Energetic studies on DNA–peptide interaction in relation to the enthalpy–entropy compensation paradox†
Robin C. K.
Yang‡
a,
Jonathan T. B.
Huang‡
a,
Shih-Chuan
Chien
a,
Roy
Huang
a,
Kee-Ching G.
Jeng
b,
Yen-Chung
Chen
a,
Mokai
Liao
a,
Jia-Rong
Wu
a,
Wei-Kang
Hung
a,
Chia-Chun
Hung
a,
Yu-Ling
Chen
a,
Michael J.
Waring
c and
Leung
Sheh
*a
aDepartment of Chemistry and Life Science Center, Tunghai Christian University, Taichung, Taiwan 407, R.O.C. E-mail: Lsheh@thu.edu.tw; Fax: +886-4-23590426; Tel: +886-4-23592674
bDepartment of Medical Research, Taichung Veterans General Hospital, R.O.C Taiwan 405
cDepartment of Pharmacology, University of Cambridge, Tennis Court Road, Cambridge CB2 1PD, England
First published on 12th September 2012
Abstract
This study aims to interpret the energetic basis of complex DNA–peptide interactions according to a novel allosteric interaction network approach. In common with other designed peptides, five new conjugates incorporating the XPRK or XHypRK motif (Hyp = hydroxyproline) attached to a N-methylpyrrole (Py) tract with a basic tail have been found to display cooperative binding to DNA involving multiple monodentate as well as interstrand bidentate interactions. Using quantitative DNase I footprinting it appears that allosteric communication via cooperative binding to multiple sites on complementary DNA strands corresponds to two different types of DNA–peptide interaction network. Temperature variation experiments using a dodecapeptide RY-12 show that lower temperature (25 °C) favor a circuit type of allosteric interaction network, whereas higher temperatures (31 and 37 °C) afford only a partial-circuit type of network. Circular dichroism studies show that our five peptides induce significant local conformational changes in DNA via the minor groove, with apparently dimeric binding stoichiometry. Isothermal titration calorimetry reveals that these peptides, together with another seven for comparison, are strongly exothermic upon binding to a model 13-mer DNA duplex, characterized by ΔH ranging from −14.7 to −74.4 kcal mol−1, and also high TΔS ranging from −6.5 to −65.9 kcal mol−1. Multiple monodentate and bidentate interactions, as well as ionic forces that mediate positive cooperativity in sequence recognition, are consistent with a dramatic decrease in entropy and a ‘tightening’ effect of DNA conformation. Distinctive enthalpy–entropy compensation (EEC) relationships are demonstrated for the interaction of all twelve designed peptides with DNA, affording a straight line of slope close to unity when ΔH is plotted versus TΔS, with a y-axis intercept (average ΔG) corresponding to −8.5 kcal mol−1, while the observed ΔG ranges from −8.2 to −9.1 kcal mol−1 for the peptides. The EEC seen with peptide RY-12 binding to the model duplex persists throughout various incubation temperatures. The net compensation of energy between the favorable negative ΔH and unfavorable negative ΔS components thus constrains the value of net binding free energy ΔG within a remarkably constant range, as is clearly visible in a 3-dimensional energetic plot. We conclude that the preservation of a rather narrowly-defined ΔG value is central to the EEC in DNA–peptide interactions, illuminating the universal EEC paradox commonly found in diverse biochemical reactions.
1 Introduction
As research on the sequence-selective binding of drugs to DNA gathers pace, it has raised increasingly sophisticated questions. After more than three decades, during which these studies have laid the foundations of various areas of structural biochemistry and have contributed much to understanding the bioorganic chemistry of nucleic acids, attention is now being turned to applications, particularly the synthesis of potentially useful drugs. Classical DNA–small molecule interaction studies include work on a large variety of small molecules that serve as effective ligands, namely antitumor antibiotics,1–4 lexitropsins,4–6 synthetic polyamides,4,7–11 heterocycles12,13 and synthetic peptides.14–18 One notable outcome is the relatively recent acknowledgment that understanding the molecular basis for the recognition of specific sequences in target DNA by small synthetic molecules will prove useful in future gene therapy.4 At the same time, efforts to investigate the molecular events underlying cooperativity between DNA–ligand binding sites are expected to provide new insight into chemical rules governing structural biochemistry.For over ten years we have been studying the sequence-selective recognition of DNA by synthetic peptides incorporating the XP(Hyp)RK motif.14–18 Peptides incorporating such motifs possess excellent DNA binding capability in the sub-micromolar concentration range. Further attachment of one or more 4-amino-1-methylpyrrole-2-carboxylic acid residues (Py) to XP(Hyp)RK peptides augments their sequence-specificity toward sequences containing consecutive arrays of A or T nucleotides. Our design of XPRK-containing peptides stems from the finding that a SPXX motif19,20 is often found in repeating sequences in histones, steroid hormone receptors, various segmentation gene products and some oncogene products. The SPXX motif assumes a β-turn stabilized by two hydrogen bonds, and the side chains of the two basic residues engage in salt bridges with the DNA phosphate groups.
It is now generally accepted that allosteric behavior of macromolecules21 such as proteins is a vital biological control process governing biochemical efficiency and energy expenditure. However, much less is known about the possible allosteric aspects of DNA–ligand binding than about the extensively studied protein–ligand interactions. Notably, over the first decade of the twenty-first century, it has been recognized that interaction networks between bio-molecules occur in many biochemical, biophysical and sub-cellular processes in nature.22,23 High-throughput techniques have helped to establish interaction maps that expose protein–protein interaction networks, metabolic networks, and transcriptional regulatory networks.22 Six years ago we proposed a network-based DNA–peptide allosteric interaction model interconnecting multiple sites in fragments of the latent membrane protein (LMP-1) gene from a pathogenic Epstein–Barr virus variant derived from nasopharyngeal carcinoma.16 Subsequently we refined the model in terms of three different types of network-based allosteric communication between synthetic peptides binding to DNA: circuit type, incomplete circuit type, and non-circuit type characterized by interstrand bidentate interactions.17,18
In this study we further explore the allosteric features of DNA–peptide molecular recognition with regard to interaction networks, choosing designed peptides that exhibit significant cooperativity in binding to DNA. Quantitative DNase I footprinting furnishes the prime methodology for studying the allosteric features of DNA interaction networks associated with the binding of two designed decapeptides incorporating the XHypRK motif: Ser-Hyp-Arg-Lys-(Py)4-Lys-Arg-NH2 (HyS-10), Glu-Hyp-Arg-Lys-(Py)4-Lys-Arg-NH2 (HyE-10), and two decapeptides incorporating a novel KPXR motif: Lys-Pro-![[M with combining low line]](https://www.rsc.org/images/entities/char_004d_0332.gif)
![[e with combining low line]](https://www.rsc.org/images/entities/char_0065_0332.gif)
![[t with combining low line]](https://www.rsc.org/images/entities/char_0074_0332.gif) -Arg-(Py)4-Lys-Arg-NH2 (PyMK-10), and Lys-Pro-
-Arg-(Py)4-Lys-Arg-NH2 (PyMK-10), and Lys-Pro-![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif)
![[l with combining low line]](https://www.rsc.org/images/entities/char_006c_0332.gif)
![[n with combining low line]](https://www.rsc.org/images/entities/char_006e_0332.gif) -Arg-(Py)4-Lys-Arg-NH2 (PyQK-10). In addition, a closely related dodecapeptide Tyr-Pro-Arg-Lys-(Py)4-Lys-Arg-Pro-Tyr-NH2 (RY-12) was also investigated. DNase footprinting experiments were carried out using a 5′-[32P]-labeled 158-mer DNA duplex and an essentially complementary 5′-[32P]-labeled 135-mer DNA duplex employed in previous work.14 Circular dichroism was also brought to bear upon the problem as a means of directly studying conformational changes induced by peptide binding as well as salt effects upon interaction between peptide HyM-10 and a 13-mer deoxyribonucleotide duplex.
