CAS.
- A. A. Isse, A. Gennaro, C. Y. Lin, J. L. Hodgson, M. L. Coote and T. Guliashvili, J. Am. Chem. Soc., 2011, 133, 6254–6264 CrossRef CAS.
- N. H. Nguyen, B. M. Rosen, X. Jiang, S. Fleischmann and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5577–5590 CrossRef CAS.
- X. Jiang, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 403–409 CrossRef CAS.
- N. H. Nguyen, M. E. Levere and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 860–873 CrossRef CAS.
- Z. B. Zhang, W. X. Wang, H. D. Xia, J. Zhu, W. Zhang and X. L. Zhu, Macromolecules, 2009, 42, 7360–7366 CrossRef CAS.
- W. X. Wang, Z. B. Zhang, J. Zhu, N. C. Zhou and X. L. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6316–6327 CrossRef CAS.
- J. Gao, Z. Zhang, N. Zhou, Z. Cheng, J. Zhu and X. L. Zhu, Macromolecules, 2011, 44, 3227–3232 CrossRef CAS.
- A. H. Soeriyadi, C. Boyer, F. Nyström, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 11128–11131 CrossRef CAS.
- B. D. Hornby, A. G. West, J. C. Tom, C. Waterson, S. Harrisson and S. Perrier, Macromol. Rapid Commun., 2010, 31, 1276–1280 CrossRef CAS.
- A. G. West, B. Hornby, J. Tom, V. Ladmiral, S. Harrisson and S. Perrier, Macromolecules, 2011, 44, 8034–8041 CrossRef CAS.
- S. Fleischmann, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1190–1196 CrossRef CAS.
- G. Lligadas and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6880–6895 CrossRef CAS.
- G. Lligadas and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2745–2754 CrossRef CAS.
- L. Wang and L. J. Broadbelt, Macromol. Theory Simul., 2011, 20, 191–204 CrossRef CAS.
- R. Wang, Y. W. Luo, B. G. Li and S. P. Zhu, AIChE J., 2007, 53, 174–186 CrossRef CAS.
- X. Y. Sun, Y. W. Luo, R. Wang, B. G. Li and S. P. Zhu, AIChE J., 2008, 54, 1073–1087 CrossRef CAS.
- Y. N. Zhou, J. J. Li and Z. H. Luo, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3052–3066 CrossRef CAS.
- H. G. Börner, D. Duran, K. Matyjaszewski, M. da Silva and S. S. Sheiko, Macromolecules, 2002, 35, 3387–3394 CrossRef.
- S. Hunig, G. Markl and J. Sauer, Arbeitsmethoden in der Organischen Chemie, Verlag Lehmanns, Berlin, 2006 Search PubMed.
- Y. K. Al-Haydari, J. M. Saleh and M. H. Matloob, J. Phys. Chem., 1985, 89, 3286–3290 CrossRef CAS.
- N. A. Dhas, C. P. Raj and A. Gedanken, Chem. Mater., 1998, 10, 1446–1452 CrossRef CAS.
- A. M. T. Sakar and S. Kapoor, J. Phys. Chem. C, 2008, 112, 3334–3340 Search PubMed.
- K. Min, J. Kwon Oh and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1413–1423 CrossRef CAS.
- Y. Zhao, Y. W. Luo, C. H. Ye, B. G. Li and S. P. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 69–79 CrossRef CAS.
- K. Nakatani, Y. Ogura, Y. Koda, T. Terashima and M. Sawamoto, J. Am. Chem. Soc., 2012, 134, 4373–4383 CrossRef CAS.
- N. H. Nguyen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4756–4765 CrossRef CAS.
- H. Fischer, Macromolecules, 1997, 30, 5666–5672 CrossRef CAS.
- H. I. Lee, K. Matyjaszewski, S. Yu and S. S. Sheiko, Macromolecules, 2005, 38, 8264–8271 CrossRef CAS.
- J. Tom, B. Hornby, A. West, S. Harrisson and S. Perrier, Polym. Chem., 2010, 1, 420–422 RSC.
- G. Lligadas, B. M. Rosen, M. J. Monteiro and V. Percec, Macromolecules, 2008, 41, 8360–8364 CrossRef CAS.
- N. H. Nguyen, B. M. Rosen, X. Jiang, S. Fleischmann and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5577–5590 CrossRef CAS.
- X. Jiang, S. Fleischmann, N. H. Nguyen, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5591–5605 CrossRef CAS.
- N. H. Nguyen, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1752–1763 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |