 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of substituted pyrenes by indirect methods
Juan M.
Casas-Solvas†‡
,
Joshua D.
Howgego†
and
Anthony P.
Davis
*
School of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: Anthony.Davis@bristol.ac.uk
First published on 14th November 2013
Abstract
The pyrene nucleus is a valuable component for materials, supramolecular and biological chemistry, due to its photophysical/electronic properties and extended rigid structure. However, its exploitation is hindered by the limited range of methods and outcomes for the direct substitution of pyrene itself. In response to this problem, a variety of indirect methods have been developed for preparing pyrenes with less usual substitution patterns. Herein we review these approaches, covering methods which involve reduced pyrenes, transannular ring closures and cyclisations of biphenyl intermediates. We also showcase the diverse range of substituted pyrenes which have been reported in the literature, and can serve as building blocks for new molecular architectures.
Juan M. Casas-Solvas obtained his first degree in chemistry at the University of Almería in 2001, where he also completed a PhD on the electrochemical characterisation of supramolecular chemistry of cyclic oligosaccharides and carbohydrate–protein binding interactions, under the supervision of Prof. A. Vargas-Berenguel. In 2010 he moved to the University of Bristol as an IEF Marie Curie Fellow under the supervision of Prof. A. P. Davis, where he worked on the design and construction of synthetic lectins for the biomimetic recognition of carbohydrates. Since May 2012 he works at Prof. Vargas-Berenguel's group on the preparation of multifunctional nanocarriers for anticancer drugs based on β-cyclodextrin, dendrimers and gold nanoparticles. |
Joshua Howgego grew up in Ipswich, UK, and completed his first degree in chemistry at the University of Reading. From there he moved to the University of Bristol to study for a PhD focussing on the design and synthesis of biomimetic receptors for carbohydrates, under the supervision of Professor Anthony Davis. Upon completion of his PhD Joshua entered the field of science journalism and in 2013 was appointed deputy news and opinions editor at SciDev.Net, a website that provides news and analysis of science as practised and applied in the developing world. |
Tony Davis gained a B.A. in Chemistry from Oxford University in 1977, then stayed for a D.Phil. under Dr G. H. Whitham and postdoctoral work with Prof. J. E. Baldwin. In 1981 he moved to the ETH Zürich as a Royal Society European Exchange Fellow working with Prof. A. Eschenmoser, then in 1982 was appointed Lecturer in Organic Chemistry at Trinity College, Dublin. In September 2000 he moved to the University of Bristol, where he is Professor of Supramolecular Chemistry in the School of Chemistry. His research interests include the supramolecular chemistry of carbohydrates and anions, and the study of steroid-based nanoporous crystals. |
1. Introduction
The pyrene nucleus is a compact polycyclic aromatic unit which is widely exploited for its electronic and photophysical properties, and for its ability to take part in non-covalent interactions. It serves as a key component in organic electronics1–4 where it has been used, for example, to make field-effect transistors5 and OLEDs.1 It is also widely utilised in supramolecular photosensors.6 It possesses an exceptionally long fluorescence lifetime, and the vibronic band structure of its emission shows a strong dependence on solvent polarity (Ham effect).7,8 Furthermore, the wavelength of the bright fluorescence of pyrene is dependent on whether it is a present as a monomer or excimer. As a result, there have been many supramolecular sensors reported in which the sensing mechanism is based on a simple monomer–excimer interconversion, triggered by the presence or absence of the guest. In other cases signalling results from guest-induced perturbation of more complicated processes such as photoinduced electron (PET) and charge (PCT) transfers, fluorescence resonance energy transfer (FRET) and chelation-enhanced fluorescence (CHEF).The pyrene unit is also valued for its binding properties. As a large aromatic surface, it is capable of taking part in π-stacking and CH–π interactions which, in water, can be reinforced by the hydrophobic effect.9,10 This has been much exploited in the noncovalent functionalisation of extended planar π-systems such as carbon nanotubes11–14 and graphene.15–17 For example, poly(pyrenebutyric acid) has been used as a non-covalent stabilising unit for single-walled carbon nanotubes, preventing formation of bundles and enabling dispersion in solvents.18 Pyrene has also found use in biological chemistry, especially in systems for binding nucleic acids,19–22 and in the design of synthetic receptors for aromatic23,24 and carbohydrate25 substrates.
Although pyrene is already used extensively, its potential appears to be under-realised. The main limitation is a lack of well-known methodology for the construction of pyrenes with diverse substitution patterns. Thus, while pyrene is easily appended to a system, the generation of architectures with pyrenyl cores is much less straightforward. This is especially relevant to supramolecular chemistry, where polysubstituted pyrenes could serve as components for a variety of structures with well-defined cavities.
The obvious starting material for substituted pyrenes is pyrene itself, which is readily accessible and inexpensive. However, as discussed in the following section, possibilities for the direct functionalisation of the parent hydrocarbon are rather limited. For this reason a range of alternatives have been developed in which groups are introduced to non-pyrene precursors, and the full aromatic system is only generated at a late stage of the synthesis. Herein we provide a survey of such “indirect” methods. We hope this review will increase awareness of the range of pyrenes which can be made, and facilitate the use of this valuable unit in supramolecular, materials and biological chemistry.
2. The direct functionalisation of pyrene – problems and limitations
To place the later sections in context, we begin with a brief account of the direct functionalisation of pyrene, i.e. those methods which involve reactions on pre-existing pyrene units. More details can be found in the recent review by Figueira-Duarte and Müllen,1 and Vollmann's classical investigations into pyrene reactivity provide a useful foundation.26 As shown in Fig. 1, pyrene 1 is most activated for electrophilic aromatic substitution at the 1, 3, 6 and 8 positions. These are the most electron-rich centres, and are predicted to be the most reactive by calculations on Wheland intermediates.27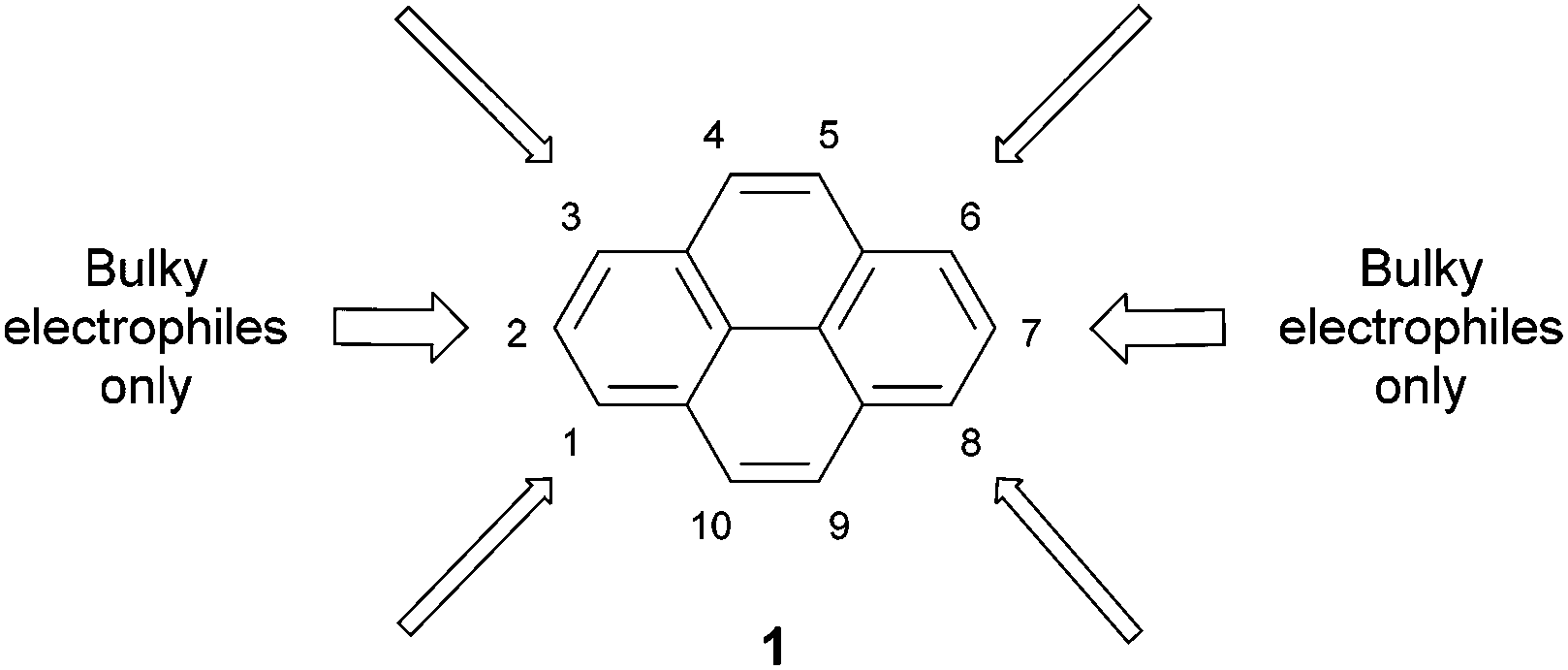 | ||
| Fig. 1 Pyrene 1 is activated towards electrophilic aromatic substitution at the 1, 3, 6 and 8 positions only. Some bulky electrophiles are forced by steric hindrance to react at the 2 and 7 positions. | ||
The synthesis of simple (tetra) 1,3,6,8-halogenated and -cyanated pyrenes was originally reported by Ogino.28 Monosubstitution has also been achieved. Further elaboration of the compounds (e.g. by Heck coupling of the bromides with styrenes)29 has been demonstrated. However, if two or three equivalents of an electrophile are used, the substitution pattern obtained is a statistical distribution of regioisomers. It is therefore difficult to prepare anything other than 1-substituted pyrenes or 1,3,6,8-tetrasubstituted pyrenes by direct electrophilic substitution. Further difficulties are caused by the fact that tetrasubstituted pyrenes are often highly insoluble. This means that purification methods are rather limited. The compounds prepared by Ogino, for example, were purified by high-temperature sublimation.
The 2 and 7 positions of pyrene are activated towards electrophilic aromatic substitution to a lesser extent than the 1, 3, 6 and 8 positions, but they can react selectively if a very bulky electrophile is employed, e.g. tert-butyl chloride.30 In a recent development, Marder has reported that the 2- and 2,7-borylated pyrenes 2 and 3 may be prepared in excellent yields via reaction with bulky iridium-boryl complexes (Scheme 1).31 Since aryl C–B bonds are useful for Suzuki–Miyaura coupling reactions as well as other transformations, this promises to be a very useful methodology. These borylated pyrenes have been used as precursors for other derivatives including alcohols, ethers, triflates, bromides and oxidative coupling products.32–36 When positions 2 and 7 are already occupied, Marder's borylation takes place at position 4.37
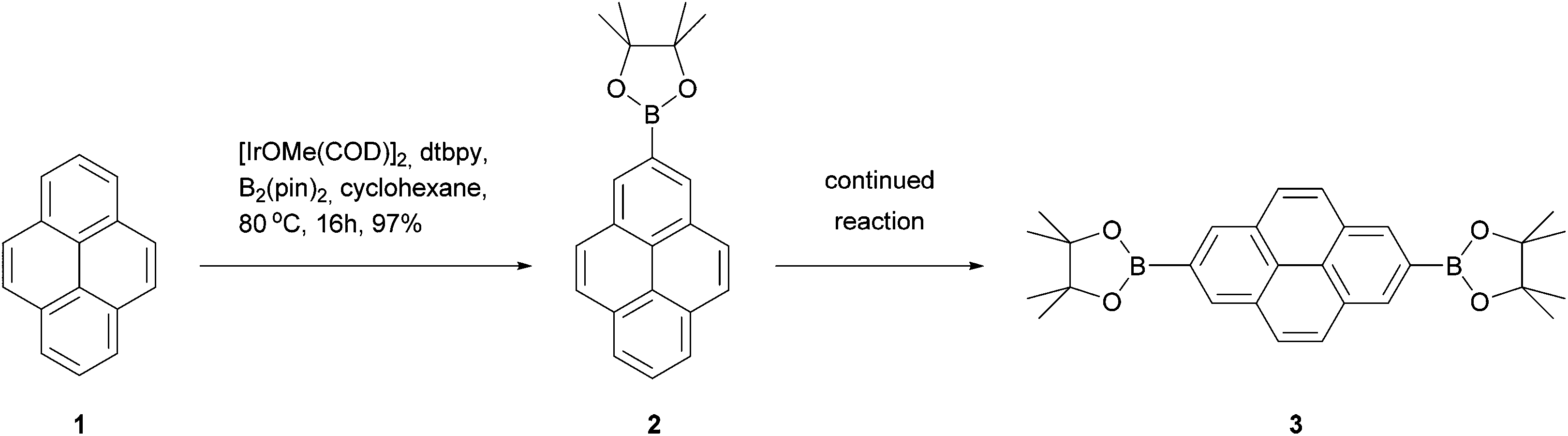 | ||
| Scheme 1 Marder's iridium-catalysed 2-borylation of pyrene. | ||
The synthesis of 2-substituted pyrenes has also been reported from the pyrene-chromium tricarbonyl (CTC) complex 4 in relatively high yields.38 The complexes can be made from native pyrene by reacting with (NH3)3Cr(CO)3 and BF3·Et2O in fair yields. The highly electron-withdrawing CTC group increases the acidity of the aromatic protons, so that treatment with a strong base (in this case LiTMP) results in deprotonation. Addition of ethyl chloroformate, B(OBu)3 (followed by oxidation) or trimethylsilyl chloride then lead to derivatives 5–7 as shown in Scheme 2. Selectivity for position 2 was generally good; only the ethoxycarbonylation gave a second product (the 1,2-disubstituted derivative). However, to our knowledge there are no examples of the procedure being used aside from the original report, which may be due to the toxicity of the chromium reagent.
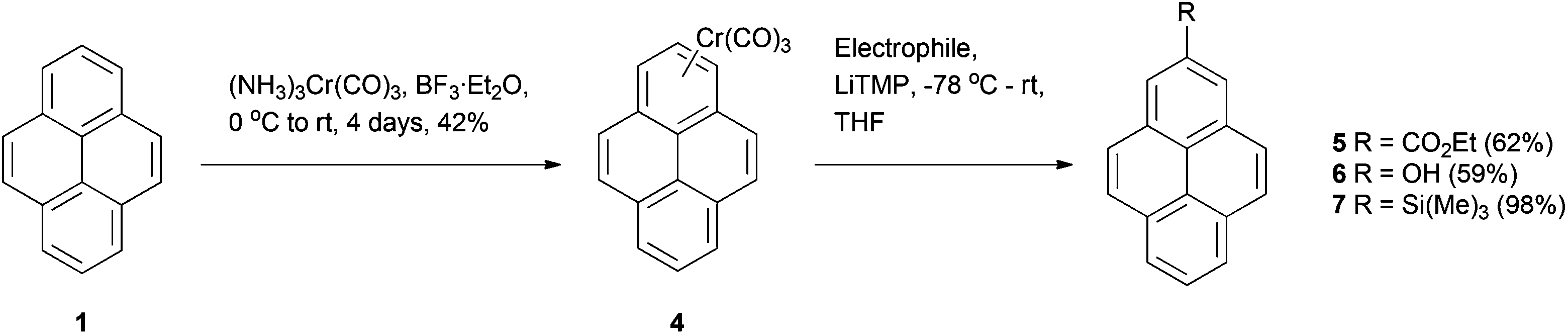 | ||
| Scheme 2 Uses of pyrene-chromium tricarbonyl (CTC) complexes. | ||
K-region39 (4,5,9,10-) substituted pyrenes are not accessible via Friedel–Crafts type reactions unless the 2 and 7 positions have been substituted with bulky tert-butyl groups which block the electronically favoured 1, 3, 6 and 8 positions. However, it is possible to prepare the tetrahydropyrene-4,5,9,10-tetraone 9via oxidation of pyrene in the presence of a ruthenium salt catalyst (Scheme 3).40,41 The 4,5-dione 8 can be prepared in fair yield (45%) under mild conditions and can be further oxidised to the tetraone 9 by increasing the concentration of oxidising agent and heating to 40 °C. Dione 8 turned out to be useful for the synthesis of desymmetrised pyrenes as it reacted selectively with bromine at the K-region to give 10 in quantitative yield,42 an important result given the difficulty in accessing these positions. Presumably the electronic effects of the carbonyl groups reduce electron density at the usually active positions and render the 9,10 positions the most electron rich in dione 8. It was also possible to reduce the carbonyls to alcohols and produce fully aromatic desymmetrised pyrene 11.
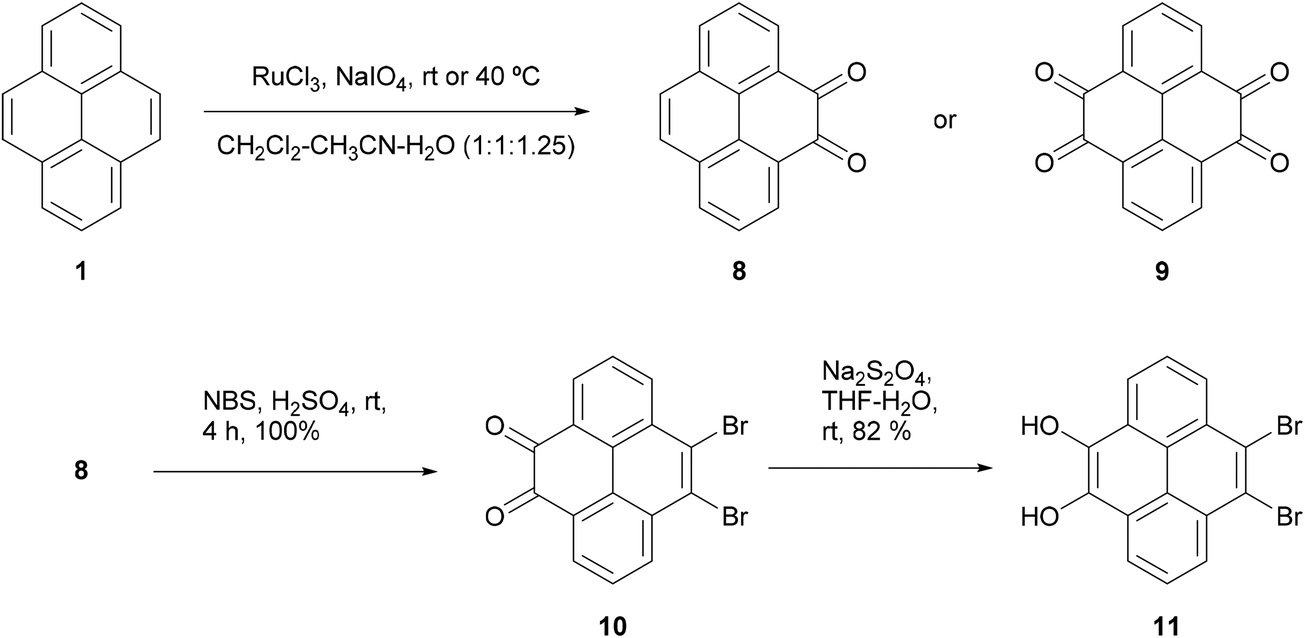 | ||
| Scheme 3 Müllen's direct 4,5-substitutions via ruthenium trichloride catalysis. | ||
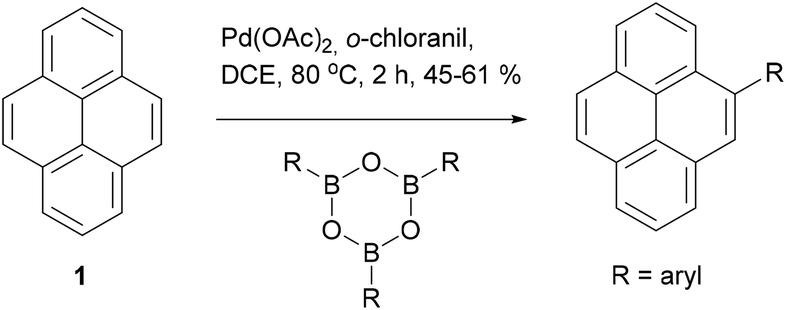
One final example of direct modification of pyrene is worth mentioning. Itami and coworkers reported that palladium catalysed arylation of pyrene with boroximes in the presence of o-chloranil takes place exclusively at the K-region (Scheme 4).43 The mechanism of this transformation is unclear but seems to be quite specific, given that alternative oxidants (such as p-chloranil) completely shut down the reaction. Several examples of aryl groups were presented and the yields of 4-arylpyrenes were fair to good (45–61%). When further equivalents of the aryl boroxime reagent were employed diarylation also took place, but there was essentially no regioselectivity; the 4,9-diarylpyrene and 4,10-diaryl pyrenes were produced in ∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Although the mechanism of the reaction is not known, the authors suggest that complexation of a Pd(II) species by the pyrene K-region may be a first step. If this is the case, further research could open up a range of other reactions making use of the C–Pd interaction as a handle for reactivity.
:1 ratio. Although the mechanism of the reaction is not known, the authors suggest that complexation of a Pd(II) species by the pyrene K-region may be a first step. If this is the case, further research could open up a range of other reactions making use of the C–Pd interaction as a handle for reactivity.
 | ||
| Scheme 4 Itami's 4-arylation of pyrene via palladium catalysis. | ||
The above summary shows that is possible to substitute pyrenes at each of its positions by direct methods. However, the methodology is not always satisfactory. Control over the degree of substitution is poor in some cases, and the range of functionality that can be introduced is often limited. Moreover the positioning of multiple substituents in specific relation to each other is often impossible. Where direct substitution cannot solve the problem, alternatives may be provided by indirect methods as described in the following sections.
3. The tetrahydropyrene (THPy) method
The first indirect method of pyrene synthesis we will consider is the tetrahydropyrene (THPy) approach. The main purpose of this strategy is to allow, effectively, the performance of electrophilic aromatic substitutions (EAS) at positions 2 and 7 on pyrene. As already described, these positions on native pyrene are accessible to only a very limited group of reagents. The THPy approach involves the reduction of pyrene to 4,5,9,10-tetrahydropyrene 12, the reactivity of which is displaced to positions 2 and 7 under EAS conditions. Subsequent re-aromatisation gives pyrene derivatives carrying substituents in either one or both extreme positions (Scheme 5).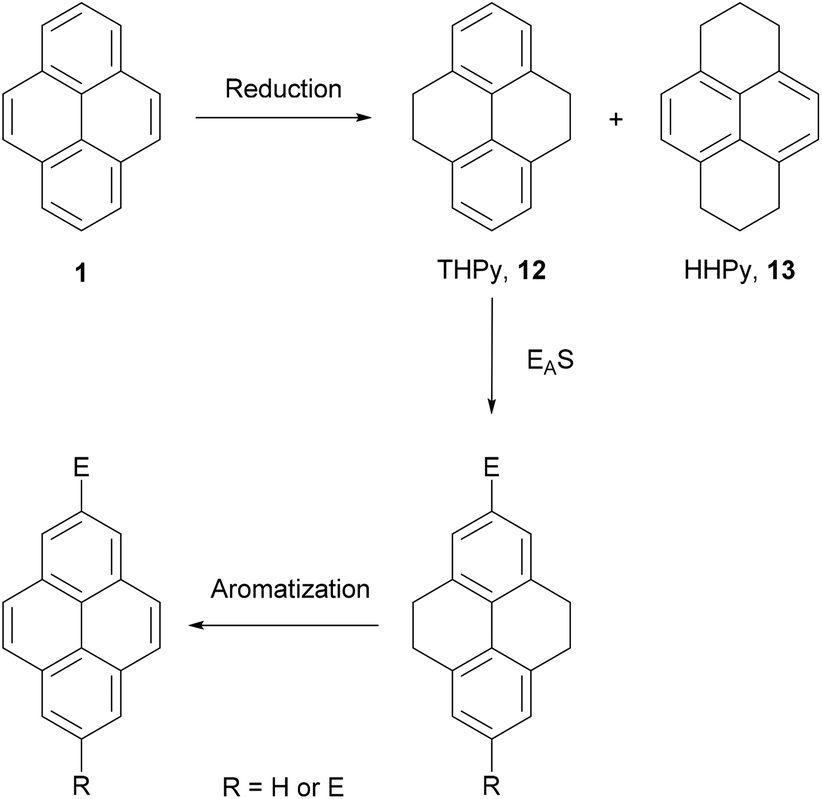 | ||
| Scheme 5 Synthesis of 2,7-substituted pyrenes via the THPy method. | ||
THPy was first identified as a minor product (ca. 10% yield) from hydrogenation of pyrene under vigorous conditions (100 bar, 400 °C) using a molybdenum–sulphur–carbon catalyst.44 However, later studies indicated that hydrogenation can be limited to the K-region by tuning conditions of the reaction.45,46 Currently there are several methods reported for the preparative synthesis of THPy using H2/Pd/C in EtOAc (Table 1).47–54 Some authors have reported that hydrogenation does not take place unless the commercial pyrene starting material is purified by column chromatography47 or desulphurisation with RANEY® nickel50,53,54 prior to the reaction. The latter method introduces traces of water (ca. 0.5%) to the reaction mixtures which are reported to help restrict over-reduction. The quality of the catalyst is also a significant factor and different commercial sources have been compared.47 In many cases hydrogenation does not go to completion in a single reaction cycle. Common steps to increase the extent of reaction are addition of a very large amount of catalyst48,54 and performing several hydrogenation cycles with fresh catalyst.49,51 More recently, higher pressures and temperatures have been applied, reducing reaction times and the amounts of impurities found in the crude products.52,53 In general, the main impurity is 1,2,3,6,7,8-hexahydropyrene (HHPy) 13, which can be removed by column chromatography on Florisil,47 silica49,51,54 or alumina,53 or by diacetylation of the crude product followed by fractional precipitation of the impurity.50
| Entry | H2 pressure (bar) | T (°C) | Time (h) | Purification | Yield (%) | Ref. |
|---|---|---|---|---|---|---|
| a Pyrene purified using Florisil column prior to reaction. b Pyrene desulphurised with RANEY® nickel prior to reaction. c No purification described. d No yield given. e Obtained as 2,7-diacetyl-THPy. | ||||||
| 1a | 3.4 | rt | 96 | Chromatography on Florisil | 95 | 47 |
| 2 | 4.1 | rt | 192 | —c | —d | 48 |
| 3 | 3.4 | rt | 24 (×3) | Chromatography on silica | 50 | 49 |
| 4b | 3.4 | rt | 65 | Acetylation and selective solubilisation | 53e | 50 |
| 5 | 3.8 | rt | 24 (×3) | Chromatography on silica | 86 | 51 |
| 6 | 140 | 90 | 7 | —c | 96 | 52 |
| 7a,b | 160 | 60 | 24 | Chromatography on alumina | 76 | 53 |
| 8b | 3.4 | rt | 72 | Chromatography on silica | —d | 54 |
Very pure THPy can be prepared on a small scale by photochemical reduction of pyrene in the presence of triphenyltin hydride. This reaction takes only 1 h and no products other than THPy are formed.55 Birch conditions with lithium reductant have also been used by some workers.56–58 Again, purification of commercial pyrene prior to the reaction seems to be essential for the reaction to be successful. The Birch reduction does not give directly the desired compound but the intermediate 4,5-dihydropyrene (DHPy), which can be subsequently hydrogenated in the presence of Pd/C to yield the desired THPy in good yields. Alternatively, the second reduction can be accomplished by applying Birch conditions again, but extremely short reaction times (2 min) must be used to avoid over-reduction. This method usually gives mixtures of DHPy and THPy which are then separated by charge-transfer chromatography (10% caffeine on silica gel).57 If sodium is employed instead of lithium, initial reduction takes place in positions 1 and 9, after which treatment with acid is required for the isomerisation to 4,5-dihydropyrene. A second reduction with sodium in xylene–ethanol yields a mixture of THPy and HHPy, from which THPy may be isolated after selective nitration of the latter.58 It is also possible to synthesise THPy via ring closure of [2,2]metacyclophane or photo-chemical cyclisation of 2,2′-divinylbiphenyl. These approaches are discussed further in sections 5 and 6.
Re-aromatisation of THPy derivatives to give the final substituted pyrenes can be achieved with a variety of conditions.59 The most popular method is the reaction with 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ).49,51,53–56,60–65 Catalytic dehydrogenation with Pd/C47,60,66 and treatment with halogens50,52,57,67,68 have also been used. There are also examples employing selenium,44 sulphur68 and o-chloranil.48,69
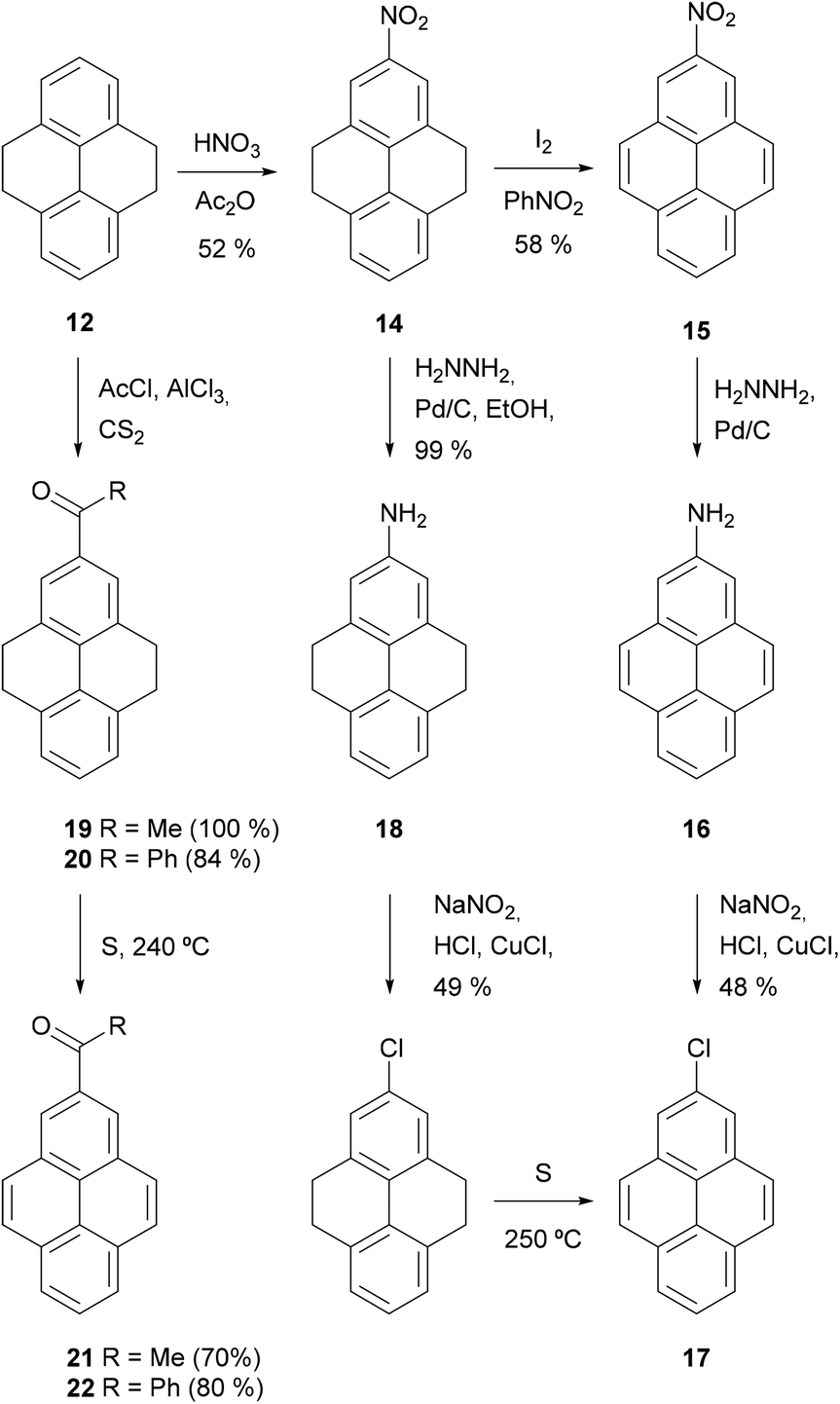
The utility of THPy as a source of 2-substituted pyrenes was first described by R. Bolton in 1964, who prepared 2-nitro- 15, 2-acetyl- 21 and 2-benzoylpyrene 22 for the first time in moderate to good yields after two steps by using this strategy (Scheme 6). Nitro derivative 15 was then used as the starting material for the synthesis of 2-aminopyrene 16 and 2-chloropyrene 17.68
 | ||
| Scheme 6 Bolton's first application of the THPy method.68 | ||
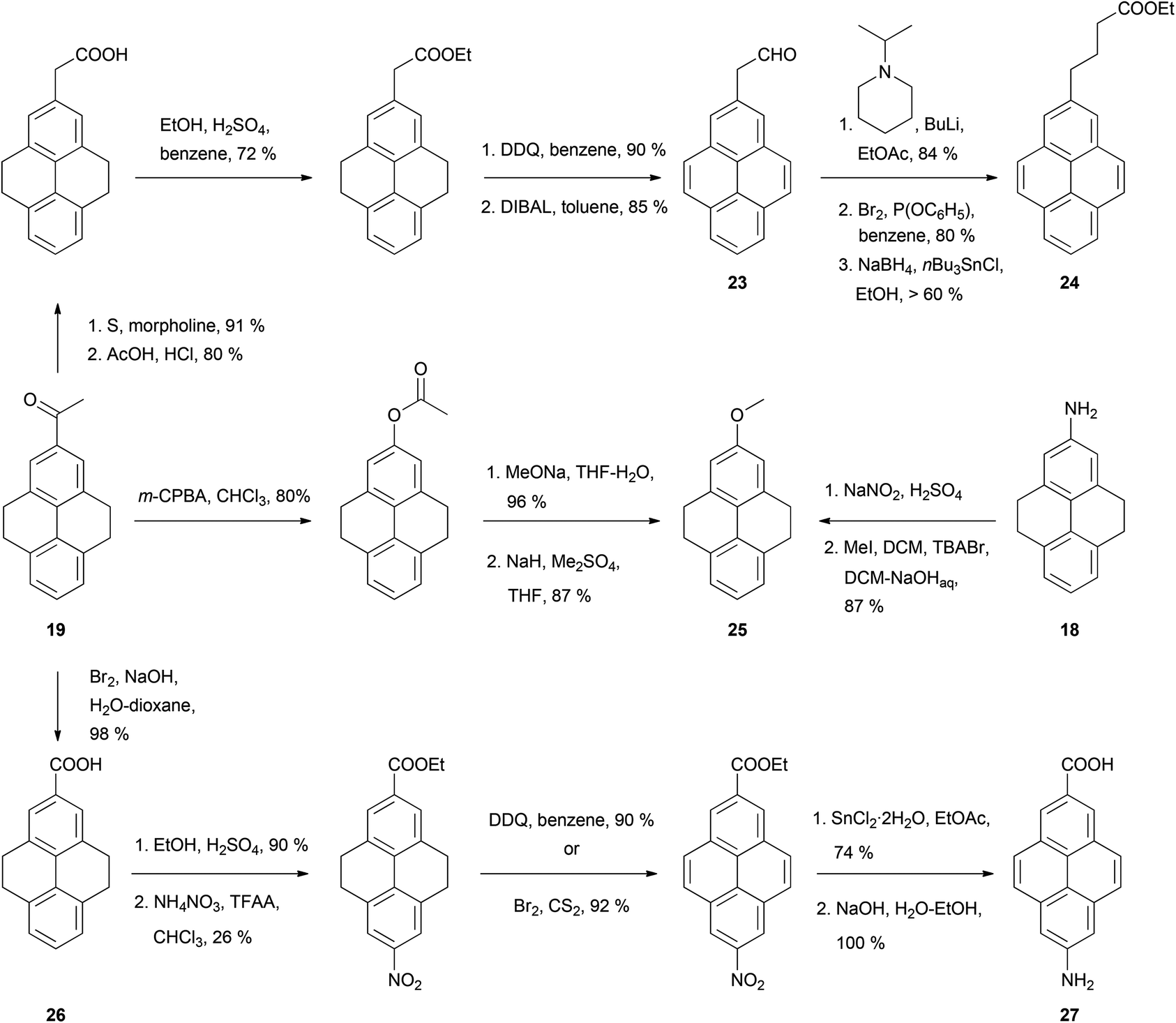
The syntheses of many of these compounds have been revisited and improved by later authors. For example, 2-nitroTHPy 14 has also been prepared by treatment of THPy with dinitrogen tetraoxide,62 or with nitrate salts in acetic or trifluoroacetic anhydride.49,51,54,55,58 The early work of Bolton had a large impact on subsequent synthetic work with pyrene, since some of the compounds initially described by this author have served as intermediates for a variety of subsequent pyrene derivatives (Schemes 7–10).