-Arg-(Py)4-Lys-Arg-NH2 (PyQK-10). In addition, a closely related dodecapeptide Tyr-Pro-Arg-Lys-(Py)4-Lys-Arg-Pro-Tyr-NH2 (RY-12) was also investigated. DNase footprinting experiments were carried out using a 5′-[32P]-labeled 158-mer DNA duplex and an essentially complementary 5′-[32P]-labeled 135-mer DNA duplex employed in previous work.14 Circular dichroism was also brought to bear upon the problem as a means of directly studying conformational changes induced by peptide binding as well as salt effects upon interaction between peptide HyM-10 and a 13-mer deoxyribonucleotide duplex.
We prefer quantitative footprinting as the main method of analysis, because it is difficult for most other techniques to discriminate multiple DNA binding sites engaged in allosteric interactions. In assessing the results, the coefficient determined by the Hill equation has been used as a comparative parameter for expressing cooperativity because of its wide applicability in research.24–26 Circular dichroism (CD) complements these techniques and is particularly useful for investigating the conformational and stoichiometric aspects of DNA–peptide binding. The influence of temperature upon the allosteric behavior of peptide RY-12 was studied by quantitative DNase I footprinting as well as CD experiments. Further valuable insight into the thermodynamic basis of the allosteric interactions has been gained using isothermal titration calorimetry (ITC).
2 Results and discussion
The major objective of the present study was two-fold: firstly, to examine the footprinting behavior of five designed peptides for the purpose of verifying our recent hypothesis that three different types of network-based allosteric communication occur when synthetic peptides bind to multiple DNA recognition sites.17,18 Secondly, to gain insight into the thermodynamic basis for the allosteric interaction of peptides incorporating the XP(Hyp)RK motifs with DNA. Hydrogen bonding, broadly categorized as monodentate or interstrand bidentate interaction,27 between the peptide moieties and DNA bases, is central to sequence-selective DNA–protein27 and DNA–small ligand14–18 recognition processes. The interactions of the Arg and Lys residues of the designed peptides with DNA bases are crucial in generating interstrand bidentate interactions as well as monodentate interactions.27 The “X” amino acid residues of these peptides, such as Ser, Glu, Gln, Tyr and Met, were chosen because their side chains could also engage in monodentate interactions with the DNA bases.27The position of interstrand bidentate interactions can be assigned by connecting bases (with pecked lines) wherever significant simultaneous DNase I blockage can be discerned on complementary strands in the differential cleavage plots (Fig. 1). The peaks of the inhibition profiles are virtually always shifted 2–3 nucleotide pairs in the 3′-direction, signifying that the ligand lies in the minor groove of the helix where DNase I cuts it.17,18
![Differential cleavage plots comparing the susceptibility of DNA fragments to DNase I cleavage after incubation with each peptide in cacodylate buffer at room temperature for 60 min. The upper traces represent the differential cleavage plot for a given peptide bound to the 5′-[32P]-labeled upper strand (158-mer) DNA fragment; the lower traces represent the corresponding plots for the 5′-[32P]-labeled lower strand (135-mer) DNA fragment. Vertical dotted lines between DNA bases represent the assignment of interstrand bidentate interactions where significant coincident H-bonding interactions occur between complementary bases in both strands. The incubation temperature for peptides HyS-10, HyE-10, PyMK-10 and PyQK-10 was 37 °C, whereas the incubation temperatures for peptide YR-12 were as stated.](/image/article/2013/OB/c2ob26320c/c2ob26320c-f1.gif) | ||
