Spotlight on ‘xeroderma pigmentosum’†
Hiva
Fassihi
*
National Xeroderma Pigmentosum Service, Department of Photodermatology, St John's Institute of Dermatology, Guy's and St Thomas’ NHS Trust, London, UK. E-mail: hiva.fassihi@gstt.nhs.uk; Fax: +44 (0)7188 1621; Tel: +44 (0)7188 6389
First published on 4th October 2012
Abstract
Xeroderma pigmentosum (XP) is a rare autosomal recessive disorder of DNA repair characterised by photosensitivity, progressive pigmentary change, and an increased incidence of ultraviolet (UV)-induced skin and mucous membrane cancers. Approximately 25% of XP patients also have progressive neurological degeneration. There are eight XP complementation groups (XP-A through to XP-G, and XP variant (XP-V)), corresponding to the affected DNA repair gene. Seven of these genes, XPA to XPG, are involved in nucleotide excision repair, removing UV-induced damage from DNA. The eighth gene, XPV (or POLH), encodes for DNA polymerase η, which is required for the replication of DNA containing unrepaired UV-induced damage. There is wide variability in clinical features both between and within XP complementation groups. The diagnosis is made clinically and confirmed by cellular tests for defective DNA repair. This is followed by identification of the defective gene (complementation analysis) and causative mutation(s). Although there is no cure, sun avoidance and regular follow-up to assess and treat any skin cancers increase life expectancy. The neurological abnormalities are progressive and result in a shortened lifespan. The study of patients with XP has highlighted the importance of nucleotide excision repair in the aetiology of skin cancers and neurological degeneration, and has solidified the link between UV exposure, DNA damage, somatic mutations and skin cancer.
Introduction
Xeroderma pigmentosum (XP) is a rare disorder of DNA repair, which manifests clinically as photosensitivity, progressive pigmentary abnormalities, and an increased incidence of ultraviolet (UV)-induced skin and mucous membrane cancers at sun-exposed sites. About two thirds of affected individuals show an exaggerated and prolonged sunburn response. The others have a normal acute response to sun exposure and only present with early ‘freckling’ at sun-exposed sites. In a minority of cases, there is also progressive neurological degeneration.1Xeroderma pigmentosum (XP) was first described in 1874 by Moriz Kaposi, the Hungarian-born professor of dermatology in Vienna. He reported four patients with xeroderma or ‘parchment skin’ in the textbook of dermatology which he wrote with his father-in-law, Professor Ferdinand Hebra.2 The term ‘pigmentosum’ was added to account for the significant pigmentary abnormalities. The first neurological problems were reported in 1883 by Albert Neisser of Breslau in Germany. He described two siblings with XP and neurological degeneration which started in the second decade.3 In 1932, the Italian physicians Carlo de Sanctis and Aldo Cacchione reported three brothers with features of XP, progressive neurological degeneration beginning at 2 years of age, and associated dwarfism and gonadal hypoplasia, and called this de Sanctis–Cacchione syndrome.4 This term is no longer in general use as it is now appreciated that XP can be associated with neurological problems of widely varying severity, and that de Sanctis–Cacchione syndrome is at the extreme end of a continuous spectrum.
DNA repair abnormalities in XP were reported by James Cleaver in 1968. He discovered deficient excision repair in cultured skin fibroblasts from XP patients.5 UV-induced DNA photoproducts were identified by Setlow6 and XP cells were found to be defective in the excision repair of these UV-induced photoproducts in vivo.7 The excision repair-proficient form of XP was first described in 1971,8 named ‘variant’ by Cleaver in 1972.9 These ‘variant’ cultured fibroblasts were able to repair UV-induced damage but were found to be defective in another DNA repair pathway, post replication repair.10 This subtype is now known as XP variant (XP-V).
Cell fusion studies carried out by de Weerd-Kastelein in 1972 demonstrated molecular heterogeneity in XP.11 Fusion of fibroblasts from different XP patients to form heterokaryons was found to correct the defect in DNA repair. This suggested that patients had different defects in nucleotide excision repair, and one defect could be corrected by the fusion of cells from a patient with a different defect because of the availability of the protein that the other was lacking. This led to the characterisation of XP into different complementation groups (XP-A through to XP-G).