 | ||
| Scheme 7 Further transformations from amine 18 and ketone 19. | ||
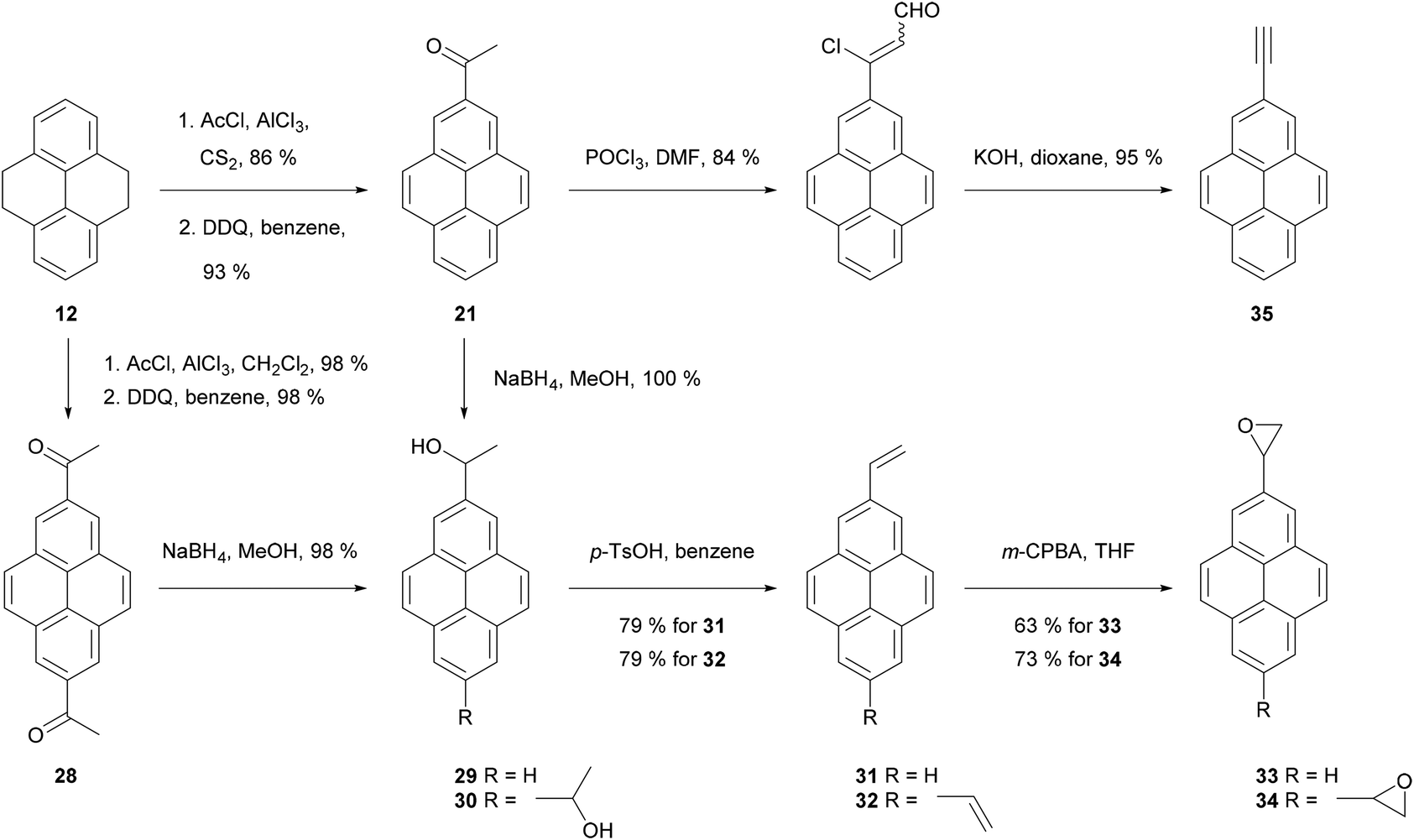 | ||
| Scheme 8 Transformations via 2-acetylpyrene 21 and 2,7-diacetylpyrene 28. | ||
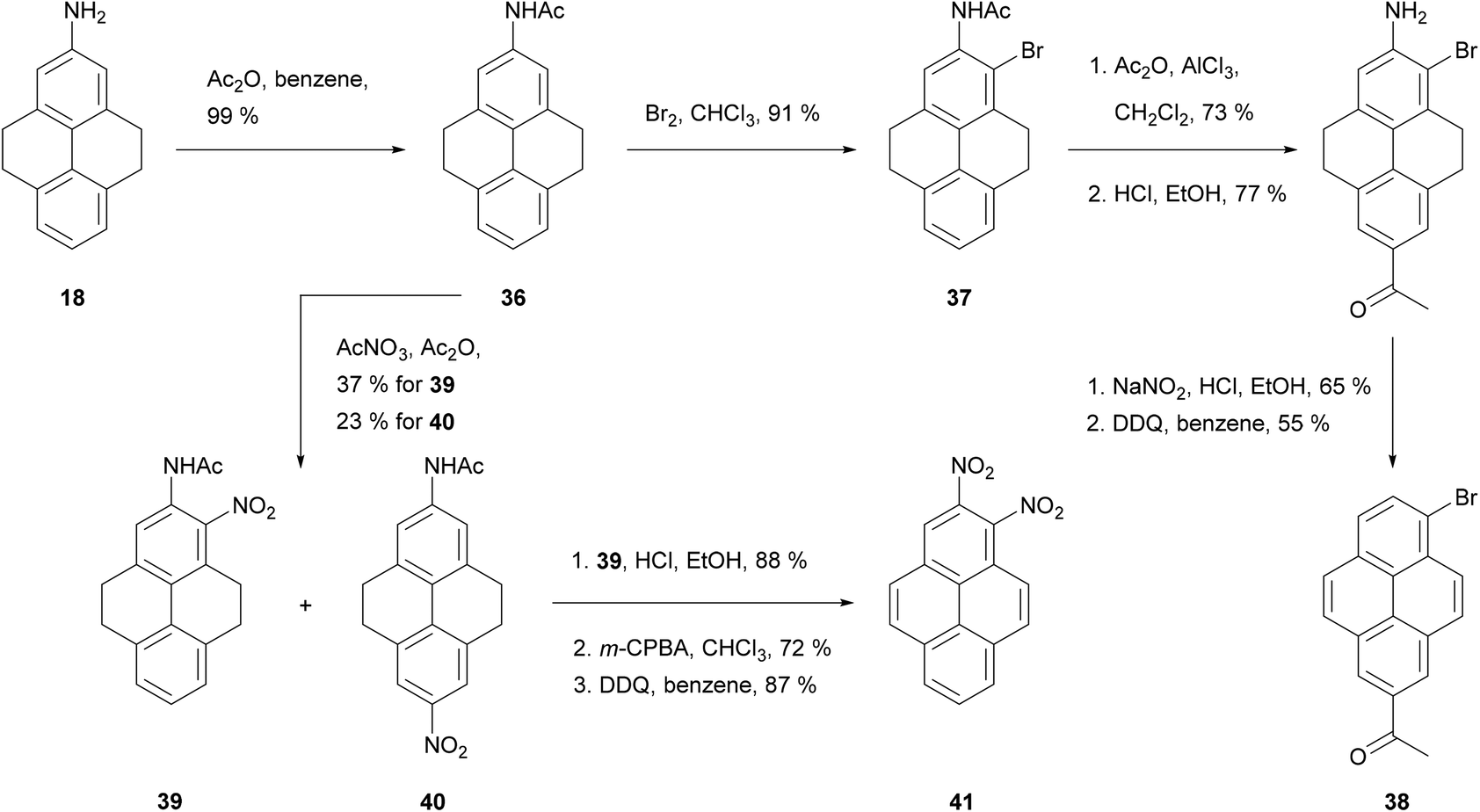 | ||
| Scheme 9 Preparation of 1,2- and 2,6-disubstituted pyrenes via 2-amino-THPy 18. | ||
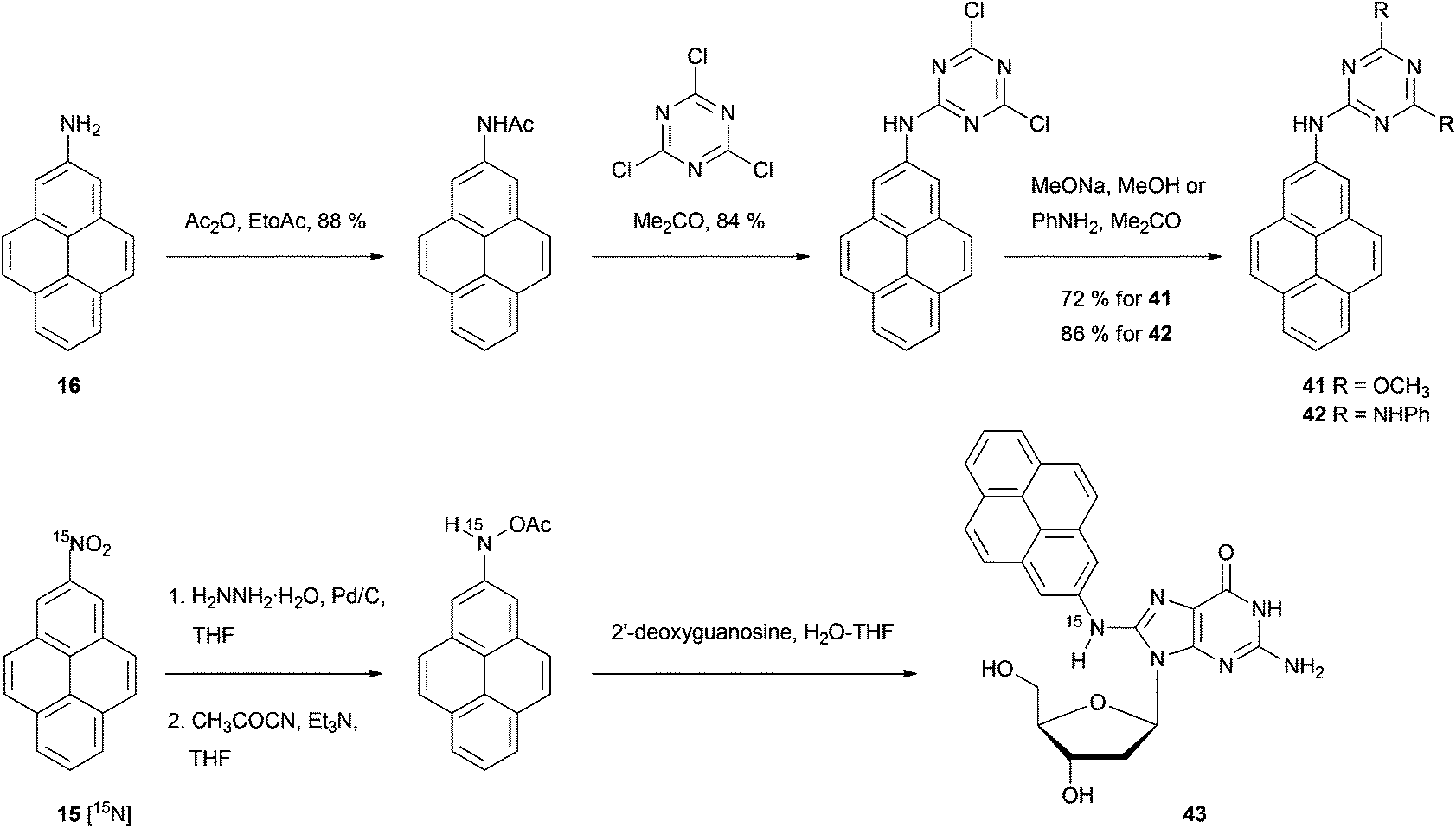 | ||
| Scheme 10 Synthesis of pyrenyltriazine derivatives 41 and 42, and pyrenyl nucleoside 43. | ||
Thus, application of the Willgerodt–Kindler method to 2-acetylTHPy 19 has allowed the preparation of 2-(formylmethyl)pyrene 23 and the butanoate derivative 24 (Scheme 7).61 2-MethoxyTHPy 25 has also been prepared from compound 19 by using Baeyer–Villiger oxidation conditions,47 or from 2-aminoTHPy 18via a diazonium salt.58 Haloform oxidation of 19 leads to the corresponding acid 26, which can be transformed into 7-amino-2-pyrenylcarboxylic acid 27 after five steps.57 Compound 19 has also been nitrated in position 7 to yield 2-acetyl-7-nitroTHPy.70
2-Acetylpyrene 21 can be reduced to the alcohol 29, dehydrated to 2-vinylpyrene 31 then transformed to the corresponding epoxide 33 (Scheme 8). A similar sequence has been performed for 2,7-disubstituted analogues (28 → 30 → 32 → 34).56 Compound 21 has also been transformed into 2-ethynyl derivative 35 using Vilsmeier–Haack–Arnold methodology followed by Bodendorf fragmentation.53 The synthesis of 35 has also been reported via other routes starting from bromopyrene derivatives (see below).
Acetylation of 2-amino-THPy 18 gives acetamide 36 (Scheme 9), which provides a point of entry to rare 1,2- and 2,6-substituted pyrenes (e.g. 2-acetyl-6-bromopyrene 38 and 1,2-dinitropyrene 41). The presence of the acetylamino group in position 2 activates the vicinal ortho position 1, which competes with position 7 depending on the electrophile. Thus, bromination of 36 takes place exclusively in position 1 to give 37, while nitration gives a mixture of 1- and 7-derivatives, 39 and 40 respectively.63
Bolton's synthetic strategy for 2-nitropyrene 15 and 2-aminopyrene 16 has been exploited for the preparation of more elaborate 2-substituted pyrenes (Scheme 10). For example, 2-aminopyrene 16 has been transformed into dimethoxy- and bis-anilino-triazines 41 and 42 in three steps.52 Zhou and Cho synthesised the 15N-labeled analogue of 15 as the starting material for the exotic nucleoside [15N](2′-deoxyguanosin-8-yl)-2-aminopyrene 43.49
THPy overreaction to give 2,7-disubstituted derivatives can be a problem, and conditions need to be tuned in order to avoid it. Disubstitution is sometimes a major outcome even when only one reagent equivalent is used, yielding complex mixtures which can be difficult to separate. This is the case for acetylation (Scheme 11). Harvey et al. have reported that the reaction can be controlled by the solvent: CS2 leads to monoacetylation while CH2Cl2 gives diacetylation,56 although temperature and reagent amount must be carefully controlled in the former case.57 2,7-Diacetyl-THPy 44 has been oxidised to THPy-2,7-dicarboxylic acid using I2 in pyridine or NaOBr in water, and subsequently esterified.50 The resulting ester 45 has been reduced to diol 46 and reacted with PBr3 to give the bis(bromomethyl) derivative 47. The latter has been conjugated with an immobilised cyclic peptide scaffold, creating a synthetic carbohydrate receptor effective in MeCN–water mixtures.25 2,7-Diacetyl-THPy 44 can also be dehydrogenated to yield 2,7-diacetylpyrene 28. Both 44 and 28 have been condensed with 8-amino-7-quinolinecarbaldehyde under Friedländer conditions to prepare the corresponding conjugates 48 and 49 carrying phenanthroline moieties.66
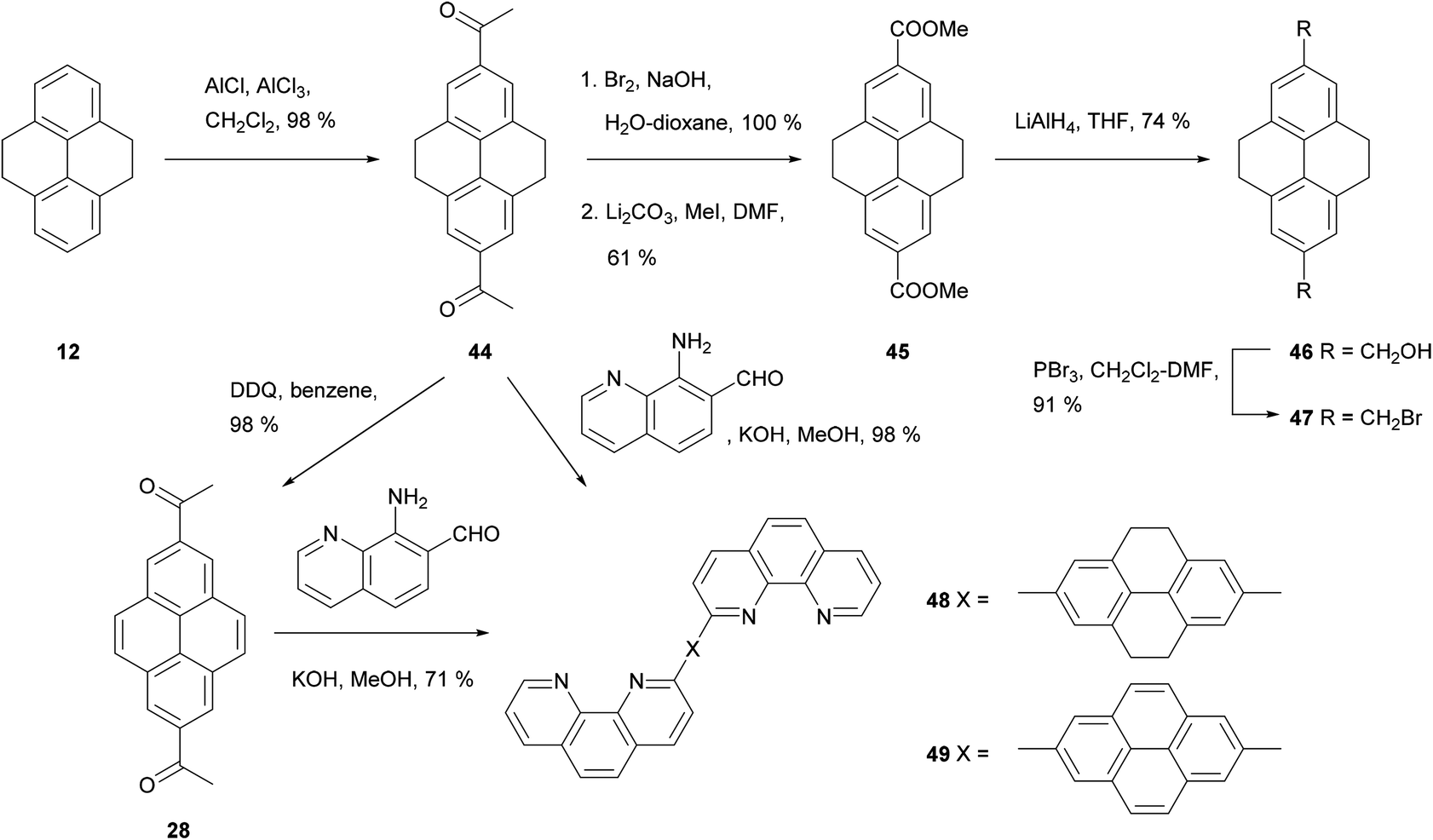 | ||
| Scheme 11 Preparation of 2,7-dibromomethyl-THPy 47 and phenanthroline derivatives 48 and 49. | ||
Disubstitution is also an issue in the case of bromination (Schemes 12 and 13). Treating THPy with bromine in the presence of FeCl3 in an aqueous medium67 or Al powder in chloroform,71 leads mainly to 2,7-dibromo derivative 50, while the monobromo analogue 51 can be formed by reaction of THPy with bromine in DMF for 6 h and then in water overnight. After dehydrogenation of compound 51, the resulting 2-bromopyrene 52 can also be transformed into the phenol 53 by reacting the corresponding Grignard reagent with diborane and alkaline peroxide,69 or into 2-ethynylpyrene 35 with trimethylsilylacetylene followed by desilylation with KOH48 or with 3-methyl-1-butyn-3-ol followed by deprotection with NaH.72 Both 2-bromopyrene 52 and 2-ethynylpyrene 35 have been conjugated with diamidopyridinyl ferrocene derivatives by Pd-catalysed coupling to prepare novel artificial nucleobase receptors 54 and 55.72 More examples of Sonogashira type reactions on bromopyrene derivatives can be found in Scheme 13. Thus, asymmetric modification of the dibromide 50 yielded bromoester 56 and thence alkynylester 57. This was transformed into the symmetric tolan 58 through a second coupling. Cyclotrimerisation of the latter in the presence of Co2(CO)8 finally gave the hexapyrenylbenzene 59.65 A Pd-catalysed coupling reaction was also used to transform 2,7-dibromopyrene 60 into the molecular rotor 63, with two axially positioned ethynyltriptycenes as paddles.73 Compound 60 may also be lithiated through reaction with n-BuLi. Subsequent reaction with CO2 gas gives the corresponding 2,7-dicarboxylic acid derivative 61 in very good yield.71
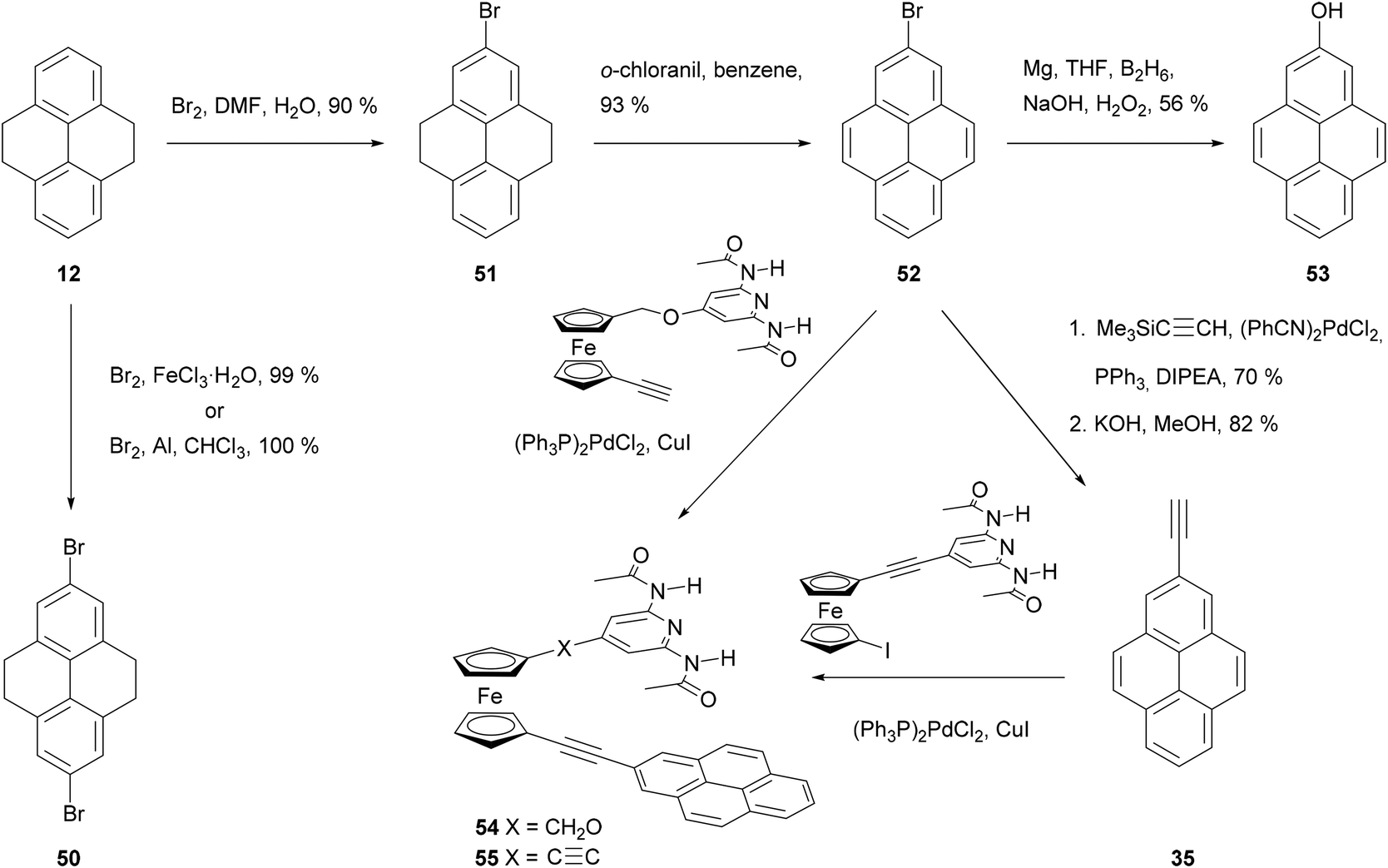 | ||
| Scheme 12 Bromination of THPy 11, and further transformations on mono-bromo derivative 51. | ||
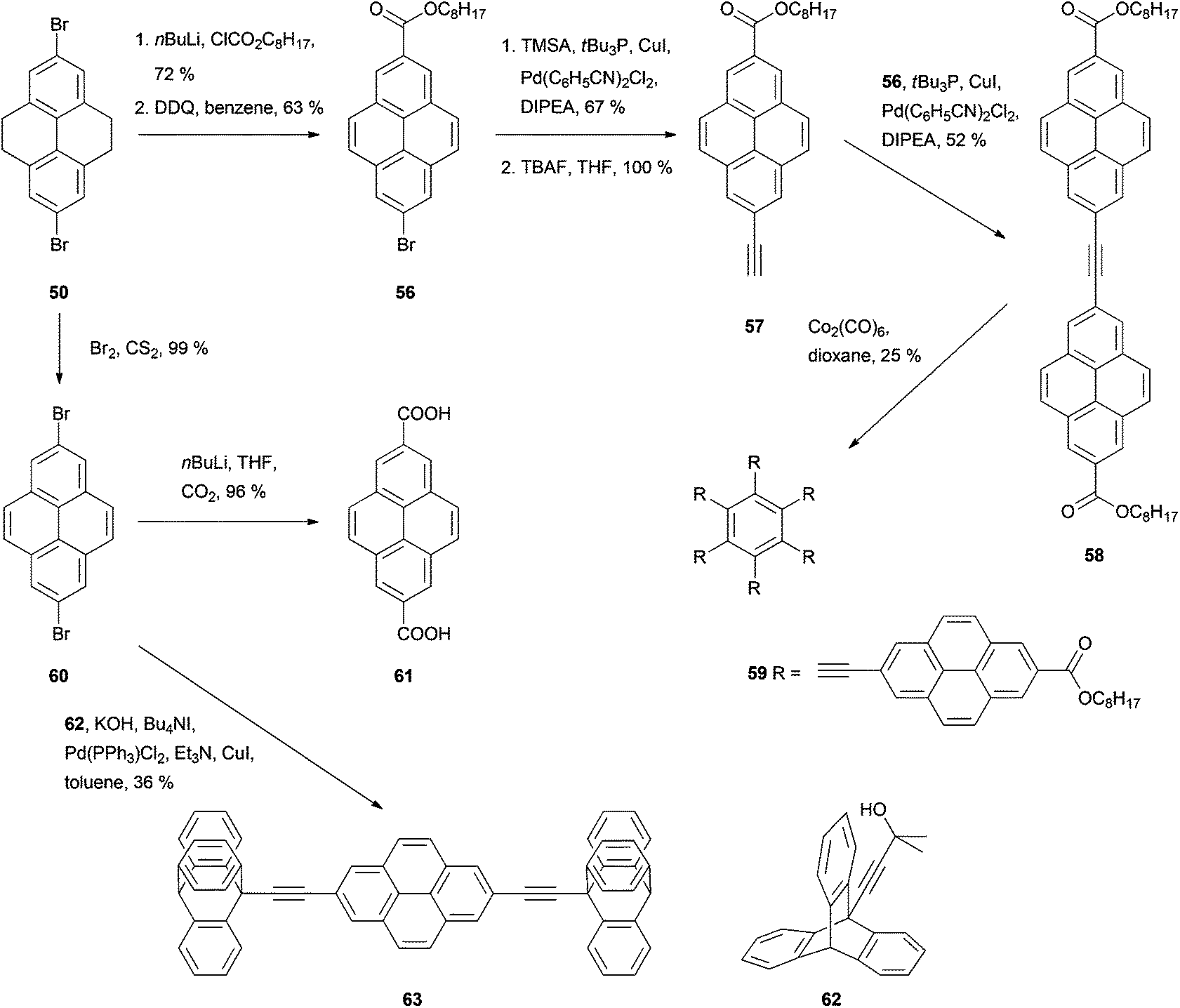 | ||
| Scheme 13 Further transformations of 2,7-dibromoTHPy 50. | ||
Routes to some fluoro- and deutero-pyrenes are shown in Scheme 14. Direct fluorination of THPy to give 64 (and thence 65) was possible only in very low yield.74 Instead, better results were obtained when 2-amino-THPy 18 was transformed into the corresponding diazonium salt and treated with NaBF4 (Scheme 14).58 This reaction has also been performed on the aromatised analogue 2-aminopyrene 16 under Balz–Schiemann conditions using t-BuONO and Et2O·BF3.51 A number of deuterated pyrenes were made via treatment of THPy 12 with P2O5 in D2O to give 66. Aromatisation–bromination–reduction then gave dideuteropyrene 67, while bromination–reduction–aromatisation gave tetradeuteriopyrene 68.64
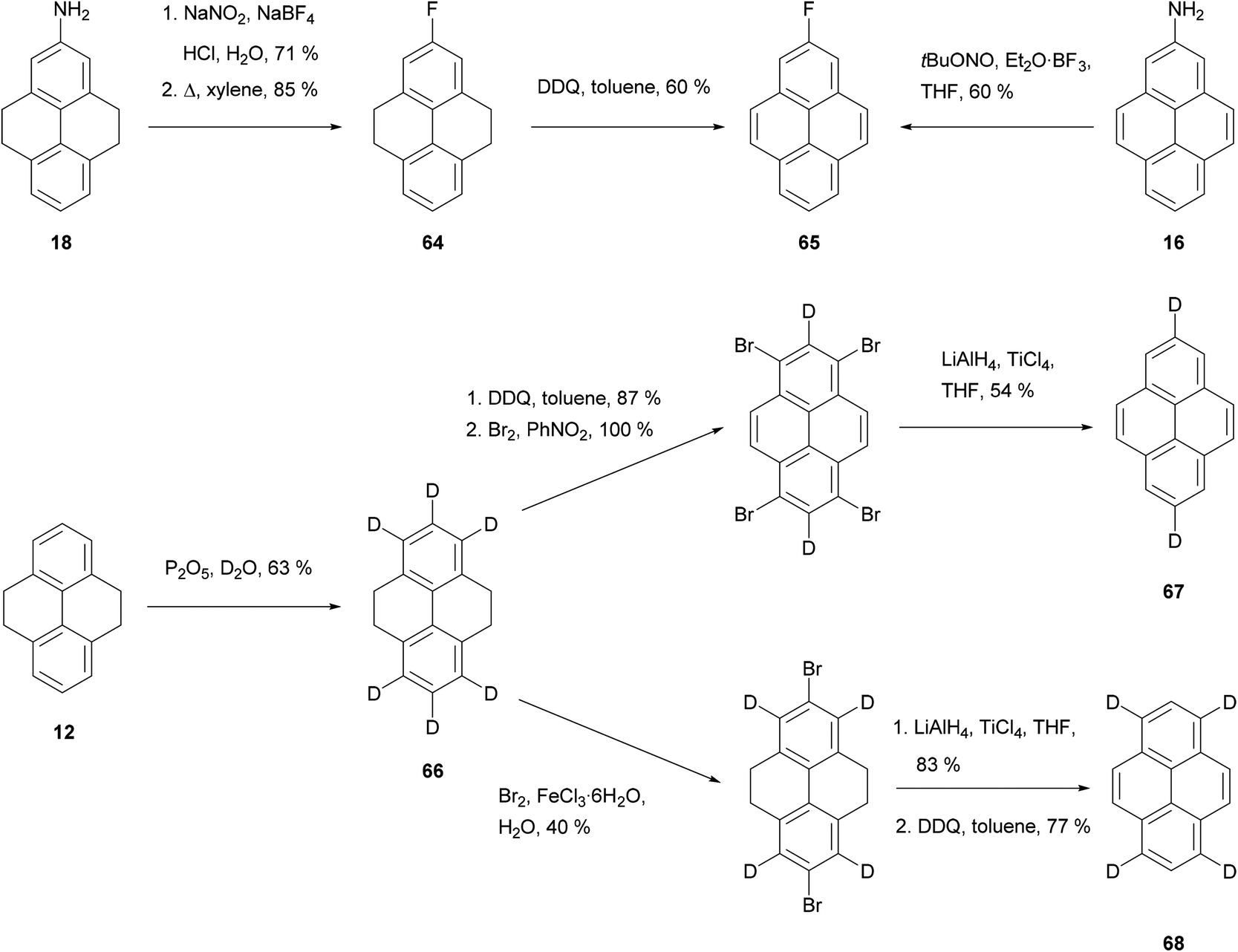 | ||
| Scheme 14 Routes to 2-fluoropyrene 65 and regioselectively deuterated pyrenes. | ||
The THPy strategy also allows the preparation of pyrene derivatives containing a new fused ring involving position 2 (Scheme 15). One approach starts with the direct modification of native pyrene on the reactive position 1. An appendage is introduced which includes a group capable of subsequent electrophilic aromatic substitution, such as an ester or an acid. This derivative is then hydrogenated to the equivalent THPy analogue in order to direct the following cyclisation to the position 2. Finally, re-aromatisation of the molecule yields the desired compound. Depending on the groups present in the new ring after the cyclisation, a careful selection of the dehydrogenation treatment may be needed.59 Lee and Harvey have employed this strategy for the preparation of 4,5,8,9,10,11-hexahydro-7-oxo-7H-cyclopenta[a]pyrene 69, where a 5-membered aliphatic ring is fused to pyrene on positions 1 and 2. Further reactions were later performed to yield 7H- and 9H-cyclopenta[a]pyrenes.60 A second strategy involves attachment of a functionalised chain to position 2 on a THPy derivative. Cyclisation either before or after dehydrogenating the THPy gives a new ring fused to positions 1 and 2. Thus, cyclisation of ethyl 4-(pyren-2-yl)butanoate 24, prepared from 2-acetyl-THPy 19 as shown in Scheme 7, was accomplished with HF. After reduction, dehydration and dehydrogenation of the new ring, benzo[a]pyrene 70 was obtained.61 Benzo[a]pyrene derivatives are compounds of particular interest because they undergo metabolic epoxidation in vivo which transforms them into highly tumorigenic agents capable of covalent conjugation with purine bases. 2-Hydroxybenzo[a]pyrene 71 was prepared in a similar way to that described for 70, although in this case cyclisation was performed before rearomatisation of the THPy core.47 Benzo[a]pyrene derivatives can also be obtained if a four-carbon alkyne chain is attached to positions 6 or 8 on a suitable pyrene derivative. Thus, 2-fluoropyrene 65, obtained from 2-aminopyrene 16 as outlined in Scheme 14, was succinoylated on position 6, after which ketone reduction with HI/P, cyclisation with HF, reduction and dehydration were also performed to yield compound 72. Further transformations yielded epoxydiols 73 and 74, which were studied as electrophiles towards water and dGMP.51
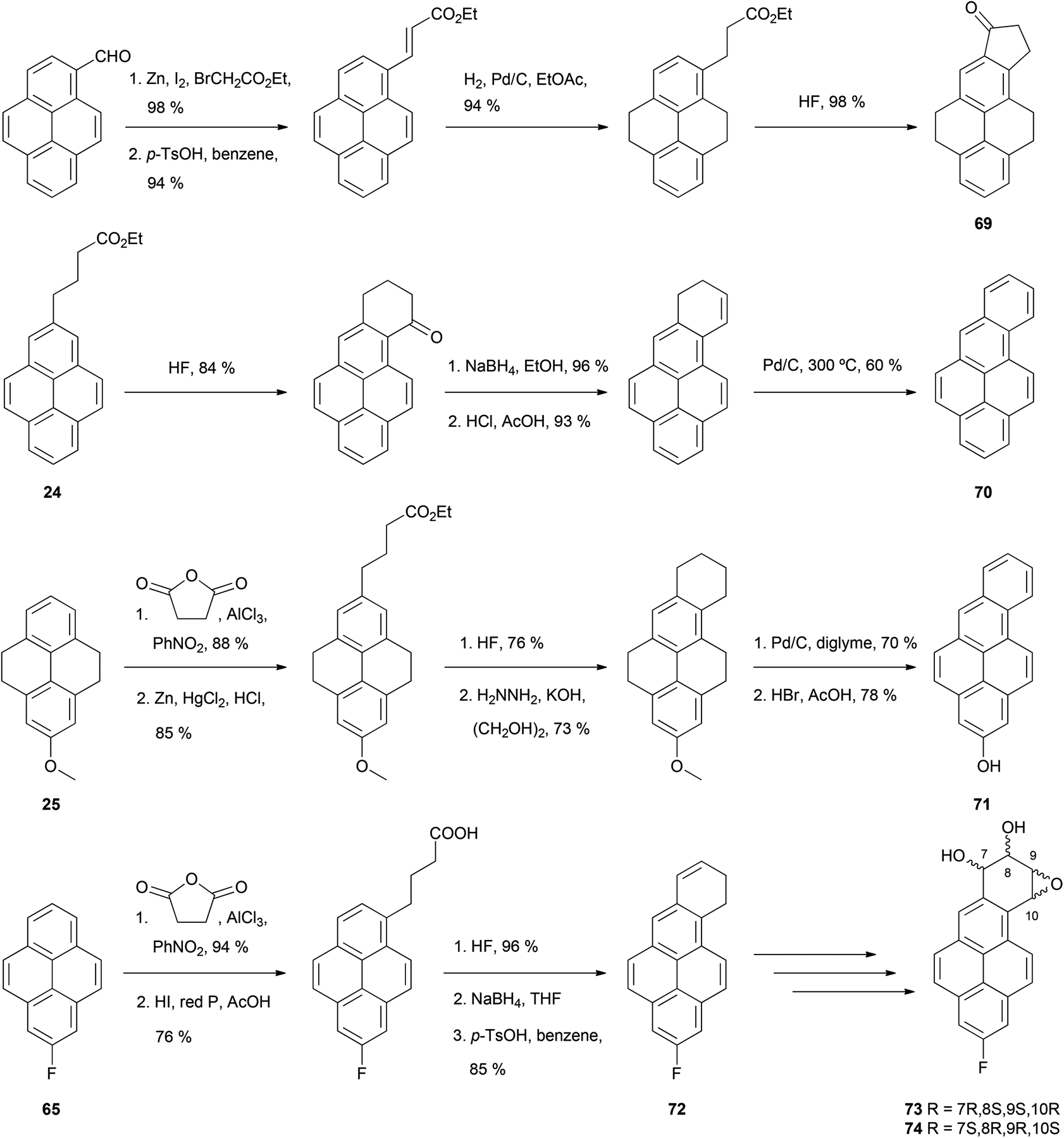 | ||
| Scheme 15 Preparation of cyclopenta[a]pyrene precursor 69 and benzo[a]pyrene derivatives 70–74. | ||
4. The hexahydropyrene (HHPy) method
As discussed in the previous section, the hydrogenation of pyrene 1 gives mainly two compounds, 4,5,9,10-tetrahydropyrene (THPy) 12 and 1,2,3,6,7,8-hexahydropyrene (HHPy) 13. The composition of the reaction mixture depends on the reduction conditions. When using Na in 1-pentanol or isoamyl alcohol the main product of the reduction is HHPy 13, which can be isolated in very pure form by a simple crystallisation from EtOH.44,55,75The reactivity of 13 towards EAS reactions is straightforward, as only one position is available for the first substitution.26 This allows the preparation of pyrene derivatives substituted on their K-region if a subsequent re-aromatisation step with o-chloranil or DDQ is applied (Scheme 16). Thus, 4-bromo-75,76,77 4-acetyl-76,78 and 4-nitropyrene5577 have been prepared in good yields following this synthetic route (Scheme 17). These compounds act as starting materials for further transformations. For example, 4-bromopyrene 75 can be treated with n-BuLi followed by CH3I or ethylene oxide to yield the corresponding 4-alkylpyrenes 78 and 79.77,79,80 Similarly, 4-acetylpyrene 76 has been converted into 4-ethynylpyrene 80 by successive Vilsmeier–Haack–Arnold reaction and Bodendorf fragmentation.53
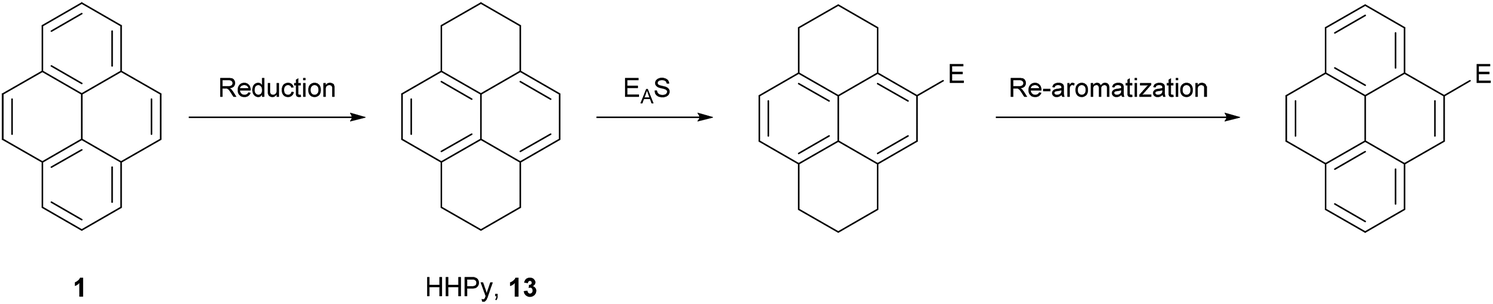 | ||
| Scheme 16 Synthesis of 4-substituted pyrenes via the HHPy method. | ||
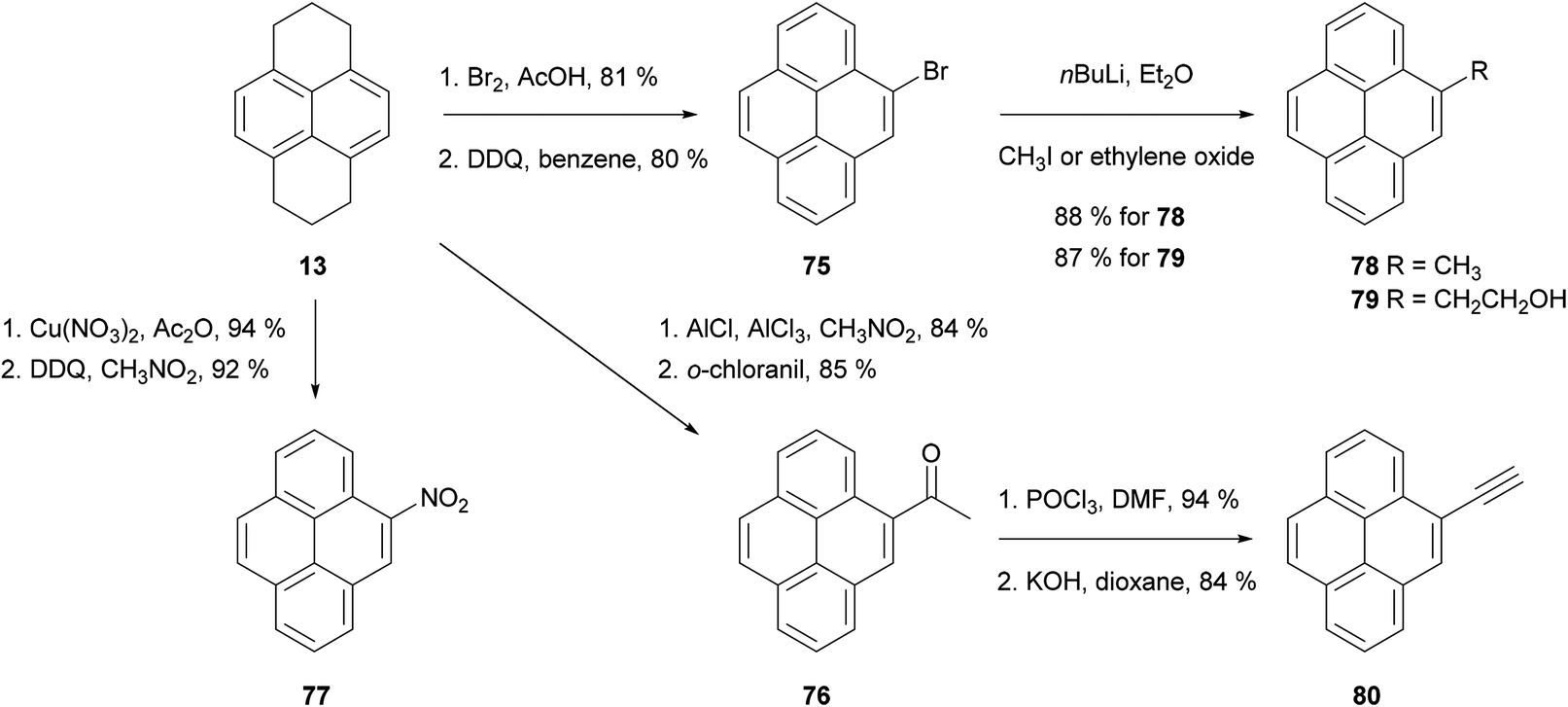 | ||
| Scheme 17 Preparation and further transformations of 4-bromo-, 4-acetyl- and 4-nitropyrene. | ||
The HHPy method has been extended to the preparation of pyrene derivatives bearing fused aromatic rings, which are of interest due to their carcinogenic nature and their presence in polluting fumes such as automobile exhausts. For instance, 1,2,3,6,7,8-hexahydro-γ-oxo-4-pyrenebutanoic acid 81, obtained by the direct Friedel–Crafts alkylation of HHPy, can undergo a Wolff–Kishner decarboxylation followed by cyclisation and oxidation to yield benzo[e]pyrene 82 (Scheme 18).75 Alternatively, compound 81 can be esterified, re-aromatised and converted into diacid 83, the double cyclisation and subsequent oxidation of which gives the methylene-bridged analogue 84.81 Extended heterocyclic pyrene derivatives 85–89, containing thiophene,82 furan83 and pyrrole84 rings fused in positions 4 and 5, have also been prepared starting from 4-bromopyrene 75 and 4-nitropyrene 77, respectively.