| Fig. 1 Differential cleavage plots comparing the susceptibility of DNA fragments to DNase I cleavage after incubation with each peptide in cacodylate buffer at room temperature for 60 min. The upper traces represent the differential cleavage plot for a given peptide bound to the 5′-[32P]-labeled upper strand (158-mer) DNA fragment; the lower traces represent the corresponding plots for the 5′-[32P]-labeled lower strand (135-mer) DNA fragment. Vertical dotted lines between DNA bases represent the assignment of interstrand bidentate interactions where significant coincident H-bonding interactions occur between complementary bases in both strands. The incubation temperature for peptides HyS-10, HyE-10, PyMK-10 and PyQK-10 was 37 °C, whereas the incubation temperatures for peptide YR-12 were as stated. | ||
The two decapeptides Ser-Hyp-Arg-Lys-(Py)4-Lys-Arg-NH2 (HyS-10) and Glu-Hyp-Arg-Lys-(Py)4-Lys-Arg-NH2 (HyE-10) were designed such that the number of amino acid residues is minimized, with four Py residues sandwiched between a XHypRK motif and a terminal dipeptide (KR) motif. A single amino acid residue variation (Ser or Glu) at the N-terminal was introduced to investigate the differences in sequence and network selectivity. Recently, we found that incorporating a new KPXR motif on the N-terminal side also affords designed peptides that bind well to DNA. Two peptide amides of this new series were synthesized, each differing only in the third amino acid residue: Lys-Pro--Arg-(Py)4-Lys-Arg-NH2 (PyMK-10), and Lys-Pro--Arg-(Py)4- Lys-Arg-NH2 (PyQK-10). Footprinting experiments show that these decapeptides bind sequence-selectively with significant positive cooperativity, as expected. The autoradiographs for the footprinting of these peptides are shown in Fig. 2 and Fig. i(a) (ESI†), and the binding parameters are shown in Table 1 (incubation temperature 37 °C).
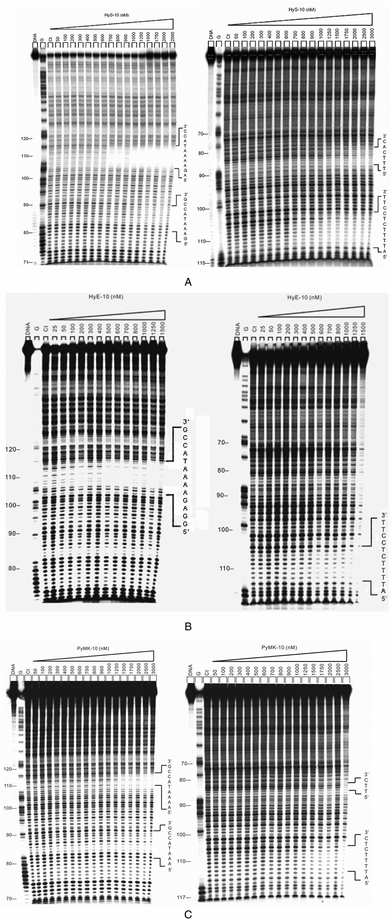 | ||
| Fig. 2 (A) Autoradiographs showing DNase I footprinting of peptide HyS-10 on DNA duplexes labeled at the 5′ end: 158-mer upper strand, left panel; 135-mer lower strand, right panel. Peptide HyS-10 was equilibrated with the DNA in 5 mM sodium cacodylate buffer, pH 6.5 at 37 °C for 60 min before DNase 1 cleavage. G represents a Maxam–Gilbert guanine sequencing track and Ct shows a DNase I digestion control lane. (B) DNase I footprinting of peptide HyE-10 on DNA duplexes labeled at the 5′ end: upper strand, left panel; lower strand, right panel. (C) DNase I footprinting of peptide PyMK-10 on DNA duplexes labeled at the 5′ end: upper strand, left panel; lower strand, right panel. | ||
| Ligand | Binding site position | Recognition sequence | K a | n H | Position of interstrand bidentate interactions | Type of interaction network |
|---|---|---|---|---|---|---|
| K a and nH are the apparent association constant and Hill coefficient determined from concentration-dependent DNase I footprinting studies, respectively. Binding sites on the upper and lower strands are identified by U and L, respectively. | ||||||
| HyS-10 | U82-90 | 5′-GAAATACCG-3′ | 1.2 × 106 | 4.0 | 82–85 | Circuit |
| U106-115 | 5′-AGAAAATACC-3′ | 1.7 × 106 | 4.5 | 106–112 | ||
| L85-80 | 5′-TTTCAC-3′ | 8.7 × 105 | 1.5 | |||
| L112-102 | 5′-ATTTTCTCCTT-3′ | 1.5 × 106 | 4.0 | |||
| HyE-10 | U104-116 | 5′-GGAGAAAATACCG-3′ | 1.1 × 107 | 1.8 | 104–112 | Partial-circuit |
| L112-102 | 5′-ATTTTCTCCTT-3′ | 3.4 × 106 | 1.7 | |||