Aetiology
Over the last 30 years the molecular pathology of XP has been better clarified. It is now known that XP is an autosomal recessive disorder with 100% penetrance and can result from mutations in any one of eight genes. The products of seven of these genes (XPA through to XPG) are involved in the repair of UV-induced photoproducts in DNA by the process of nucleotide excision repair (NER) (Fig. 1).12 This is a complex process involving a number of proteins, by which DNA damage, caused by UV-induced photoproducts and similar chemically-induced products, is recognised and repaired. It is made up of two pathways: global genome repair (GGR) in which damage to DNA not undergoing transcription is repaired, and transcription-coupled repair (TCR) in which damage in transcribed regions of DNA is repaired.13 In GGR, the photoproducts are recognised by the proteins, XPC and XPE. In TCR, the lesions appear to block the progress of RNA polymerase II in a process involving two proteins not involved in XP, CSA and CSB. Following this initial damage recognition, the pathways converge. XPB and XPD are part of a protein complex TFIIH, which opens up the structure of the DNA around the site of the photoproduct. XPA protein verifies that the proteins are in the correct position and then the endonucleases, XPG and XPF, cut the DNA on either side of the damage, so that the damaged section, in a fragment of about 30 nucleotides, can be removed. The gap is filled in by de novo DNA synthesis and the new stretch of DNA is finally joined up to the pre-existing strand. Mutations in any of the seven genes encoding for these proteins result in abnormal NER and therefore XP.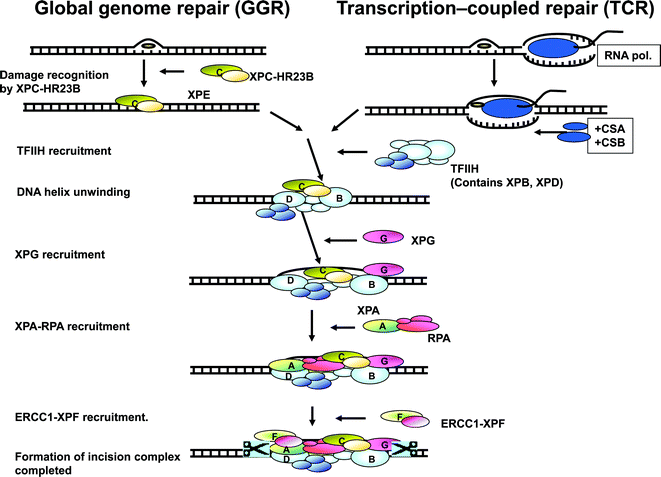 | ||
| Fig. 1 Nucleotide excision repair. Modified from Volker et al., Mol. Cell, 2001, 8, 213–224. | ||
Defects in the eighth XP gene do not affect NER. About 20% of patients with XP, the so-called XP variants (XP-V), have problems replicating DNA containing UV-induced damage.14 They have defects in the XPV (or POLH) gene, which encodes for DNA polymerase η, one of the specialised enzymes required for replication past damaged sites (translesion synthesis (TLS)).10,15
The different XP complementation groups (XP-A through to XP-G and XP-V) correspond to the affected DNA repair gene. Table 1 summarises the genes, chromosomal locations, protein products, and the main clinical features in different XP complementation groups.
| XP complementation group | Gene | No. of exons | Chromosomal location | Protein size (aa) | Protein function | Defective pathway | Clinical features |
|---|---|---|---|---|---|---|---|
| NER: nucleotide excision repair; GGR: global genome repair subpathway; TLS: translesion synthesis. | |||||||
| XP-A | XPA | 6 | 9q22.23 | 273 | Damage verification | NER | Exaggerated sunburn |
| Mild to severe neurological abnormalities | |||||||
| XP-B | XPB (ERCC3) | 15 | 2q14.3 | 782 | Helicase | NER | Exaggerated sunburn |
| Mild neurological abnormalities | |||||||
| XP-C | XPC | 16 | 3p25.1 | 940 | Damage recognition | NER (GGR) | No abnormal sunburn reaction |
| No neurological abnormalities | |||||||
| XP-D | XPD (ERCC2) | 23 | 19q13.32 | 760 | Helicase | NER | Exaggerated sunburn |
| No neurology to severe neurological abnormalities | |||||||
| XP-E | XPE (DDB2) | 10 | 11p11.2 | 427 | Damage recognition | NER (GGR) | No abnormal sunburn reaction |
| No neurological abnormalities | |||||||
| XP-F | XPF (ERCC4) | 11 | 16p13.12 | 916 | Nuclease | NER | Exaggerated sunburn |
| No neurology to severe neurological abnormalities | |||||||
| XP-G | XPG (ERCC5) | 15 | 13q33.1 | 1186 | Nuclease | NER | Exaggerated sunburn |
| No neurology to severe neurological abnormalities | |||||||
| XP-V | XPV (POLH) | 11 | 6p21.1 | 713 | Polymerase | TLS | No abnormal sunburn reaction |
| No neurological abnormalities | |||||||
Epidemiology
XP has been described in all continents and across all racial groups with males and females equally affected. Estimates from the 1970s suggested an incidence in the USA of 1 in 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 00016 and in Japan of 1 in 80000.17 A more recent survey in Western Europe suggests approximately 2.3 per million live births.18 The prevalence is higher in North Africa and the Middle East, especially in communities in which consanguinity is common.
