A review of UVB-mediated photosensitivity disorders†
F.
Kiss
ab and
A. V.
Anstey
*ac
aAneurin Bevan Health Board, St Woolos Hospital, Stow Hill, Newport, UK. E-mail: alex.anstey@wales.nhs.uk
bDepartment of Dermatology, University of Debrecen, Medical and Health Science Centre, Nagyerdei boulevard 98, Debrecen 4032, Hungary
cDepartment of Wound Healing and Dermatology, School of Medicine, Cardiff University, Heath Park, Cardiff CF14 4XN, UK
First published on 1st October 2012
Abstract
Photosensitivity to UVB is a prominent feature of a small number of congenital skin disorders. In addition UVB may also contribute to the action spectrum of a number of acquired photosensitivity syndromes. The gene mutations underlying the genetically inherited disorders have largely been identified and have provided insights into DNA repair pathways. The pathomechanisms for the acquired disorders are still largely undefined. Few therapeutic options are available so management of all these disorders still relies on rigorous photo-protection.
Introduction
The adverse effects of UVB on human skin are familiar to those with fair-skin. Sun burn following excess sun exposure is painful and distressing. Severe sun burn can lead to blistering followed by skin exfoliation, and may take a number of days to resolve. In view of the potency of these UVB-mediated effects, it is perhaps surprising that photosensitivity mediated by UVB alone is very rare. More common are photosensitivity conditions in which the action spectrum includes UVB in addition to UVA and sometimes visible light.There are a number of inflammatory skin conditions which may be worsened, termed “photo-aggravated”, by excess sun exposure. Paradoxically, some of these same inflammatory skin disorders are also improved, termed “photoresponsive”, following small repeated doses of UVB administered over a period of a few weeks. This review will not include these photoaggravated disorders, but focuses instead on the primary photosensitivity disorders in which sensitivity to UVB can be demonstrated in some or most cases by objective, standardised phototesting. The gold-standard objective test for establishing the action spectrum of clinical photosensitivity reactions is monochromator light testing. This test procedure uses equipment consisting of a Xenon-arc lamp, focusing lenses, a closed system of running water (to remove the infra-red), a monochromator (consisting of an enclosed optical box containing a diffraction grating and system of lenses and reflecting mirrors), and an exit port to which is attached a liquid light guide. The liquid light guide delivers the UV or visible light to the patient's skin. The equipment is calibrated to a traceable source. The output of the equipment is determined by a thermopile. Doses are then calculated on the basis of time as recorded by a stop-watch operated by the test technician. Testing involves exposing small areas of skin on the lower back to a dose sequence for a selected range of wavelengths in the ultraviolet and visible light spectra. Knowledge of normal ranges in the local population allows for the identification of abnormal reactions from this test system.
Few centres have monochromatic light testing facilities and most use broad spectrum UVA, UVB and visible radiation sources to assess how the skin responds.
Methods
The following search strategy was used:Medline, Embase, PubMed and the Cochrane Library were searched for the following terms during the period of January 2005–April 2012;
Photosensitiv*
UVB or ultraviolet light/radiation or sunlight
Xeroderma pigmentosum
Cockayne syndrome
UV sensitive syndrome
Trichothiodystrophy
Rothmund–Thomson syndrome
Bloom's syndrome
Kindler syndrome
Polymorphic light eruption/polymorphous light eruption
Chronic actinic dermatitis
Solar urticaria
Actinic prurigo
Lupus erythematosus (LE)/systemic LE/subacute cutaneous LE/discoid LE/LE tumidus
Photoprotection/UV(B) protection/sun protection
Congenital photosensitivity syndromes
Xeroderma pigmentosum
Xeroderma pigmentosum (XP) is probably the best known UVB-mediated photosensitivity syndrome on account of the striking clinical phenotype and often poor prognosis. XP is a congenital photosensitivity syndrome in which DNA (nucleotide) excision repair (NER) is defective; affected individuals show marked hypersensitivity to ultraviolet (UV) irradiation. XP is transmitted in an autosomal recessive manner affecting both sexes equally.1 It is rare, with an estimated prevalence of 1 in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 population in Europe and North America, and moderately less rare in Japan, and the Middle East.2 A founder effect is believed to explain the unexpectedly high prevalence of XPC in the Maghreb countries of North Africa.3 About eighty percent of patients suffer from one of the seven subtypes (A–G OMIM 278700, 610651, 278720, 278730, 278740, 278760, 278280) of “classic” XP each caused by a distinctive mutation in genes involved in one of the first three steps of the NER pathway (Fig. 1); the other 20% are classified as XP-variant (OMIM 278750) where the defect is in a post-replication step caused by a mutation in the DNA polymerase eta gene.4
000000 population in Europe and North America, and moderately less rare in Japan, and the Middle East.2 A founder effect is believed to explain the unexpectedly high prevalence of XPC in the Maghreb countries of North Africa.3 About eighty percent of patients suffer from one of the seven subtypes (A–G OMIM 278700, 610651, 278720, 278730, 278740, 278760, 278280) of “classic” XP each caused by a distinctive mutation in genes involved in one of the first three steps of the NER pathway (Fig. 1); the other 20% are classified as XP-variant (OMIM 278750) where the defect is in a post-replication step caused by a mutation in the DNA polymerase eta gene.4
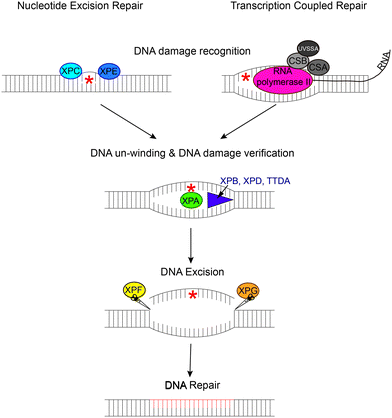 | ||
| Fig. 1 Genes causing congenital photosensitivity syndromes involved in the NER and TCR Pathways. The nucleotide excision repair pathway corrects UV-induced mutations in the genome. If transcription occurs before the NER has repaired the mutation then CSB protein binds stably to RNA polymerase II which then causes other proteins to bind such as CSA and UVSSA. Both pathways share the DNA unwinding, DNA excision and repair steps. | ||
Early onset chronic photodamage and the development of multiple skin cancers due to UV sensitivity are typical in XP. Involvement of the eyes is also frequent resulting in photophobia and inflammation of the anterior structures (lids, conjunctiva and cornea).1 The risk of basal and squamous cell carcinoma and melanoma is greatly elevated (perhaps 1000-fold) in XP patients at sun-exposed areas such as the skin and eyes.5 The onset of cancer formation without UV protection from a very early age is generally in teenage years and early twenties. An exaggerated sunburn response following minimal sun exposure is present in about 60% of XP patients; it may result in skin blistering and is typically a presenting feature within the first two years of life.6 In XP patients where tanning appears to be normal, intense freckling usually develops on sun-exposed sites.7 A recently published article reports on the pathophysiological role of oxidized glycerophosphocholines and platelet-activating factor in the induction of photosensitivity associated with cases where the protein product of the XPA gene is deficient.8 Solar keratoses and dry skin are often prominent features of XP. Neurological abnormalities are common in complementation groups A, B, D and G; these include low intelligence, impaired hearing, microcephaly and peripheral neuropathy. Neuronal degeneration is believed to form the basis for some of these features. The life expectancy of XP patients is shortened in most cases, but may approach normal in those with milder disease where photo-protection has been rigorous and persistent from an early age.
