 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent trends in Pd-catalyzed remote functionalization of carbonyl compounds
Ivan
Franzoni
and
Clément
Mazet
*
Organic Chemistry Department, University of Geneva, 30 quai Ernest Ansermet, Geneva 1204-CH, Switzerland. E-mail: clement.mazet@unige.ch; Fax: (+41)22 379 32 15; Tel: (+41)22 379 62 88
First published on 6th November 2013
Abstract
Recent advances in the palladium-catalyzed remote functionalization of carbonyl derivatives are highlighted in this review. The structure of the article is based on the three strategies that have emerged in recent years as the most viable tactics to construct C(sp3)–C, C(sp3)–N or C(sp3)–O bonds at a distance of at least three carbon atoms from the carbonyl functionality. The specific aspects of each of these approaches are discussed in detail (reaction conditions, substrate scope and limitations). As the reading progresses, their complementarity should also appear clearly.
 Ivan Franzoni | Ivan Franzoni was born in Edolo (Italy) in 1986. He studied chemistry at the University of Milano (Italy) and received his M.Sc. in Chemistry in October 2011. He subsequently enrolled as a graduate student in the group of Prof. Clément Mazet in the Organic Chemistry Department of the University of Geneva. His research project focuses on the development of novel catalytic methods for the vicinal and distal stereoselective arylation of aldehydes. |
 Clément Mazet | Clément Mazet received his PhD from the University of Strasbourg under the supervision of Prof. L. H. Gade (2002). After postdoctoral stays with Prof. A. Pfaltz (University of Basel – 2003–2005) and Prof. E. N. Jacobsen (Harvard University – 2006–2007) he joined the University of Geneva to establish his independent research program. His interests include mechanistic and synthetic chemistry with particular emphasis on asymmetric catalysis. He recently received the Zasshikai Lectureship Award from the University of Tokyo (2012) and the Werner Prize from the Swiss Chemical Society (2013). |
1. Introduction
The ability of certain enzymes to interact with a functional group and to selectively modify a remote C–H or C–C bond has been a continuous source of inspiration for the synthetic chemist. The introduction of the concept of remote functionalization in chemical synthesis can be traced back to the 1970s with the emergence of biomimetic approaches.1 Since then, numerous effective synthetic methods have been developed to selectively activate and modify a distal position from a functional handle.2 Even more remarkably, cases of remote stereochemical control have also been reported, in particular with the recent explosion of organocatalytic methods.3,4 As many of these achievements have already been reviewed, for the sake of clarity and brevity, they will not be discussed again in this perspective. Instead, emphasis will be specifically placed on the most recent and important tactics to remotely functionalize carbonyl derivatives (or surrogates thereof) using Pd catalysis and that generate C(sp3) centers. The structure of this article is articulated around the three strategies that have been dominating over the last two decades: (i) the remote functionalization of carbonyls by direct C–H activation of C(sp3)–H bonds; (ii) the direct arylation of extended enolates (or enolate equivalents); (iii) the Heck arylation of isomerizable double bonds remote from a carbonyl or an alcohol.Furthermore, as the notion of ‘remote functionalization’ might be somewhat subjective, a distance of at least 3 carbon atoms between the carbonyl group and the position being modified (γ position or further) was used as the threshold for our selection.
2. C–H activation of C(sp3)–H bonds
An ever increasing number of methods based on the regioselective functionalization of C(sp2)–H bonds in heteroarenes and arenes have been reported in recent years.5 In comparison, less progress has been made for the selective modification of C(sp3)–H bonds, in particular for remote aliphatic positions.6 A common aspect of these approaches is the use of directing groups to facilitate C–H activation. Nonetheless, from a practical point of view, this imposes that, beyond its efficiency, the prerequisite auxiliary must be easily installed and equally easily removed.Daugulis and co-workers first demonstrated the ability of picolinamide (PA) to facilitate the Pd-catalyzed direct C–H arylation of remote aliphatic positions.7 However, this was achieved at the cost of relatively demanding reaction conditions (5 mol% Pd(OAc)2, 4 equiv. aryliodide, 1 equiv. AgOAc, 130–150 °C). Inspired by this precedent, the Chen group has elaborated milder and more general methods for the remote functionalization of γ C(sp3)–H bonds (5 mol% Pd(OAc)2, 1.5 equiv. aryliodide, 1.0 equiv. Ag2CO3, 80 °C).8 Electron-rich, electron-neutral, electron-poor as well as ortho-substituted iodoarenes were coupled with cyclic PA-modified amino esters in moderate to excellent yields (47–85%) (Scheme 1A). In contrast, linear substrates such as the threonine methyl ester shown in Scheme 1(B) were usually obtained in reduced yields. Both an increase in temperature and the use of benzoquinone (BQ, 10 mol%) allowed γ-alkenylation to occur selectively on cyclic systems; albeit in more modest yields (12–69%). The removal of the PA directing group was found to be challenging. The use of an ortho methyloxysilyl analogue proved to be a viable alternative as it could be cleaved under acidic conditions (Scheme 1D). Preliminary mechanistic investigations were supportive of a C–H activation/arylation sequence.
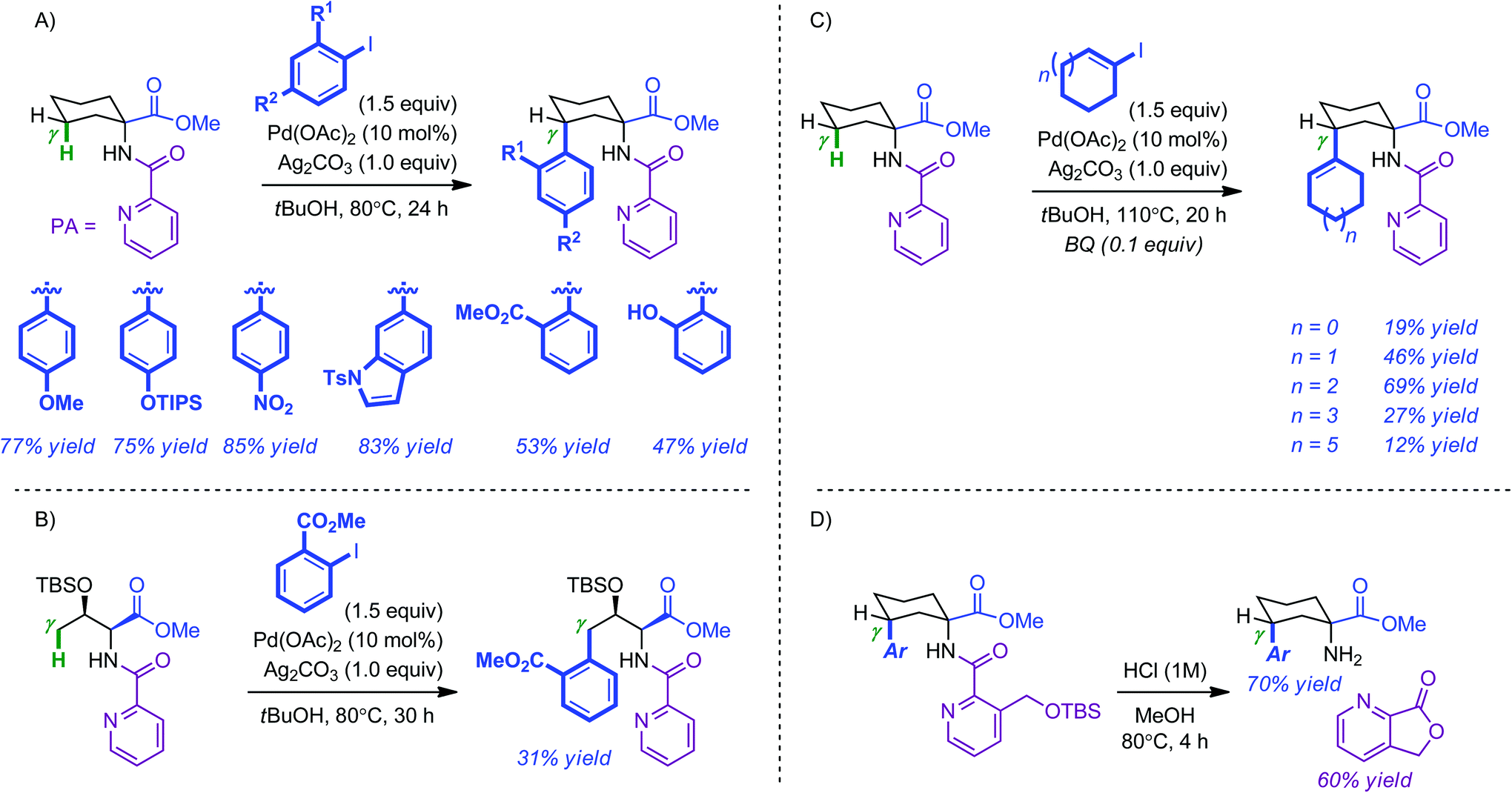 | ||
| Scheme 1 Pd-catalyzed picolinamide-directed C(sp3)-H activation of the γ position of modified-amino esters. (A) Arylation of cyclic systems. (B) Arylation of linear systems. (C) Vinylation of cyclic systems. (D) Directing-group removal and recovery. | ||
Having laid the fundamental basis in the remote functionalization of C(sp3)–H bonds, the same group next developed a set of Pd-catalyzed directed intramolecular amination reactions leading to azetidines, pyrrolidines, indolines and pyrrolidones by simply changing the oxidant (PhI(OAc)2, 2.5 equiv.).9,10 Representative examples for each compound are shown in Scheme 2 (respectively in eqn (1)–(4)). Yields and diastereoselectivities were usually high when 2.0 equivalents of AcOH were used. Of note, access to pyrrolidinones required the use of 8-aminoquinoline (AQ) as the directing group. Additional studies showed that 8-methyl-5-methoxyquinoline (MQ) not only provided enhanced performances in the cyclization reactions but was also best suited in view of post-reaction ablation (3 equiv. CAN in CH3CN–H2O at room temperature).11
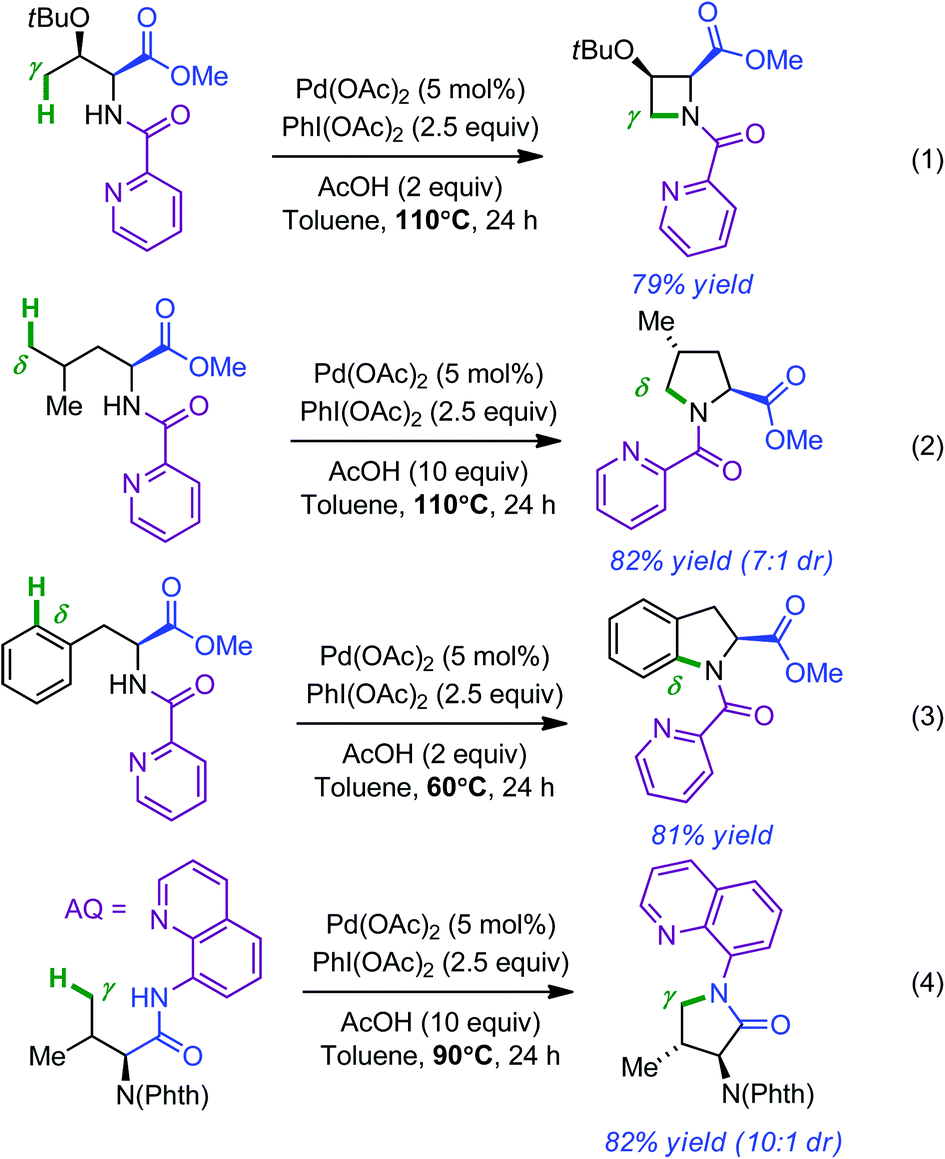 | ||
| Scheme 2 Pd-catalyzed directed azetidines (eqn (1)), pyrrolidines (eqn (2)), indolines (eqn (3)) and pyrrolidinones (eqn (4)) formation. | ||
The observation that acetoxylation took place preferentially over intramolecular amination when AcOH was used as a solvent prompted the authors to develop an intermolecular Pd-catalyzed PA-directed remote alkoxylation.12 The isolated yields were usually high and the reaction featured a broad substrate scope by simply changing the nature of the alcohol. Typical products obtained by this method are shown in Scheme 3.
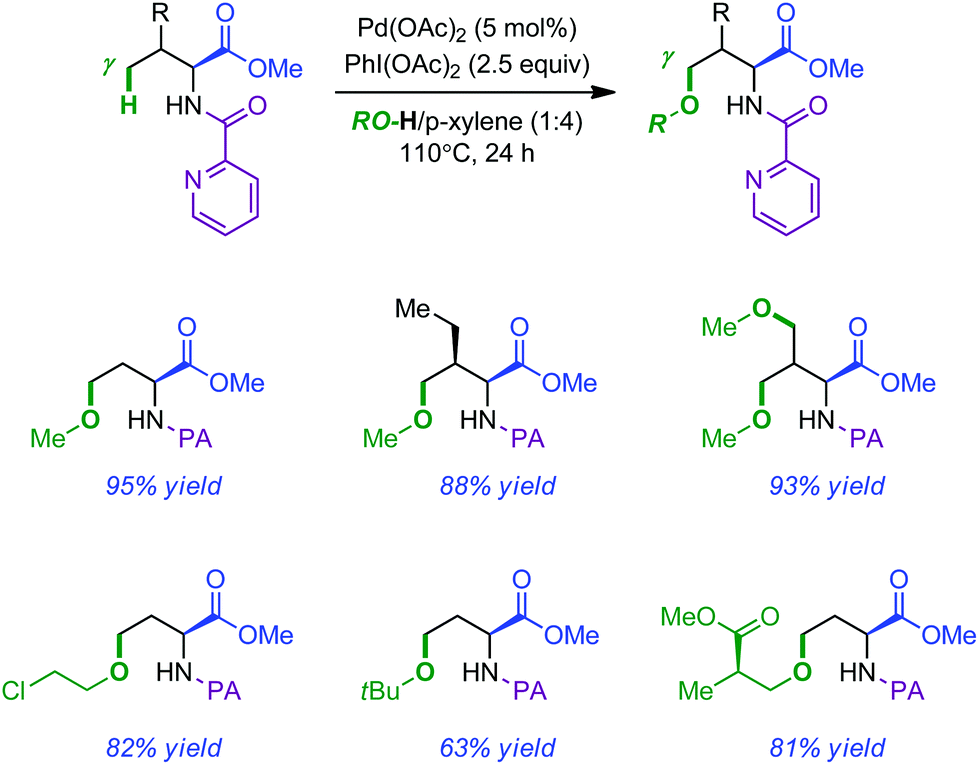 | ||
| Scheme 3 Intermolecular alkoxylation of PA-modified amino esters. | ||
The Carretero group has recently introduced 2-pyridyl-sulfonyl (PySO2) as an easily installable and removable directing group in a variety of Pd-catalyzed C(sp2)–H bond activation protocols.13 As a further extension of this work, they recently demonstrated that PySO2 was also efficient in the remote activation of C(sp3)–H bonds of amino ester derivatives.14 Their optimized conditions for the γ-arylation of L-valine methyl ester required elevated temperature (150 °C) and the use of 10 mol% of Pd(OAc)2, 2.5 equivalents of iodo arene, 1.5 equivalent of oxidant (Ag2CO3) and 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP), a privileged solvent in C–H activation processes.15 Although the desired mono-arylated product could be isolated in appreciable yield, its formation was often plagued by the presence of the bis-arylated compound. Consequently, the scope of the reaction was mostly centred on PySO2-modified amino esters with sterically distinct γ and γ′ positions (Scheme 4). The corresponding mono-arylated products were usually isolated in good yields (60–72%). Further investigations established that electron-rich and electron-poor iodo arenes were equally tolerated (average yield of 72% with allo-isoleucine). Removal of the 2-pyridylsulfonyl auxiliary was achieved by treatment of the arylated product with Zn in THF–NH4Cl(aq) for 16 h at 60 °C without any erosion of the enantiopurity (73% yield).
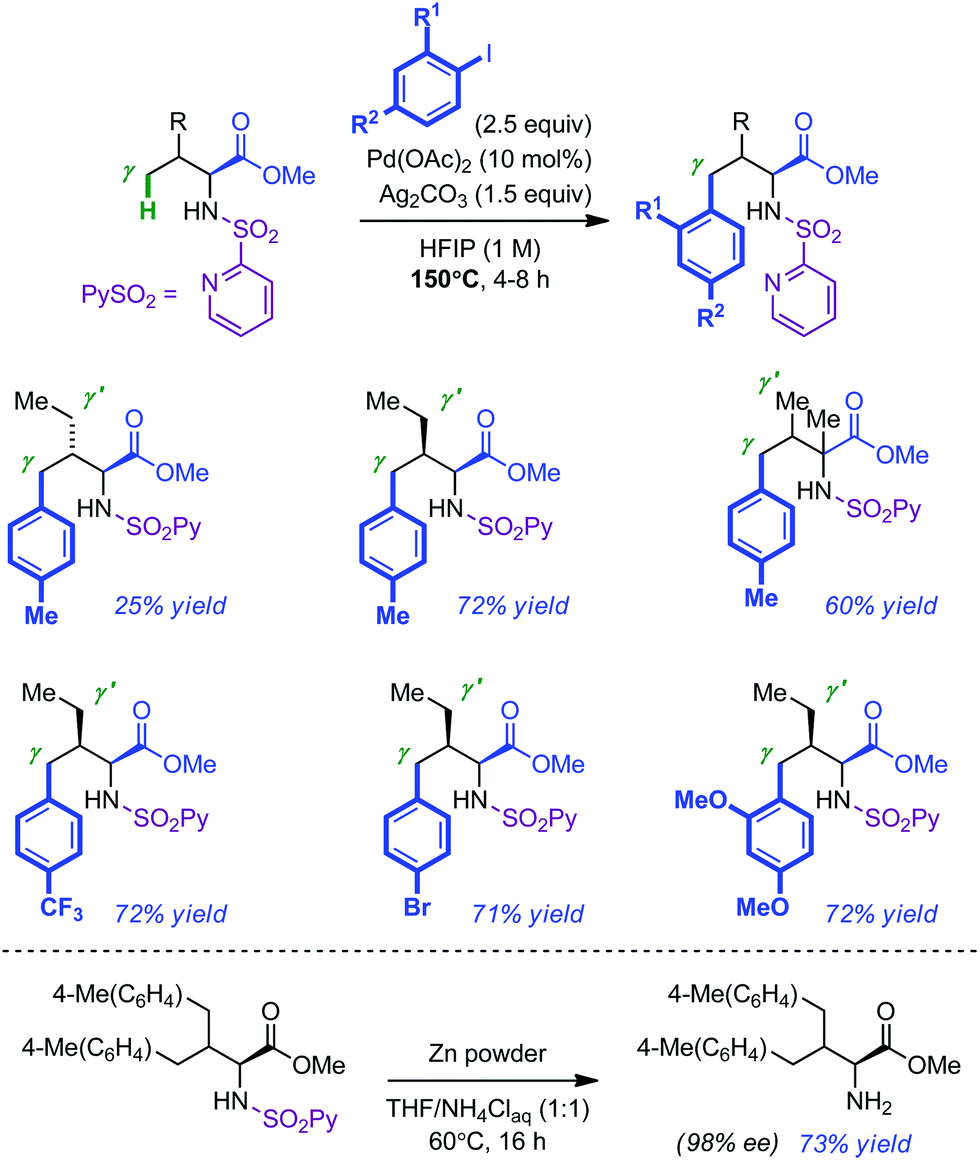 | ||
| Scheme 4 Pd-catalyzed PySO2-directed intermolecular arylation of amino esters. | ||
A related study was reported almost concomitantly by Fan and Ma.16 Their approach for the Pd-catalyzed γ-arylation of amino esters consisted in demonstrating the superior utility of 2-methoxyiminoacetyl (MIA) as a directing group for C(sp3)–H bond activation. Whereas Pd(OAc)2 and HFIP proved to be the catalyst and the solvent of choice respectively, a combination of AgOAc and PivOH as additives was found to be optimal (100 °C, 24 h). Under these conditions, a wide number of iodoarenes (2.5 equiv.) were efficiently introduced in the γ position of MIA-modified homoalanine (see Scheme 5 for representative examples). Substituted amino esters were usually arylated at the less hindered γ position. Consistent with the observation of the Carretero group, the L-valine derivative resulted in a mixture of mono and bis-arylated products (58% and 19% respectively). The MIA directing group was found to be easily removed by simple hydrolysis of a variety of arylation products with 1 M KOH in dioxane at room temperature (the corresponding amino acids were usually directly re-protected using (Boc)2O: 5 examples, 65–82% yield; no erosion of the enantiopurity).
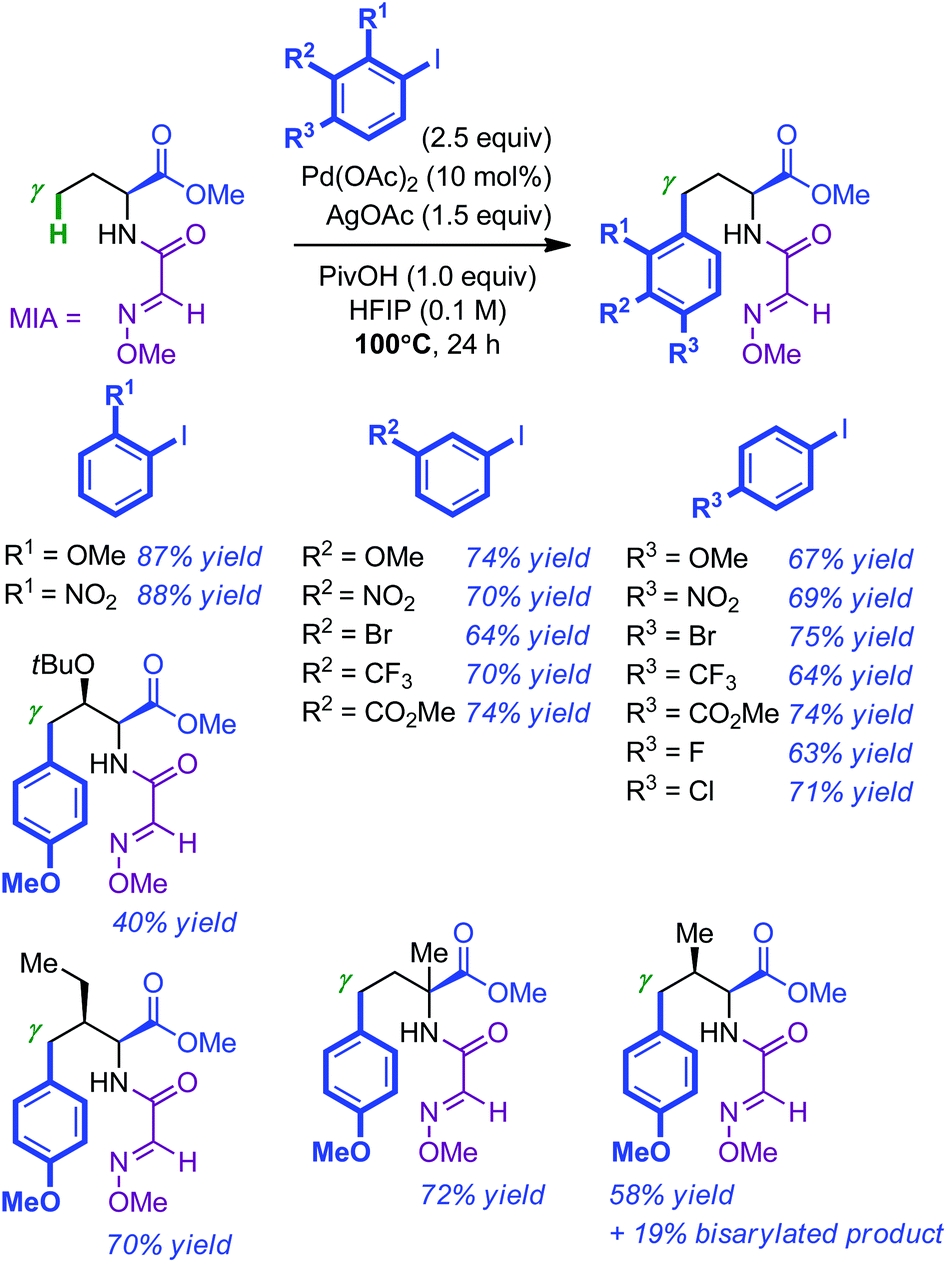 | ||
| Scheme 5 Pd-catalyzed MIA-directed intermolecular arylation of amino esters. | ||
3. Arylations of extended enolates
The Pd-catalyzed arylation of enolates with aryl halides or aryl halide equivalents has progressed at an impressive pace over the last decade,17 culminating with the development of enantioselective α-arylations of amides,18 esters,19 ketones20 and even of the more sensitive aldehydes.21 Yet, the corresponding vinylogous coupling of α,β-unsaturated carbonyl derivatives has not reached the same level of refinement.22 A first explanation for this discrepancy is certainly, the regioselectivity issues associated with the development of such coupling methods (α vs. β vs. γ arylation).Miura and co-workers first disclosed that the Pd-catalyzed arylation of enals and enones can occur with perfect γ regioselectivity under conditions similar to those developed for the α-arylation of ketones or aldehydes.23 With a combination of PPh3 and Pd(OAc)2 (5 mol% and 10 mol% respectively), temperatures ranging from 60 to 120 °C, DMF and Cs2CO3 as the solvent and the base of choice, they were able to couple electron-rich and electron-neutral bromoarenes with linear aldehydes and cyclic ketones. A representative selection of the products formed is displayed in Scheme 6. Most of them were obtained in moderate isolated yields (40–64% yield). The authors noticed that the presence of an α-blocking substituent in the linear aldehydes was required to favor perfect γ-selective arylation.