00016 and in Japan of 1 in 80000.17 A more recent survey in Western Europe suggests approximately 2.3 per million live births.18 The prevalence is higher in North Africa and the Middle East, especially in communities in which consanguinity is common.
Clinical findings and basic cell biology
There is wide variability in clinical features both between and within XP complementation groups. The clinical manifestations, severity of disease, and the age of onset are in part dependent on exposure to sunlight, the complementation group, and the precise nature of the pathogenic mutation(s).1,16,19Skin and mucous membrane cancers
Epidemiological studies in the normal population demonstrate a link between sunlight and development of skin cancer. The study of XP patients solidifies this relationship even further with early onset and increased frequency of both non-melanoma and melanoma skin cancers following UV exposure. Absorption of UV by DNA results in the formation of cyclobutane pyrimidine dimers (CPDs) and pyrimidine (6-4) pyrimidone photoproducts (6-4PPs), which are recognised and repaired by NER. If left unrepaired, they can result in the classic ‘UVB signature’ mutations found in skin cancers.20,21 The molecular defects in XP cells result in abnormal recognition and repair of UV-induced DNA damage, and subsequent significantly increased induction of ‘UVB signature’ mutations in the sun-exposed skin of affected individuals. This increased mutation frequency is likely to account for the pigmentary changes and the skin cancers in XP patients. Indeed examination of mutations in the p53 gene in tumours from XP patients revealed mutations characteristic of UV exposure in the majority of tumours.22Patients with XP have a greater than 10000-fold increased risk of non-melanoma skin cancer (NMSC), with a median age of first cancer at 9 years (compared to 67 years in the general population). The median age at onset of the first XP melanoma is 22 years (compared to 55 years in the general population) and there is a 2000-fold increased risk of melanoma under the age of 20 when compared to the general population.19 XP patients also have a greatly increased frequency of cancer of the oral cavity, particularly squamous cell carcinoma of the tip of the tongue, a presumed sun-exposed area.12,23 In contrast to patients with XP complementation group A to G who generally develop skin cancer before the age of 20, XP-V patients develop their first skin cancers in the second or third decade of life.
The increased frequency of skin and mucous membrane cancer in XP highlights the important role of DNA repair after UV exposure in protection against cancer. In addition, the reversal in median age at onset of NMSC and melanoma in XP when compared to the general population suggests a greater role of sun exposure in the development of NMSC. Covered, UV protected areas of the skin show a lower frequency of cancer, thus demonstrating the importance of UV protection in the prevention of sun induced DNA damage and malignancy.