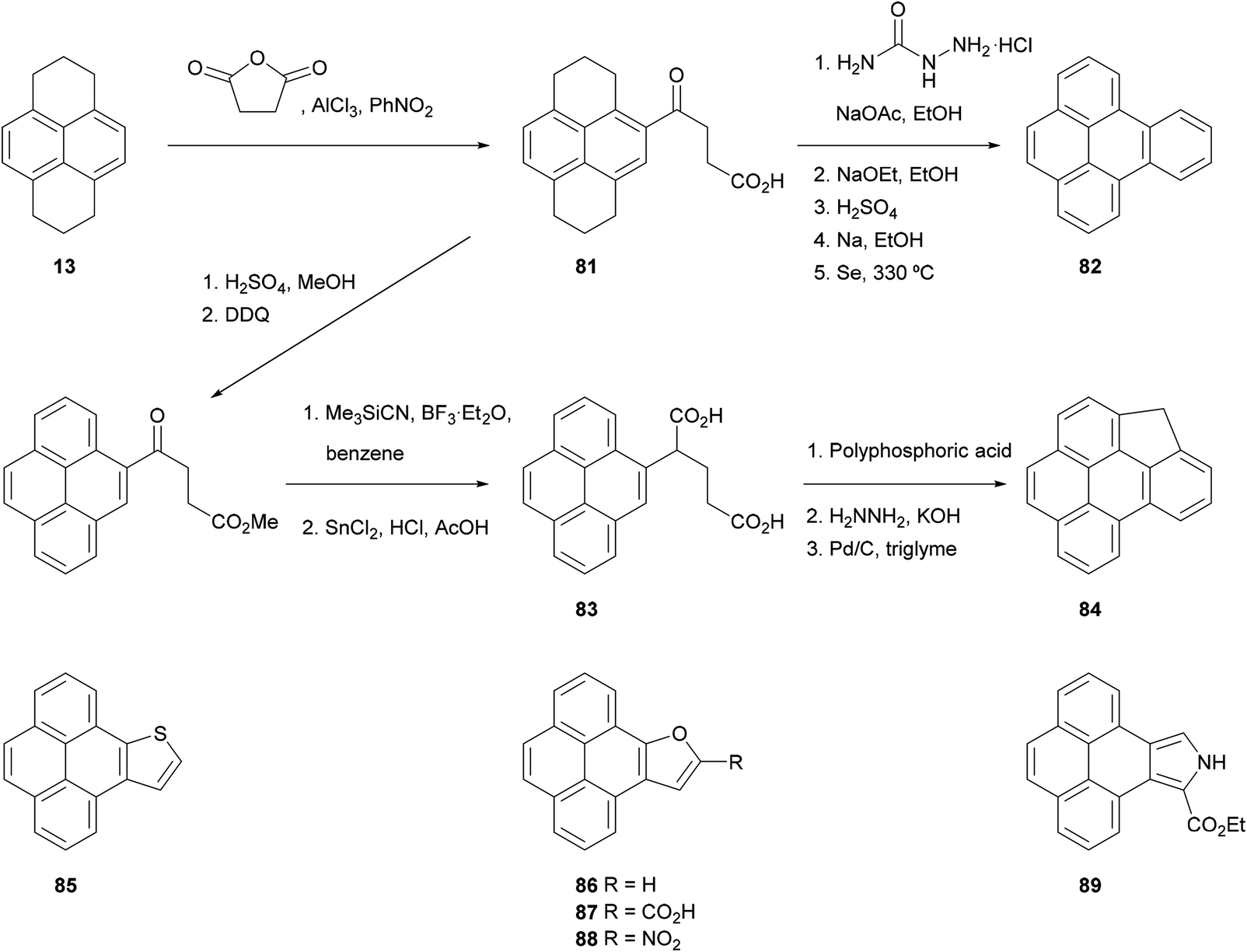 | ||
| Scheme 18 Benzo[e]pyrenes and analogous heterocycles prepared via the HHPy strategy. | ||
Among the fused aromatic ring pyrene analogues reported, cyclopenta[c,d]pyrene (CPP) 90 is probably the one whose synthesis has received the most attention. The key compound for this synthesis is 4-pyrenylacetic acid 91, which has been prepared in several ways starting from HHPy or its derivatives (Scheme 19). For example, HHPy was reacted with methyl (methylsulfinyl)acetate to give 92, rearomatised to 93 then desulfurised with zinc–copper.85 Alternatively, two routes used 4-acetylpyrene 76 as starting material: (i) Willgerodt oxidation to 4-pyrenylthioacetamide, followed by treatment with HCl–AcOH,78 and (ii) oxidation with thallium trinitrate.86 As a further option, 4-methylpyrene 78 was brominated to give 94 and cyanated to give 95 before obtaining the desired acid by simple hydrolysis.79 Finally, 2-(4-pyrenyl)ethanol 79 was oxidised in two steps employing the N-chlorosuccinimide/Me2S method followed by Ag2O.80 CPP 90 has also been prepared following a slightly different strategy by oxidizing HHPy at position 1 and condensing the ketone with the Wittig reagent triethyl phosphonoacetate. The resulting product was subsequently hydrogenated, cyclised, decarboxylated and finally rearomatised (Scheme 20).87
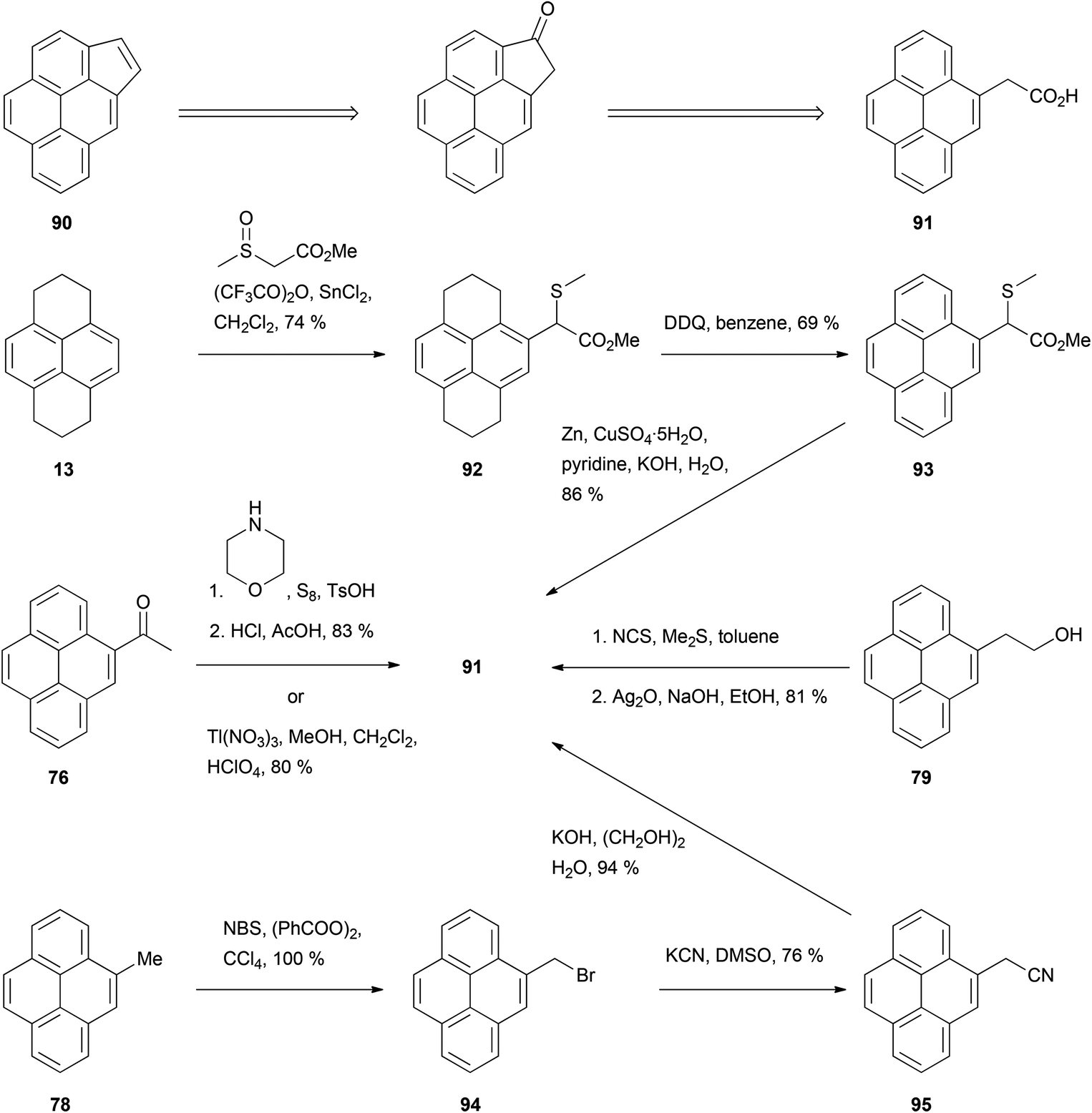 | ||
| Scheme 19 Methods for the preparation of 4-pyrenylacetic acid 91, en route to cyclopenta[c,d]pyrene 90. | ||
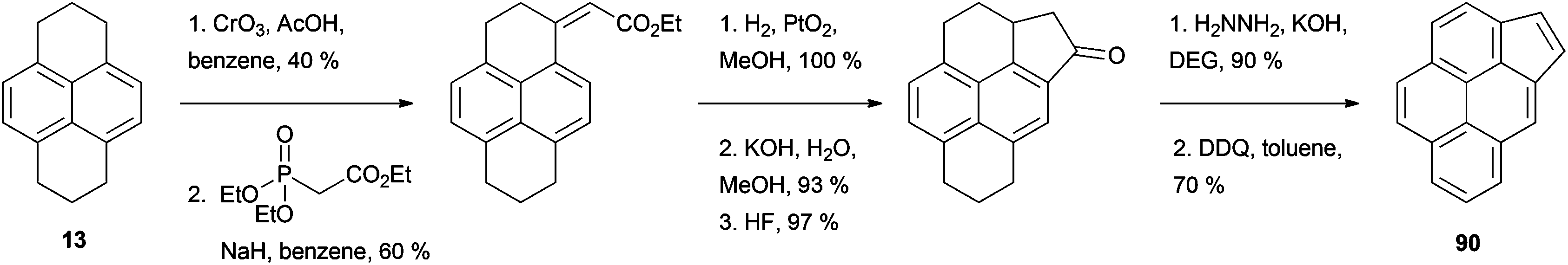 | ||
| Scheme 20 Alternative route to cyclopenta[c,d]pyrene 90. | ||
HHPy may also be dibrominated to give selectively the 4,9-dibromide 96. Recently, this has been used to synthesise the phosphoramidite ligand 97, employed in a gold(I)-catalysed enantioselective cyclisation of allenenes (Scheme 21).88
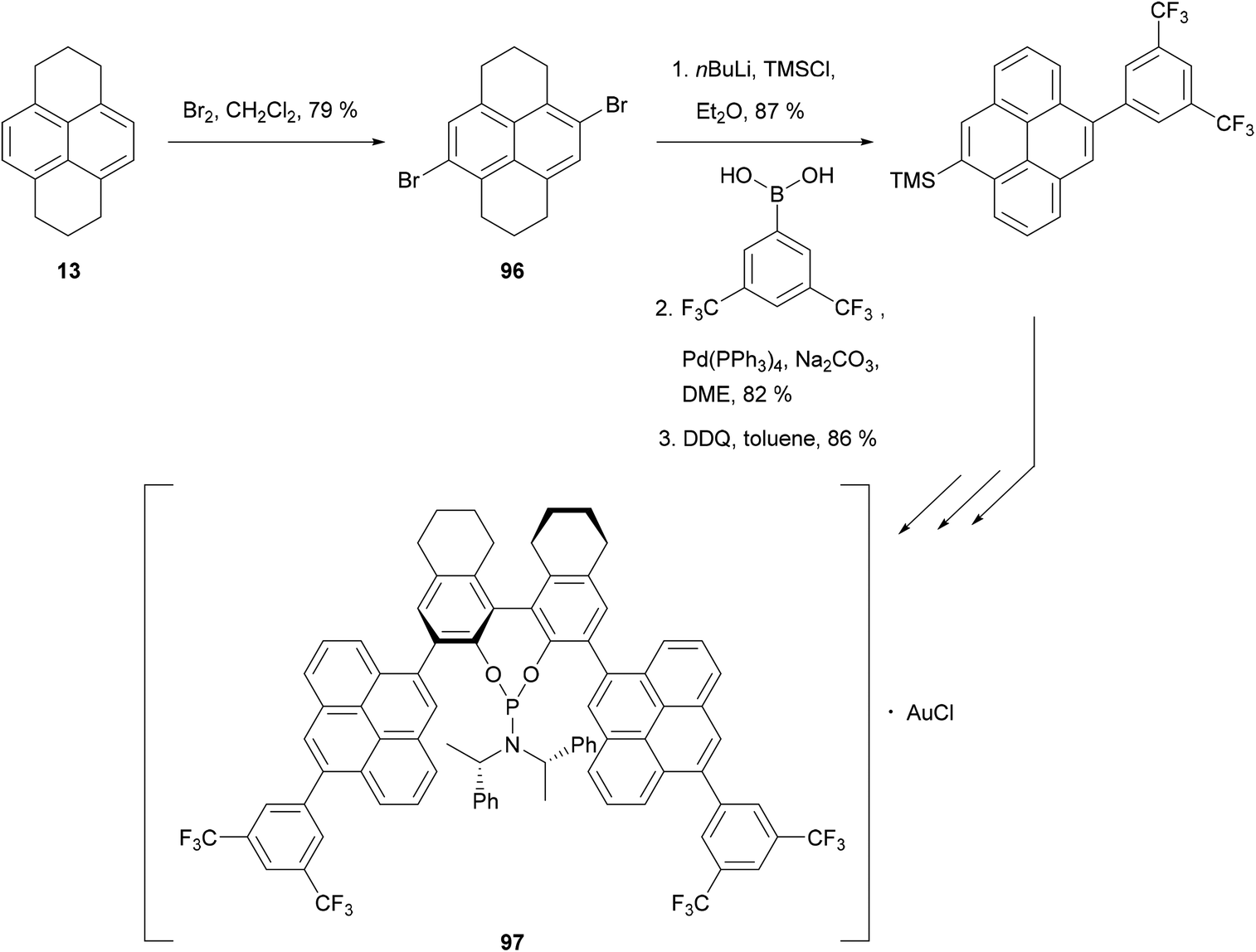 | ||
| Scheme 21 A ligand for gold catalysis incorporating 4,9-disubstituted pyrene units. | ||
5. Transannular ring closure of [2,2]metacyclophanes
It is well known that [2,2]metacyclophanes such as 98 can undergo transannular ring closure to yield products with the 4,5,9,10-tetrahydropyrene (THPy) skeleton (e.g.12),89 which may then be dehydrogenated to pyrenes as discussed in section 3 (Scheme 22). The transannular ring closing reaction can be triggered by the presence of an electrophilic reagent which can also be incorporated into the THPy product. If there is no substituent present on the starting [2,2]metacyclophane derivative, this electrophile is generally incorporated at position 5 (position 2 of the THPy product; for cyclophane numbering, see Scheme 22). Otherwise, other positions can be attacked. For example, when [2,2]metacyclophane 98 is treated with dilute HNO3 in AcOH, 2-nitro-THPy 14 is obtained in 83% (Scheme 23). On the other hand, the 5,13-dimethyl analogue 99 leads to the THPy derivative 100 nitrated in position 1.89 Bromine also effects the cycloisomerisation of 98. Depending on the amount of halogen added, the final product can be 2-bromo-THPy 51 or dibromide 101.90 Interestingly, when the latter reaction is carried out in the presence of iron, no bromination of the final THPy is observed. Pyridinium hydrobromide perbromide (Py·HBr3) also gives the same result.91 Transannular ring closure of 98 to 12, without incorporating a substituent, can be achieved under ultraviolet irradiation in the presence of I2. However this method is not general, and only a low yield (26%) can be achieved for the “dimethyl-analogue” 99 → 102.92 An improved method for the latter transformation employed FeCl3 as oxidant, giving 102 in 98% yield.93 Electrochemical oxidation has also been proposed as a cyclisation method, effective for 98 and 99 as well as the variously substituted derivatives 103–107 (giving 108–112 respectively).94 DDQ was employed for the transannular ring closure of diamino-[2,2]metacyclophanes 113 and 114, giving 115 and 116 respectively.95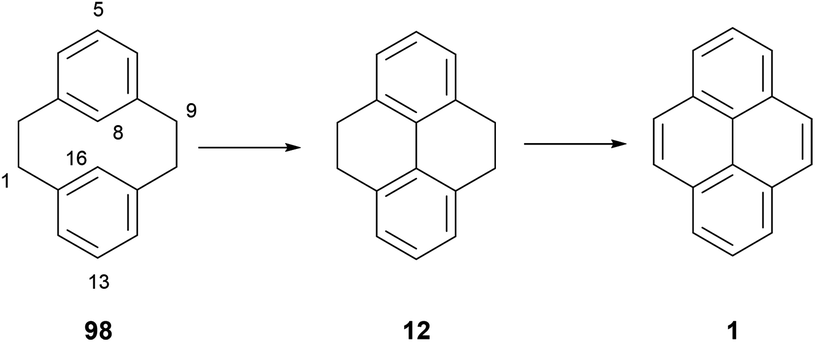 | ||
| Scheme 22 Transannular ring closure of [2,2]metacyclophane to THPy and pyrene. | ||
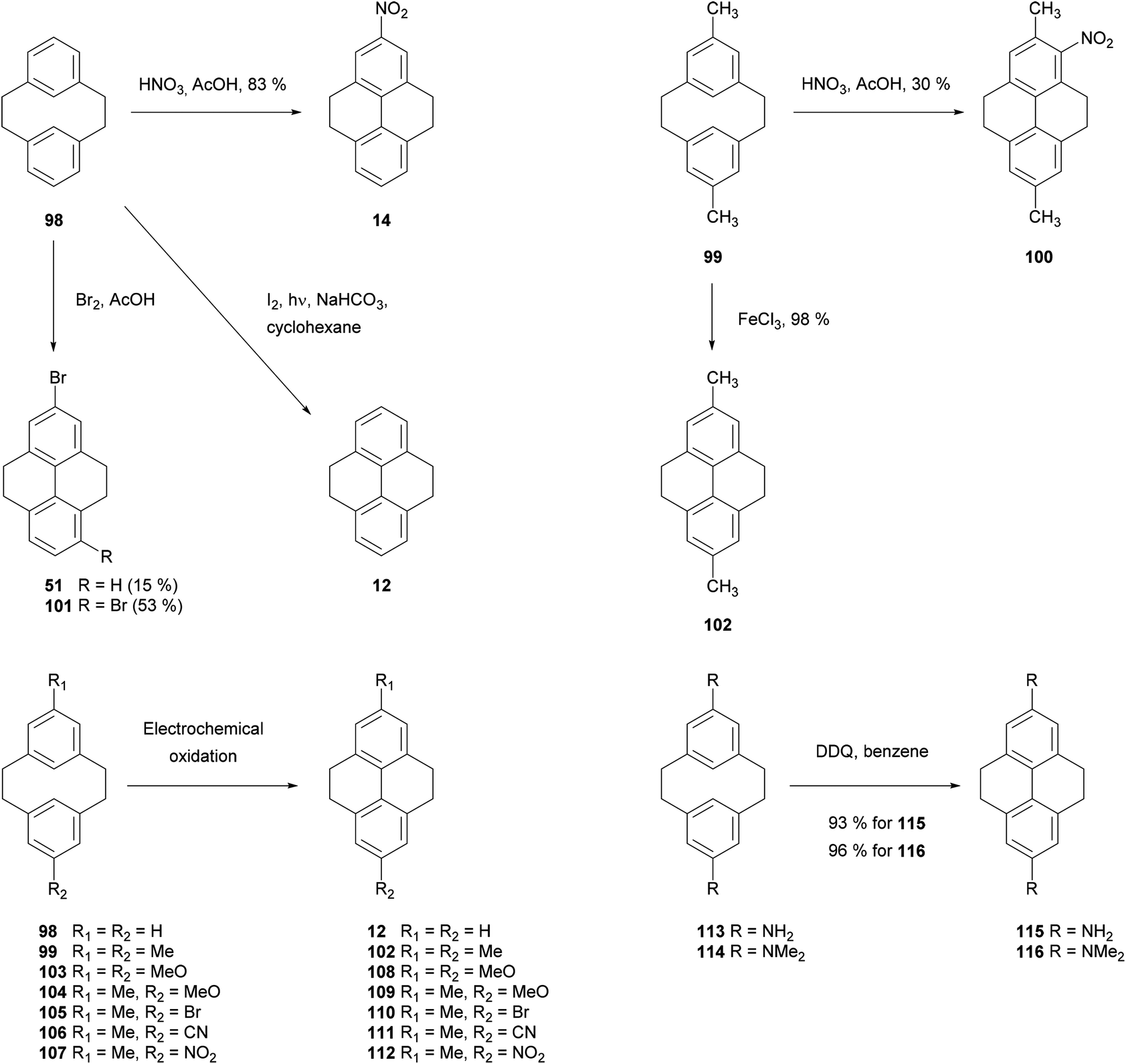 | ||
| Scheme 23 THPy syntheses via [2,2]metacyclophane transannular ring closure. | ||
Transannular ring closing reactions can be achieved even in the presence of substituent groups at carbons 8 and 16 of the [2,2]metacyclophane skeleton (Scheme 24; for numbering see Scheme 22). For instance, when alkyl groups are placed at those positions (as in 117), Br2–Fe or FeBr3 promote THPy formation with alkyl migration to give 2,7-disubstituted products such as 118 and 119. Bromination of some other positions is also observed when using the former reagent.96 Cyclisation can also take place when the group in position 8 is methoxy, although in this case the MeO group is generally lost (e.g.120 → 121, 122 → 123; see Scheme 24).97,98
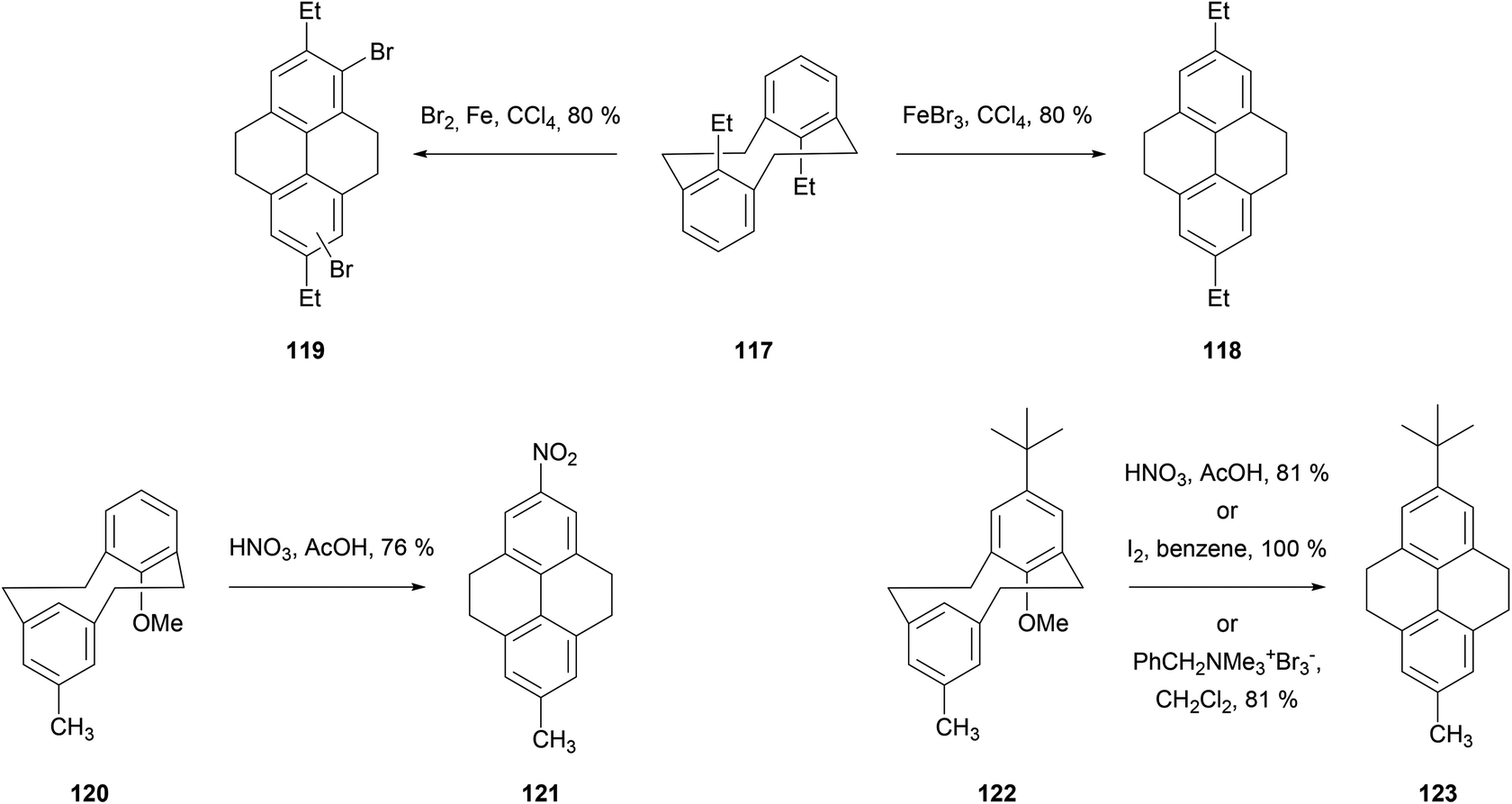 | ||
| Scheme 24 Transannular ring closures of 8,16-disubstituted [2,2]metacyclophanes. | ||