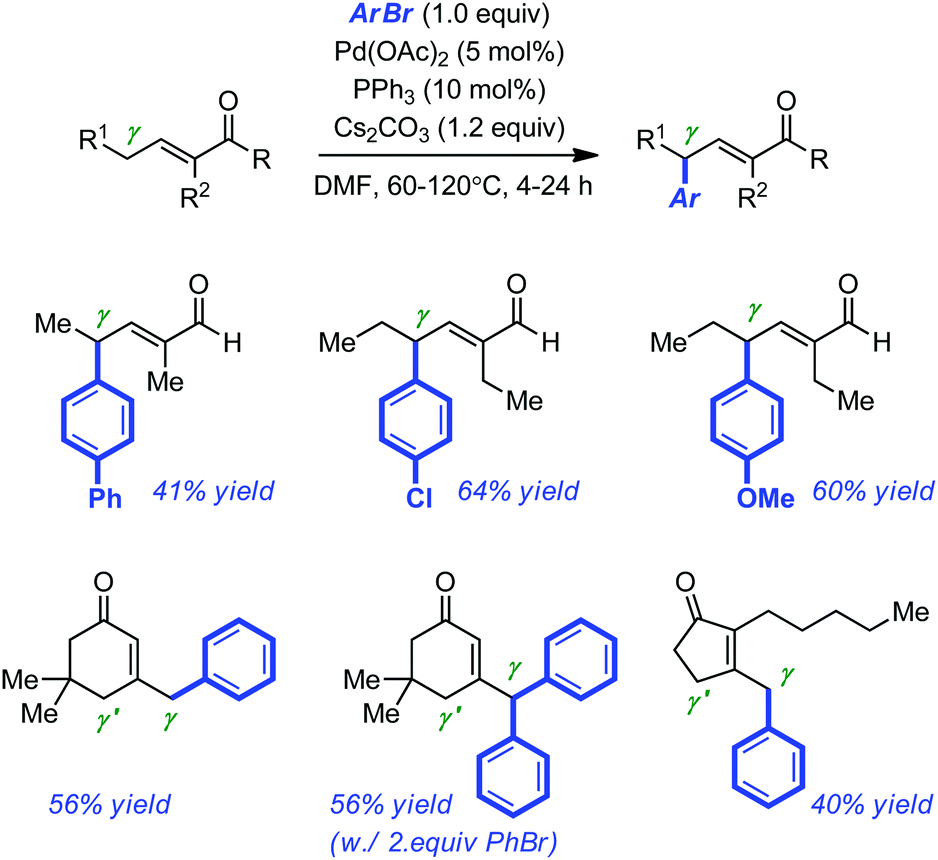 | ||
| Scheme 6 Pd-catalyzed γ-arylation of α,β-unsaturated aldehydes and ketones. | ||
Huang and Hartwig have developed an efficient Pd-catalyzed γ-arylation and γ-vinylation of α,β-unsaturated esters starting from the corresponding preformed silyl ketene acetals.24 The reaction proceeds at room temperature with low loadings of palladium (typically 1 mol%). Optimization of the catalytic system led to the identification of three different sets of reaction conditions (see Scheme 7 for details). However, the beneficial use of a triethylsilyl group (TES) on the ketene acetals, of ZnCl2 as the additive and of tBu3P as the ligand, was found to be a common feature of these arylations. Impressively, more than 30 different bromoarenes were evaluated to delineate the scope of the electrophilic component of the reaction. Noticeably, the average yield is approaching 80% and functional groups such as amine, ether, ketone, ester, cyano, indole, pyridine, quinoline, and thiophene were all tolerated. Introduction of a substituent on either the α or the β position of the silyl ketene acetals resulted in the formation of stereoisomeric mixtures of the corresponding arylated products. Although the exact role of ZnCl2 in these reactions is not fully understood, preliminary investigations suggested that it did not only serve as a source of chlorine but also facilitated labilization of the halide on palladium.
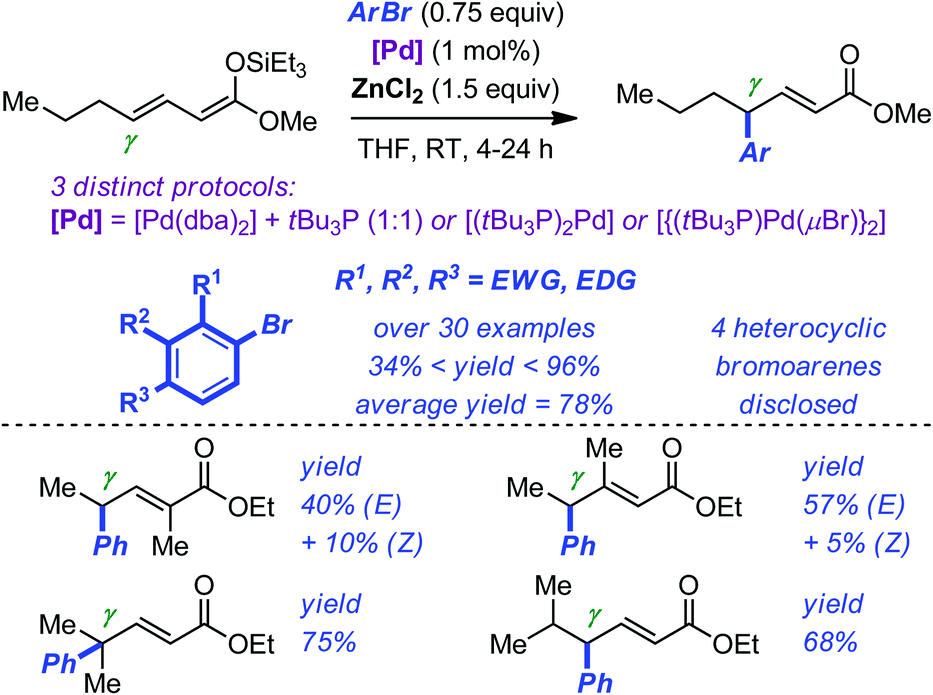 | ||
| Scheme 7 Pd-catalyzed γ-arylation of silyl ketene acetals. | ||
As part of their program aimed at the enantioselective arylation of aldehydes, Mazet and co-workers recently developed an efficient Pd-catalyzed γ-arylation of γ-branched α,β-unsaturated aldehydes.25 The precedent from Miura with linear substrates favoured rapid optimization of the reaction conditions and led to the identification of a robust protocol that allowed quaternary centers to be forged exclusively at the γ position without any detectable traces of the α- or β-arylation products (Pd(OAc)2 (1 mol%), tBu3P (2 mol%), Cs2CO3 (1.2 equiv.), DMF, 110 °C).26 The reaction proved very general with respect to the electrophile as electron-rich, electron-poor and sterically demanding aryl bromides were coupled in usually practical yields (19–87%, average = 68% yield). A slight erosion of the geometrical purity of the substrates was observed upon coupling, although the E/Z ratio was still relatively high (typically >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The orthogonal reactivity of the α,β-unsaturated aldehydes obtained was demonstrated through a series of standard derivatizations and – more remarkably – without any optimization of the initial protocol the ε arylation of α,β,δ,γ-unsaturated was achieved and the corresponding product isolated in a promising 43% yield (Scheme 8).
:1). The orthogonal reactivity of the α,β-unsaturated aldehydes obtained was demonstrated through a series of standard derivatizations and – more remarkably – without any optimization of the initial protocol the ε arylation of α,β,δ,γ-unsaturated was achieved and the corresponding product isolated in a promising 43% yield (Scheme 8).
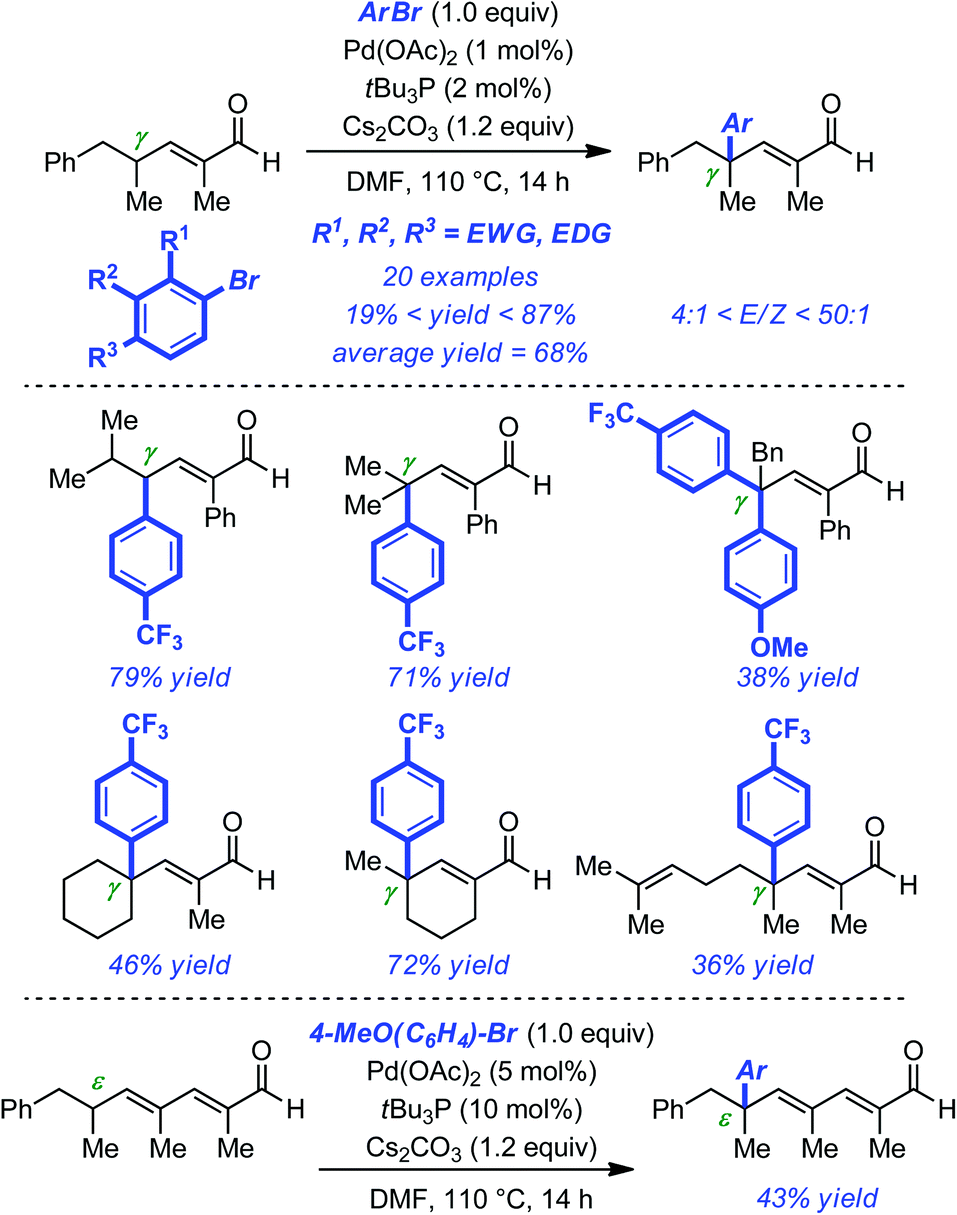 | ||
| Scheme 8 Pd-catalyzed γ-arylation of α,β-unsaturated γ-branched aldehydes and ε-arylation of α,β,γ,δ-unsaturated ε-branched aldehydes. | ||
The Pd-catalyzed intermolecular migrative β-arylation of ester lithium enolates was recently reported by Baudoin and co-workers as a viable alternative to directed C–H activation strategies for the functionalization of carbonyl compounds.27 This original approach relies on the rearrangement of congested palladium enolates to palladium homoenolates. Detailed theoretical mechanistic investigations have lent support on the β-H elimination, rotation and re-insertion sequence initially proposed.28 Although the primary system, focused on carboxylic esters, was limited to aryl halides with an electron-withdrawing substituent in the ortho position, a much more general β-arylation was subsequently reported for benzyl-protected alanine esters.29 During these investigations, the authors were also able to extend the arylations to the terminal position of amino esters with longer alkyl chains. Even if, in these cases again, only aryl halides with at least one fluorinated withdrawing ortho substituent were tolerated and the yields of the products decreased as the alkyl chain length increased, it is remarkable that arylation occurred exclusively at the terminal position of the carbon chain (as far as 6 carbon atoms away from the carbonyl functionality, see Scheme 9).
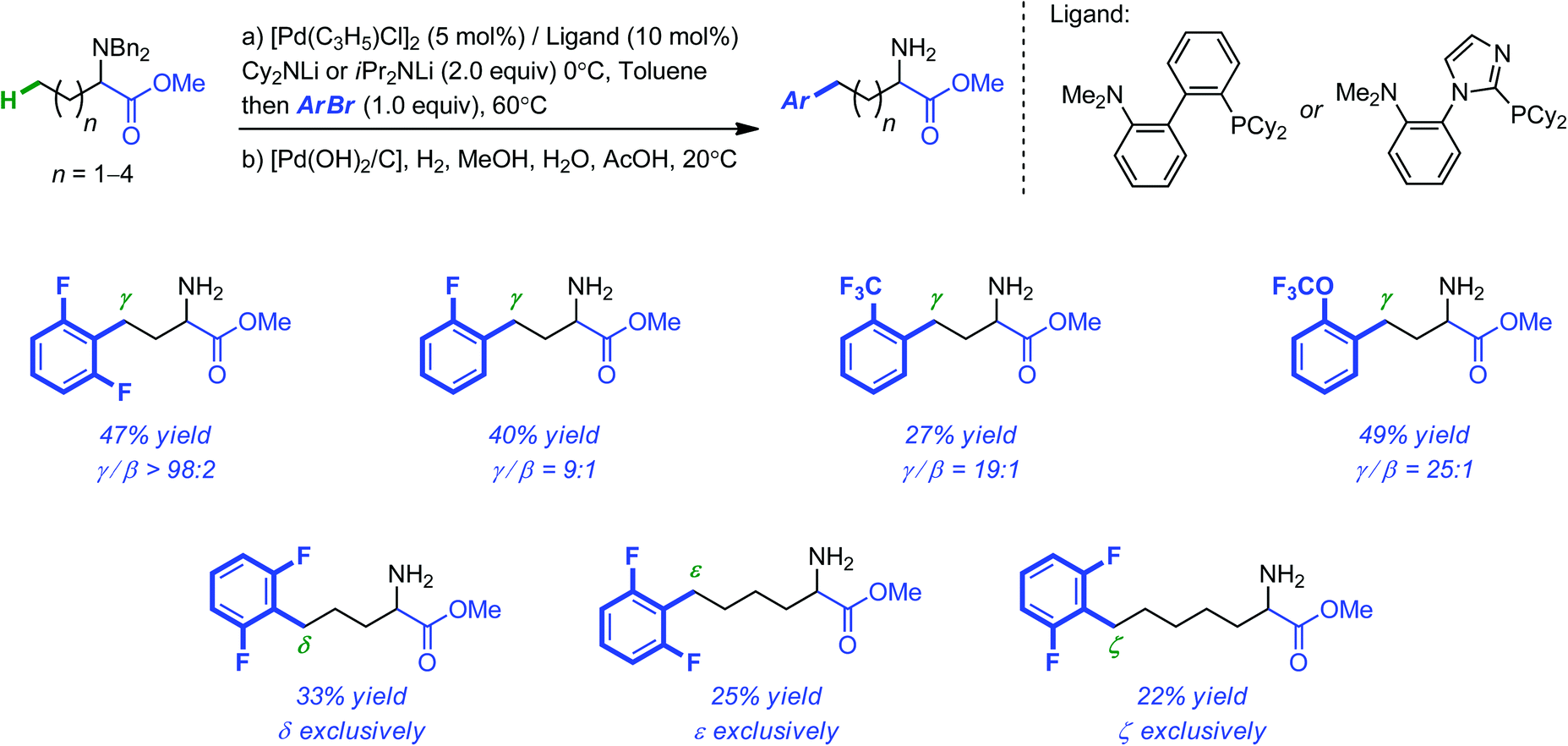 | ||
| Scheme 9 Selective and remote arylation of linear amino esters using ortho-substituted electron-deficient aryl bromides. | ||
4. Heck-type
Buchwald and Hyde recently reported the Pd-catalyzed γ-arylation of γ-substituted β,γ-unsaturated linear and cyclic ketones.30 Although this reaction could have well been categorized as a direct arylation of dienolates, we arbitrarily decided to discuss it in the ‘Heck-type’ section. Our decision stemmed essentially from the striking difference observed between the γ-arylation of α,β-unsaturated and β,γ-unsaturated ketones. In the optimized system, a ligand as simple as dppe (dppe = diphenylphosphinoethane; 4 mol%) in combination with Pd(OAc)2 (2 mol%) Cs2CO3 (1.5 equiv.) proved general for a variety of vinyl and aryl bromides (toluene, 100 °C, 8 h) and the products possessing a congested γ quaternary center were systematically obtained in good to excellent yield (50–81% yield; Scheme 10A). Subsequently, the use of ortho-bromoaniline derivatives allowed the efficient one-pot preparation of several ketoindolines by a γ-arylation/conjugate addition sequence. Further refinement of the system led to the identification of DTBM-Segphos as a highly selective ligand for the asymmetric variant of this transformation. Although the yields of the product were modest, the enantioselectivity levels were very high (Scheme 10B). Later, the same authors reported the regioselective γ-arylation of Angelica lactone and some analogues using a related procedure (2 mol% [Pd2(dba)3], 4 mol% Me-Phos, 1.2 equiv. K2CO3, toluene–t-amyl alcohol (2:1), 100 °C, 12 h), thus providing a straightforward access to 5,5-disubstituted butenolides – a ubiquitous structural motif in the synthesis of biologically active compounds (Scheme 10C).31,32