Exaggerated sunburn reactions and pigmentary change
Approximately 65% of XP patients have exaggerated and prolonged sunburn reactions from the first few weeks of life. These reactions often take weeks to resolve,19 and may be blamed on neglect or labelled wrongly as impetigo. These patients gradually develop marked ‘freckling’ at the sites of UV exposure and sunburn. It is patients with XP-A, XP-B, XP-D, XP-F and XP-G who often have these sunburn reactions. In contrast, patients in groups XP-C, XP-E and XP-V do not show any abnormal sunburn and present only with gradual ‘freckling’ at sun exposed sites, typically from the age of 2–3. In time, all patients develop dry skin with hypo- and/or hyperpigmentation. All the skin changes are the result of exposure to UV, mainly from the sun, therefore the severity of these changes is totally dependent on the amount of sun-exposure and the degree of UV protection. Delayed diagnosis and poor sun protection will exacerbate the cutaneous features, resulting in significant pigmentary changes, multiple skin cancers and a worse prognosis.In the general population chronic cumulative UV-exposure has been associated with the development of squamous cell carcinomas and, to a lesser degree, basal cell carcinomas. This is in contrast to severe sunburn with blistering in childhood which has been implicated in the early development of melanoma. In XP, paradoxically, those individuals who have severe and exaggerated sunburn reactions in childhood have a lower frequency of skin cancer than those children who did not sunburn and only presented with progressive ‘freckling’. This is probably the result of earlier diagnosis of the former group and a disinclination of this group to go out in sunlight without protection, therefore experiencing less cumulative sun exposure.19 In XP patients, acute sunburn is not necessary for the development of skin cancer, and the study of these patients separates acute sunburn from the pathogenesis of UV-induced carcinogenesis. It also highlights the fact that different abnormalities in DNA repair result in an increased frequency of cancer in all XP cases, but acute photosensitivity in only some patients.
These exaggerated and prolonged sunburn reactions in some XP complementation groups may be a consequence of cell killing. The molecular defects in XP result in increased UV-induced lethality, which varies substantially between individuals. The level of cell killing is less in individuals with mutations in the XPC and XPE genes (and with some hypomorphic mutations in other XP genes), because of residual functional DNA repair. This may explain the lack of exaggerated sunburn reactions in these complementation groups. Patients with XP-C and XP-E are deficient in the GGR pathway of NER but the TCR pathway remains active, and so does the ability to selectively remove photoproducts from the transcribed strand of active genes with kinetics similar to those observed in normal cells.24 From previous XPC knockout mice model studies,25,26 it has been hypothesized that the preservation of TCR in XP-C patients is the mechanism behind reduced severity of sunburn reactions compared to other XP genotypes (where both GGR and TCR are affected).
Ocular abnormalities
Ocular abnormalities are limited to the anterior, UV-exposed structures of the eye including the lids, cornea, and conjunctiva. Without adequate ocular UV protection, severe keratitis resulting in corneal opacification and vascularization may develop. Photophobia is common. There is a 1000-fold increase in cancer of the sun-exposed tissues of the eyes with ocular squamous cell carcinoma and melanoma described in the literature.12,19 The lens is a barrier to the penetration of UV radiation thus protecting the deeper structures of the eye.XP and other malignancies
DNA damage caused by carcinogens in cigarette smoke (chemical compounds such as benzo-a-pyrene) is also repaired by NER in normal cells27 and therefore patients with XP are at higher risk of smoking-induced cancers.23XP patients under the age of 20 also have an approximately 50-fold increase in the cancers of the brain including medulloblastoma,23 glioblastoma, spinal cord astrocytomas,28 and schwannoma. These malignancies are not UV-related and the precise relationship to DNA damage is unknown. High dose X-ray irradiation can be used for the treatment of some of these brain tumours as the pathways involved in the repair of DNA damage by X-rays are normal in patients with XP.28,29
Interestingly, heterozygous mice who are carriers of XP mutations have an increase in both skin and internal cancers.30 If this was extrapolated to human, it would suggest that unaffected carriers of XP disease-causing mutations may be at increased risk of cancer. However mice are not perfect models of this disease and further careful assessment of unaffected carriers is needed before any conclusions are made.
Neurological abnormalities
Neurological abnormalities of varying severity have been reported in about 25% of XP cases.16,19,31 The age at onset and rate of progression of the neurological abnormalities are variable between and within different complementation groups, and even when comparing individuals with the same pathogenic mutations. Patients with neurological abnormalities often have defects in XPA, XPB, XPD, XPF, or XPG. They are usually born with normal size and weight, and gradually develop progressive degeneration of cerebellar neurons, microcephaly, diminished or absent deep tendon stretch reflexes, progressive sensorineural hearing loss, spasticity, ataxia, seizures and progressive cognitive impairment.1 MRI imaging demonstrates thinning of the cortex of the brain with concomitant dilation of the ventricles, and a secondary thickening of the skull bones. The presence of neurological abnormalities is associated with a worse prognosis.The causes of the neurological abnormalities are poorly understood. They are clearly not connected with exposure to UV radiation. Current theories suggest that oxidative DNA damage is generated during normal metabolism in the central nervous system, and that some types of this damage must be repaired by NER.32 In the absence of functional repair, the lesions persist and result in neuronal death. Interestingly, it is patients with XP-C, XP-E and XP-V, without exaggerated and prolonged sunburn reactions, who also do not develop neurological abnormalities. This suggests that the TCR pathway may also play an important role in the pathogenesis of the neurodegeneration seen in XP.