A process closely related to the transannular ring closure of [2,2]metacyclophanes is the valence isomerisation undergone by [2,2]metacyclophane-1,9-dienes such as 124 (Scheme 25). This reaction, which may be thermal or photochemical, transforms the starting material into trans-15,16-dihydropyrenes such as 127. The latter compound is readily converted to pyrene through exposure to UV light or oxygen.99 Halogenated analogues such as 125 and 126 undergo similar ring closures, to 128 and 129 respectively. In these cases aromatisation occurs thermally with migration of a halogen atom to position 1, giving 131 and 132 respectively.100
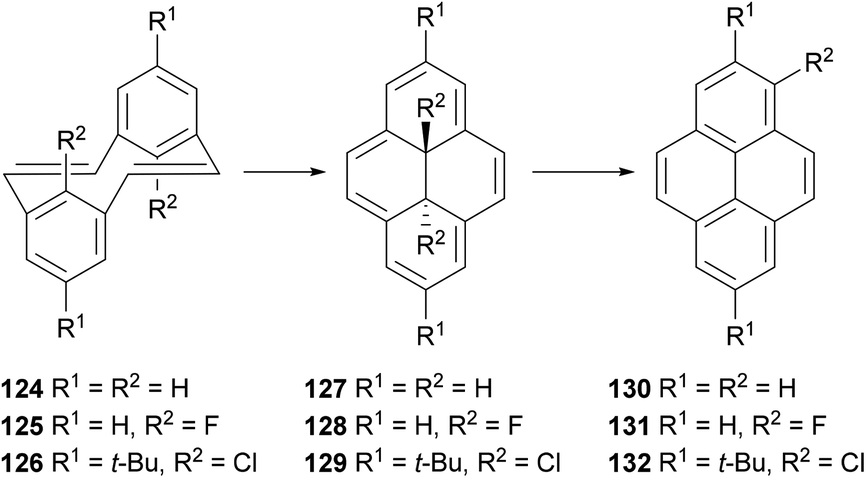 | ||
| Scheme 25 Isomerisation of [2,2]metacyclophane-1,9-dienes to pyrene derivatives. | ||
Although attractive in some respects, these cyclisation methods are limited by the requirement to synthesize the starting macrocycles. Methods for preparing [2,2]metacyclophane and [2,2]metacyclophane-1,9-diene derivatives are not especially convenient. Typically, m-di(bromomethyl)- and m-di(mercaptomethyl)benzenes are combined under high dilution to give dithiamacrocycles, which are then desulfurised by oxidation/pyrolysis94,97,98 or S-methylation/rearrangement/elimination99,100 (Scheme 26). Reagents and conditions tend to be vigorous, and overall yields are often low. Furthermore, in the case of the [2,2]metacyclophane route, the conditions for the transannular ring closure do not seem to be general and require careful selection depending on substitution pattern. In addition, hydropyrenes other than THPy can be formed as by-products, and may even be obtained as the major products under certain conditions.101,102
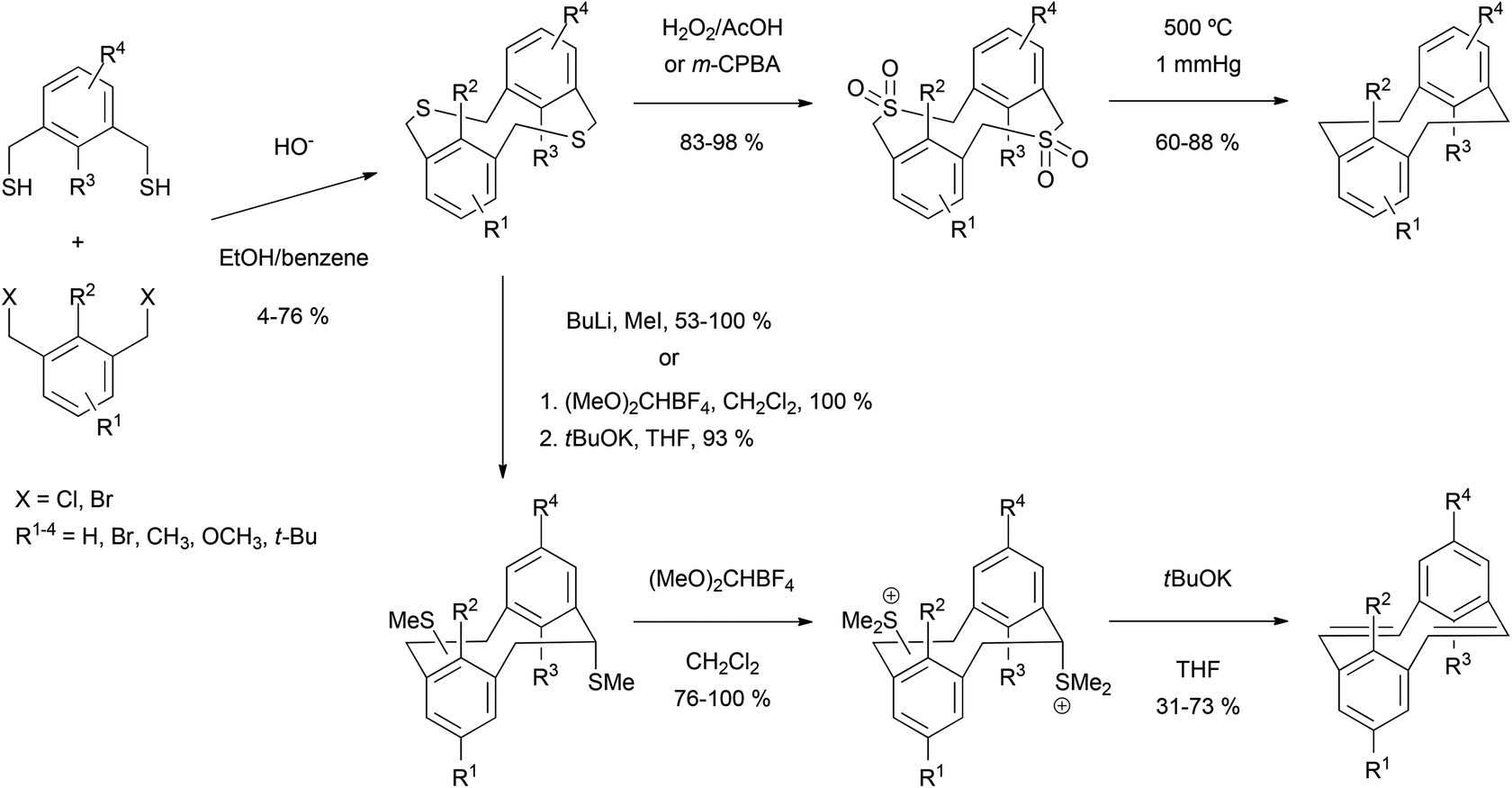 | ||
| Scheme 26 Routes to [2,2]metacyclophane and [2,2]metacyclophane-1,9-diene derivatives. | ||
The cycloisomerisation of [2,2]metacyclophanes has found its main synthetic applications in the preparation of [2,2]pyrenophanes such as 133 (Scheme 27).103–105 Meanwhile, the valence isomerisation/dehydrogenation of [2,2]metacyclophane-1,9-dienes has proved especially useful for the synthesis of pyrenophanes containing strained pyrene moieties, of general form 134. As shown in Scheme 27 the method allows the framework of the pyrenophane to be constructed via unstrained macrocyclic precursors. The aromatisation is delayed until the final step, where it compensates for the strain generated. The method has been used quite extensively; pyrenophanes incorporating a range of tethers including alkyl, ether, phenyl and polyphenyl chains have been reported in the last decade.106–109
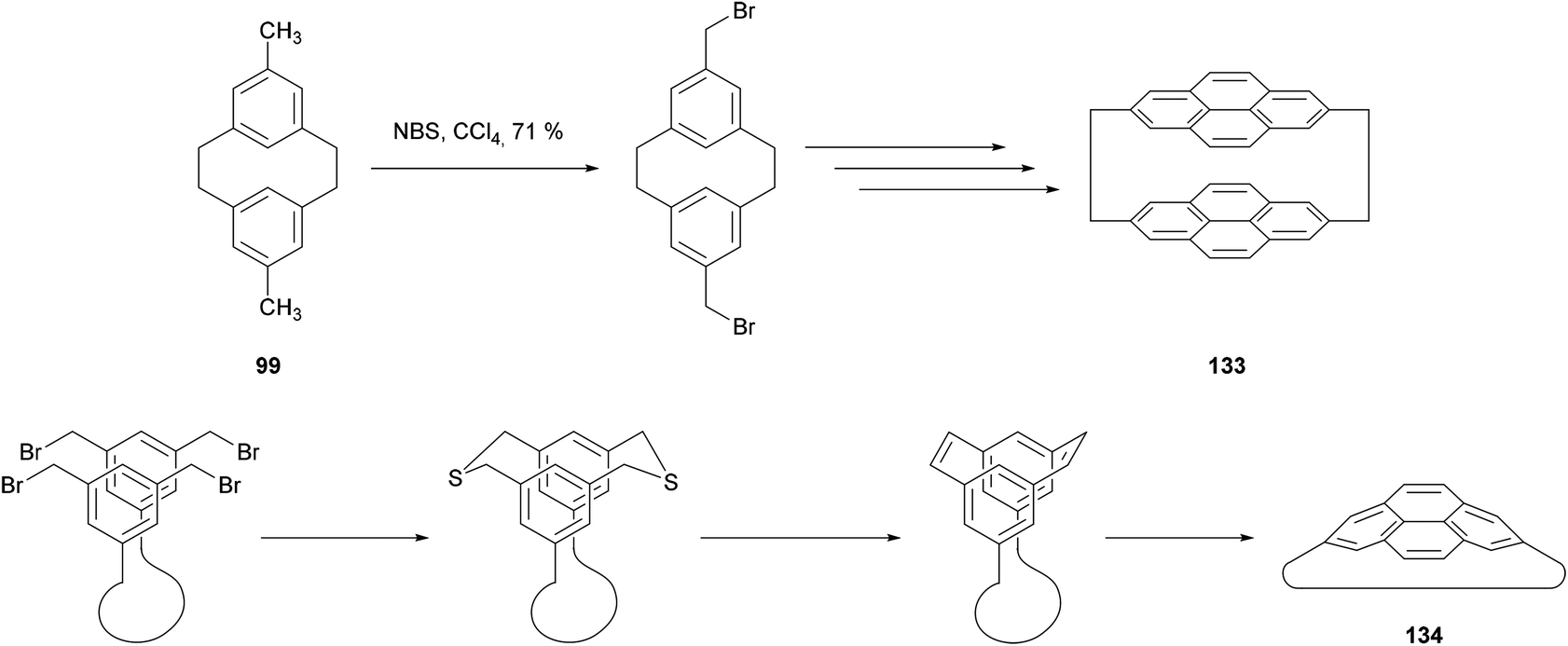 | ||
| Scheme 27 Synthesis of pyrenophanes via [2,2]metacyclophanes. | ||
6. Biphenyl annulations
The construction of pyrenes from biphenyl cores is an attractive option, given that the starting materials are readily available through modern coupling methodology. One possibility is the photocyclisation of 2,2′-divinyl biphenyls 135 as shown in Scheme 28. The method was first demonstrated by Laarhoven for the case of styrenyl biphenyls (where R = Ph).110 Later, the strategy was successfully applied to the synthesis of several 4,9-disubstituted THPy derivatives (Table 2).111,112 The efficiency of the cyclisation was found to be dependent on the substituent on the vinyl group and the conditions used (particularly λ). In particular, it is important to take account of a competing [2 + 2] intramolecular cycloaddition reaction, which can be reversed by using λ = 300 nm or lower.111 A mixture of stereoisomers results at the THPy stage, but if the objective is a pyrene derivative then oxidation of all the products leads to the same aromatic product. Müllen has extended this reaction to the preparation of 4,9-dialkylTHPy derivatives with n-octyl or n-pentyl substituents, yields for which were 80 and 85%, respectively.113,114 The long chain 4,9-dialkylpyrenes obtained by oxidising these products were found to be highly soluble. Although the method has not been used very widely, it seems to be versatile and fairly general, and could probably be extended beyond the simple systems that have been reported so far.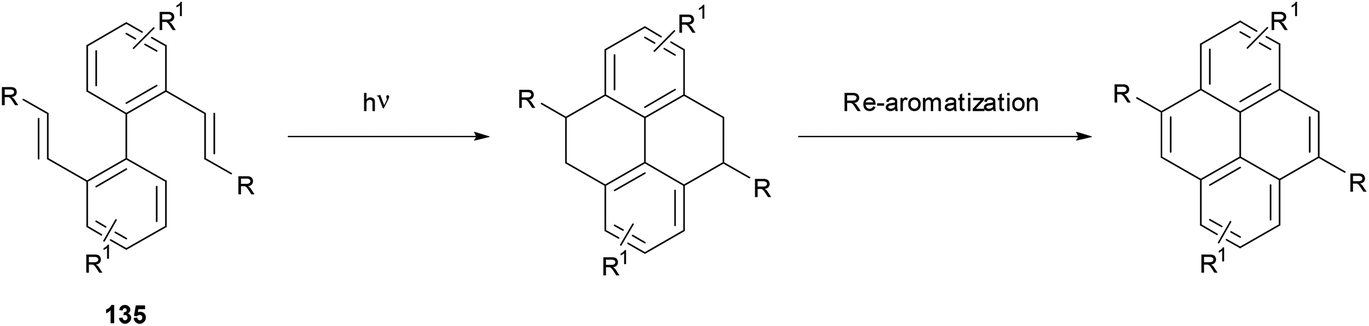 | ||
| Scheme 28 Synthesis of 4,10-disubstituted pyrenes via photocyclisation of divinylbiphenyls. | ||
A second method employing biphenyl starting materials involves the expulsion of sulphur from ortho-thiocarbonyl substituents. The transformation was originally demonstrated in a phenanthrene synthesis due to Wang and Zhang.115 As shown in Scheme 29, biphenyl-based polymeric ketones 136 were treated with thionating agents (either Lawesson's reagent or boron sulfide, formed in situ from [(C6H11)Sn]2S and BCl3) which converted the ketones to the corresponding thiones. Refluxing in trichloroethane was required to effect this transformation, and once the thiones were formed they immediately decomposed with expulsion of molecular sulphur to give phenanthrenes, possibly via four-membered cyclic disulphide units as in 137.