| PyMK-10 | U83-90 | 5′-AAATACCG-3′ | 8.4 × 105 | 2.4 | 83–84 | Circuit |
| U108-116 | 5′-AAAATACCG-3′ | 6.4 × 105 | 4.3 | 108–110 | ||
| L84-82 | 5′-TTC-3′ | 6.5 × 105 | 3.2 | |||
| L110-105 | 5′-ATTTTCTC-3′ | 9.6 × 105 | 5.4 | |||
| PyQK-10 | U85-90 | 5′-ATACCG-3′ | 9.1 × 105 | 2.5 | 107–112 | Partial-circuit |
| U107-117 | 5′-GAAAATACCG-3′ | 9.9 × 105 | 3.1 | |||
| L112-107 | 5′-ATTTTC-3′ | 1.0 × 106 | 3.1 | |||
To explore the temperature-dependence of sequence preference in the XHypRK series of peptides, we went on to examine DNase I footprinting with a dodecapeptide RY-12 using the same DNA fragments at three incubation temperatures (Fig. 1 and Fig. i(b)–i(d), ESI†). RY-12 was designed with its C-terminal tetrapeptide sequence reversed compared to the parent peptide PyPro-1218 and has two tyrosine residues substituted for histidine. At the usual incubation temperature of 37 °C, like HyE-10, dodecapeptide RY-12 seems to bind at only two strong sites: on the upper strand around position U108-115, corresponding to the sequence 5′-AAAATACC-3′ and showing weak positive cooperativity (nH = 1.4), and on the lower strand around position L112-105 comprising the sequence 5′-ATTTTCTC-3′ with more significant positive cooperativity (nH = 2.4). Weak DNase 1 blockages are also observed in position U84-90. Interstrand bidentate interactions are therefore assigned around position 108–112. At 31 °C, the sequence preference of RY-12 remains much the same (Fig. 1 and Fig. i(c), ESI†), with significant positively cooperative binding at position U106-116 (nH = 2.8) and position L112-105 (nH = 2.2) indicating interstrand bidentate interactions around position 106–112.
When the incubation temperature is lowered to 25 °C, significant changes occur (Fig. 1 and Fig. i(b), ESI†). Peptide RY-12 now picks up the same four sites as peptides HyM-10, HyQ-10 and HyS-10, namely two on the upper strand, both displaying strong cooperativity: U84-90 (nH = 3.4) and U107-116 (nH = 2.9) together with two on the lower strand, L86-80 (nH = 3.4) and L112-105 (nH = 2.7). Interstrand bidentate interactions are assigned around positions 84–86 and 107–112.
Based on the footprinting results, network models17,18 were constructed for each peptide in an effort to interpret the complex communication between binding sites (Fig. 3A and B). It is clear that a broadly similar model applies to all of them, suggesting network-based allosteric communication between practically all recognition sites. Conformational changes at particular binding loci must be readily transmitted to adjacent binding sites and also to sites on the complementary strand, facilitating (or possibly impeding) the binding of further peptide molecules. In this way, occupation of almost any binding locus can be signaled to adjacent sub-binding sites and also to sites on the complementary strand, doubtless via interstrand bidentate interactions. Allosteric communications may occasionally be transmitted to a more remote binding site on the same DNA strand, spanning as many as 12–16 base pairs. Recent footprinting experiments18 show that the nH values for peptides PyHyp-12 and PyHyp-9 binding to 81-mer duplexes (S-81) containing a single d(AAAA)-d(TTTT) binding locus are significantly higher than those of the 158-mer and 135-mer duplexes, which contain two binding loci d(AAAA)-d(TTTT) and d(AAA)-d(TTT). This finding suggests that the allosteric relay of peptide binding to neighboring d(AAA)-d(TTT) and d(AAAA)-d(TTTT) positions spanning an intervening sequence of 12–14 base pairs on the same DNA strand may be mediated via a negative cooperative effect.