The diagnosis of XP is usually clinical, based on a detailed and careful history and physical examination. An exaggerated sun-burn response from first sun exposure, early onset cutaneous photodamage, and consanguineous marriage of the parents are positive features that raise the possibility of this diagnosis. Monochromator light testing usually shows low minimal erythema dose (MED) to UVB irradiation, with delayed onset and prolonged erythema lasting for several days; blistering may develop at sites of UVB irradiation. UV exposure cell survival studies using cultured fibroblasts from a small punch biopsy of skin may confirm the diagnosis. Decreased cell survival after UV exposure is characteristic; fibroblasts derived from patients with XP-variant show impaired daughter strand repair. Molecular genetic testing is also available in specialized laboratories and may be used to confirm the diagnosis.
XP requires rigorous sun-avoidance, protection by clothing, sunglasses and sunscreens (UVB and broad spectrum UVA protection) applied several times daily.6 Modification of the patients’ home and school or work environment with UV protective film and enhanced shade is also necessary.6 Dermatological surveillance for skin malignancies is needed in XP patients with significant photodamage, at a frequency determined by clinical need.9 Skin cancers may be treated by a variety of techniques including surgical techniques,7 photodynamic therapy,10,11 topical 5-fluorouracil and topical imiquimod.12 Cryotherapy may be used for the treatment of premalignant lesions such as solar keratoses. Dermabrasion, chemical peels and high-dose oral retinoids may be advantageous in preventing new skin cancer formation in some patients. Clinical trials with a bacterial DNA repair enzyme called T4 endonuclease V, decreased the incidence of basal cell carcinoma and solar keratoses in XP patients.13 Gene therapy, currently in research phase,14 is a possible future option in XP therapy by complementation of defective genes. However, significant issues need to be resolved before such therapy is a realistic option.
Cockayne syndrome
Cockayne syndrome (CS) is an autosomal recessive congenital photosensitivity syndrome; it is even rarer than XP. CS type A (OMIM 216400) and type B (OMIM 133540) result from a defect in transcription-coupled repair (TCR). TCR overlaps with the NER pathway and occurs when a mutation that hasn't been repaired by NER is encountered by RNA polymerase II during transcription (Fig. 1). Mutations occur most commonly in genes involved in the TCR pathway such as ERCC8 (CKN1 or CSA) or the ERCC6 (CSB) genes or very rarely in XP genes which are involved in both pathways such as XPB, XPD and XPG producing a CS phenotype (XP–CS complex). This defect impairs recovery from blocked transcription leading to DNA damage and cell death.The most common clinical feature of CS is failure to thrive due to a halt in growth and development; this usually commences at the start of the first year of life and leads to dwarfism.15 As with XP, the range of clinical phenotype and disease severity for CS is wide. Early onset, progressive neurodegeneration leads to hearing loss, visual defects, cataracts, spasticity, peripheral neuropathy, sensorineural hearing loss, and intellectual and behavioural decline. Photosensitivity and a prematurely senile appearance are characteristic of CS; in contrast to XP the risk of cutaneous malignancies is not elevated. Some patients show a prominent exaggerated sunburn response. Photo-testing in two Japanese brothers with CS revealed sensitivity to UVB.16 Life expectancy in CS shortened; death usually occurs in the first or second decade.
The diagnosis of CS is usually established on the basis of these characteristic clinical features,17 and confirmed by DNA repair studies on cultured skin fibroblasts; these show UV sensitivity and impaired RNA synthesis recovery following UV irradiation. In atypical cases, gene mutational analysis may be of benefit, but is not routinely available.
Patient-tailored multidisciplinary management and surveillance is advisable including physical therapy, neurological and audiometric examinations, sun protection and educational programs. A small case series from Boston including three CS patients with motor complications reported beneficial effects for carbidopa–levodopa on the patients’ daily activities.18
UV sensitive syndrome
In 1994 Itoh and co-workers from Japan coined the term “UV-sensitive syndrome” (UVSS) for a condition characterized by mild photosensitivity and defective DNA repair.19 The condition is extremely rare; less than 10 patients have been reported in the literature. UVSS is a genetically heterogeneous syndrome caused by mutations in the genes CSB (OMIM 600630) and CSA (OMIM 614621) or a recently identified gene UVSSA (OMIM 614631) coding for a protein that interacts with CSB (Fig. 1).20–22 Following UV radiation, RNA synthesis is decreased. According to complementation studies UVSS is different from all other known XP and CS groups.Moderate photosensitivity and mild freckling without cancer predisposition is typical of UVSS.19,20 The life span is not shortened and no other abnormalities have been reported in affected individuals. Because of the mild clinical manifestations the frequency of the condition is probably underestimated.
Lack of diagnostic criteria make UVSS a diagnosis by exclusion; affected patients have a DNA-repair deficiency but do not fit into the complementation groups of XP or CS.23 Diagnosis is therefore difficult. The condition should be suspected in patients with mild congenital photosensitivity and normal development without cutaneous malignancies. Laboratory tests including RNA-synthesis studies, cell response to oxidative stress and molecular genetic analysis are needed to rule out other inherited NER deficiency syndromes. Appropriate sun protection is recommended for UVSS patients. Long-term dermatological surveillance is recommended in order to gather more clinical data on treatment, outcome and prognosis for this very unusual and rare condition.
Trichothiodystrophy
Trichothiodystrophy (TTD; OMIM 601675) is another very rare geno-photodermatosis with autosomal recessive transmission. Mutations occur in one of four genes, XPB, XPD and TTDA, all involved in the DNA unwinding step of the NER/TCR pathways (Fig. 1) or TTDN1, a gene of unknown function.24 Sulphur-deficient sparse, brittle hair is characteristic of the condition and may lead to alopecia.25 The pathogenesis of TTD is not clearly understood; an embryonic defect leads to complications during pregnancy for both mother and foetus.26 UVB-induced XP-like photosensitivity is observed in about 50% of TTD patients with a mutation in one of the NER system genes. However, the risk of cutaneous malignancy in these patients is not increased.27 Clinical features of TTD include neurological defects, growth retardation, recurrent infections and cutaneous manifestations. Skin findings comprise ichthyosis, occasionally collodion membrane at birth and dry skin.28 The nails in TTD may be fragile, hypoplastic and generally dystrophic. Mortality for this condition is about 20-fold higher than normal.Brittle hair or hair shaft anomalies observed at physical examination are often helpful clinical clues for the diagnosis of TTD. Hair studies with polarised microscopy reveal “tiger tail banding” pattern with alternating light and dark bands; biochemical analysis of hair reveals decreased sulphur content and a reduced cysteine/cystine ratio.29 DNA-repair abnormalities can be measured and molecular biologic testing may also be performed. Prenatal diagnosis is possible using these laboratory techniques. A Canadian group reported three unrelated TTD patients with photosensitivity in whose fibroblast cell lines showed decreased cell survival following UVB exposure.30
The management of TTD is multidisciplinary. Generally paediatricians are in charge of long-term follow-up. Appropriate photo-protection is necessary in photosensitive patients.