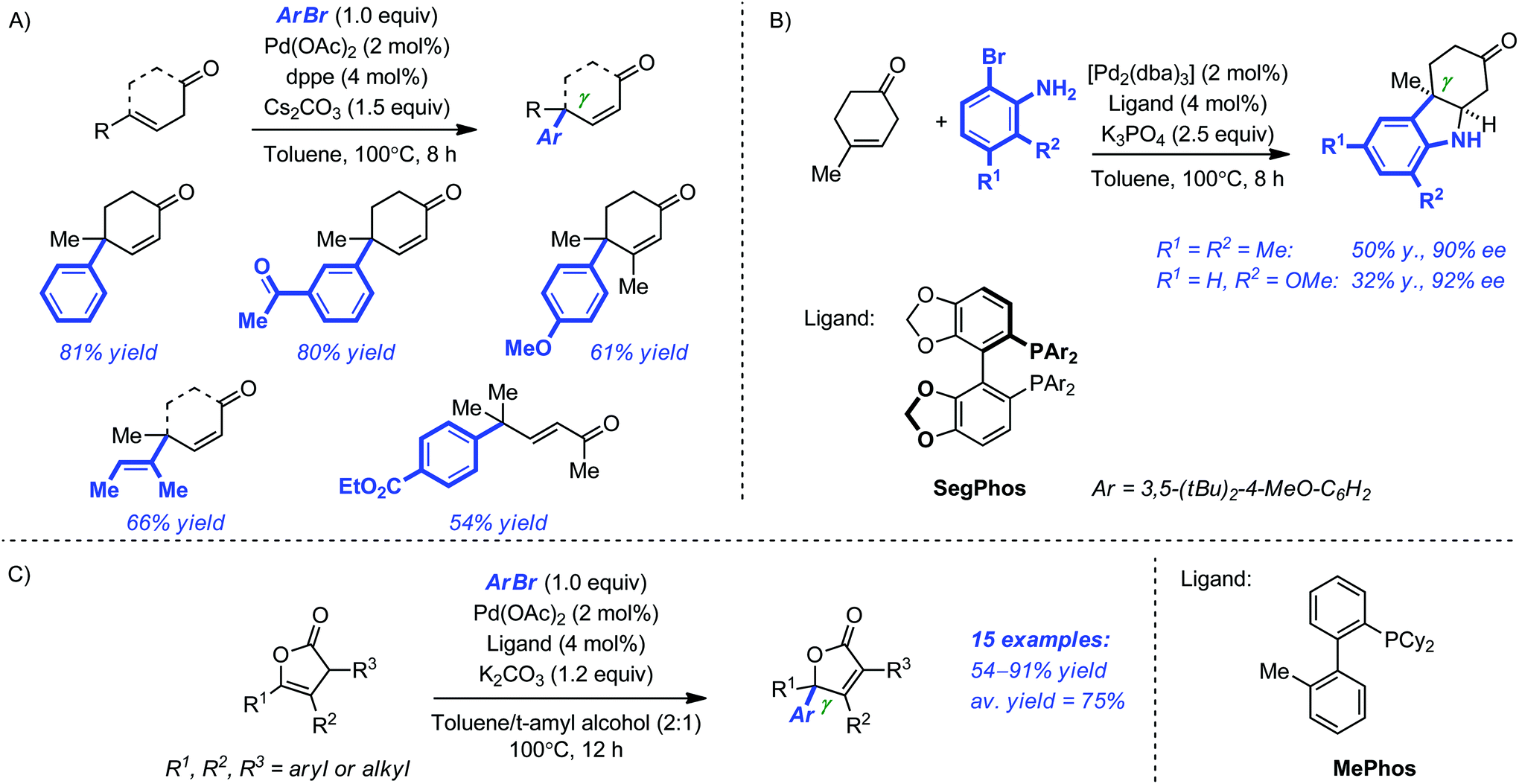 | ||
| Scheme 10 γ-Arylation of γ-substituted cyclic ketones (A) and Angelica lactone derivatives (C). Enantioselective synthesis of ketoindolines by a γ-arylation conjugate/addition sequence (B). | ||
Distinct from the above-described approach, Sigman and co-workers have recently reported a Pd-catalyzed enantioselective redox-relay Heck reaction of acyclic alkenyl 1° and 2° alcohols where aryl addition is followed by olefin migration through the carbon chain and eventually oxidation of the alcohol.33 The authors capitalized on previous works which described similar outcome when allylic alcohols were employed.34 Key to the success of this approach was the use of an electrophilic catalyst which favoured migration (i.e. chain walking) over dissociation from the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. This was achieved by combining aryldiazonium salts with a chiral pyridine–oxazoline ligand. The overall results are spectacular as chiral centers were installed with almost perfect stereocontrol not only at the β but also at the γ and δ positions of the resulting carbonyl group. In the latter cases, site selectivity remains to be improved. Of additional note, higher yields were obtained when starting from (Z) rather than (E) alkenes. A selection of the aldehydes and ketones that were obtained by this method is represented in Scheme 11.
C bond. This was achieved by combining aryldiazonium salts with a chiral pyridine–oxazoline ligand. The overall results are spectacular as chiral centers were installed with almost perfect stereocontrol not only at the β but also at the γ and δ positions of the resulting carbonyl group. In the latter cases, site selectivity remains to be improved. Of additional note, higher yields were obtained when starting from (Z) rather than (E) alkenes. A selection of the aldehydes and ketones that were obtained by this method is represented in Scheme 11.
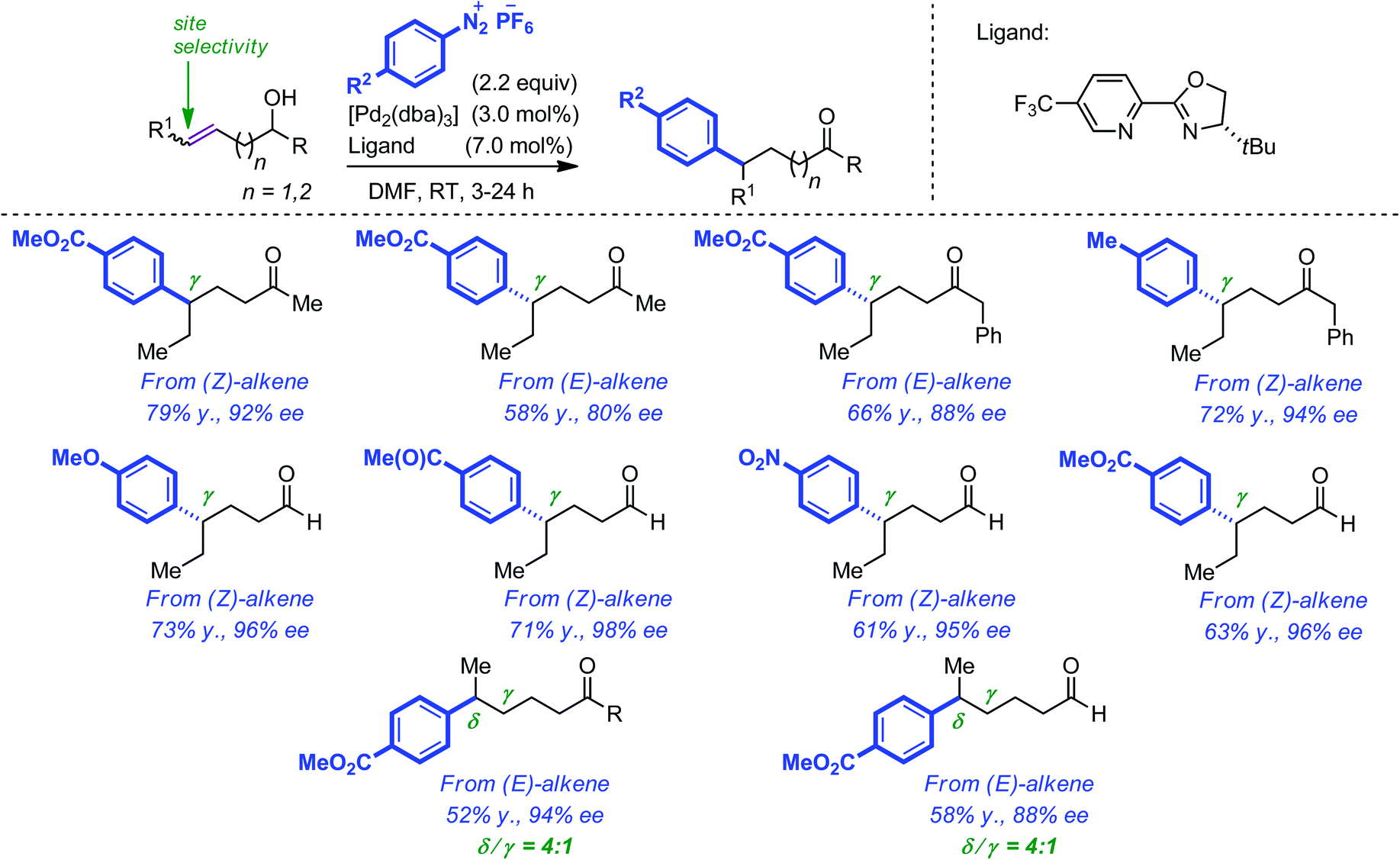 | ||
| Scheme 11 Pd-catalyzed enantioselective redox-relay Heck reaction of alkenyl primary and secondary alcohols. | ||
To circumvent some of the limitations of their initial system, Sigman and co-workers have next developed an enantioselective redox-relay Heck arylation of linear alkenyl alcohols using boronic acids (i.e. oxidative Heck).35 In addition to the coupling partners, the optimized protocol required the combination of [Pd(CH3CN)2(OTs)2] (6 mol%), [Cu(OTf)2] (6 mol%), the chiral ligand (13 mol%), O2 and 3 Å molecular sieves in DMF. The reaction for homoallylic alcohols turned out to be very general as electron-rich or electron-deficient aryl boronic acids were equally tolerated, producing the corresponding γ-arylated aldehydes and ketones in high yield, excellent enantioselectivity and much improved regioselectivity. Extension to alkenols with longer carbon chains proved also feasible, with the more distant C(sp2) being systematically preferred for aryl insertion. While the yields and enantioselectivities remained very high, the regioselectivity of arene insertion was found to decrease as the length of the alkyl chain increased (Scheme 12).
