An efficient organocatalytic enantioselective synthesis of spironitrocyclopropanes†
Utpal
Das
,
Yi-Ling
Tsai
and
Wenwei
Lin
*
Department of Chemistry, National Taiwan Normal University, 88, Section 4, Tingchow Road, Taipei 11677, Taiwan, R.O.C. E-mail: wenweilin@ntnu.edu.tw; Fax: (+886) 229324249; Tel: (+886) 277346131
First published on 22nd October 2012
Abstract
An organocatalytic asymmetric synthesis of spironitrocyclopropanes has been demonstrated starting from 2-arylidene-1,3-indandiones and bromonitroalkanes catalyzed by a cinchona-derived bifunctional organocatalyst. The products were obtained with excellent enantioselectivities, diastereoselectivities and with good yields.
Several biologically active compounds and natural products are known to have cyclopropane rings as important structural subunits.1 Among them, nitrocyclopropanes are of special interest because they can be found in various biologically active natural products.2 Moreover, they serve as the key intermediates for many useful synthetic transformations.3 As a result, asymmetric cyclopropanations have been well explored, including enantioselective versions of Simmons–Smith reactions4 and transition metal catalyzed reactions using carbene intermediates.5 An alternative path for catalytic cyclopropanation was introduced by Aggarwal,6 Gaunt7 and MacMillan8 involving ylides9 as intermediates.
In recent years, organocatalytic asymmetric cyclopropanation reactions were found to be an attractive alternative, and many elegant strategies were reported.10 However, the direct organocatalytic synthesis of spirofused nitrocyclopropanes is very limited10a,d and that employing 2-arylidene-1,3-indandiones (1) has not yet been reported. The substrates 1 are known as a restricted class of trisubstituted electron poor alkenes and have rarely been used in organocatalysis.11 Very recently, Lattanzi and co-workers11a described an elegant Michael-initiated cascade reaction to prepare spirocyclopropanes starting from 1. However, their attempted base-catalyzed cyclopropanation of 1 with bromonitromethane (2) was not successful. As evident in the literature,10a,d cinchona alkaloid derived thioureas12 are well known for their bifunctional behavior to activate electron poor alkenes and pronucleophiles simultaneously. Accordingly, we planned to use bifunctional catalysts (I–IV) for our study of nitrocyclopropanation. Thiourea catalysts V–VI derived from cyclohexane diamine were also included for our study. In a continuation of our recent efforts,13 herein we wish to report our attempts to prepare enantioenriched spironitrocyclopropane derivatives.14 We began our study by using 1a (alkene component) and bromonitromethane (2a, pronucleophile) as the model reaction partners in the presence of quinidine derived bifunctional tertiary amine-thiourea catalyst I. Na2CO3 was used to neutralize the liberated HBr. However, our initial attempt at the predicted nitrocyclopropanation was unsuccessful, and only a trace amount of the expected product was detected (Table 1, entry 1). We reasoned that this failure was due to a lack of hydrogen-bond interactions between the N–H bonds of thiourea and the 1,3-dicarbonyl groups of 1a. Water is well known for its ability to participate in hydrogen bonding interactions and many organocatalytic reactions are known to proceed in water.15 Unfortunately, we did not observe any conversion of starting material when the reaction was conducted in water (entry 2). To our surprise, we noted that precise quantities of water were critical for the reaction to proceed. In the presence of one equivalent of water the reaction proceeded smoothly to furnish the nitrocyclopropanation adduct (3a) in 49% isolated yield in just two hours with 75% ee at room temperature (entry 3). Lowering of the reaction temperature to 0 °C was beneficial as product 3a was obtained in 76% yield with a diasteremeric ratio of 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 91% enantiomeric excess (entry 4).
:1 and 91% enantiomeric excess (entry 4).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Solvent | t (h) | 3a (%) | drc | eed (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (1.5 equiv.), cat. (20 mol%), base (1 equiv.) in 0.5 mL anhydrous solvent. b Isolated yield. c Diastereomeric ratio was determined by 1H NMR spectroscopic analysis of the crude reaction mixture. d Enantiomeric excess was determined by HPLC analysis. e At room temperature. f No water was used as an additive. g NaHCO3 was used. h The reaction was performed at −20 °C in 0.2 mL toluene. i 10 mol% I was used. | ||||||
| 1e,f | I | Toluene | 24 | — | — | — |
| 2e | I | H2O | 24 | — | — | — |
| 3e | I | Toluene | 2 | 49 | 6.5:1 |
75 |
| 4 | I | Toluene | 5 | 76 | 11:1 |
91 |
| 5 | II | Toluene | 5 | 69 | 11:1 |
−91 |
| 6 | III | Toluene | 5 | 65 | 10:1 |
−88 |
| 7 | IV | Toluene | 6 | 79 | 12:1 |
−75 |
| 8 | V | Toluene | 48 | 49 | — | ± |
| 9 | VI | Toluene | 30 | 49 | — | ± |
| 10 | I | CH2Cl2 | 6 | 78 | 6.5:1 |
87 |
| 11 | I | THF | 6 | 64 | 4.3:1 |
67 |
| 12g | I | Toluene | 48 | 49 | 11:1 |
90 |
| 13h | I | Toluene | 6 | 79 |
19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
94 |
| 14h,i | I | Toluene | 14 | 79 | 16:1 |
88 |
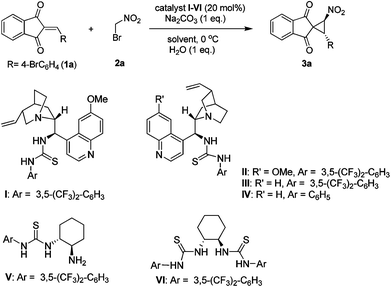
Encouraged by this promising output, we also evaluated other cinchona alkaloid derived catalysts (II–IV) and catalysts V–VI, and the results are presented in Table 1 (entries 5–9). Although all the catalysts (I–IV) could promote the reaction, catalyst I was found to be better in terms of chemical yield and enantioselectivity. However catalysts V and VI turned to be very poor, as not only did the reaction take longer (<70% conversion in the indicated time), but also the obtained products were racemic. The organocatalytic nitrocyclopropanation reaction for the formation of 3a was then further optimized. A number of different solvents and bases were screened (entries 10–13, for details see ESI†), and the results indicate that proper choice of solvent and base play an important role. Adduct 3a was afforded in reduced diastereoselectivity and enantioselectivity when the reaction was conducted in dichloromethane or tetrahydrofuran (entries 10 and 11). Product 3a could be formed with a similar product profile (entry 12) using the weaker sodium bicarbonate base, but the progress of reaction was too slow. When the reaction temperature was further lowered to −20 °C, 3a was furnished in 94% ee coupled with 19:1 diastereoselectivity and 79% yield (entry 13). It is worth mentioning that both the diastereoselectivity and enantioselectivity of 3a dropped upon lowering of catalyst loading (10 mol%) under these reaction conditions (entry 14).
With an effective protocol for the enantioselective formation of spironitrocyclopropane in hand, the substrate scope and generality of the methodology were examined. A variety of substituents on 2-arylidene-1,3-indandiones (1a–l) and different bromonitroalkanes (2a–c) could be employed to provide spironitrocyclopropanes (3a–n) in good yields (up to 88%), excellent diastereoselectivities (up to 19:1) and enantioselectivities (up to 98%) (Table 2). The electronic nature, bulkiness or positions of substituents in the aryl group seems to have little effect on the results (entries 1–10). Slightly reduced enantioselectivities were observed in the cases of 4-nitrophenyl and 3-fluorophenyl substituted arylidene-1,3-indandiones (1c and 1f). We have also evaluated substituted bromonitroalkanes (2b and 2c) as pronucleophile (entries 11–14) for synthesis of the products 3k–n bearing a quaternary stereocenter, which is considered a challenging task in organic synthesis.16 Thus the reaction of 1-bromo-1-nitroethane (2b) with substituted 2-arylidene-1,3-indandiones having different electronic natures proceeded smoothly to give the desired products (3k–m) in good yields, excellent diastereoselectivities and enantioselectivities (entries 11–13). It is noteworthy that the reaction between 1-bromo-1-nitropropane (2c) and 1a was slower and the product (3n) was obtained in moderate enantioselectivity (56%) albeit with good yield and diastereoselectivity (entry 14 vs. 1 and 11). Less reactive cyclohexyl substituted alkylidene-1,3-indandiones were also examined with 2a. However, a poor result was obtained under our reaction conditions (entry 15).17 Additionally, cyclopropanation of 4-methoxyphenyl substituted arylidene-1,3-indandione (1l) and 2a did not take place under identical reaction conditions (entry 16).18 The absolute configuration of 3a and 3k were determined by single crystal X-ray data analyses and those of the others were assigned by analogy.19
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | t (h) | 3,b (%) | drc | eed (%) |
| a Reaction conditions: 1a–l (0.1 mmol), 2a–c (1.5 equiv.), I (20 mol%), Na2CO3 (1 equiv.) in 0.2 mL anhydrous toluene. b Isolated yield. c Determined by 1H NMR spectroscopic analysis of the crude reaction mixture. d ee for the major isomer (ee after single recrystallization). e A mixture of three compounds (70% ee, 87% ee and 72% ee) were obtained (please also see ESI†). | ||||||
| 1 | 4-BrC6H4 | H | 6 | 3a, 79 | 19:1 |
96 (>99) |
| 2 | 4-ClC6H4 | H | 8 | 3b, 68 | 19:1 |
95 |
| 3 | 4-NO2C6H4 | H | 8 | 3c, 68 | 18:1 |
86 |
| 4 | 4-CNC6H4 | H | 12 | 3d, 78 | 19:1 |
92 |
| 5 | 2-BrC6H4 | H | 8 | 3e, 68 | 19:1 |
96 (>99) |
| 6 | 3-FC6H4 | H | 7 | 3f, 71 | 16:1 |
86 |
| 7 | C6H5 | H | 10 | 3g, 78 | 18:1 |
96 |
| 8 | 4-MeC6H4 | H | 5 | 3h, 68 | 18:1 |
96 |
| 9 | 2-MeC6H4 | H | 5 | 3i, 65 | 18:1 |
96 |
| 10 | 4-tBuC6H4 | H | 6 | 3j, 63 | 18:1 |
98 |
| 11 | 4-BrC6H4 | Me | 12 | 3k, 81 | 19:1 |
96 (>99) |
| 12 | C6H5 | Me | 24 | 3l, 88 | 19:1 |
93 |
| 13 | 4-MeC6H4 | Me | 36 | 3m, 78 | 19:1 |
96 |
| 14 | 4-BrC6H4 | Et | 72 | 3n, 65 | 16:1 |
56 |
| 15e | Cyclohexyl | H | 8 | — | — | — |
| 16 | 4-OMeC6H4 | H | 24 | — | — | — |
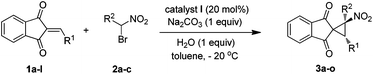
We believe that the reaction proceeds through simultaneous activation of the pronucleophile (bromonitroalkane) and electrophilic 2-arylidene-1,3-indandiones via water assisted H-bonding interaction by the bifunctional catalyst. Nucleophilic addition of bromonitroalkane and subsequent intramolecular cyclisation furnished the desired spirocyclopropane products (see ref. 10d).
Furthermore, we envisioned that spirocyclopropane product (3g) can also be accessed by employing bromonitrostyrene, such as 4, as a dielectrophilic component10a,20 and 1,3-indandione (5) as a dinucleophile in presence of appropriate catalyst. However, our initial attempt to use the present optimized reaction conditions was not very successful and the adduct ent-3g was obtained with only 35% ee and moderate yield (57%) (Scheme 1).
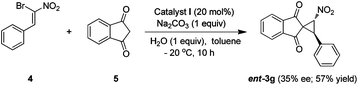 | ||
| Scheme 1 Synthesis of ent-3g starting from 4 and 5. | ||
In summary, we have developed an efficient asymmetric pathway for the preparation of spironitrocyclopropanes catalyzed by cinchona-derived bifunctional organocatalysts. To the best of our knowledge, this is the first asymmetric route to prepare spironitrocyclopropanes starting from 2-arylidene-1,3-indandiones and bromonitroalkanes. The products were obtained in good yields (up to 88%), excellent enantioselectivities (up to 98%) and diastereoselectivities (up to 19:1). Further investigation of the synthetic route described in Scheme 1, biological evaluation of spironitrocyclopropanes and investigation of the reaction mechanism are currently underway in our laboratory.
Acknowledgements
The authors thank the National Science Council of the Republic of China (NSC 99-2119-M-003-004-MY2) and National Taiwan Normal University (NTNU100-D-06) for financial support.Notes and references
- (a) D. Y.-K. Chen, R. H. Pouwer and J.-A. Richard, Chem. Soc. Rev., 2012, 41, 4631 RSC; (b) F. Brackmann and A. de Meijere, Chem. Rev., 2007, 107, 4493 CrossRef CAS; (c) J. A. Thomson and R. B. Perni, Curr. Opin. Drug Discovery Dev., 2006, 9, 606 CAS; (d) W. A. Donaldson, Tetrahedron, 2001, 57, 8589 CrossRef CAS; (e) J. Pietruszka, Chem. Rev., 2003, 103, 1051 CrossRef CAS; (f) R. Faust, Angew. Chem., Int. Ed., 2001, 40, 2251 CrossRef CAS; (g) A. de Meijere, Top. Curr. Chem., 2000, 207, 1 CrossRef.
- (a) R. Ballini, A. Palmieri and D. Fiorini, ARKIVOC, 2007, 7, 172 Search PubMed; (b) B. D. Zlatopolskiy, K. Loscha, P. Alvermann, S. I. Kozhushkov, S. V. Nikolaev, A. Zeeck and A. de Meijere, Chem.–Eur. J., 2004, 10, 4708 CrossRef CAS; (c) J. Zindel and A. de Meijere, J. Org. Chem., 1995, 60, 2968 CrossRef CAS.
- For reviews on nitrocyclopropanes, see: (a) B. Moreau, D. Alberico, V. N. G. Lindsay and A. B. Charette, Tetrahedron, 2012, 68, 3487 CrossRef CAS; (b) E. B. Averina, N. V. Yashin, T. S. Kuznetsova and N. S. Zefirov, Russ. Chem. Rev., 2009, 78, 887 CrossRef CAS ; for selected examples, see: ; (c) S. S. So, T. J. Auvil, V. J. Garza and A. E. Mattson, Org. Lett., 2012, 14, 444 CrossRef CAS; (d) V. N. G. Lindsay, C. Nicolas and A. B. Charette, J. Am. Chem. Soc., 2011, 133, 8972 CrossRef CAS; (e) O. Lifchits, D. Alberico, I. Zakharian and A. B. Charette, J. Org. Chem., 2008, 73, 6838 CrossRef CAS; (f) E. M. Budynina, O. A. Ivanova, E. B. Averina, T. S. Kuznetsova and N. S. Zefirov, Tetrahedron Lett., 2006, 47, 647 CrossRef CAS; (g) R. P. Wurz and A. B. Charette, J. Org. Chem., 2004, 69, 1262 CrossRef CAS.
- For reviews, see: (a) H. Pellissier, Tetrahedron, 2008, 64, 7041 CrossRef CAS; (b) H. Lebel, J. F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977 CrossRef CAS ; for recent examples, see: ; (c) S. R. Goudreau and A. B. Charette, J. Am. Chem. Soc., 2009, 131, 15633 CrossRef CAS; (d) L. E. Zimmer and A. B. Charette, J. Am. Chem. Soc., 2009, 131, 15624 CrossRef CAS.
- For selected recent examples see: (a) X. Xu, H. Lu, J. V. Ruppel, X. Cui, S. L. de Mesa, L. Wojtas and X. P. Zhang, J. Am. Chem. Soc., 2011, 133, 15292 CrossRef CAS; (b) J.-i. Ito, S. Ujiie and H. Nishiyama, Chem.–Eur. J., 2010, 16, 4986 CrossRef CAS; (c) S. Zhu, X. Xu, J. A. Perman and X. P. Zhang, J. Am. Chem. Soc., 2010, 132, 12796 CrossRef CAS; (d) S. Chuprakov, S. W. Kwok, L. Zhang, L. Lercher and V. V. Fokin, J. Am. Chem. Soc., 2009, 131, 18034 CrossRef CAS; (e) M. Ichinose, H. Suematsu and T. Katsuki, Angew. Chem., Int. Ed., 2009, 48, 3121 CrossRef CAS.
- (a) S. L. Riches, C. Saha, N. F. Filgueira, E. Grange, E. M. McGarrigle and V. K. Aggarwal, J. Am. Chem. Soc., 2010, 132, 7626 CrossRef CAS; (b) V. K. Aggarwal, E. Alonso, G. Fang, M. Ferrara, G. Hynd and M. Porcelloni, Angew. Chem., Int. Ed., 2001, 40, 1433 CrossRef CAS.
- (a) C. C. C. Johansson, N. Bremeyer, S. V. Ley, D. R. Owen, S. C. Smith and M. J. Gaunt, Angew. Chem., Int. Ed., 2006, 45, 6024 CrossRef CAS; (b) N. Bremeyer, S. C. Smith, S. V. Ley and M. J. Gaunt, Angew. Chem., Int. Ed., 2004, 43, 2681 CrossRef CAS.
- (a) R. K. Kunz and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 3240 CrossRef CAS; (b) for a related example, see: A. Hartikka and P. I. Arvidsson, J. Org. Chem., 2007, 72, 5874 CrossRef CAS.
- For recent selected examples in asymmetric cyclopropanations, see: (a) A. Biswas, S. De Sarkar, L. Tebben and A. Studer, Chem. Commun., 2012, 48, 5190 RSC; (b) L. Gao, G.-S. Hwang and D. H. Ryu, J. Am. Chem. Soc., 2011, 133, 20708 CrossRef CAS; (c) B.-H. Zhu, R. Zhou, J.-C. Zheng, X.-M. Deng, X.-L. Sun, Q. Shen and Y. Tang, J. Org. Chem., 2010, 75, 3454 CrossRef CAS.
- (a) X. Dou and Y. Lu, Chem.–Eur. J., 2012, 18, 8315 CrossRef CAS; (b) C. D. Fiandra, L. Piras, F. Fini, P. Disetti, M. Moccia and M. F. A. Adamo, Chem. Commun., 2012, 48, 3863 RSC; (c) M. Rueping, H. Sundén, L. Hubener and E. Sugiono, Chem. Commun., 2012, 48, 2201 RSC; (d) F. Pesciaioli, P. Righi, A. Mazzanti, G. Bartoli and G. Bencivenni, Chem.–Eur. J., 2011, 17, 2842 CrossRef CAS; (e) Y. Cheng, J. An, L.-Q. Lu, L. Luo, Z.-Y. Wang, J.-R. Chen and W.-J. Xiao, J. Org. Chem., 2011, 76, 281 CrossRef CAS; (f) V. Terrasson, A. Lee, R. M. Figueiredo and J. M. Campagne, Chem.–Eur. J., 2010, 16, 7875 CrossRef CAS; (g) Y. Xuan, S. Nie, L. Dong, J. Zhang and M. Yan, Org. Lett., 2009, 11, 1583 CrossRef CAS; (h) J. Lv, J. Zhang, Z. Lin and Y. Wang, Chem.–Eur. J., 2009, 15, 972 CrossRef CAS; (i) T. Inokuma, S. Sakamoto and Y. Takemoto, Synlett, 2009, 1627 CAS; (j) I. Ibrahem, G.-L. Zhao, R. Rios, J. Vesely, H. Sunden, P. Dziedzic and A. Cordova, Chem.–Eur. J., 2008, 14, 7867 CrossRef CAS; (k) R. Rios, H. Sunden, J. Vesely, G.-L. Zhao, P. Dziedzic and A. Cordova, Adv. Synth. Catal., 2007, 349, 1028 CrossRef CAS; (l) H. Xie, L. Zu, H. Li, J. Wang and W. Wang, J. Am. Chem. Soc., 2007, 129, 10886 CrossRef CAS; (m) S. H. McCooey, T. McCabe and S. J. Connon, J. Org. Chem., 2006, 71, 7494 CrossRef CAS.
- (a) A. Russo, S. Meninno, C. Tedesco and A. Lattanzi, Eur. J. Org. Chem., 2011, 5096 CrossRef CAS; (b) A. Russo and A. Lattanzi, Org. Biomol. Chem., 2010, 8, 2633 RSC; (c) D. B. Ramachary, N. S. Chowdari and C. F. Barbas III, Synlett, 2003, 1910 CAS.
- For reviews, see: (a) W.-Y. Siau and J. Wang, Catal. Sci. Technol., 2011, 1, 1298 RSC; (b) S. J. Connon, Chem. Commun., 2008, 2499 RSC; (c) X. Yu and W. Wang, Chem.–Asian J., 2008, 3, 516 CrossRef CAS; (d) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS; (e) T. Akiyama, J. Itoh and K. Fuchibe, Adv. Synth. Catal., 2006, 348, 999 CrossRef CAS; (f) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520 CrossRef CAS.
- (a) U. Das, C.-H. Huang and W. Lin, Chem. Commun., 2012, 48, 5590 RSC; (b) S.-e. Syu, T.-T. Kao and W. Lin, Tetrahedron, 2010, 66, 891 CrossRef CAS.
- Spirocyclopropane can be found in biologically active compounds such as iludins, see: (a) N. Rasool, M. A. Rashid, H. Reinke, C. Fischer and P. Langer, Tetrahedron, 2008, 64, 3246 CrossRef CAS; (b) N. Rasool, M. A. Rashid, M. Adeel, H. Görls and P. Langer, Tetrahedron Lett., 2008, 49, 2254 CrossRef CAS; (c) M. C. Pirrung and H. Liu, Org. Lett., 2003, 5, 1983 CrossRef CAS.
- For recent reviews on organocatalytic reactions in water, see: (a) J. G. Hernández and E. Juaristi, Chem. Commun., 2012, 48, 5396 RSC; (b) S. Bhowmick and K. C. Bhowmick, Tetrahedron: Asymmetry, 2011, 22, 1945 CrossRef CAS; (c) N. Mase and C. F. Barbas III, Org. Biomol. Chem., 2010, 8, 4043 RSC; (d) M. Raj and V. K. Singh, Chem. Commun., 2009, 6687 RSC.
- For selected recent reviews, see: (a) J. P. Das and I. Marek, Chem. Commun., 2011, 47, 4593 RSC; (b) M. Shimizu, Angew. Chem., Int. Ed., 2011, 50, 5998 CrossRef CAS; (c) C. Hawner and A. Alexakis, Chem. Commun., 2010, 46, 7295 RSC; (d) M. Bella and T. Gasperi, Synthesis, 2009, 1583 CrossRef CAS.
- Preparation of primary or tertiary alkyl substituted alkylidene-1,3-indandiones was not successful, and therefore a secondary alkyl substituted alkylidene-1,3-indandione such as 1k was examined for our study. However, a mixture of three compounds were obtained, and their polarities are too close to be separated by flash column chromatography.
- No product formation was also observed in the case of using heteroaromatic residue (e.g. 2-furyl, 2-thienyl) as R1 in our preliminary study.
- CCDC 892806 (3a) and 892808 (3k) contain supplementary crystallographic data for this paper (please see ESI†).
- M. Rueping, A. Parra, U. Uria, F. Besselievre and E. Merino, Org. Lett., 2010, 12, 5680 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, spectral data of new compounds. CCDC 892806 (3a) and 892808 (3k). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ob26943k |
| This journal is © The Royal Society of Chemistry 2013 |