Rothmund–Thomson syndrome
The Rothmund–Thomson syndrome (RTS; OMIM 268400) is a rare autosomal recessive congenital syndrome. Homozygous or compound heterozygous mutations occur in many but not all RTS patients in the RECQL4 (RECQ4) gene.31 The protein product of this gene takes part in DNA repair after UV damage. Other syndromes associated with RECQL4 mutations include RAPADILINO (Radial and patellar aplasia; OMIM 266280)32 and Baller–Gerold syndrome (radial aplasia and craniosynostosis; OMIM 218600).33 Defective function of this gene results in chromosomal instability; complete loss of function is lethal in humans. In some RTS patients the responsible gene/genes is/are yet unidentified.An erythematous rash starts flourishing on sun-exposed sites in infancy gradually developing into a persistent poikiloderma characterized by hypo- and hyperpigmentation, telangiectases and atrophy. Prominent features include short stature, skeletal dysplasia, cataracts, sparse hair and an elevated risk of malignancy.34 Dental and nail abnormalities and warty hyperkeratosis may also occur. Progressive photodamage and an exaggerated sunburn response have been reported in several patients exhibiting the effect of UVB irradiation in the cutaneous features of RTS. The severity of symptoms may vary greatly between patients. The most frequent neoplasm is osteosarcoma with an estimated prevalence of 30% in RTS;34 it is especially common in patients with RECQL4 mutations. Cutaneous malignancies such as Bowen's disease, BCC and SCC are also not uncommon with an estimated prevalence of 5% in RTS, usually occurring in adulthood.34
Diagnosis of RTS is not easy; it relies mainly on the above mentioned clinical features, especially the early onset of poikiloderma in sun-exposed areas. Other disorders associated with childhood poikiloderma should be considered in the differential diagnosis, including Kindler syndrome. Molecular genetic testing of the RECQL4 gene is recommended in RTS patients.
The management of RTS patients is multidisciplinary and should involve a paediatrician, dermatologist, oncologist, ophthalmologist, dentist and an orthopaedic surgeon. Sun-avoidance and photo-protection are important. Pulsed dye laser therapy has been reported to improve the telangiectatic component of cutaneous lesions.35
Bloom's syndrome
Bloom's syndrome (OMIM 210900) is a rare inherited condition characterized by photosensitivity associated with telangiectasia, pigmentary abnormalities, growth retardation and impaired immune function. The latter leads to recurrent infections and susceptibility to childhood malignancies. The mode of inheritance is autosomal recessive; mutations in the BLM/RecQL gene result in errors during DNA replication.36 The disease is most common in the Ashkenazi Jewish population.Following sunlight exposure, erythema can be observed on the exposed skin. The facial skin is erythematous due to telangiectasia and becomes progressively redder with repeated sun exposure.37 Vesicle and crust formation alongside bleeding may also be recognized at sun-exposed sites. Cutaneous changes and photosensitivity have a tendency to improve with time.
Gene mutation analysis can confirm the diagnosis of Bloom's syndrome. A Japanese group reported two patients whose peripheral blood mononuclear cells were more sensitive to UVB irradiation compared to controls after 3 and 7 days of culture.38 In a few cases, monochromator light testing of patients suffering from Bloom's syndrome has demonstrated UVB sensitivity. However, photosensitivity in Bloom's syndrome is still poorly defined; furthermore, UV sensitivity is not a universal feature of cells derived from all patients with the condition.
The management of patients with Bloom's syndrome is multidisciplinary. Regular follow-ups and screening for neoplasms are essential. Infections require appropriate antibacterial therapy. Skin care requires rigorous photo-protection with clothing and sun-avoidance combined with the use of high SPF sunscreens to sun-exposed skin. Genetic counselling is available for carriers and family members.
Kindler syndrome
Kindler syndrome (KS; OMIM 173650) is a very rare congenital autosomal recessive photosensitivity syndrome. Acral blistering shortly after birth, generalised and progressive poikiloderma, skin fragility, photosensitivity and diffuse skin atrophy are typical in the condition caused by mutations in the KIND1 gene encoding the protein kindlin-1.39 Mucosal involvement leading to urethral, anal and oesophageal stenosis is not uncommon. Gingivitis and periodontitis are also frequent oral features of KS. The sexes are affected equally and the anticipated life span is normal in patients.Patients with KS are usually prone to sunburn. Photosensitivity tends to improve gradually with age, as does blistering. Cutaneous changes are most prominent on sun-exposed areas such as the dorsum of the hands and forearms. Early onset solar keratoses may occur, further emphasizing sensitivity to UV irradiation.40 Cosmetic deformity may result in significant morbidity in patients suffering from Kindler syndrome. The diagnosis is generally based on clinical phenotype and is confirmed by gene mutation analysis. Photo-testing may be helpful; some patients show reduced MEDs to irradiation with UVA or with UVB.41 Management of the disease is multidisciplinary including paediatricians, surgeons, dentists, psychologists and dermatologist. Pulsed dye laser treatment may be beneficial for telangiectasias. Appropriate sun protection is necessary by use of sunscreens, clothing and UV protective film.
Acquired photosensitivity syndromes
Chronic actinic dermatitis
Chronic actinic dermatitis (CAD) is often a severe photosensitivity condition categorized among the idiopathic acquired photosensitivity syndromes.42 The age of onset for CAD is typically between 40 and 80 years; men are more frequently affected than women. Other dermatological conditions such as allergic contact dermatitis, atopic dermatitis or drug-induced photoallergy sometimes precede CAD.It has been suggested that CAD is an immune-mediated disease generated by photo-induced endogenous cutaneous allergens exhibiting a type IV hypersensitivity reaction.43 CAD has also been reported to occur following the administration of a new medication and drug-induced photosensitivity in some patients which persists despite the withdrawal of the provoking agent.44 The exact pathomechanism of the condition is still unclear despite the considerable amount of research in the field. The disorder can impair the quality of life of patients greatly.
An observation of the changing contact allergens in association with CAD was reported in a recent UK study by comparison of 50 patients patch tested between 2000 and 2005 and 86 between 1987 and 1992.45 The most common contact allergen was found to be sesquiterpene lactone demonstrated by a positive reaction in approximately 20% of cases in this study.45 In India airborne contact dermatitis caused by the commonly growing plant parthenium is a relevant provocation factor for CAD.46
Clinical manifestations include severe, persistent erythema or eczema of sun-exposed and sometimes also non-exposed sites. Histology reveals a spectrum of pathological changes ranging from chronic eczematous reaction to the appearance of cutaneous T-cell lymphoma-like changes. Monochromator light testing typically demonstrates marked photosensitivity with reduced MEDs to UVB, UVA and/or visible light. Patch and photo-patch testing may reveal relevant contact allergens but not always.
CAD is a chronic condition that has a tendency to persist for many years; resolution was 50% after 15 years according to one study47 and 20% after 10 years according to another study.48 After the diagnosis has been established, CAD usually ameliorates in severity in response to a strict photo-protection regime. Appropriate protection against the confirmed activating wavelengths (UVB, UVA and/or visible light) is essential in the treatment of the disorder. In most cases topical steroids and topical calcineurin inhibitors are of therapeutic use.49 Systemic immunosuppression using corticosteroids, azathioprine, cyclosporine or mycophenolate mofetil50 may be helpful in more severe cases when the above mentioned management strategies are not enough to control the disease. Desensitization with systemic phototherapy (PUVA or NB-UVB) has been tried with success with extreme caution.51,52 Avoidance of known contact allergens is another essential element of the management strategy of the condition.
Solar urticaria
Solar urticaria (SU) is another idiopathic acquired photosensitivity syndrome also belonging to the physical urticarias. Cutaneous hives (wheals or urticaria) occur typically after a few minutes of sun exposure53 and sometimes angio-oedema (facial swelling) can also be observed.54 SU is a rare condition affecting women slightly more frequently than men. The activating wavelengths in case of SU usually include UVA and occasionally UVB and/or visible light as well.The condition is postulated to be an IgE-mediated immunological reaction to a yet unidentified light-induced allergen provoking mast cell degranulation; although the exact mechanism is still unclear.55 The urticarial reaction characteristically arises within minutes of sun exposure lasting for a few hours and is typically present on sun-exposed skin or rarely underneath thin layers of clothing. The hives are intensely itchy. After conditional accumulative sun exposure tolerance to sunlight might develop, although spontaneous regression was only 26% at 10 years according to one group53 and somewhat higher 57% at 5 years according to another series.56
The diagnosis of SU is based upon a detailed and careful history from the patient, physical examination or photographs revealing the characteristic lesions and monochromator light testing determining the action spectrum of the triggering light. The quality of life of SU patients may be markedly impaired as demonstrated by the Dermatology Life Quality Index (DLQI). According to a study the DLQI was more than 10 (this is considered to be a significant effect on the quality of patients’ lives) in more than one third of patients suffering from SU.57
The first line treatment of SU consists of long-acting non-sedating antihistamines and proper sun protection. Sometimes desensitization by phototherapy (PUVA or NB-UVB) with extreme caution or systemic therapy is administered. Intravenous immunoglobulin (IVIG), an IgG antibody preparation, has been tried in several severe SU patients with some success but not in all patients.58–60 Plasmapheresis is another expensive treatment option that has been tried in a few centres in severe SU patients.61 The anti-IgE monoclonal antibody omalizumab has also been tried recently in a few cases with some success.62,63 A brand new therapeutic option in SU still in clinical trial phase is an implant of afamelanotide, an agonist analogue of α-melanocyte stimulating hormone. In a study from Manchester five SU patients were treated with a single dose of afamelanotide (16 mg subcutaneous implant applied in winter) and the minimal urtication dose (MUD) was reported to be reduced by greater than twofold at day 60 (300–600 nm) and a significant reduction in wheal area was also observed.64
Actinic prurigo
Actinic prurigo (AP) is a rare acquired, idiopathic photosensitivity disorder. Although the pathogenesis of the syndrome remains unclear, an immune-mediated reaction is hypothesized in the background against sunlight induced autoantigens in genetically susceptible individuals. AP was first described among North American Indians and Central and South American mestizos while it is less common in Asians and Caucasians. The condition affects more women then men (ratio of 2–4:1.4) and the typical age of onset is in childhood or in young adults. HLA typing revealed a strong association with an elevated incidence of HLA DR4, especially the DRB1*0407 subtype.65–67
Profoundly itchy papules and nodules are the typical lesions of AP localized mainly on sun exposed sites, although covered areas may be affected as well. Symptoms are generally worse in spring and summer especially in temperate climates however the disease may be perennial, particularly in tropical climates. Mucosal involvement is fairly common; the lower lip is affected mainly. Spontaneous regression frequently occurs over years, although the disorder may be chronic with remissions and relapses.
The diagnosis of AP is based on the history and clinical features suggesting the disorder. Monochromator light testing may be negative in some cases but more than half of the patients exhibit a positive reaction to UVA and/or UVB. Photoprovocation testing is positive in about two thirds of patients, therefore it can aid the diagnosis. HLA typing for subtype DRB1*0407 is recommended in cases where the diagnosis is not clear.
In the management of AP photo-protection is crucial by behavioural changes, properly covering clothing, sunscreens,68 sun-glasses and window protection. Potent topical corticosteroids69 and calcineurin inhibitors can result in marked improvement of symptoms. Short courses of systemic steroids may also be helpful in acute relapses. Desensitization phototherapy with NB-UVB70 or PUVA71 is sometimes beneficial in resistant cases; repetitive courses may be required. Thalidomide was reported as highly effective in several severe cases.72 Other systemic therapeutic options for resistant cases include β-carotene, antimalarials, pentoxifylline,73 azathioprine,74 cimetidine, methotrexate, cyclosporine or oral antibiotics such as tetracycline and vitamin E.75
Polymorphic light eruption
Polymorphic light eruption (PLE) is a very common, acquired, idiopathic photosensitivity disorder. According to a recent multicentre European study the estimated lifetime prevalence of PLE is approximately 18%.76 Typical age of onset is around the 2nd–3rd decade of life. Women are more frequently affected then men (ratio of 3:1). A delayed-type (type IV) hypersensitivity reaction against a cutaneous light induced neoantigen in susceptible individuals is suggested primarily as the cause of the disease77 although insufficient immunosuppressive effect of UV irradiation is also suspected in the background. Genetic studies demonstrate an inherited component as well.
PLE lesions are highly variable between patients, presenting as itchy papules, vesicles, plaques or even pustules on sun-exposed sites with a typical sparing of the face and the dorsum of the hands which are exposed to UVR perennially. Clinical features usually develop in a cyclic manner mainly in spring and summer. The latent period between sun exposure and the development of cutaneous lesions generally ranges from a few hours to several days with a usual duration of the rash between 1–14 days. Spontaneous remission is characteristic in autumn. Repeated sun exposure can generate induced tolerance of the skin.
The diagnosis of this condition is generally straightforward based on the typical history and clinical manifestations. Further tests are rarely needed to confirm the diagnosis. If the diagnosis is uncertain a few screening tests such as porphyrin screen or lupus serology may be advantageous by excluding other dermatoses characterized by photosensitivity. Cutaneous photo-testing, if performed, is usually negative although photoprovocation using broad-band UVA or a NB-UVB light source may often show a positive reaction. The action spectrum of PLE is mainly in the UVA and sometimes in the UVB spectrum.
In the management of the condition prevention of the rash, if possible, is important and generally sufficient using appropriate photo-protection by broad-spectrum sunscreen,78,79 clothing and sun avoidance. Potent topical corticosteroids are usually effective and are capable to induce remission in many PLE patients with mild to moderate symptoms although no clinical study has been performed to support this observation. In more severe cases a short course of systemic steroids is generally beneficial.80 Cautious desensitizing phototherapy generally administered in the early springtime by NB-UVB or PUVA can be advantageous in more difficult cases although symptoms may be provoked by this technique.81 Other systemic therapeutic options available for recalcitrant cases include hydroxychloroquine,82 azathioprine83 and cyclosporine.84
Lupus erythematosus
Lupus erythematosus (LE) is a chronic, multisystem, autoimmune disorder with a multifactorial and polygenic background. Skin manifestations (cutaneous lupus erythematosus: CLE) can be detected in about 5–85% of systemic LE (SLE) patients during the whole course of their disease.85 The systemic disease is approximately ten times more frequent in women while the more localized forms of LE such as subacute cutaneous LE (SCLE) and discoid LE (DLE) although are still more common in females but to a lesser extent. Cutaneous manifestations can be subclassified as specific and non-specific lesions.Photosensitivity is one of the diagnostic criteria for SLE and is frequent in all types of cutaneous LE as abnormal photoreactivity is often present. A clear-cut relationship exists between sun exposure and LE skin lesions affecting light-exposed body sites. UVR is known to play an influential role in the pathogenesis of LE by induction and exacerbation of skin lesions in many patients. Under experimental circumstances photoprovocation by both UVB and UVA is capable of induction and therefore reproduction of skin lesions in numerous LE patients.86 The appearance of skin manifestations following UV irradiation is typically prolonged taking a few days or sometimes even weeks; thereafter lesions tend to remain for months.87 According to a study from the United States 68% of a group of patients suffering from various forms of LE reported some form of photosensitivity and these individuals had more severe cutaneous disease as well as significant functional impairments compared to the non-photosensitive group.88 Several reactions involved in photosensitivity are identified; however the exact cause of abnormal susceptibility to UVR still remains unclear. Possible suggested mechanisms include UV-mediated initiation of apoptosis through autoantigens in apoptotic blebs, modulation of antibody location (from the nucleus to the cell surface), upregulation of several adhesion molecules, cytokines and chemokines, cytotoxic effects, UV-induced antigenic DNA and expression of inducible nitric oxide synthase.87,89 According to a German study where 405 LE patients were included more individuals reacted with skin lesions to UVB irradiation (42%) than to UVA irradiation (34%).86
Skin manifestations of LE lesions are usually asymptomatic and frequently present on light-exposed body sites. An erythematous rash typically localized on the cheeks in a butterfly distribution is the most characteristic cutaneous feature of acute cutaneous LE. Subacute cutaneous LE lesions are mainly present on the anterior chest, upper back, shoulders and neck either as scaly “psoriasiform” papules or in an annular configuration. Discoid LE is by far the most frequent form of chronic cutaneous LE (CCLE) and is characterized by thick, scaly, scarring, sharply delineated, hypo- or hyperpigmented plaques on the scalp, face and ears. Lupus erythematosus tumidus subclassified into the intermittent cutaneous LE group is the most photosensitive LE variant, while SCLE and DLE patients are photosensitive to a lesser extent.87
Characteristic skin lesions and a history of light sensitivity may suggest the possibility of LE. In order to establish a correct diagnosis and subclassification various other tests are performed. Serologic studies screen for the presence of autoantibodies such as anti-Ro, anti-La, antinuclear and anti-double strand-DNA antibodies. Histological tests include common histopathology revealing epidermal atrophy, vacuolar degeneration of the basal keratinocytes, thickening of the basement membrane and inflammatory – mainly lymphocytic – infiltrate in the dermis and direct immunofluorescence studies searching for immunoglobulin and/or complement deposition at the dermo-epidermal junction. The lupus band test (immunoreactants in the basement membrane zone) is typically positive in SLE, although it is not considered either specific or sensitive. Light-testing with UVB, UVA and sometimes visible light may be beneficial in patients. Photoprovocation is particularly advantageous in patients by triggering characteristic skin lesions. MED values may be decreased in LE patients when compared to controls.90
Numerous topical and systemic agents have been used in LE patients for skin manifestations with different efficacy.85,91 Because of favourable cost–benefit ratio topical corticosteroids and calcineurin inhibitors are administered most commonly. For more severe cases systemic treatment such as hydroxychloroquine or other antimalarial medications may be necessary. Other treatment options comprise oral steroids, dapsone – especially beneficial in bullous cases-, retinoids – for hyperkeratotic lesions-, thalidomide – most commonly administered for refractory DLE-, methotrexate, azathioprine and mycophenolate mofetil. Recently new immunomodulatory and biological treatments such as anti-TNFα agents, monoclonal anti-CD20 (rituximab) or efalizumab have also been tried. Lasers may be helpful for the treatment of vascular LE lesions. Various trials reported UVA1 as an effective therapeutic option resulting in decreased disease activity in several SLE patients.92,93 Favourable results were observed with UVB “hardening” phototherapy in several patients.94 Preventive sun protection is essential in photosensitive individuals.95
UVB protection
The management of all the above mentioned photosensitivity conditions requires appropriate photo-protection. It is easier to protect against UVB than UVA irradiation since the shorter wavelengths of UVB are only capable of superficial penetration in the skin and the ozone layer already absorbs about 90% of these rays.Photo-protection may be a prophylactic and also a therapeutic measure against the harmful effects of UVR.96 Behavioural changes (limited sun exposure), the use of protective clothing and sunscreens all result in various degrees of photo-protection. Clothing (including gloves) – especially made of tightly woven, thick and dark fibres-, hats and sunglasses are all recommended and feasible for photo-protection. According to a 2007 Lancet review an average cotton T shirt provides a protection equivalent to an SPF10 value.97 It is noteworthy that there are several specially UV protective clothing lines commercially available.
Sunscreens are cosmetic formulations containing filters: such molecules that are capable of absorbing, scattering or reflecting UVR. Sunscreens are reviewed thoroughly in Expert Reviews of Dermatology 2011 volume six, issue 5 and only a brief summary is given here. Photo-protective agents first became regulated by the FDA in the 1940s. Sunscreens may contain inorganic (physical blocker) and/or organic (chemical absorber) UV filters. Sun protection factor (SPF) is a number describing the effectiveness of sunscreens in relation to UVB irradiation.96,98 Inorganic filters form a physical barrier on the skin surface, they include iron and zinc oxide, titanium dioxide;98,99 currently the latter two are mainly used in nanoparticle (smaller than 100 nm) preparations creating less viscous sunscreens. Organic UVB filters comprise 4-amino-benzoic acid, cinnamates and salicylates. Sunscreens should be applied often and generously in patients with photosensitivity.97
Conclusion
Photosensitivity to UVB alone is very rare, and usually manifests as a form of congenital photosensitivity with presentation following first sun exposure in infancy. Acquired photosensitivity to UVB is more commonly seen, but typically also includes photosensitivity to UVA, and occasionally visible light too. These disorders, whilst very rare, are of great interest to photobiologists, providing as they do, opportunities to study the diversity of mechanisms involved in mediating photosensitivity in these conditions. This in turn gives us important insights into the multiple mechanisms involved in protecting the skin from UV-induced damage.References
- K. H. Kraemer, M. M. Lee and J. Scotto, Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases, Arch. Dermatol., 1987, 123, 241–250 CAS.
- Y. Doubaj, F. Z. Laarabi and S. C. Elalaoui, et al. Carrier frequency of the recurrent mutation c.1643_1644delTG in the XPC gene and birth prevalence of the xeroderma pigmentosum in Morocco, J. Dermatol., 2012, 39, 382–384 CrossRef CAS.
- N. Soufir, C. Ged and A. Bourillon, et al. A prevalent mutation with founder effect in xeroderma pigmentosum group C from north Africa, J. Invest. Dermatol., 2010, 130, 1537–1542 CrossRef CAS.