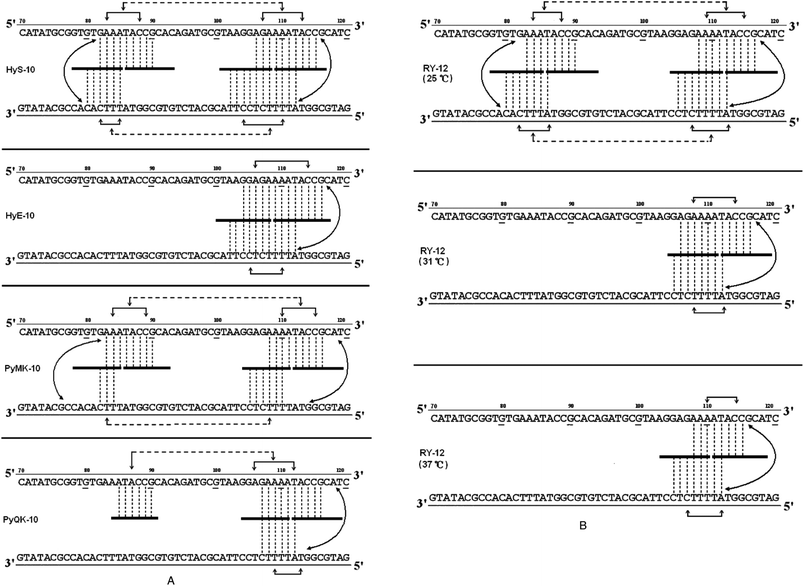 | ||
| Fig. 3 (A) Proposed allosteric interaction network models for cooperative binding of peptides HyS-10, HyE-10, PyMK-10, and PyQK-10 to pBR322 fragments based on quantitative footprinting experiments. The portion of the ligand binding to each DNA site/sub-site is represented by a thick horizontal line. Monodentate interactions and interstrand bidentate interactions are represented by vertical broken lines. The solid horizontal arrow lines represent communication of the allosteric interaction between DNA sub-binding sites. Broken horizontal arrow lines between neighboring binding loci some 12–16 nucleotides apart are intended to represent moderate or weak cooperative communication. (B) Proposed allosteric interaction network models for the cooperative binding of peptide RY-12 to pBR322 fragments at different incubation temperatures based on quantitative footprinting results. Representations as in the legend to (A). | ||
The allosteric interaction network models support the idea that binding of decapeptides HyS-10 and PyMK-10 to pBR322 fragments possessing multiple sites proceeds via a circuit type17,18 of process within a network system: the communications form a closed circuit (Fig. 3A). In the circuit type of interaction network, strong intrastrand allosteric communications are conveyed by conformational changes between two peptide molecules bound to two sub-sites of a wide binding site (Fig. 3A and B). Allosteric communications can also be relayed between complementary sites from one strand to another through interstrand bidentate interactions. Weak intrastrand allosteric communications may persist between two widely separated binding sites spanning 12–15 base pairs as, for example, the binding of peptide HyS-10 to two neighboring DNA loci: 5′-AGAAAATACC-3′′ and 5′-GAAATACCG-3′ at positions U106-115 and U82-90, respectively (Fig. 3A). On the other hand, peptide HyE-10 fails to bind to the d(AAA)-d(TTT) site, and peptide PyQK-10 refuses to bind the d(TTT) site in the lower strand, both resulting in a partial or incomplete circuit network.17,18
It is interesting that temperature has a notable effect on the allosteric interaction behavior of peptide RY-12 (Fig. 3B). At 25 °C, the peptide engages in a circuit type of network interaction. At 31 °C and 37 °C, it switches to a partial-circuit type of network in which the allosteric communications form a partial or incomplete circuit. By contrast, previous studies revealed that the interaction network of peptides (HPRK)3NH2 [HR-12]16 and (SPRK)3NH2 [SP-12]16 binding to DNA fragments is devoid of interstrand bidentate interactions and consequently is referred to as a non-circuit type of allosteric communication. Thus, quantitative footprinting results in this work as well as other studies further support our recent hypothesis17,18 of three different types of allosteric interaction networks in peptide–DNA molecular recognition.
To gain further evidence that conformational changes are involved in the molecular recognition process, we employed circular dichroism spectroscopy (CD) to investigate changes in the helical structure associated with peptide–DNA binding. A 13-mer deoxyribonucleotide duplex was prepared, d(TAGGAGAAAATAC)-d(GTATTTTCTCCTA) (U4A-L4T), in which 12 base pairs correspond to the complementary recognition site of the 158-mer fragment at positions 104–114. With this duplex a negative CD band occurs around 248 nm and a positive band around 276 nm (Fig. 4A). Peptide HyS-10 produces a dose-dependent negative CD enhancement around 248 nm as well as two strong positive CD enhancement bands around 276 nm and 328 nm (Fig. 4B). In the difference spectra the negative band around 248 nm can be seen to be red-shifted to 267 nm and the two strong positive bands appear around 288 nm and 332 nm (Fig. 4C). A near-isoelliptic point occurs at 278 nm, consistent with a two-component binding process. The induced positive band around 332 nm is peptide concentration-dependent and free from interference by native DNA and peptide, so it can be used for plotting binding isotherms. The significant enhancement of CD around 332 nm by HyS-10 and other peptides17,18 indicates that the principle interaction lies in the minor groove of DNA.11–13
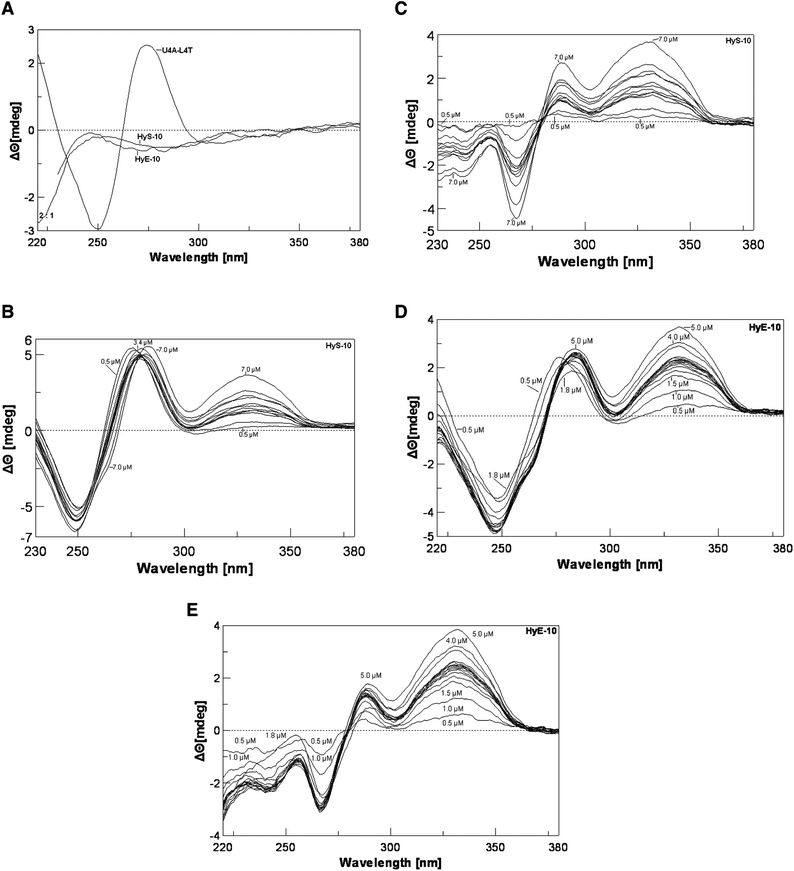 | ||
| Fig. 4 Panel A: CD spectra of DNA duplex U4A-L4T alone and peptide alone at 37 °C. Panel B: titration of duplex U4A-L4T versus peptide HyS-10 at peptide concentrations of 0.5, 1.0, 2.2, 2.4, 2.6, 2.8, 3.0, 3.2, 3.4, 4.0, 5.0, 7.0 μM at 37 °C. Panel C: corresponding CD difference spectra with the contribution of free duplex and peptide HyS-10 subtracted. Panel D: titration of U4A-L4T versus peptide HyE-10 at peptide concentrations of 0.5, 1.0, 1.5, 1.8, 2.1, 2.2, 2.3, 2.4, 2.5, 2.6, 2.7, 2.8, 3.0, 3.5, 4.0, 5.0 μM at 37 °C. Panel E: corresponding difference spectra with the contribution of free duplex and peptide HyE-10 subtracted. | ||