Diagnosis
Diagnosis is based on clinical features and is confirmed by robust cellular tests for defective DNA repair that are available in several countries around the world. In the UK, these tests are conducted in Professor Alan Lehmann's laboratory at the Genome Damage and Stability Centre, University of Sussex in Brighton. The most commonly used test is the measurement of unscheduled DNA synthesis in cultured skin fibroblasts. After DNA damage has been removed, a patch of newly-synthesised DNA replaces the damaged section. Synthesis of this new DNA is referred to as unscheduled DNA synthesis or UDS.The diagnostic technique involves a skin biopsy, from an unexposed area of skin, establishment of fibroblast cultures, and irradiation with UV. UDS can be measured as incorporation of nucleotides, incubated with 3H-thymidine, a DNA radioactive precursor, into DNA of the irradiated cells either by autoradiography,33 liquid scintillation counting,34 or more recently using a fluorescence assay.35 A reduced level of UDS confirms the diagnosis of XP.
XP variants do not show this defect in UDS, as NER is unaffected in these patients. Fibroblast cells from XP-V patients are not hypersensitive to killing by UV radiation. However it was found empirically that caffeine specifically sensitises XP-V cells to killing by UV.36 To diagnose XP-V cells, fibroblast cultures are exposed to UV, incubated in caffeine for a few days and their viability compared to that of normal cells. Specific sensitivity to UV in the presence of caffeine together with normal UDS confirms the diagnosis of XP-V.37
Further analysis can identify the defective gene in patients (complementation analysis)38 and the causative mutation(s). This can give further insight into genotype–phenotype correlations in XP.
More recently, the lack of immunohistochemistry staining for the XPC protein in skin biopsies from patients with XP-C has shown promising results as a diagnostic technique.39 As XP-C is the most common complementation group, with over 30% of all mutations in the XPC gene, this new diagnostic methodology, which is fast, cost effective and widely available, is potentially very useful as an initial screening tool in patients suspected of having XP.
Genetic counselling
Genetic counselling and psychological support are an essential part of the management of patients and families with XP. This allows discussion around the aetiology and inheritance of disease, probability of occurrence in future pregnancies, and increased likelihood of occurrence in consanguineous marriages.Antenatal diagnosis
Prenatal diagnosis is possible through measurement of UDS in cultured chorionic villus cells or amniocytes in affected families, or through analysis of DNA after chorionic villus sampling if the mutations in the proband have been identified.Management and treatment
In the UK, St John's Institute of Dermatology at Guy's and St Thomas’ Hospital has been designated the national centre for treating children and adults with XP. The aim of the National Multidisciplinary XP Service is to offer expertise for patients with XP from many specialities including dermatology, neurology, ophthalmology, neuropsychology and clinical genetics.Although no cure is available, the skin problems can be dramatically ameliorated by appropriate sun protection. As the skin changes are all caused by UV light, total protection from exposure to UV can prevent the pigmentary changes and skin cancers. Protective measures include restricting outdoor activities to night time. If outdoors during the day, photoprotection with appropriate clothing, hats, UV-protective sunglasses, custom-made face shields and protective UV-resistant film on windows should be considered. Frequent application of high-factor sunscreen and lip balm is recommended. All patients should be examined by a dermatologist three monthly for skin cancer surveillance, so that any pre-cancerous lesions can be removed as early as possible. Photographs can be helpful in monitoring lesions. Annual ophthalmology examinations are recommended as well as regular neurological review and hearing tests to serve as a screen for the presence of XP-associated neurological abnormalities. Vitamin D deficiency as a result of rigorous sun-protection should be corrected.
Psychosocial issues including social isolation from peers at school and at home, limited career prospects and the impact of meticulous photoprotection on the patient’s quality of life need to be addressed. There are XP support groups in the UK,40 USA,41 France42 and Germany.43 They offer a wealth of advice and help.