- J. H. Hoeijmakers, DNA damage, aging, and cancer, N. Engl. J. Med., 2009, 361, 1475–1485 CrossRef CAS.
- I. Rapin, Y. Lindenbaum and D. W. Dickson, et al. Cockayne syndrome and xeroderma pigmentosum, Neurology, 2000, 55, 1442–1449 CrossRef CAS.
- A. R. Lehmann, D. McGibbon and M. Stefanini, Xeroderma pigmentosum, Orphanet J. Rare Dis., 2011, 6, 70 CrossRef.
- K. H. Kraemer, N. J. Patronas and R. Schiffmann, et al. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype–phenotype relationship, Neuroscience, 2007, 145, 1388–1396 CrossRef CAS.
- Y. Yao, K. A. Harrison and M. Al-Hassani, et al. Platelet-activating factor receptor agonists mediate xeroderma pigmentosum A photosensitivity, J. Biol. Chem., 2012, 287, 9311–9321 CrossRef CAS.
- S. Moriwaki and K. H. Kraemer, Xeroderma pigmentosum—bridging a gap between clinic and laboratory, Photodermatol., Photoimmunol. Photomed., 2001, 17, 47–54 CrossRef CAS.
- D. M. Larson and B. B. Cunningham, Photodynamic therapy in a teenage girl with xeroderma pigmentosum type C, Pediatr. Dermatol., 2012, 29, 373–374 CrossRef.
- S. Segura, S. Puig and C. Carrera, et al. Non-invasive management of non-melanoma skin cancer in patients with cancer predisposition genodermatosis: a role for confocal microscopy and photodynamic therapy, J. Eur. Acad. Dermatol. Venereol., 2011, 25, 819–827 CrossRef CAS.
- N. K. Weisberg and M. Varghese, Therapeutic response of a brother and sister with xeroderma pigmentosum to imiquimod 5% cream, Dermatol. Surg., 2002, 28, 518–523 CrossRef.
- D. Yarosh, J. Klein and A. O'Connor, et al. Effect of topically applied T4 endonuclease V in liposomes on skin cancer in xeroderma pigmentosum: a randomised study. Xeroderma Pigmentosum Study Group, Lancet, 2001, 357, 926–929 CrossRef CAS.
- E. Warrick, M. Garcia and C. Chagnoleau, et al. Preclinical corrective gene transfer in xeroderma pigmentosum human skin stem cells, Mol. Ther., 2012, 20, 798–807 CrossRef CAS.
- M. A. Nance and S. A. Berry, Cockayne syndrome: review of 140 cases, Am. J. Med. Genet., 1992, 42, 68–84 CrossRef CAS.
- H. Miyauchi, T. Horio and T. Akaeda, et al. Cockayne syndrome in two adult siblings, J. Am. Acad. Dermatol., 1994, 30, 329–335 CrossRef CAS.
- E. Ozdirim, M. Topcu and A. Ozon, et al. Cockayne syndrome: review of 25 cases, Pediatr. Neurol., 1996, 15, 312–316 CrossRef CAS.
- E. G. Neilan, M. R. Delgado and M. A. Donovan, et al. Response of motor complications in Cockayne syndrome to carbidopa–levodopa, Arch. Neurol., 2008, 65, 1117–1121 CrossRef.
- T. Itoh, T. Ono and M. Yamaizumi, A new UV-sensitive syndrome not belonging to any complementation groups of xeroderma pigmentosum or Cockayne syndrome: siblings showing biochemical characteristics of Cockayne syndrome without typical clinical manifestations, Mutat. Res., 1994, 314, 233–248 CrossRef CAS.
- Y. Nakazawa, K. Sasaki and N. Mitsutake, et al. Mutations in UVSSA cause UV-sensitive syndrome and impair RNA polymerase IIo processing in transcription-coupled nucleotide-excision repair, Nat. Genet., 2012, 44, 586–592 CrossRef CAS.
- G. Spivak, UV-sensitive syndrome, Mutat. Res., 2005, 577, 162–169 CrossRef CAS.
- X. Zhang, K. Horibata and M. Saijo, et al. Mutations in UVSSA cause UV-sensitive syndrome and destabilize ERCC6 in transcription-coupled DNA repair, Nat. Genet., 2012, 44, 593–597 CrossRef CAS.
- T. Itoh, Y. Fujiwara and T. Ono, et al. UVs syndrome, a new general category of photosensitive disorder with defective DNA repair, is distinct from xeroderma pigmentosum variant and rodent complementation group I, Am. J. Hum. Genet., 1995, 56, 1267–1276 CAS.
- M. Stefanini, E. Botta and M. Lanzafame, et al. Trichothiodystrophy: from basic mechanisms to clinical implications, DNA Repair (Amst), 2010, 9, 2–10 CrossRef CAS.
- P. H. Itin, A. Sarasin and M. R. Pittelkow, Trichothiodystrophy: update on the sulfur-deficient brittle hair syndromes, J. Am. Acad. Dermatol., 2001, 44, 891–920 CrossRef CAS , quiz 1–4.
- D. Tamura, M. Merideth and J. J. DiGiovanna, et al. High-risk pregnancy and neonatal complications in the DNA repair and transcription disorder trichothiodystrophy: report of 27 affected pregnancies, Prenat. Diagn., 2011, 31, 1046–1053 CrossRef CAS.
- J. J. DiGiovanna and K. H. Kraemer, Shining a light on xeroderma pigmentosum, J. Invest. Dermatol., 2012, 132, 785–796 CrossRef CAS.
- S. Faghri, D. Tamura and K. H. Kraemer, et al. Trichothiodystrophy: a systematic review of 112 published cases characterises a wide spectrum of clinical manifestations, J. Med. Genet., 2008, 45, 609–621 CrossRef CAS.
- S. Cheng, J. Stone and D. de Berker, Trichothiodystrophy and fragile hair: the distinction between diagnostic signs and diagnostic labels in childhood hair disease, Br. J. Dermatol., 2009, 161, 1379–1383 CrossRef CAS.
- C. McCuaig, D. Marcoux and J. E. Rasmussen, et al. Trichothiodystrophy associated with photosensitivity, gonadal failure, and striking osteosclerosis, J. Am. Acad. Dermatol., 1993, 28, 820–826 CrossRef CAS.
- L. Larizza, G. Roversi and L. Volpi, Rothmund–Thomson syndrome, Orphanet J. Rare Dis., 2010, 5, 2 CrossRef.
- H. A. Siitonen, O. Kopra and H. Kaariainen, et al. Molecular defect of RAPADILINO syndrome expands the phenotype spectrum of RECQL diseases, Hum. Mol. Genet., 2003, 12, 2837–2844 CrossRef CAS.
- L. Van Maldergem, H. A. Siitonen and N. Jalkh, et al. Revisiting the craniosynostosis-radial ray hypoplasia association: Baller–Gerold syndrome caused by mutations in the RECQL4 gene, J. Med. Genet., 2006, 43, 148–152 CrossRef CAS.
- L. L. Wang, M. L. Levy and R. A. Lewis, et al. Clinical manifestations in a cohort of 41 Rothmund–Thomson syndrome patients, Am. J. Med. Genet., 2001, 102, 11–17 CrossRef CAS.
- J. R. Potozkin and R. G. Geronemus, Treatment of the poikilodermatous component of the Rothmund–Thomson syndrome with the flashlamp-pumped pulsed dye laser: a case report, Pediatr. Dermatol., 1991, 8, 162–165 CrossRef CAS.
- N. A. Ellis and J. German, Molecular genetics of Bloom's syndrome, Hum. Mol. Genet., 1996, 5 Spec No, 1457–1463 CAS.
- H. Kaneko and N. Kondo, Clinical features of Bloom syndrome and function of the causative gene, BLM helicase, Expert Rev. Mol. Diagn., 2004, 4, 393–401 CrossRef CAS.
- T. Ozawa, N. Kondo and F. Motoyoshi, et al. Preferential damage to IgM production by ultraviolet B in the cells of patients with Bloom's syndrome, Scand. J. Immunol., 1993, 38, 225–232 CrossRef CAS.
- J. E. Lai-Cheong and J. A. McGrath, Kindler syndrome, Dermatol. Clin., 2010, 28, 119–124 CrossRef CAS.
- M. A. D'Souza, R. M. Kimble and J. R. McMillan, Kindler syndrome pathogenesis and fermitin family homologue 1 (kindlin-1) function, Dermatol. Clin., 2010, 28, 115–118 CAS.
- H. Penagos, M. Jaen and M. T. Sancho, et al. Kindler syndrome in native Americans from Panama: report of 26 cases, Arch. Dermatol., 2004, 140, 939–944 Search PubMed.
- L. M. Yap, P. Foley and R. Crouch, et al. Chronic actinic dermatitis: a retrospective analysis of 44 cases referred to an Australian photobiology clinic, Australas. J. Dermatol., 2003, 44, 256–262 CrossRef.
- H. Menage, J. S. Ross and P. G. Norris, et al. Contact and photocontact sensitization in chronic actinic dermatitis: sesquiterpene lactone mix is an important allergen, Br. J. Dermatol., 1995, 132, 543–547 CrossRef CAS.
- S. A. Holme, A. D. Pearse and A. V. Anstey, Chronic actinic dermatitis secondary to simvastatin, Photodermatol., Photoimmunol. Photomed., 2002, 18, 313–314 CrossRef CAS.
- A. L. Chew, S. J. Bashir and J. L. Hawk, et al. Contact and photocontact sensitization in chronic actinic dermatitis: a changing picture, Contact Dermatitis, 2010, 62, 42–46 CrossRef CAS.
- V. K. Sharma and G. Sethuraman, Parthenium dermatitis, Dermatitis, 2007, 18, 183–190 CrossRef.
- M. Trakatelli, S. Charalampidis and L. B. Novakovic, et al. Photodermatoses with onset in the elderly, Br. J. Dermatol., 2009, 161(Suppl 3), 69–77 CrossRef.
- R. S. Dawe, I. K. Crombie and J. Ferguson, The natural history of chronic actinic dermatitis, Arch. Dermatol., 2000, 136, 1215–1220 CAS.
- Y. Ma and Z. Lu, Treatment with topical tacrolimus favors chronic actinic dermatitis: a clinical and immunopathological study, J. Dermatol. Treat., 2010, 21, 171–177 CrossRef CAS.
- M. A. Thomson, D. G. Stewart and H. M. Lewis, Chronic actinic dermatitis treated with mycophenolate mofetil, Br. J. Dermatol., 2005, 152, 784–786 CrossRef CAS.
- A. Khaled, N. Kerkeni and D. Baccouche, et al. Chronic actinic dermatitis: two patients with successful management using narrowband ultraviolet B phototherapy with systemic steroids, Thérapie, 2011, 66, 453–457 CrossRef.
- H. C. Nousari, G. J. Anhalt and W. L. Morison, Mycophenolate in psoralen-UV-A desensitization therapy for chronic actinic dermatitis, Arch. Dermatol., 1999, 135, 1128–1129 CAS.
- P. E. Beattie, R. S. Dawe and S. H. Ibbotson, et al. Characteristics and prognosis of idiopathic solar urticaria: a cohort of 87 cases, Arch. Dermatol., 2003, 139, 1149–1154 Search PubMed.
- P. Calzavara-Pinton, R. Sala and M. Venturini, et al. Local angioedema following sun exposures: a report of five cases, Int. Arch. Allergy Immunol., 2010, 153, 315–320 CrossRef.
- E. Smith, F. Kiss and R. M. Porter, et al. A review of UVA-mediated photosensitivity disorders, Photochem. Photobiol. Sci., 2012, 11, 199–206 CAS.
- G. Monfrecola, E. Masturzo and A. M. Riccardo, et al. Solar urticaria: a report on 57 cases, Am. J. Contact Dermatitis, 2000, 11, 89–94 CrossRef CAS.
- C. T. Jong, A. Y. Finlay and A. D. Pearse, et al. The quality of life of 790 patients with photodermatoses, Br. J. Dermatol., 2008, 159, 192–197 CrossRef CAS.
- H. Adamski, C. Bedane and A. Bonnevalle, et al. Solar urticaria treated with intravenous immunoglobulins, J. Am. Acad. Dermatol., 2011, 65, 336–340 CrossRef CAS.
- R. Hughes, C. Cusack and G. M. Murphy, et al. Solar urticaria successfully treated with intravenous immunoglobulin, Clin. Exp. Dermatol., 2009, 34, e660–e662 CrossRef CAS.
- M. Llamas-Velasco, D. D. Argila and C. Eguren, et al. Solar urticaria unresponsive to intravenous immunoglobulins, Photodermatol., Photoimmunol. Photomed., 2011, 27, 53–54 CrossRef.
- V. Leenutaphong, E. Holzle and G. Plewig, et al. Plasmapheresis in solar urticaria, Dermatologica, 1991, 182, 35–38 CrossRef CAS.
- O. Guzelbey, E. Ardelean and M. Magerl, et al. Successful treatment of solar urticaria with anti-immunoglobulin E therapy, Allergy, 2008, 63, 1563–1565 CrossRef CAS.
- K. H. Waibel, D. A. Reese and R. G. Hamilton, et al. Partial improvement of solar urticaria after omalizumab, J. Allergy Clin. Immunol., 2010, 125, 490–491 CrossRef CAS.
- A. K. Haylett, Z. Nie and M. Brownrigg, et al. Systemic photoprotection in solar urticaria with alpha-melanocyte-stimulating hormone analogue [Nle4-D-Phe7]-alpha-MSH, Br. J. Dermatol., 2011, 164, 407–414 CrossRef CAS.
- R. S. Dawe, P. Collins and J. Ferguson, et al. Actinic prurigo and HLA-DR4, J. Invest. Dermatol., 1997, 108, 233–234 CAS.
- H. d. Menage, R. W. Vaughan and C. S. Baker, et al. HLA-DR4 may determine expression of actinic prurigo in British patients, J. Invest. Dermatol., 1996, 106, 362–367 Search PubMed.
- A. Suarez, M. C. Valbuena and M. Rey, et al. Association of HLA subtype DRB10407 in Colombian patients with actinic prurigo, Photodermatol., Photoimmunol. Photomed., 2006, 22, 55–58 CrossRef CAS.
- R. M. Fusaro and J. A. Johnson, Topical photoprotection for hereditary polymorphic light eruption of American Indians, J. Am. Acad. Dermatol., 1991, 24, 744–746 CrossRef CAS.
- P. R. Lane, A. A. Moreland and D. J. Hogan, Treatment of actinic prurigo with intermittent short-course topical 0.05% clobetasol 17-propionate. A preliminary report, Arch. Dermatol., 1990, 126, 1211–1213 CAS.
- P. Collins and J. Ferguson, Narrow-band UVB (TL-01) phototherapy: an effective preventative treatment for the photodermatoses, Br. J. Dermatol., 1995, 132, 956–963 CrossRef CAS.
- P. M. Farr and B. L. Diffey, Treatment of actinic prurigo with PUVA: mechanism of action, Br. J. Dermatol., 1989, 120, 411–418 CrossRef CAS.
- C. R. Lovell, J. L. Hawk and C. D. Calnan, et al. Thalidomide in actinic prurigo, Br. J. Dermatol., 1983, 108, 467–471 CrossRef CAS.
- B. Torres-Alvarez, J. P. Castanedo-Cazares and B. Moncada, Pentoxifylline in the treatment of actinic prurigo. A preliminary report of 10 patients, Dermatology, 2004, 208, 198–201 CrossRef CAS.
- D. Lestarini, L. S. Khoo and C. L. Goh, The clinical features and management of actinic prurigo: a retrospective study, Photodermatol., Photoimmunol. Photomed., 1999, 15, 183–187 CrossRef CAS.
- M. M. Duran, C. P. Ordonez and J. C. Prieto, et al. Treatment of actinic prurigo in Chimila Indians, Int. J. Dermatol., 1996, 35, 413–416 CrossRef CAS.
- L. E. Rhodes, M. Bock and A. S. Janssens, et al. Polymorphic light eruption occurs in 18% of Europeans and does not show higher prevalence with increasing latitude: multicenter survey of 6,895 individuals residing from the Mediterranean to Scandinavia, J. Invest. Dermatol., 2010, 130, 626–628 CrossRef CAS.
- P. G. Norris and J. L. Hawk, Polymorphic light eruption, Photodermatol., Photoimmunol. Photomed., 1990, 7, 186–191 CAS.
- C. M. Proby, C. S. Baker and O. Morton, et al. New broad-spectrum sunscreen for polymorphic light eruption, Lancet, 1993, 341, 1347–1348 CrossRef CAS.
- V. Schleyer, O. Weber and A. Yazdi, et al. Prevention of polymorphic light eruption with a sunscreen of very high protection level against UVB and UVA radiation under standardized photodiagnostic conditions, Acta Derm. Venereol., 2008, 88, 555–560 CAS.
- D. C. Patel, G. J. Bellaney and P. T. Seed, et al. Efficacy of short-course oral prednisolone in polymorphic light eruption: a randomized controlled trial, Br. J. Dermatol., 2000, 143, 828–831 CrossRef CAS.
- D. Bilsland, S. A. George and N. K. Gibbs, et al. A comparison of narrow band phototherapy (TL-01) and photochemotherapy (PUVA) in the management of polymorphic light eruption, Br. J. Dermatol., 1993, 129, 708–712 CrossRef CAS.
- G. M. Murphy, J. L. Hawk and I. A. Magnus, Hydroxychloroquine in polymorphic light eruption: a controlled trial with drug and visual sensitivity monitoring, Br. J. Dermatol., 1987, 116, 379–386 CrossRef CAS.
- P. G. Norris and J. L. Hawk, Successful treatment of severe polymorphous light eruption with azathioprine, Arch. Dermatol., 1989, 125, 1377–1379 CAS.
- O. Lasa, I. Trebol and J. Gardeazabal, et al. Prophylactic short-term use of cyclosporin in refractory polymorphic light eruption, J. Eur. Acad. Dermatol. Venereol., 2004, 18, 747–748 CrossRef CAS.
- H. W. Walling and R. D. Sontheimer, Cutaneous lupus erythematosus: issues in diagnosis and treatment, Am. J. Clin. Dermatol., 2009, 10, 365–381 CrossRef.
- A. Kuhn, M. Sonntag and D. Richter-Hintz, et al. Phototesting in lupus erythematosus: a 15-year experience, J. Am. Acad. Dermatol., 2001, 45, 86–95 CrossRef CAS.
- P. Lehmann and B. Homey, Clinic and pathophysiology of photosensitivity in lupus erythematosus, Autoimmun. Rev., 2009, 8, 456–461 CrossRef CAS.
- K. Foering, R. Goreshi and R. Klein, et al. Prevalence of self-report photosensitivity in cutaneous lupus erythematosus, J. Am. Acad. Dermatol., 2012, 66, 220–228 CrossRef.
- N. Scheinfeld and V. A. Deleo, Photosensitivity in lupus erythematosus, Photodermatol., Photoimmunol. Photomed., 2004, 20, 272–279 CrossRef.
- A. Kuhn, A. Wozniacka and J. C. Szepietowski, et al. Photoprovocation in cutaneous lupus erythematosus: a multicenter study evaluating a standardized protocol, J. Invest. Dermatol., 2011, 131, 1622–1630 CrossRef CAS.
- A. Kuhn, F. Ochsendorf and G. Bonsmann, Treatment of cutaneous lupus erythematosus, Lupus, 2010, 19, 1125–1136 CrossRef CAS.
- H. McGrath Jr., Ultraviolet-A1 irradiation decreases clinical disease activity and autoantibodies in patients with systemic lupus erythematosus, Clin. Exp. Rheumatol., 1994, 12, 129–135 CAS.
- H. McGrath, P. Martinez-Osuna and F. A. Lee, Ultraviolet-A1 (340–400 nm) irradiation therapy in systemic lupus erythematosus, Lupus, 1996, 5, 269–274 CrossRef CAS.
- C. J. Sanders, H. Y. Lam and C. A. Bruijnzeel-Koomen, et al. UV hardening therapy: a novel intervention in patients with photosensitive cutaneous lupus erythematosus, J. Am. Acad. Dermatol., 2006, 54, 479–486 CrossRef.
- A. Kuhn, K. Gensch and M. Haust, et al. Photoprotective effects of a broad-spectrum sunscreen in ultraviolet-induced cutaneous lupus erythematosus: a randomized, vehicle-controlled, double-blind study, J. Am. Acad. Dermatol., 2011, 64, 37–48 CrossRef CAS.
- S. Gonzalez, M. Fernandez-Lorente and Y. Gilaberte-Calzada, The latest on skin photoprotection, Clin. Dermatol., 2008, 26, 614–626 CrossRef.
- S. Lautenschlager, H. C. Wulf and M. R. Pittelkow, Photoprotection, Lancet, 2007, 370, 528–537 CrossRef CAS.
- T. Maier and H. C. Korting, Sunscreens – which and what for?, Skin Pharmacol. Physiol., 2005, 18, 253–262 CrossRef CAS.
- U. Osterwalder, B. Herzog and S. Q. Wang, Advance in sunscreens to prevent skin cancer, Expert Rev. Dermatol., 2011, 6, 479–491 CrossRef.
Footnote |
| † This article is published as part of a themed issue on current topics in photodermatology. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2013 |