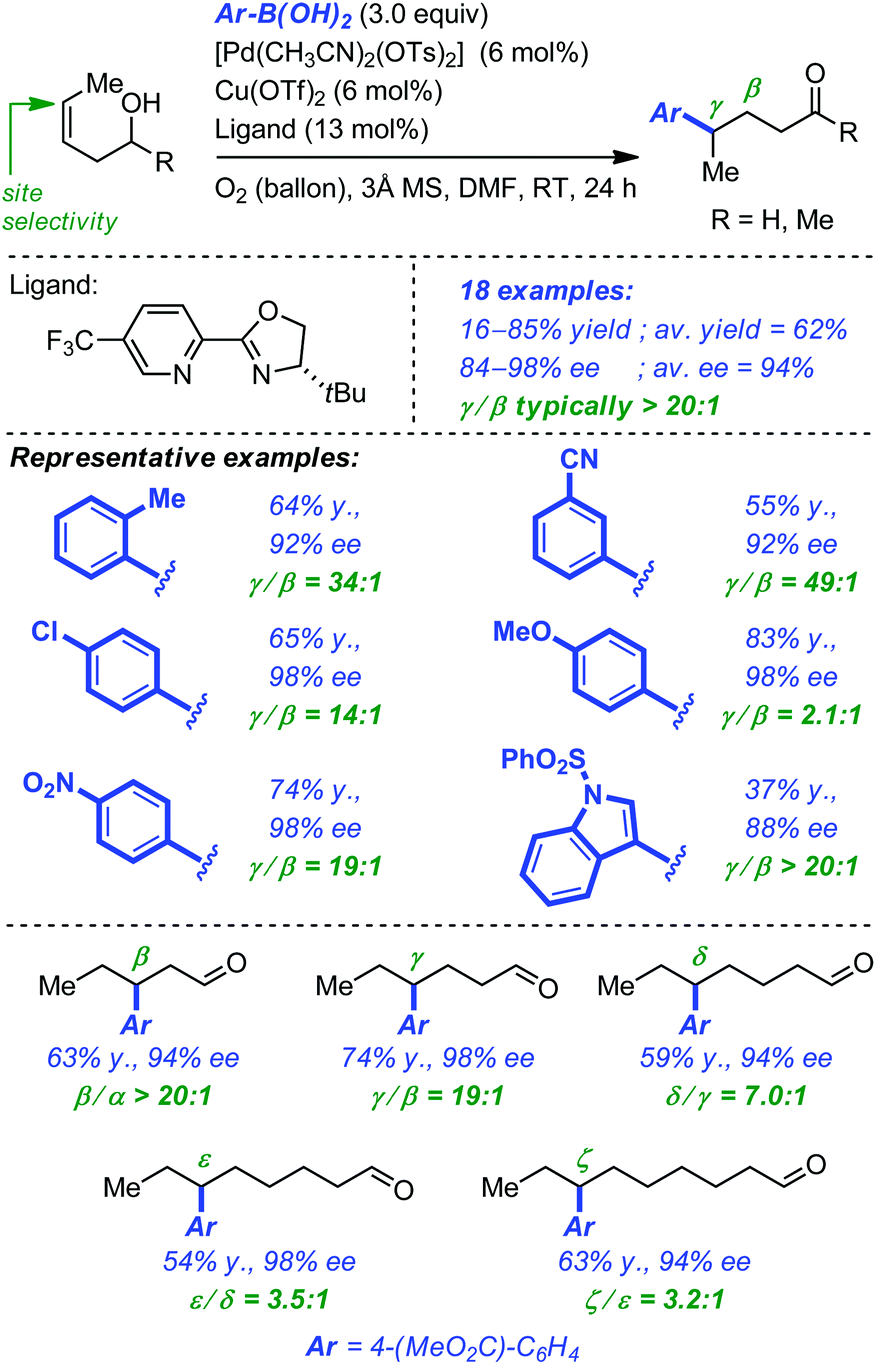 | ||
| Scheme 12 Pd-catalyzed oxidative redox-relay Heck reaction of alkenyl alcohols. | ||
5. Conclusions
The ability to selectively functionalize carbonyl derivatives at more and more distant positions from the CO bond using Pd catalysis has attracted increased interest in recent years. Progress in the directing group remote functionalization strategy has been mainly focused on elaborating more efficient auxiliaries that can not only facilitate the C(sp3)–H bond functionalization but can also be installed and removed easily. In addition to the C–C bond forming process, intra and intermolecular C–N and C–O bond forming reactions leading to ubiquitous heterocyclic structures have also been developed. As most of the approaches reported permit functionalization at the γ position, it would be interesting to develop novel directing groups to functionalize efficiently more remote C–H bonds. The vinylogous coupling of enolates now allows to introduce indifferently electron-rich, electron-poor or even congested aryl halides with high levels of efficiency at the γ position of α,β-unsaturated carbonyl derivatives. Generalization of these arylations to more remote positions is certainly an important objective. Furthermore, additional studies are expected to be focused on the development of the asymmetric versions of these reactions. The formidable achievements in the enantioselective redox-relay Heck and oxidative Heck reactions of alkenyl alcohols developed by Sigman and co-workers have paved the way for further studies. The scope of these reactions certainly needs to be extended. For instance generating quaternary stereocenters at remote positions from the carbonyl group produced upon isomerization of the CC bond would constitute an important achievement.
Acknowledgements
The authors thank the University of Geneva and the Swiss National Foundation (Project PP00P2_133482) for financial support.Notes and references
- (a) R. Breslow, Chem. Soc. Rev., 1972, 1, 553 RSC; (b) R. Breslow, Acc. Chem. Res., 1980, 13, 170 CrossRef CAS.
- (a) H. Schwarz, Acc. Chem. Res., 1989, 22, 282 CrossRef CAS; (b) H. Sailes and A. Whiting, J. Chem. Soc., Perkin Trans. 1, 2000, 1785 RSC; (c) J. S. Johnson and D. A. Evans, Acc. Chem. Res., 2000, 33, 325 CrossRef CAS PubMed; (d) K. Mikami, M. Shimizu, H.-C. Zhang and B. E. Maryanoff, Tetrahedron, 2001, 57, 2917 CrossRef CAS.
- (a) J. Clayden and N. Vassiliou, Org. Biomol. Chem., 2006, 4, 2667 RSC; (b) J. Clayden, Chem. Soc. Rev., 2009, 38, 817 RSC.
- For recent reviews on organocatalytic methods: (a) D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef CAS PubMed; (b) S. Bertelsen and K. A. Jørgensen, Chem. Soc. Rev., 2009, 38, 2178 RSC; (c) K. L. Jensen, G. Dickmeiss, H. Jiang, Ł. Albrecht and K. A. Jørgensen, Acc. Chem. Res., 2012, 45, 248 CrossRef CAS PubMed; (d) E. Arceo and P. Melchiorre, Angew. Chem., Int. Ed., 2012, 51, 5290 CrossRef CAS PubMed; (e) J.-L. Li, T.-Y. Liu and Y.-C. Chen, Acc. Chem. Res., 2012, 45, 1491 CrossRef CAS PubMed; (f) I. Kumar, P. Ramaraju and N. A. Mir, Org. Biomol. Chem., 2013, 11, 709 RSC; (g) H. Jiang, Ł. Albrecht and K. A. Jørgensen, Chem. Sci., 2013, 4, 2287 RSC.
- For general reviews on C–H functionalization see: (a) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS PubMed; (b) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (c) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (d) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (e) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC ; for a recent special issue, see: ; (f) Acc. Chem. Res, 2012, 6, 777–958. For a review on C(sp2)–H functionalization, see: Search PubMed; (g) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS PubMed ; for reviews on C(sp3)–H functionalization, see: ; (h) R. Jazzar, J. Hitce, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Chem.–Eur. J., 2010, 16, 265 CrossRef PubMed; (i) O. Baudoin, Chem. Soc. Rev., 2011, 40, 4902 RSC.
- The Yu group has recently reported a fascinating Pd-catalyzed meta functionalization of aromatic substrates using a quite elaborated directing group: D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518 CrossRef CAS PubMed . Upon unmasking a carbonyl group is potentially revealed. As this is a formal C(sp2)–H bond activation, this paper will not be discussed here. For a recent highlight, see: T. Truong and O. Daugulis, Angew. Chem., Int. Ed., 2012, 51, 11677 CrossRef PubMed.
- V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed.
- G. He and G. Chen, Angew. Chem., Int. Ed., 2011, 50, 5192 CrossRef CAS PubMed.
- G. He, Y. Zhao, S. Zhang, C. Lu and G. Chen, J. Am. Chem. Soc., 2012, 134, 3 CrossRef CAS PubMed.
- G. He, S.-Y. Zhang, W. A. Nack, Q. Li and G. Chen, Angew. Chem., Int. Ed., 2013, 52, 11124 CrossRef CAS PubMed.
- For other examples of Pd-catalyzed AQ-directed C–H functionalization see: (a) B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391 CrossRef CAS PubMed; (b) Y. Feng and G. Chen, Angew. Chem., Int. Ed., 2010, 49, 958 CrossRef CAS PubMed; (c) Y. Feng, Y. Wang, B. Landgraf, S. Liu and G. Chen, Org. Lett., 2010, 12, 3414 CrossRef CAS PubMed; (d) Y. Ano, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2011, 133, 12984 CrossRef CAS PubMed; (e) W. R. Gutekunst, R. Gianatassio and P. S. Baran, Angew. Chem., Int. Ed., 2012, 51, 7507 CrossRef CAS PubMed.