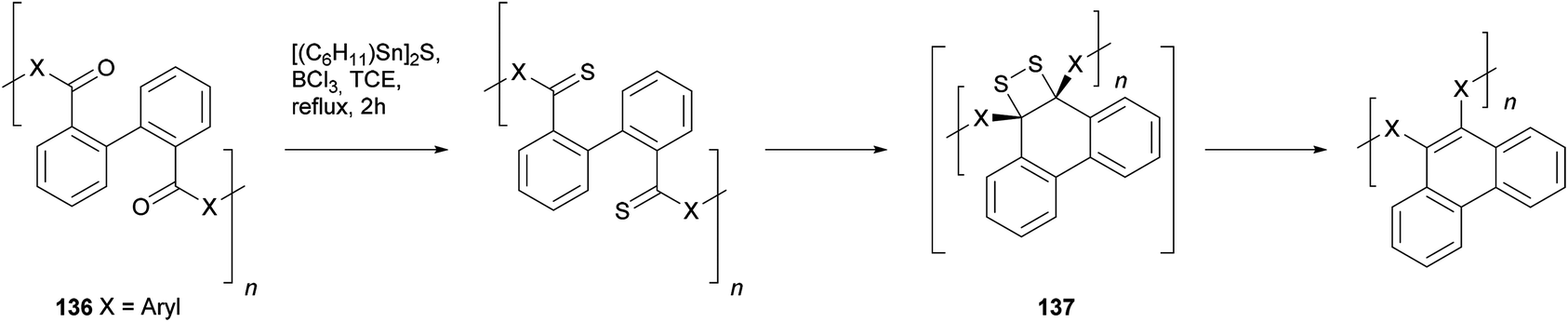 | ||
| Scheme 29 Wang and Zhang's polymeric phenanthrene synthesis via spontaneous thermal annulation of dithione units and subsequent molecular sulphur extrusion. | ||
Clearly there was scope for extending this strategy to pyrene synthesis, as recognised by Müllen, Baumgarten and coworkers (Scheme 30). After constructing the tetraketone precursor 138 these workers applied similar thionating conditions but at room temperature in CH2Cl2. 4,5,9,10-tetraphenylpyrene 140 was formed directly, in 54% yield. This methodology also allowed the preparation of the more complicated derivative 141, which was subsequently polymerised.116
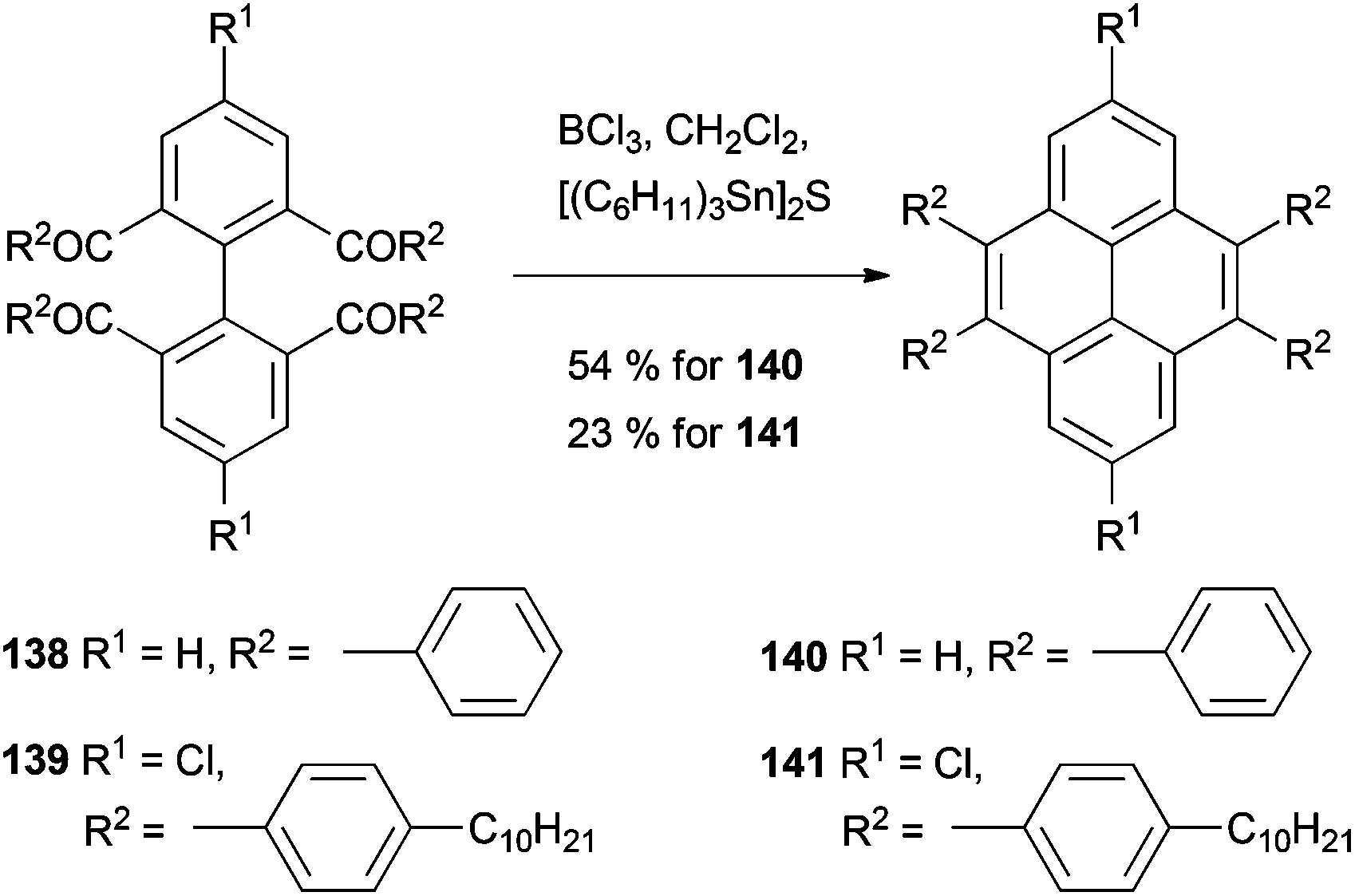 | ||
| Scheme 30 Müllen's synthesis of 4,5,9,10-tetraarylpyrenes by thermal annulation of tetrathiones. | ||
A third annulation approach is based on the cyclisation of alkynyl substituents. This type of reaction has been known for some time as a method for appending rings to aromatic nuclei (Scheme 31). An early example due to Barluenga employed I(py)2+ as an electrophilic initiator (142 → 143),117 and this approach was later extended by Swager118 and Larock119 to make a variety of phenanthrenes (e.g.144 → 145). Meanwhile Fürstner showed that cyclisation could also occur under less acidic conditions, induced by transition metal salts.120 PtCl2 was effective for simple alkynes (146 → 147), while AuCl was employed for iodoalkynes (148 → 149). Surprisingly the latter reaction entailed a 1,2-iodine migration, away from the carbon involved in ring closure.
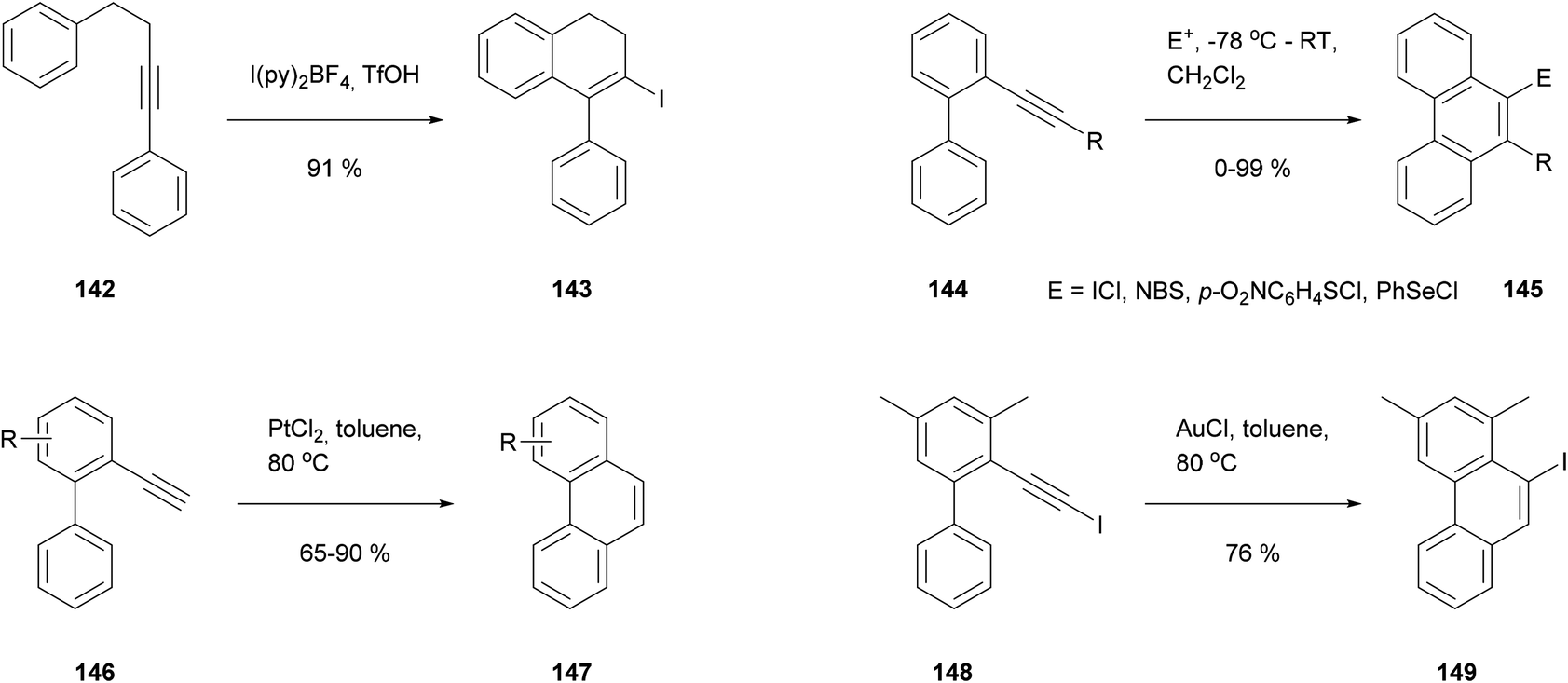 | ||
| Scheme 31 Alkyne cyclisations applied to the synthesis of polycyclic aromatics. | ||
Our group in Bristol saw the opportunity to extend these methods to prepare a wide range of pyrenes. As shown in Scheme 32, the starting materials could be prepared from boronates 150 and iododibromides 151, followed by Sonogashira coupling to give bis-alkynes 152. Bis-cyclisation and further transformations could then give pyrenes 153. In common with other biphenyl-based approaches, the versatility of the initial Suzuki coupling would allow various options for substitution outside the K-region (i.e. R1/R2 in 150–153). The sequence was followed for a number of cases, employing Fürstner's two cyclisation protocols (146 → 147, 148 → 149) for the final steps. Compounds 154–156 serve as examples of highly-substituted pyrenes prepared by this approach.121 Similar methodology has subsequently been employed by other workers to prepare 157–159.122,123 The electrophile-initiated cyclisations (142 → 143, 144 → 145) could probably be used to extend the scope of this approach even further, but as far as we know this has not yet been attempted.124
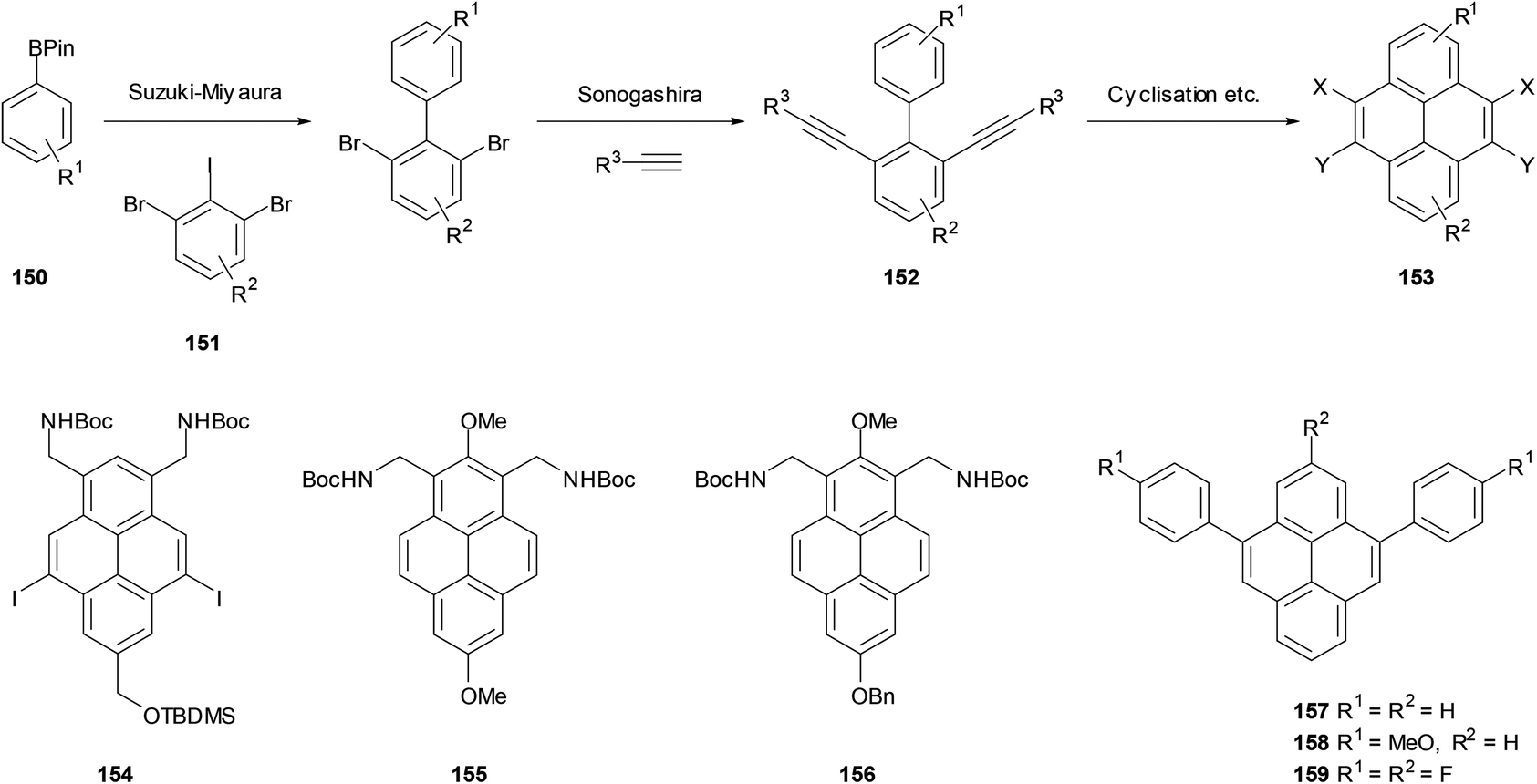 | ||
| Scheme 32 Top: synthesis of pyrenes via cyclisation of bis-alkynylbiphenyls. Bottom: examples of targets prepared using this strategy. | ||
7. Summary and outlook
The indirect methods described in this article provide access to a range of substituted pyrenes which would be difficult or impossible to prepare via direct substitutions. We hope that this compilation will facilitate the use of these methods, and thus encourage the design of new systems which exploit the optical, electronic and structural properties of the pyrene nucleus. There is clearly scope for further methodology development, and we believe the approach of “biphenyl annulation” is especially promising. As progress continues, one may hope that pyrene can play an increasingly important role as both a functional component and scaffold in materials, supramolecular and biological chemistry.Acknowledgements
Financial support from the EPSRC (studentship to J.H.) and the European Commission (Marie Curie Intra-European Fellowship to J.M.C.S.) is gratefully acknowledged. We also thank Dr D. B. Walker for key conceptual and experimental contributions to our work in this area.Notes and references
- T. M. Figueira-Duarte and K. Müllen, Chem. Rev., 2011, 111, 7260–7314 CrossRef CAS PubMed.
- J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452–483 CrossRef CAS PubMed.
- Y. Shirota and H. Kageyama, Chem. Rev., 2007, 107, 953–1010 CrossRef CAS PubMed.
- J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed.
- S. Diring, F. Camerel, B. Donnio, T. Dintzer, S. Toffanin, R. Capelli, M. Muccini and R. Ziessel, J. Am. Chem. Soc., 2009, 131, 18177–18185 CrossRef CAS PubMed.
- For a recent review on sensors and biological probes based on pyrene, see: S. Karuppannan and J.-C. Chambron, Chem.–Asian J., 2011, 6, 964–984 CrossRef CAS PubMed.
- I. B. Berlman, Handbook of Fluorescence Spectra of Aromatic Molecules, Academic Press, New York, 1971 Search PubMed.
- F. M. Winnik, Chem. Rev., 1993, 93, 587–614 CrossRef CAS.
- L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed.
- M. Nishio, Y. Umezawa, K. Honda, S. Tsuboyama and H. Suezawa, CrystEngComm, 2009, 11, 1757–1788 RSC.
- D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105–1136 CrossRef CAS PubMed.
- N. Karousis, N. Tagmatarchis and D. Tasis, Chem. Rev., 2010, 110, 5366–5397 CrossRef CAS PubMed.
- A. Le Goff, K. Gorgy, M. Holzinger, R. Haddad, M. Zimmerman and S. Cosnier, Chem.–Eur. J., 2011, 17, 10216–10221 CrossRef CAS PubMed.
- Y. Chen, B. Zhu, Y. Han and Z. Bo, J. Mater. Chem., 2012, 22, 4927–4931 RSC.
- Y. Xu, H. Bai, G. Lu, C. Li and S. Shi, J. Am. Chem. Soc., 2008, 130, 5856–5857 CrossRef CAS PubMed.
- V. K. Kodali, J. Scrimgeour, S. Kim, J. H. Hankinson, K. M. Carroll, W. A. de Heer, C. Berger and J. E. Curtis, Langmuir, 2011, 27, 863–865 CrossRef CAS PubMed.
- J. A. Mann, J. Rodríguez-López, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2011, 133, 17614–17617 CrossRef CAS PubMed.
- X. F. Wu and G. Q. Shi, J. Mater. Chem., 2005, 15, 1833–1837 RSC.
- J. C. Wu, Y. Zou, C. Y. Li, W. Sicking, I. Piantanida, T. Yi and C. Schmuck, J. Am. Chem. Soc., 2012, 134, 1958–1961 CrossRef CAS PubMed.
- M. Printz and C. Richert, Chem.–Eur. J., 2009, 15, 3390–3402 CrossRef CAS PubMed.
- M. Endo and H. Sugiyama, ChemBioChem, 2009, 10, 2420–2443 CrossRef CAS PubMed.
- I. V. Astakhova, V. A. Korshun and J. Wengel, Chem.–Eur. J., 2008, 14, 11010–11026 CrossRef CAS PubMed.
- M. Inouye, K. Fujimoto, M. Furusyo and H. Nakazumi, J. Am. Chem. Soc., 1999, 121, 1452–1458 CrossRef CAS.
- H. Abe, Y. Mawatari, H. Teraoka, K. Fujimoto and M. Inouye, J. Org. Chem., 2004, 69, 495–504 CrossRef CAS PubMed.
- J. M. Benito and M. Meldal, QSAR Comb. Sci., 2004, 23, 117–129 CAS.
- H. Vollmann, H. Becker, M. Corell and H. Streeck, Liebigs Ann., 1937, 531, 1–159 CrossRef CAS.
- M. J. S. Dewar and R. D. Dennington, J. Am. Chem. Soc., 1989, 111, 3804–3808 CrossRef CAS.
- K. Ogino, S. Iwashima, H. Inokuchi and Y. Harada, Bull. Chem. Soc. Jpn., 1965, 38, 473–477 CrossRef CAS.
- M. Sharif, S. Reimann, K. Wittler, L. R. Knopke, A. E. Surkus, C. Roth, A. Villinger, R. Ludwig and P. Langer, Eur. J. Org. Chem., 2011, 5261–5271 CrossRef CAS.
- T. Yamato, A. Miyazawa and M. Tashiro, J. Chem. Soc., Perkin Trans. 1, 1993, 3127–3137 RSC.
- D. N. Coventry, A. S. Batsanov, A. E. Goeta, J. A. K. Howard, T. B. Marder and R. N. Perutz, Chem. Commun., 2005, 2172–2174 RSC.
- H. M. Colquhoun, Z. X. Zhu, D. J. Williams, M. G. B. Drew, C. J. Cardin, Y. Gan, A. G. Crawford and T. B. Marder, Chem.–Eur. J., 2010, 16, 907–918 CrossRef CAS PubMed.
- A. G. Crawford, Z. Liu, I. A. I. Mkhalid, M.-H. Thibault, N. Schwarz, G. Alcaraz, A. Steffen, J. C. Collings, A. S. Batsanov, J. A. K. Howard and T. B. Marder, Chem.–Eur. J., 2012, 18, 5022–5035 CrossRef CAS PubMed.
- H. Chen, X. Hu and S.-C. Ng, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5562–5569 CrossRef CAS.
- Y. Qiao, J. Zhang, W. Xu and D. Zhu, Tetrahedron, 2011, 67, 3395–3405 CrossRef CAS.
- O. P. Lee, A. T. Yiu, P. M. Beaujuge, C. H. Woo, T. W. Holcombe, J. E. Millstone, J. D. Douglas, M. S. Chen and J. M. J. Fréchet, Adv. Mater., 2011, 23, 5359–5363 CrossRef CAS PubMed.
- Z. Liu, Y. Wang, Y. Chen, J. Liu, Q. Fang, C. Kleeberg and T. B. Marder, J. Org. Chem., 2012, 77, 7124–7128 CrossRef CAS PubMed.
- J. A. Morley and N. F. Woolsey, J. Org. Chem., 1992, 57, 6487–6495 CrossRef CAS.
- Benzenoid polycyclic aromatic hydrocarbons can be visualised as a group of benzene rings annulated to each other through exposed outer π-bonds called the K-region. Since their π-electrons cannot take part in the benzene sextet π system, these bonds show a considerable degree of double bond character ( R. G. Harvey, Polycyclic Aromatic Hydrocarbons, Wiley-VCH, New York, 1997). In the case of pyrene, K-region denotes 4,5- and 9,10-bonds Search PubMed.
- J. Hu, D. Zhang and F. W. Harris, J. Org. Chem., 2005, 70, 707–708 CrossRef CAS PubMed.
- L. Zöphel, V. Enkelmann, R. Riege and K. Müllen, Org. Lett., 2011, 13, 4506–4509 CrossRef PubMed.
- L. Zöphel, D. Beckmann, V. Enkelmann, D. Chercka, R. Rieger and K. Müllen, Chem. Commun., 2011, 6960–6962 RSC.
- K. Mochida, K. Kawasumi, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2011, 113, 10716–10719 CrossRef PubMed.
- E. A. Coulson, J. Chem. Soc., 1937, 1298–1305 RSC.
- P. P. Fu and R. G. Harvey, Tetrahedron Lett., 1977, 18, 415–418 CrossRef.
- P. P. Fu, H. M. Lee and R. G. Harvey, J. Org. Chem., 1980, 45, 2797–2803 CrossRef CAS.
- M. Bukowska and R. G. Harvey, Polycyclic Aromat. Compd., 1992, 2, 223–228 CrossRef CAS.
- M. Foroozesh, G. Primrose, Z. Guo, L. C. Bell, W. L. Alworth and F. P. Guengerich, Chem. Res. Toxicol., 1997, 10, 91–102 CrossRef CAS PubMed.
- L. Zhou and B. P. Cho, Chem. Res. Toxicol., 1998, 11, 35–43 CrossRef CAS PubMed.
- D. M. Connor, S. D. Allen, D. M. Collard, C. L. Liotta and D. A. Schiraldi, J. Org. Chem., 1999, 64, 6888–6890 CrossRef CAS PubMed.
- T. Yang, Y. Huang and B. P. Cho, Chem. Res. Toxicol., 2006, 19, 242–254 CrossRef CAS PubMed.
- P. Šoustek, M. Michl, N. Almonasy, O. Machalický, M. Dvořák and A. Lyčka, Dyes Pigm., 2008, 78, 139–147 CrossRef.
- V. V. Filichev, I. V. Astakhova, A. D. Malakhov, V. A. Korshun and E. B. Pedersen, Chem.–Eur. J., 2008, 14, 9968–9980 CrossRef CAS PubMed.
- E. F. Plaza-Medina, W. Rodríguez-Córdoba and J. Peon, J. Phys. Chem. A, 2011, 115, 9782–9789 CrossRef CAS PubMed.
- A. M. van der Braken-van Leersum, C. Tintel, M. van't Zelfde, J. Cornelisse and J. Lugtenburg, Recl. Trav. Chim. Pays-Bas, 1987, 106, 120–128 CrossRef.
- R. G. Harvey, M. Konieczny and J. Pataki, J. Org. Chem., 1983, 48, 2930–2932 CrossRef CAS.
- A. Musa, B. Sridharan, H. Lee and D. L. Mattern, J. Org. Chem., 1996, 61, 5481–5484 CrossRef CAS.
- L. Rodenburg, M. Floor, A. Lefeber, J. Cornelisse and J. Lugtenburg, Recl. Trav. Chim. Pays-Bas, 1988, 107, 1–8 CrossRef CAS.
- For a general review on dehydrogenation of polycyclic hydroaromatic compounds, see: P. P. Fu and R. G. Harvey, Chem. Rev., 1978, 78, 317–361 CrossRef CAS.
- H. Lee and R. G. Harvey, J. Org. Chem., 1982, 47, 4364–4367 CrossRef CAS.
- S. E. Klassen, G. H. Daub and D. L. Van der Jagt, J. Org. Chem., 1983, 48, 4361–4366 CrossRef CAS.
- D. W. Miller, D. Herrero-Saenz, K. H. Huang, T. M. Heinze and P. P. Fu, J. Org. Chem., 1992, 57, 3746–3748 CrossRef CAS.
- M. Minabe, H. Mochizuki, M. Yoshida and T. Toda, Bull. Chem. Soc. Jpn., 1989, 62, 68–72 CrossRef CAS.
- O. Hammerich, M. F. Nielsen, H. Zuilhof, P. P. J. Mulder, G. Lodder, R. C. Reiter, D. E. Kage, C. V. Rice and C. D. Stevenson, J. Phys. Chem., 1996, 100, 3454–3462 CrossRef CAS.
- D. Rausch and C. Lambert, Org. Lett., 2006, 8, 5037–5040 CrossRef CAS PubMed.
- L. Chouai, F. Wu, Y. Jang and R. R. Thummel, Eur. J. Inorg. Chem., 2003, 2774–2782 CrossRef CAS.
- H. Lee and R. G. Harvey, J. Org. Chem., 1986, 51, 2847–2848 CrossRef CAS.
- R. Bolton, J. Chem. Soc., 1964, 4637–4638 CAS.
- R. G. Harvey, S. Schmolka, C. Cortez and H. Lee, Synth. Commun., 1988, 18, 2207–2209 CrossRef CAS.
- K. K. Laali, M. A. Arrica, T. Okazaki and S. D. Bunge, Eur. J. Org. Chem., 2008, 6093–6105 CrossRef CAS.
- H. Dang, T. Maris, J.-H. Yi, F. Rosei, A. Nanci and J. D. Wuest, Langmuir, 2007, 23, 11980–11985 CrossRef CAS PubMed.
- M. Inouye, Y. Hyodo and H. Nakazumi, J. Org. Chem., 1999, 64, 2704–2710 CrossRef CAS PubMed.
- C. E. Godinez, G. Zepeda and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2002, 124, 4701–4707 CrossRef CAS PubMed.
- K. K. Laali, M. Tanaka, F. Forohar, M. Cheng and J. C. Fetzer, J. Fluorine Chem., 1998, 91, 185–190 CrossRef CAS.
- J. W. Cook, C. L. Hewett and I. Hieger, J. Chem. Soc., 1933, 395–405 RSC.
- A. Streitwieser Jr., R. G. Lawler and D. Schwaab, J. Org. Chem., 1965, 30, 1470–1473 CrossRef.
- R. Lapouyade, J. Pereyre and P. Garrigues, C. R. Acad. Sci., Ser. II: Mec., Phys., Chim., Sci. Terre Univers, 1986, 303, 903–906 CAS.
- A. Gold, J. Schultz and E. Eisenstadt, Tetrahedron Lett., 1978, 46, 4491–4494 CrossRef.
- M. Konieczny and R. G. Harvey, J. Org. Chem., 1979, 44, 2158–2160 CrossRef CAS.
- Y. Sahali, P. L. Skipper and R. Tannenbaum, J. Org. Chem., 1990, 55, 2918–2920 CrossRef CAS.
- R. J. Young and R. G. Harvey, Tetrahedron Lett., 1989, 30, 6603–6606 CrossRef CAS.
- R. Pratap, Y. Tominaga, M. L. Lee and R. N. Castle, J. Heterocycl. Chem., 1981, 18, 973–975 CrossRef CAS.
- P. Demerseman, B. Tric, H. Strapélias and R. Royer, J. Heterocycl. Chem., 1985, 22, 1137–1139 CrossRef.
- V. Gandhi, M. L. Thompson and T. D. Lash, Tetrahedron, 2010, 66, 1787–1799 CrossRef CAS.
- S. Veeraraghavan, S. Jostmeyer, J. Myers and J. C. Wiley Jr., J. Org. Chem., 1987, 52, 1355–1357 CrossRef CAS.
- R. Sangaiah and A. Gold, J. Org. Chem., 1988, 53, 2620–2622 CrossRef CAS.
- R. Sangaiah and A. Gold, J. Org. Chem., 1991, 56, 6717–6720 CrossRef CAS.
- A. Z. González, D. Benitez, E. Tkatchouk, W. A. Goddard III and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 5500–5507 CrossRef PubMed.
- F. Vögtle and P. Neumann, Angew. Chem., Int. Ed. Engl., 1972, 11, 73–158 CrossRef.
- N. L. Allinger, B. J. Gorden, S.-E. Hu and R. A. Ford, J. Org. Chem., 1967, 32, 2272–2278 CrossRef CAS.
- T. Umemoto, T. Kawashima, Y. Sakata and S. Misumi, Tetrahedron Lett., 1975, 16, 1005–1006 CrossRef.
- T. Sato, S. Akabori, S. Muto and K. Hata, Tetrahedron, 1968, 24, 5557–5567 CrossRef CAS.
- T. Koening and M. Tuttle, J. Org. Chem., 1974, 39, 1308–1311 CrossRef.
- T. Sato, K. Torizuka, R. Komaki and H. Atobe, J. Chem. Soc., Perkin Trans. 2, 1980, 561–568 RSC.
- N. Ueda, B. Natsume, K. Yanagiuchi, Y. Sakata, T. Enoki, G. Saito, H. Inokuchi and S. Misumi, Bull. Chem. Soc. Jpn., 1983, 56, 775–779 CrossRef CAS.
- T. Yamato, J.-i. Matsumoto, K. Tokuhisa, M. Shigekuni, K. Suehiro and M. Tashiro, J. Org. Chem., 1992, 57, 395–396 CrossRef CAS.
- M. Tashiro, S. Mataka, Y. Takezaki, M. Takeshita, T. Arimura, A. Tsuge and T. Yamato, J. Org. Chem., 1989, 54, 451–458 CrossRef CAS.
- T. Yamato, S. Ide, K. Tokuhisa and M. Tashiro, J. Org. Chem., 1992, 57, 271–275 CrossRef CAS.
- R. H. Mitchell and V. Boekelheide, J. Am. Chem. Soc., 1970, 92, 3510–3512 CrossRef CAS.
- T. Yamato, A. Miyazawa and M. Tashiro, J. Org. Chem., 1992, 57, 266–270 CrossRef CAS.
- T. Yamagishi, K. Torizuka and T. Sato, Bull. Chem. Soc. Jpn., 1982, 55, 1140–1143 CrossRef CAS.
- T. Sato, K. Nishiyama, A. Morita and Y. Iitaka, Bull. Chem. Soc. Jpn., 1985, 58, 2366–2369 CrossRef CAS.
- T. Umemoto, S. Satni, Y. Sakata and S. Misumi, Tetrahedron Lett., 1975, 16, 3159–3162 CrossRef.
- R. H. Mitchell, R. J. Carruthers and C. M. Zwinkels, Tetrahedron Lett., 1976, 17, 2585–2588 CrossRef.
- T. Kawashima, T. Otsubo, Y. Sakata and S. Misumi, Tetrahedron Lett., 1978, 19, 5115–5118 CrossRef.
- For a recent review, see T. Yao, H. Yu, R. J. Vermeij and G. J. Bodwell, Pure Appl. Chem., 2008, 80, 533–546 CrossRef CAS and references therein.
- B. L. Merner, L. N. Dawe and G. J. Bodwell, Angew. Chem., Int. Ed., 2009, 48, 5487–5491 CrossRef CAS PubMed.
- Y. Yang, M. R. Mannion, L. N. Dawe, C. M. Kraml, R. A. Pascal Jr. and G. J. Bodwell, J. Org. Chem., 2012, 77, 57–67 CrossRef CAS PubMed.
- P. R. Nandaluru, P. Dongare, C. M. Kraml, R. A. Pascal Jr., L. N. Dawe, D. W. Thompson and G. J. Bodwell, Chem. Commun., 2012, 48, 7747–7749 RSC.
- W. H. Laarhoven and Th. J. H. M. Cuppen, J. Chem. Soc., Perkin Trans. 1, 1972, 2074–2079 RSC.
- P. H. G. op het Veld and W. H. Laarhoven, J. Chem. Soc., Perkin Trans. 2, 1977, 269–273 Search PubMed.
- A. Padwa, C. Doubleday and A. Mazzu, J. Org. Chem., 1977, 42, 3271–3279 CrossRef CAS.
- M. Kreyenschmidt, M. Baumgarten, N. Tyutyulkov and K. Müllen, Angew. Chem., Int. Ed. Engl., 1994, 33, 1957–1959 CrossRef.
- M. Kreyenschmidt, F. Uckert and K. Müllen, Macromolecules, 1995, 28, 4577–4582 CrossRef CAS.
- Z. Y. Wang and C. Zhang, Macromolecules, 1992, 25, 5851–5854 CrossRef CAS.
- S. Kawano, C. Yang, M. Ribas, S. Baluschev, M. Baumgarten and K. Müllen, Macromolecules, 2008, 41, 7933–7937 CrossRef CAS.
- J. Barluenga, J. M. Gonzalez, P. J. Campos and G. Asensio, Angew. Chem., Int. Ed. Engl., 1988, 27, 1546–1547 CrossRef.
- M. B. Goldfinger, K. B. Crawford and T. M. Swager, J. Am. Chem. Soc., 1997, 119, 4578–4593 CrossRef CAS.
- T. L. Yao, M. A. Campo and R. C. Larock, J. Org. Chem., 2005, 70, 3511–3517 CrossRef CAS PubMed.
- V. Mamane, P. Hannen and A. Fürstner, Chem.–Eur. J., 2004, 10, 4556–4575 CrossRef CAS PubMed.
- D. B. Walker, J. Howgego and A. P. Davis, Synthesis, 2010, 3686–3692 CAS.
- T. Matsuda, T. Moriya, T. Goya and M. Murakami, Chem. Lett., 2011, 40, 40–41 CrossRef CAS.
- M. M. Machuy, C. Würtele and P. R. Schreiner, Synthesis, 2012, 1405–1409 CAS.
- However, see A. Mukherjee, K. Pati and R. S. Liu, J. Org. Chem., 2009, 74, 6311–6314 CrossRef CAS PubMed , for a related cyclisation of ethynylphenanthrenes.
Footnotes |
| † J. M. Casas-Solvas and J. D. Howgego contributed equally to this work. |
| ‡ Current address: Department of Chemistry and Physics, University of Almería, Crta. de Sacramento s/n, 04120, Almería, Spain. |
| This journal is © The Royal Society of Chemistry 2014 |