Prognosis
There is no cure for XP. The overall median age of death is reported as 32 years,19 with skin cancer and neurological degeneration being the main causes of death. Sun avoidance and regular follow-up to assess and treat any skin cancers increase life expectancy. For those without neurological disease and rigorous UV protection, the prognosis is good. However the neurological abnormalities are progressive and the median age at death in these patients (29 years) is significantly younger than in XP patients without neurological degeneration (37 years).19Conclusions
The study of patients with XP has highlighted the importance of nucleotide excision repair in the aetiology of skin cancers and neurological degeneration, and has solidified the link between UV exposure, DNA damage, somatic mutations and skin cancer. Although there is no cure, early diagnosis, instigation of meticulous protection from UV, and careful patient management can dramatically improve the quality of life and life expectancy of affected individuals. As yet there are a number of unanswered questions including why some patients present with severe and exaggerated sunburn reactions and others do not. In addition, at present there are no treatment options for the progressive neurological disease which is associated with a poor prognosis. We need to increase our understanding of the aetiology of the neurological abnormalities, and why some complementation groups are at risk and others not. This will enable the development of treatments to alleviate the neurological degeneration.Acknowledgements
The National Multidisciplinary XP Clinic in the UK is funded by the National Commissioning Group of the UK National Health Service.Thanks to Professor Alan Lehmann at the Genome Damage and Stability Centre, University of Sussex in Brighton, and Dr Robert Sarkany, Head of Photodermatology, St John's Institute of Dermatology, Guy's and St Thomas’ NHS Trust, London for all their help and support.
References
- K. H. Kraemer, M. M. Lee and J. Scotto, Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases, Arch. Dermatol., 1987, 123, 241–250 CAS.
- F. Hebra and M. Kaposi, On diseases of the skin including exanthemata, volume III, New Sydenham Soc., 1874, 61, 252–258 Search PubMed.
- A. Neisser, Ueber das ‘Xeroderma pigmentosum’ (Kaposi): Lioderma essentialis cum melanosi et telangiectasia, Vierteljahrschr. Dermatol. Syphil., 1883, 47–62 CrossRef.
- C. de Sanctis and A. Cacchione, L'idiozia xerodermica, Riv. Sper. Freniatr., 1932, 56, 269–292 Search PubMed.
- J. E. Cleaver, Defective repair replication of DNA in xeroderma pigmentosum, Nature, 1968, 218, 652–656 CrossRef CAS.
- R. B. Setlow and J. K. Setlow, Evidence that ultraviolet-induced thymine dimers in DNA cause biological damage, Proc. Natl. Acad. Sci. U. S. A., 1962, 48, 1250–1257 CrossRef CAS.
- J. H. Epstein, K. Fukuyama, W. B. Reed and W. L. Epstein, Defect in DNA synthesis in skin of patients with xeroderma pigmentosum demonstrated in vivo, Science, 1970, 168, 1477–1478 CAS.
- P. G. Burk, M. A. Lutzner, D. D. Clarke and J. H. Robbins, Ultraviolet-stimulated thymidine incorporation in xeroderma pigmentosum lymphocytes, J. Lab Clin. Med., 1971, 77, 759–767 CAS.
- J. E. Cleaver, Xeroderma pigmentosum: variants with normal DNA repair and normal sensitivity to ultraviolet light, J. Invest. Dermatol., 1972, 58, 124–128 CAS.
- A. R. Lehmann, S. Kirk-Bell, C. F. Arlett, M. C. Paterson, P. H. Lohman, E. A. de Weerd-Kastelein and D. Bootsma, Xeroderma pigmentosum cells with normal levels of excision repair have a defect in DNA synthesis after UV-irradiation, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 219–223 CrossRef CAS.
- E. A. de Weerd-Kastelein, W. Keijzer and D. Bootsma, Genetic heterogeneity of xeroderma pigmentosum demonstrated by somatic cell hybridization, Nat. New Biol., 1972, 238, 80–83 CAS.
- M. Stefanini and K. H. Kraemer, Xeroderma pigmentosum, in Neurocutaneous Diseases, ed. M. Ruggieri, I. Pascual-Castroviejo and C. Di Rocco, 2008, ch. 51, 771–792 Search PubMed.
- J. E. Cleaver, E. T. Lam and I. Revet, Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity, Nat. Rev. Genet., 2009, 10, 756–768 CrossRef CAS.
- E. C. Friedberg, DNA damage and repair, Nature, 2003, 421, 436–440 CrossRef.
- C. Masutani, R. Kusumoto, A. Yamada, N. Dohmae, M. Yokoi, M. Yuasa, M. Araki, S. Iwai, K. Takio and F. Hanaoka, The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta, Nature, 1999, 399, 700–704 CrossRef CAS.
- J. H. Robbins, K. H. Kraemer, M. A. Lutzner, B. W. Festoff and H. G. Coon, Xeroderma pigmentosum: an inherited disease with sun-sensitivity, multiple cutaneous neoplasms, and abnormal DNA repair, Ann. Intern. Med, 1974, 80, 221–248 CAS.
- H. Takebe, Y. Miki, T. Kozuka, J. I. Furuyama, K. Tanaka, M. S. Sasaki, Y. Fujiwara and H. Akiba, DNA repair characteristics and skin cancers of xeroderma pigmentosum patients in Japan, Cancer Res., 1977, 37, 490–495 CAS.
- W. J. Kleijer, V. Laugel, M. Berneburg, T. Nardo, H. Fawcett, A. Gratchev, N. G. Jaspers, A. Sarasin, M. Stefanini and A. R. Lehmann, Incidence of DNA repair deficiency disorders in western Europe: xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy, DNA Repair (Amst), 2008, 7, 744–750 CrossRef CAS.
- P. T. Bradford, A. M. Goldstein, D. Tamura, S. G. Khan, T. Ueda, J. Boyle, K. S. Oh, K. Imoto, H. Inui, S. I. Moriwaki, S. Emmert, K. M. Pike, A. Raziuddin, T. M. Plona, J. J. Digiovanna, M. A. Tucker and K. H. Kraemer, Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair, J. Med. Genet., 2010, 48, 168–176 CrossRef.
- A. Ziegler, A. Jonason, J. Simon, D. Leffell and D. E. Brash, Tumor suppressor gene mutations and photocarcinogenesis, Photochem. Photobiol., 1996, 63, 432–435 CrossRef CAS.
- E. D. Pleasance and R. K. Cheetham, et al., A comprehensive catalogue of somatic mutations from a human cancer genome, Nature, 2010, 463, 191–197 CrossRef CAS.
- L. Daya-Grosjean and A. Sarasin, The role of UV induced lesions in skin carcinogenesis: an overview of oncogene and tumor suppressor gene modifications in xeroderma pigmentosum skin tumors, Mutat. Res., 2005, 571, 43–56 CrossRef CAS.
- K. H. Kraemer, M. M. Lee, A. D. Andrews and W. C. Lambert, The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm, Arch. Dermatol., 1994, 130, 1018–1021 CAS.
- J. Venema, A. van Hoffen, V. Karcagi, A. T. Natarajan, A. A. van Zeeland and L. H. Mullenders, Xeroderma pigmentosum complementation group C cells remove pyrimidine dimers selectively from the transcribed strand of active genes, Mol. Cell Biol., 1991, 11, 4128–4134 CAS.
- R. J. Berg, H. J. Ruven, A. T. Sands, F. R. de Gruijl and L. H. Mullenders, Defective global genome repair in XPC mice is associated with skin cancer susceptibility but not with sensitivity to UVB induced erythema and edema, J. Invest. Dermatol., 1998, 110, 405–409 CrossRef CAS.
- J. Garssen, H. van Steeg, F. de Gruijl, J. de Boer, G. T. van der Horst, H. van Kranen, H. van Loveren, M. van Dijk, A. Fluitman, G. Weeda and J. H. Hoeijmakers, Transcription-coupled and global genome repair differentially influence UV-B-induced acute skin effects and systemic immunosuppression, J. Immunol., 2000, 164, 6199–6205 CAS.
- V. M. Maher, J. D. Patton, J. L. Yang, Y. Y. Wang, L. L. Yang, A. E. Aust, N. Bhattacharyya and J. J. McCormick, Mutations and homologous recombination induced in mammalian cells by metabolites of benzo[a]-pyrene and 1-nitropyrene, Environ. Health Perspect., 1987, 76, 33–39 CrossRef CAS.
- J. J. DiGiovanna, N. Patronas, D. Katz, D. Abangan and K. H. Kraemer, Xeroderma pigmentosum: spinal cord astrocytoma with 9-year survival after radiation and isotretinoin therapy, J. Cutan. Med. Surg., 1998, 2, 153–158 CAS.
- F. Giannelli, J. Avery, P. E. Polani, C. Terrell and V. Giammusso, Xeroderma pigmentosum and medulloblastoma: chromosomal damage to lymphocytes during radiotherapy, Radiat. Res., 1981, 88, 194–208 CrossRef CAS.
- D. L. Cheo, L. B. Meira, D. K. Burns, A. M. Reis, T. Issac and E. C. Friedberg, Ultraviolet B radiation-induced skin cancer in mice defective in the Xpc, Trp53, and Apex (HAP1) genes: genotype-specific effects on cancer predisposition and pathology of tumors, Cancer Res., 2000, 60, 1580–1584 CAS.
- A. D. Andrews, S. F. Barrett and J. H. Robbins, Xeroderma pigmentosum neurological abnormalities correlate with colony-forming ability after ultraviolet radiation, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 1984–1988 CrossRef CAS.
- P. J. Brooks, The 8,5′-cyclopurine-2′-deoxynucleosides: candidate neurodegenerative DNA lesions in xeroderma pigmentosum, and unique probes of transcription and nucleotide excision repair, DNA Repair (Amst), 2008, 7, 1168–1179 CrossRef CAS.
- M. Stefanini, W. Keijzer, L. Dalpra, R. Elli, M. N. Porro, B. Nicoletti and F. Nuzzo, Differences in the levels of UV repair and in clinical symptoms in two sibs affected by xeroderma pigmentosum, Hum. Genet., 1980, 54, 177–182 CrossRef CAS.
- A. R. Lehmann and S. Stevens, A rapid procedure for measurement of DNA repair in human fibroblasts and for complementation analysis of xeroderma pigmentosum cells, Mutat. Res., 1980, 69, 177–190 CrossRef CAS.
- S. Limsirichaikul, A. Niimi, H. Fawcett, A. Lehmann, S. Yamashita and T. Ogi, A rapid non-radioactive technique for measurement of repair synthesis in primary human fibroblasts by incorporation of ethynyl deoxyuridine (EdU), Nucl. Acids Res., 2009, 37, e31 CrossRef.
- C. F. Arlett, S. A. Harcourt and B. C. Broughton, The influence of caffeine on cell survival in excision-proficient and excision-deficient xeroderma pigmentosum and normal human cell strains following ultraviolet light irradiation, Mutat. Res., 1975, 33, 341–346 CrossRef CAS.
- B. C. Broughton, A. Cordonnier, W. J. Kleijer, N. G. Jaspers, H. Fawcett, A. Raams, V. H. Garritsen, A. Stary, M. F. Avril, F. Boudsocq, C. Masutani, F. Hanaoka, R. P. Fuchs, A. Sarasin and A. R. Lehmann, Molecular analysis of mutations in DNA polymerase eta in xeroderma pigmentosum-variant patients, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 815–820 CrossRef CAS.
- F. Chavanne, B. C. Broughton, D. Pietra, T. Nardo, A. Browitt, A. R. Lehmann and M. Stefanini, Mutations in the XPC gene in families with xeroderma pigmentosum and consequences at the cell, protein and transcript levels, Cancer Res., 2000, 60, 1974–1982 CAS.
- S. de Feraudy, I. Boubakour-Azzouz, S. Fraitag, M. Berneburg, L. Chan, K. Chew, C. L. Clericuzio, B. Cunningham, W. D. Tope and J. E. Cleaver, Diagnosing xeroderma pigmentosum group C by immunohistochemistry, Am. J. Dermatopathol., 2010, 32, 109–117 CrossRef.
- Xeroderma pigmentosum support group [http://joomla.xpsupportgroup.org.uk].
- XP family support group [http://www.xpfamilysupport.org] and xeroderma pigmentosum society [http://www.xps.org/].
- Enfants de la lune [http://asso.orpha.net/AXP].
- XP Freu(n)de [http://www.xerodermapigmentosum.de].
Footnote |
| † This article is published as part of a themed issue on current topics in photodermatology. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2013 |