- S.-H. Yu, G. He, Y. Zhao, K. Wright, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2012, 134, 7313 CrossRef PubMed.
- (a) A. García-Rubia, R. Gómez Arrayás and J. C. Carretero, Angew. Chem., Int. Ed., 2009, 48, 6511 CrossRef PubMed; (b) A. García-Rubia, B. Urones, R. Gómez Arrayás and J. C. Carretero, Chem.–Eur. J., 2010, 16, 9676 CrossRef PubMed; (c) A. García-Rubia, B. Urones, R. Gómez Arrayás and J. C. Carretero, Angew. Chem., Int. Ed., 2011, 50, 10927 CrossRef PubMed.
- C. N. Rodríguez, J. A. Romero-Revilla, M. A. Fernández-Ibáñez and J. C. Carretero, Chem. Sci., 2013, 4, 175 RSC.
- M. Ochiai, K. Miyamoto, T. Kaneaki, S. Hayashi and W. Nakanishi, Science, 2011, 332, 448 CrossRef CAS PubMed.
- M. Fan and D. Ma, Angew. Chem., Int. Ed., 2013, 52, 12152 CrossRef CAS PubMed.
- For recent reviews on enolates α-arylation (a) D. A. Culkin and J. F. Hartwig, Acc. Chem. Res., 2003, 36, 234 CrossRef CAS PubMed; (b) A. C. B. Bortoloso, Synlett, 2009, 320 CrossRef PubMed; (c) C. C. C. Johansson and T. J. Colacot, Angew. Chem., Int. Ed., 2010, 49, 676 CrossRef CAS PubMed; (d) F. Bellina and R. Rossi, Chem. Rev., 2010, 110, 1082 CrossRef CAS PubMed; (e) C. Mazet, Synlett, 2012, 1999 CrossRef CAS PubMed.
- For recent examples of enantioselective α-arylations of amides: (a) S. Lee and J. F. Hartwig, J. Org. Chem., 2001, 66, 3402 CrossRef CAS PubMed; (b) T. Aaro, K. Kondo and T. Aoyama, Tetrahedron Lett., 2006, 47, 1417 CrossRef PubMed; (c) E. P. Kündig, T. M. Seidel, Y.-X. Jia and G. Bernardinelli, Angew. Chem., Int. Ed., 2007, 46, 8484 CrossRef PubMed; (d) X. Luan, R. Mariz, C. Robert, M. Gatti, S. Blumentritt, A. Linden and R. Dorta, Org. Lett., 2008, 10, 5569 CrossRef CAS PubMed; (e) S. Würtz, C. Lohre, R. Fröhlich, K. Bergander and F. Glorius, J. Am. Chem. Soc., 2009, 131, 8344 CrossRef PubMed; (f) X. Luan, L. Wu, E. Drinkel, R. Mariz, M. Gatti and R. Dorta, Org. Lett., 2010, 12, 1912 CrossRef CAS PubMed; (g) Y.-X. Jia, D. Katayev, G. Bernardinelli, T. M. Seidel and E. P. Kündig, Chem.–Eur. J., 2010, 16, 6300 CrossRef CAS PubMed; (h) L. Liu, N. Ishida, S. Ashida and M. Murakami, Org. Lett., 2011, 13, 1666 CrossRef CAS PubMed.
- For recent examples on the asymmetric α-arylation of esters: (a) R. Galarini, A. Musco, R. Pontellini and R. Santi, J. Mol. Catal., 1992, 72, L11 CrossRef CAS; (b) D. J. Spielvogel and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 3500 CrossRef CAS PubMed; (c) D. J. Spielvogel, W. M. Davis and S. L. Buchwald, J. Am. Chem. Soc., 2002, 121, 3833 Search PubMed; (d) X. Liu and J. F. Hartwig, J. Am. Chem. Soc., 2004, 126, 5182 CrossRef CAS PubMed.
- For recent examples of the enantioselective α-arylation of ketones: (a) J. M. Fox, X. Huang, A. Chieffi and S. L. Buchwald, J. Am. Chem. Soc., 2000, 122, 1360 CrossRef CAS; (b) T. Hamada, A. Chieffi, J. Åhman and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 1261 CrossRef CAS PubMed; (c) G. Chen, F. Y. Kwong, H. O. Chan, W.-Y. Yu and A. S. C. Chan, Chem. Commun., 2006, 1413 RSC; (d) X. Liao, Z. Weng and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 195 CrossRef CAS PubMed; (e) S. Ge and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 1633 CrossRef PubMed.
- For the enantioselective α-arylations of aldehydes: (a) J. Garcia-Fortanet and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 8108 CrossRef CAS PubMed; (b) P. Nareddy, L. Mantilli, L. Guénée and C. Mazet, Angew. Chem., Int. Ed., 2012, 51, 3826 CrossRef CAS PubMed; (c) N. Humbert, E. Larionov, L. Mantilli, P. Nareddy, C. Besnard, L. Guénée and C. Mazet, Chem.–Eur. J., 2013 DOI:10.1002/chem.201303146.
- For reviews on reactions based on the vinylogous analogy: (a) R. C. Fuson, Chem. Rev., 1935, 16, 1 CrossRef CAS; (b) G. Casiraghi, F. Zanardi, G. Appendino and G. Rassu, Chem. Rev., 2000, 100, 1929 CrossRef CAS PubMed; (c) S. F. Martin, Acc. Chem. Res., 2002, 35, 895 CrossRef CAS PubMed; (d) S. E. Denmark, J. R. Heemstra Jr. and G. L. Beutner, Angew. Chem., Int. Ed., 2005, 44, 4682 CrossRef CAS PubMed.
- (a) Y. Terao, T. Satoh, M. Miura and M. Nomura, Tetrahedron Lett., 1998, 39, 6203 CrossRef CAS; (b) Y. Terao, T. Satoh, M. Miura and M. Nomura, Bull. Chem. Soc. Jpn., 1999, 72, 2345 CrossRef CAS.
- D. S. Huang and J. F. Hartwig, Angew. Chem., Int. Ed., 2010, 49, 5757 CrossRef CAS PubMed.
- I. Franzoni, L. Guénée and C. Mazet, Chem. Sci., 2013, 4, 2619 RSC.
- (a) Quaternary Stereocentres: Challenges and Solutions for Organic Synthesis, ed. J. Christoffers and A. Baro, Wiley-VCH, Weinheim, 2005 Search PubMed; (b) K. Fuji, Chem. Rev., 1993, 93, 2037 CrossRef CAS; (c) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945 CrossRef CAS PubMed; (d) M. Bella and T. Gasperi, Synlett, 2009, 1583 CAS; (e) I. Marek and G. Sklute, Chem. Commun., 2007, 1683 RSC; (f) C. Hawner and A. Alexakis, Chem. Commun., 2010, 46, 7295 RSC.
- (a) A. Renaudat, L. Jean-Gérard, R. Jazzar, C. E. Kefalidis, E. Clot and O. Baudoin, Angew. Chem., Int. Ed., 2010, 49, 7261 CrossRef CAS PubMed . For a seminal contribution, see: ; (b) M. Jørgensen, S. Lee, X. Liu, J. P. Wolkowski and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 12557 CrossRef PubMed.
- P. Larini, C. E. Kefalidis, R. Jazzar, A. Renaudat, E. Clot and O. Baudoin, Chem.–Eur. J., 2012, 18, 1932 CrossRef CAS PubMed.
- S. Aspin, A.-S. Goutierre, P. Larini, R. Jazzar and O. Baudoin, Angew. Chem., Int. Ed., 2012, 124, 10966 CrossRef.
- A. M. Hyde and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 177 CrossRef CAS PubMed.
- A. M. Hyde and S. L. Buchwald, Org. Lett., 2009, 11, 2663 CrossRef CAS PubMed.
- (a) B. Figadère, Acc. Chem. Res., 1995, 28, 359 CrossRef; (b) M. F. Braña, M. L. García, B. López, B. de Pascual-Teresa, A. Ramos, J. M. Pozuelo and M. T. Domínguez, Org. Biomol. Chem., 2004, 2, 1864 RSC; (c) J. Dogné, C. T. Supuran and D. J. Pratico, J. Med. Chem., 2005, 48, 2251 CrossRef PubMed.
- E. W. Werner, T.-S. Mei, A. J. Burckle and M. S. Sigman, Science, 2012, 338, 1455 CrossRef CAS PubMed.
- (a) R. C. Larock, W.-Y. Leung and S. Stolz-Dunn, Tetrahedron Lett., 1989, 30, 6629 CrossRef CAS; (b) K. Yonehara, K. Mori, T. Hashizume, K.-G. Chung, K. Ohe and S. Uemura, J. Organomet. Chem., 2000, 603, 40 CrossRef CAS; (c) S. Bouquillon, B. Ganchegui, B. Estrine, F. Hénin and J. Muzart, J. Organomet. Chem., 2001, 634, 153 CrossRef CAS; (d) M. Quadir, R. E. Priestley, T. W. D. F. Rising, T. Gelbrich, S. J. Coles, M. B. Hursthouse, P. W. Shedrake, N. Whittall and K. K. (Mimi) Hii, Tetrahedron Lett., 2003, 44, 3675 CrossRef; (e) F. Berthiol, H. Doucet and M. Santelli, Tetrahedron, 2006, 62, 4372 CrossRef CAS PubMed.
- T.-S. Mei, E. W. Werner, A. J. Burckle and M. S. Sigman, J. Am. Chem. Soc., 2013, 135, 6830 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |