Copper(0)-mediated radical polymerisation in a self-generating biphasic system†
Cyrille
Boyer
a,
Amir
Atme
a,
Christopher
Waldron
b,
Athina
Anastasaki
b,
Paul
Wilson
b,
Per B.
Zetterlund
a,
David
Haddleton
*b and
Michael R.
Whittaker
*a
aCentre for Advanced Macromolecular Design (CAMD), School of Chemical Engineering, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: mikey.whittaker@unsw.edu.au
bDepartment of Chemistry, University of Warwick, Coventry, UK. E-mail: D.M.Haddleton@warwick.ac.uk
First published on 12th September 2012
Abstract
Herein, we demonstrate the synthesis of well-defined poly(n-alkyl acrylate)s via copper(0)-mediated radical polymerisation in a self-generating biphasic system. During the polymerisation of n-butyl acrylate in DMSO, the polymer phase separates to yield a polymer-rich layer with very low copper content (ICP-MS analysis: 0.016 wt%). The poly(n-butyl acrylate) has been characterized by a range of techniques, including GPC, NMR and MALDI-TOF, to confirm both the controlled character of the polymerisation and the end group fidelity. Moreover, we have successfully chain extended poly(n-butyl acrylate) in this biphasic system several times with n-butyl acrylate to high conversion without intermediate purification steps. A range of other alkyl acrylates have been investigated and the control over the polymerisation is lost as the hydrophobicity of the polymer increases due to the increase in alkyl chain length indicating that it is important for the monomer to be soluble in the polar solvent.
Introduction
A range of transition metal-mediated radical polymerisation (ATRP) techniques,1 including initiators for continuous activator regeneration (ICAR),2–6 activators regenerated by electron transfer (ARGET),7–9 and activators generated by electron transfer (AGET),10 have been developed which can control the polymerisation of a large range of monomers and allow routine access to complex polymer architectures such as multi-block copolymers, star polymers and variations thereof. Recently, it has been demonstrated that copper(0)-mediated radical polymerisation in polar solvents (also referred to as SET-LRP) results in extremely high end-group fidelity, an indication of excellent livingness, to very high monomer conversion in the case of acrylates.11–18 A main drawback of conventional transition metal-mediated polymerisation techniques in general (including Cu(0)/SET-LRP) is the presence of metal–copper complexes in the final polymer. Traces of metal salts could result in polymer degradation due to potential catalytic oxidation,19,20 contribute to the presence of residual colour and may even contribute to biological toxicity.21 A variety of different routes have been explored to decrease the amount of metallic residues.5,22,23 A range of methods has been reported which keeps the catalyst insoluble in the polymerisation phase including the use of fluorous biphase systems,24–27 supported catalysts28–31 and ionic liquids.32–35 A solid supported catalyst can be easily removed,36,37 but diffusion of reactants to the catalytic site can present a problem.38,39 One of us has also explored the use of [N-alkyl-(2-pyridyl)methanimine]copper(I) complexes which had tuneable solubility in certain organic solvents used in this type of polymerisation.40 Other homogeneous approaches have included the reduction of the amount of copper employed during the polymerisation (ARGET and ICAR),4,5,41 the use of a redox system (e.g. ascorbic acid or radical), which have made it possible to decrease the amount of copper to ppm levels.5In the present work, we report a convenient polymerisation methodology using a self-generating, surfactant-free biphasic system, which allows efficient separation and removal of almost all copper catalysts from the polymer product without complex purification procedures. In this initial work, the method has been successfully applied to the polymerisation of n-butyl acrylate (BA) in DMSO using Cu(0)-mediated radical polymerisation at ambient temperature. The polymer starts to phase separate from the reaction mixture after reaching a molecular weight of ∼2500 g mol−1, corresponding to approximately 50% conversion under the conditions studied, to give a biphasic system comprising an upper polymer-rich phase and a lower DMSO-rich phase. Surprisingly, this phase separation does not significantly affect the polymerisation process in terms of control/livingness as evidenced by both narrow polydispersity (PDI) and high end group fidelity.
Experimental part
Materials
Ethyl 2-bromoisobutyrate (EbiB, Aldrich, 98%), copper(II) bromide (CuBr2, Sigma-Aldrich, 99%), tetrahydrofuran (THF, Sigma, 99%) and dimethyl sulphoxide (DMSO, UNIVAR, AR) were all used as received. Copper wire (diameter = 1.25 mm) was activated by washing with sulfuric acid for 10 min. Tris(2-(dimethylamino)ethyl)amine (Me6TREN) was synthesised according to the literature procedure42 and stored under nitrogen prior to use. Monomers, such as n-butyl acrylate (BA, Aldrich, 99%), lauryl acrylate, isobutyl acrylate, tert-butyl acrylate and 2-ethylhexyl acrylate were de-inhibited by percolating over a column of basic alumina (Ajax, AR).Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ethyl acetate:hexane to give white crystals (8.2 g, 70.3%) 1H NMR (CDCl3): δ (ppm) 4.44 (t, 4H), 1.94 (s, 12H).
:1 ethyl acetate:hexane to give white crystals (8.2 g, 70.3%) 1H NMR (CDCl3): δ (ppm) 4.44 (t, 4H), 1.94 (s, 12H).
For the chain extension, a further 2.4 mL of a degassed monomer in DMSO (50 vol%) was then carefully added via a gas tight syringe and again the solution was allowed to polymerize at ambient temperature for another 24 h with stirring.
Samples of the reaction mixture and polymer were removed for 1H NMR, GPC and MALDI-TOF MS analysis. The samples for 1H NMR analysis were simply diluted with CDCl3, while the samples for GPC were diluted with CHCl3, then passed over an alumina column to remove metal salts.
Results and discussion
Cu(0)-mediated radical polymerisation of BA has been carried out using ethyl bromoisobutyrate, CuBr2 and Me6TREN in DMSO at ambient temperature. The polymerisation proceeds rapidly with full monomer conversion (>95%) achieved in less than 3 h, as determined by 1H NMR analysis. The monomer conversion is calculated using the signals at 5.5–6.5 ppm and 4.0 ppm, which are attributed to acrylic and CH2O groups, respectively. The polymerisation fulfils all the criteria of a controlled/living radical polymerisation. Interestingly, ln[M]t/[M]0 increases linearly with time up until one hour (56% conversion) consistent with a constant concentration of propagating radicals at the polymerisation loci. After one hour, the polymerisation rate increases significantly. Finally, over 90% monomer conversion is observed in less than 120 min (Fig. 1A). When PBA reaches a molecular weight in the range 2000–3000 g mol−1 (∼50% conversion), the system becomes biphasic due to the low solubility of PBA in DMSO (the phase separation is difficult to detect unless the magnetic stirring is stopped), resulting in an upper polymer-rich phase and a lower DMSO-rich phase which contains monomer and catalyst. From this point onwards in the reaction, further chain extension (i.e. polymerisation) is confined to the polymer-rich phase. After phase separation has occurred, the system comprises one phase consisting mainly of PBA with low amounts of DMSO (<5 wt%) and BA monomer (<5 wt%) at 80% monomer conversion as determined by 1H NMR analysis, and another second phase comprising DMSO, BA, copper (copper wire) and copper complex(es) (ligand–Cu(II)) as seen by a green colouration. The formation of the second phase in the polymerisation mixture is accompanied by a distinct increase in the polymerisation rate (ln[M]t/[M]0vs. time shows a step change in the gradient whilst remaining linear). Remarkably, the polymerisation proceeds with good control under these biphasic conditions as indicated by the low PDI values (approx. 1.20–1.30). The PDI values decrease from 1.23 to 1.13 in the first phase of the polymerisation (until 1 hour, corresponding to the homogenous system), and after this critical point, the PDI values increase slightly to approximately 1.32. The good correlation between the experimental molecular weight and the theoretical values (Fig. 1B) also confirms the controlled/living character of the polymerisation. The theoretical values were calculated using the equation: Mn,theo. = α × ([M]0/[Init]0) × MBAw + MInitw. Throughout the polymerisation, GPC chromatograms indicate a mono-modal distribution shifting to higher molecular weights with increasing conversion (Fig. 1C), with no apparent influence of the phase separation.![Cu(0)-mediated radical polymerisation of n-butyl acrylate in DMSO at ambient temperature. (A) Monomer conversion and ln([M]0/[M]) vs. time (h); (B) theoretical (full line) and experimental (squares) molecular weights and PDI values vs. monomer conversion; (C) w(log M) versus log M at different monomer conversions (inset: picture of the mixture after polymerisation); (D) MALDI-TOF spectrum of poly(n-butyl acrylate) obtained at >99% monomer conversion (inset: magnified MALDI-TOF spectrum). Note: [BA]0 : [Initiator]0 = 39.0 : 1.0.](/image/article/2013/PY/c2py20560b/c2py20560b-f1.gif) | ||
| Fig. 1 Cu(0)-mediated radical polymerisation of n-butyl acrylate in DMSO at ambient temperature. (A) Monomer conversion and ln([M]0/[M]) vs. time (h); (B) theoretical (full line) and experimental (squares) molecular weights and PDI values vs. monomer conversion; (C) w(log M) versus log M at different monomer conversions (inset: picture of the mixture after polymerisation); (D) MALDI-TOF spectrum of poly(n-butyl acrylate) obtained at >99% monomer conversion (inset: magnified MALDI-TOF spectrum). Note: [BA]0:[Initiator]0 = 39.0:1.0. | ||
Following polymerisation, the polymer is present as a clearly distinguishable colourless top layer (inset in Fig. 1C), while the bottom DMSO layer contains the vast majority of the copper–ligand complexes as indicated by the green colour from Cu(II) complexes (as well as the Cu wire). After separation of the top layer at >99% monomer conversion, the polymer was analyzed by a range of techniques, including NMR, MALDI-TOF and GPC. 1H NMR analysis of the top layer (polymer phase without any purification) reveals the absence of monomer and the presence of pure polymer with a small peak of DMSO. Interestingly, the presence of a low intensity peak of DMSO observed by 1H NMR analysis suggests that the polymer is swelling. Another important observation is that the polymer phase remains colourless after prolonged exposure to air. This observation suggests the absence of copper(I) species in this phase as trace amounts of copper(I) should quickly oxidize to Cu(II) species with a typical green/blue colour characteristic of d9 complexes. To confirm this assumption, the polymer phase was analyzed by XPS (Fig. S1 in the ESI†) and ICP-MS. XPS fails to detect the presence of copper in this phase, confirming that the catalyst (copper–ligand) is indeed confined to the bottom layer, or it is present in the upper layer in a quantity of less than 0.05 atomic% (XPS detection limit) (Table S1 in the ESI†). ICP-MS confirms that the amount of copper is 0.016 wt% in the polymer (determined using two different samples). This value is extraordinarily low as conventional ATRP usually produces polymers with approximately 1 wt% of copper.26
It has been reported previously that Cu(0)-mediated radical polymerisation allows the preservation of very high end group fidelity to high conversion in polar solvents.14,43–46 MALDI-TOF analyses before and after phase separation during the polymerisation confirm “perfect” end group fidelity under these polymerisation conditions. Two major distributions separated by 128 Daltons are observed, attributed to PBA-Br/Na+ and PBA-Br/K+ species, respectively (Table S2 in the ESI†). It is very surprising and quite remarkable that the polymerisation proceeds with excellent control/livingness considering that the Cu wire and the Cu–ligand complexes are (primarily) located in the lower DMSO-rich phase as opposed to in the polymer-rich phase (where polymerisation must occur).
The PBA synthesized was systematically chain extended using our previously developed iterative polymerisation methodology.43,46 At full monomer conversion (>99%), a fresh degassed solution of BA in DMSO was added to the polymerisation mixture. It is important to note that the reaction system remains fully biphasic. The conversion and evolution of molecular weight were monitored by NMR and GPC, respectively (Fig. 2A). Interestingly, we observe that the polymerisation rate is lower in this second chain extension, as the polymerisation requires 5 hours to reach 90% monomer conversion. This effect can be attributed to the dilution of the reaction mixture due to the addition of monomer and DMSO. In the biphasic systems, PBA is successfully chain extended with good control over the molecular weight and PDI (Fig. 2B). Again, this result is quite remarkable as one would expect limited accessibility of the monomer to the locus of polymerisation (the polymer-rich phase where the propagating radicals must reside) in this biphasic system. GPC chromatograms confirm the absence of low molecular weight populations and coupling products (Fig. 2A). MALDI-TOF spectroscopy after one chain extension confirms a single population (Fig. S2 in the ESI†) corresponding to bromide terminated polymer confirming high end group fidelity.
![Chain extension of poly(n-butyl acrylate) (using poly(n-butyl acrylate) from >99% in Fig. 1 as macroinitiator) (PBA) in DMSO by Cu(0)-mediated polymerisation using PBA as a macroinitiator. (A) Monomer conversion (filled symbols) and ln([M]0/[M]t) (open symbols) vs. time (h); (B) theoretical (full line) and experimental (squares) molecular weight and PDI values vs. monomer conversion; (C) w(log M) vs. log M at different monomer conversions.](/image/article/2013/PY/c2py20560b/c2py20560b-f2.gif) | ||
| Fig. 2 Chain extension of poly(n-butyl acrylate) (using poly(n-butyl acrylate) from >99% in Fig. 1 as macroinitiator) (PBA) in DMSO by Cu(0)-mediated polymerisation using PBA as a macroinitiator. (A) Monomer conversion (filled symbols) and ln([M]0/[M]t) (open symbols) vs. time (h); (B) theoretical (full line) and experimental (squares) molecular weight and PDI values vs. monomer conversion; (C) w(log M) vs. log M at different monomer conversions. | ||
Polymers could be chain extended in an iterative fashion at least 4 times (each time reaching near full conversion) in DMSO without significant loss of control/livingness. The molecular weights determined by GPC analysis are in near perfect agreement with the theoretical values for each chain extension (Fig. 3A). The mass fraction of living polymers chains was determined by deconvolution of the GPC detector response vs. time traces, revealing in excess of 95 wt% living polymer after four chain extensions. This result (as well as the fact that the first stage proceeds with control/livingness) is extremely surprising as the catalyst (copper wire and copper complex–ligand) is not located in the same phase as the polymer. It would appear that an interfacial mechanism is likely to be involved in the activation/deactivation process, although mechanistic details remain to be elucidated. The evolution of the PDI (Fig. 3A) and molecular weight suggests that the activation and deactivation reactions are not adversely affected by the biphasic system during the successive chain extensions. The copper content in the polymer after 4 chain extensions (before purification) was determined by ICP-MS to be 0.009 wt%.
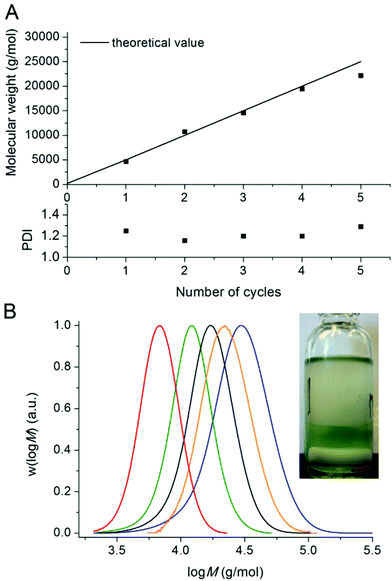 | ||
| Fig. 3 Number-average molecular weights, PDI values and molecular weight distributions for five successive chain extensions of poly(n-butyl acrylate) >99% conversion in Fig. 1 with n-butyl acrylate by use of Cu(0)-mediated radical polymerisation in DMSO at ambient temperature. | ||
In addition, we have also applied this methodology using a difunctional initiator, ethylene bis(2-bromoisobutyrate) (2F-BiB). During these experiments the effects of steric requirements of the monomer and the alkyl chain length of the monomer were also investigated.
In an analogous way to the polymerisation of n-butyl acrylate above, polymerisation from 2F-BiB also resulted in the same phase separation. A complete monomer conversion, as determined by 1H NMR, is achieved in 3 hours to yield a polymer with a PDI = 1.08 (Fig. 4), again showing that the phase separation does not negatively impact the control of the polymerisation.
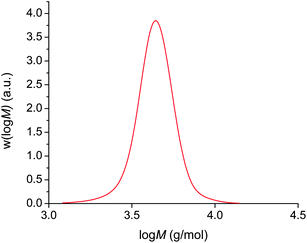 | ||
| Fig. 4 Cu(0)-mediated radical polymerisation of n-butyl acrylate using a bifunctional initiator in DMSO at ambient temperature. | ||
MALDI-TOF MS analysis of this polymer shows two major distributions, unlike the monomodal distribution seen using GPC. The main distribution corresponds to the desired bifunctional polymer, the end group fidelity of which is confirmed by both the m/z values which are in close agreement with theoretical values and the isotopic pattern which indicate the presence of two bromide terminal groups (Fig. 5). The second, lower molecular weight distribution is that of polymer which has been terminated at one end via loss of a bromide but continued to grow; this is again elucidated by the agreement with theoretical mass values and splitting patterns. We have attributed this effect to chain transfer to ligand in the early stages of the reaction, where the ligand can react with the initiator at a comparable rate to the monomer. Further studies have shown that this second distribution can be increased or suppressed depending on the level of ligand in the system (i.e. increased ligand content gives increased termination), however, this work will be presented in a future publication as it is outside the focus of this current work.
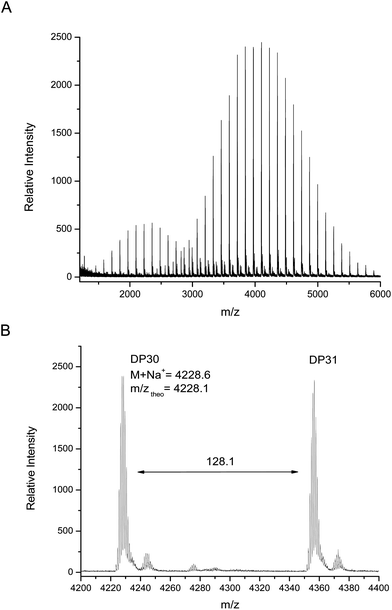 | ||
| Fig. 5 (A) MALDI-TOF MS spectra obtained at 99% monomer conversion and (B) expansion from 4200–4400 m/z. | ||
The polymerisation was repeated but with isomers of n-butyl acrylate (iso- and tert-) (Table 1). Phase separation is also observed and the polymerisations proceed with good control, however, a small increase in the PDI can be observed. We have attributed this effect simply due to the increase in steric bulk from n- to iso- to tert-.
| Monomer | Target Mn (g mol−1) | M n (GPC) | PDI |
|---|---|---|---|
| n-Butyl acrylate | 3000 | 4200 | 1.10 |
| iso-Butyl acrylate | 3000 | 4300 | 1.16 |
| tert-Butyl acrylate | 3000 | 2900 | 1.42 |
| nBA high Mn | 12000 |
15000 |
1.24 |
| nBA high Mn chain extension | 25000 |
21500 |
1.55 |
| Lauryl acrylate | 3000 | 22300 |
2.23 |
| 2-Ethylhexyl acrylate | 3000 | 4500 | 5.75 |
We were interested in achieving higher molecular weight poly(acrylates), and thus poly(n-butyl acrylate) of higher molecular weight (12000 g mol−1) was targeted and subsequently chain extended via addition of extra monomers in DMSO upon reaching complete monomer conversion. We were able to obtain a polymer with Mn = 15000 g mol−1 and PDI = 1.24, demonstrating that the good control is not just limited to polymers with a low target molecular weight. The subsequent chain extension to 21500 g mol−1 was successful, although an increase in the PDI was observed (Fig. 6). The large PDI is attributed to the presence of dead poly(n-butyl acrylate)s obtained during the initial polymerization.
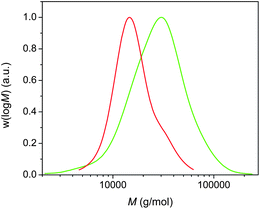 | ||
| Fig. 6 Molecular weight distributions for (i) poly(n-butyl acrylate) with Mn = 15000 g mol−1 (red) and (ii) after subsequent chain extension using a biphasic process. | ||
Finally, we examined the effect of some more hydrophobic acrylates (lauryl and 2-ethyl hexyl acrylate) (Table 1). As these monomers are insoluble in DMSO at ambient temperature, the reaction mixture was in two phases at the beginning of the polymerisation. Polymerisation was achieved as indicated by a high monomer conversion (>90%), however, the large molecular weights and high PDI achieved indicate that control of the reaction has been lost. Thus, it is important for the monomers to be soluble in the polar solvent whilst the polymer becomes insoluble.
Conclusions
In summary, we have developed a self-generating biphasic polymerisation system that is compatible with Cu(0)-mediated radical polymerisation at ambient temperature. This approach provides a facile method to remove residual copper from the polymer without complex purification procedures – the polymer phase contains only 0.016 wt% copper by ICP-MS analysis. The method has been exploited to conduct the first ever Cu(0)-mediated radical polymerisation of n-butyl acrylate with a very good control of the molecular weight. We explored this biphasic approach for the synthesis of a range of poly(acrylate)s with longer aliphatic chains. However, for these more hydrophobic acrylates, we observed that the polymerization is not well controlled.Acknowledgements
CB is thankful for an APD Fellowship (Discovery Grant DP1092640). Equipment used in this research was partly funded through Advantage West Midlands (AWM) Science City Initiative and partly funded by the ERDF. DMH is a Royal Society/Wolfson Fellow. The authors thank the Mark Wainwright Analytical Centre for NMR analysis and ICP-MS measurement. Funding from Lubrizol (AA, CW) and Syngenta (PW) is gratefully acknowledged.References
- T. Noda, A. J. Grice, M. E. Levere and D. M. Haddleton, Eur. Polym. J., 2007, 43, 2321 CrossRef CAS.
- D. R. D'Hooge, D. Konkolewicz, M.-F. Reyniers, G. B. Marin and K. Matyjaszewski, Macromol. Theory Simul., 2012, 21, 52 CrossRef CAS.
- L. Mueller, W. Jakubowski, W. Tang and K. Matyjaszewski, Macromolecules, 2007, 40, 6464 CrossRef CAS.
- L. Mueller and K. Matyjaszewski, Macromol. React. Eng., 2010, 4, 180 CrossRef CAS.
- T. Pintauer and K. Matyjaszewski, Chem. Soc. Rev., 2008, 37, 1087 RSC.
- N. V. Tsarevsky and W. Jakubowski, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 918 CrossRef CAS.
- M. Kamigaito, Organomet. News, 2009, 66 CAS.
- D.-L. Popescu and N. V. Tsarevsky, Macromol. Rapid Commun., 2012, 33, 869 CrossRef CAS.
- W. Jakubowski and K. Matyjaszewski, Angew. Chem., Int. Ed., 2006, 45, 4482 CrossRef CAS.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015 CrossRef CAS.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156 CrossRef CAS.
- N. H. Nguyen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4241 CAS.
- N. H. Nguyen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4227 CAS.
- N. H. Nguyen, M. E. Levere, J. Kulis, M. J. Monteiro and V. Percec, Macromolecules, 2012, 45, 4606 CrossRef CAS.
- N. H. Nguyen, M. E. Levere and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 860 CrossRef CAS.
- N. H. Nguyen, M. E. Levere and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 35 CrossRef CAS.
- M. E. Levere, I. Willoughby, S. O'Donohue, P. M. Wright, A. J. Grice, C. Fidge, B. C. Remzi and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1753 CrossRef CAS.
- P. M. Wright, G. Mantovani and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7376 CrossRef CAS.
- M. S. Silverstein, Y. Najary, G. S. Grader and G. E. Shter, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 1023 CrossRef CAS.
- S. M. Grimes, H. Lateef, A. J. Jafari and L. Mehta, Polym. Degrad. Stab., 2006, 91, 3274 CrossRef CAS.
- F. Perreault, A. Oukarroum, S. P. Melegari, W. G. Matias and R. Popovic, Chemosphere, 2012, 87, 1388 CrossRef CAS.
- K. Matyjaszewski, Isr. J. Chem., 2012, 52, 206 CrossRef CAS.
- Q. Lou and D. A. Shipp, ChemPhysChem DOI:10.1002/cphc.201200166.
- M. Contel, C. Izuel, M. Laguna, P. R. Villuendas, P. J. Alonso and R. H. Fish, Chem.–Eur. J., 2003, 9, 4168 CrossRef CAS.
- G. Ragagnin, B. Betzemeier, S. Quici and P. Knochel, Tetrahedron, 2002, 58, 3985 CrossRef CAS.
- T. Pintauer and K. Matyjaszewski, Chem. Soc. Rev., 2008, 37, 1087 RSC.
- D. M. Haddleton, S. G. Jackson and S. A. F. Bon, J. Am. Chem. Soc., 2000, 122, 1542 CrossRef CAS.
- D. M. Haddleton, D. J. Duncalf, D. Kukulj and A. P. Radigue, Macromolecules, 1999, 32, 4769 CrossRef CAS.
- G. Kickelbick, H.-J. Paik and K. Matyjaszewski, Macromolecules, 1999, 32, 2941 CrossRef CAS.
- D. Haddleton, D. Kukulj and A. Radigue, Chem. Commun., 1999, 99 RSC.
- S. Faucher and S. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 553 CrossRef CAS.
- T. Sarbu and K. Matyjaszewski, Macromol. Chem. Phys., 2001, 202, 3379 CrossRef CAS.
- T. Biedroń and P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2799 CrossRef.
- A. J. Carmichael, D. M. Haddleton, S. A. F. Bon and K. R. Seddon, Chem. Commun., 2000, 1237 RSC.
- S. K. Mohan, in Green Solvents I, ed. A. Mohammad, SpringerNetherlands, 2012, p. 251 Search PubMed.
- Y. Shen, S. Zhu, F. Zeng and R. H. Pelton, Macromolecules, 2000, 33, 5427 CrossRef CAS.
- S. Ding, Y. Xing, M. Radosz and Y. Shen, Macromolecules, 2006, 39, 6399 CrossRef CAS.
- S. C. Hong and K. Matyjaszewski, Macromolecules, 2002, 35, 7592 CrossRef CAS.
- B. Otazaghine, C. Boyer, J.-J. Robin and B. Boutevin, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2377 CrossRef CAS.
- D. M. Haddleton, D. J. Duncalf, D. Kukulj, M. C. Crossman, S. G. Jackson, S. A. F. Bon, A. J. Clark and A. J. Shooter, Eur. J. Inorg. Chem., 1998, 1998, 1799 CrossRef.
- D. Konkolewicz, A. J. D. Magenau, S. E. Averick, A. Simakova, H. He and K. Matyjaszewski, Macromolecules, 2012, 45, 4461 CrossRef CAS.
- M. Ciampolini and N. Nardi, Inorg. Chem., 1966, 5, 41 CrossRef CAS.
- C. Boyer, A. Derveaux, P. B. Zetterlund and M. R. Whittaker, Polym. Chem., 2012, 3, 117 RSC.
- A. H. Soeriyadi, C. Boyer, F. Nystrom, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 11128 CrossRef CAS.
- F. Nystroem, A. H. Soeriyadi, C. Boyer, P. B. Zetterlund and M. R. Whittaker, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5313 CrossRef CAS.
- C. Boyer, A. H. Soeriyadi, P. B. Zetterlund and M. R. Whittaker, Macromolecules, 2011, 44, 8028 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, Fig. S1 and S2 and Tables S1 and S2. See DOI: 10.1039/c2py20560b |
| This journal is © The Royal Society of Chemistry 2013 |