The CD spectra seen with decapeptides HyE-10 (Fig. 4D and E), PyMK-10 and PyQK-10 (Fig. ii(a–d), ESI†) are similar to that of peptide HyS-10. All four peptides induce a strong positive CD band around 330 nm, which is correlated with peptide–DNA binding stoichiometry. Fig. 5A and B show the variation of CD with [peptide]/[duplex 4A-4T] ratio. Peptide HyE-10 induces the greatest ellipticity change among the three peptides, suggesting that more drastic conformational changes in the DNA minor groove accompany the binding of this peptide. At peptide/DNA ratios between 0.5 and 2.0 it is likely that one molecule of each peptide binds to the d(AAAA)-d(TTTT) locus of the minor groove, as suggested in some plots by a hint of a plateau. At [peptide]/[duplex] ratios between 2 and 3 small plateau regions do appear in some of the titration curves, indicating that two peptide molecules bind in a dimeric fashion to the binding site in the minor groove. As the [peptide]/[duplex] ratio rises above 3, progressive, usually slower, increases occur in Δθ, suggesting that further peptide molecules start to bind non-specifically—probably to the wide major groove, which can loosely accommodate more peptide molecules than the minor groove (Fig. 5A and B). The CD observations concur with the DNase I footprinting results in suggesting that dimeric peptide binding to the d(AAAA)–d(TTTT) locus is often favored, consistent with the very long binding sites spanning over 9–14 base pairs. Subsequent isothermal titration calorimetric experiments also agree that dimeric peptide binding to the d(AAAA)–d(TTTT) locus may be favored, as described below. The dramatic drop in Δθ after the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 peptide/DNA molar ratio induced by peptide PyMK-10 is unusual, and might indicate dissociation of this peptide from the minor groove at higher concentrations (Fig. 5B).
:1 peptide/DNA molar ratio induced by peptide PyMK-10 is unusual, and might indicate dissociation of this peptide from the minor groove at higher concentrations (Fig. 5B).
![Panel A: plot of CD intensity at 322 nm versus [peptide]/[duplex] for the titration of duplex U4A-L4T with peptides HyS-10, HyE-10, and RY-12 at 37 °C. The proposed stoichiometric binding ratios are as indicated, with binding <2 : 1 considered to be predominantly 1 : 1. Panel B: plot of CD at 322 nm versus [peptide]/[duplex] for PyMK-10 and PyQK-10. Panel C: plot of CD at 322 nm versus [RY-12]/[duplex] at incubation temperatures of 25 °C, 31 °C and 37 °C. Panel D: plot of CD intensity at 322 nm versus [peptide]/[duplex] at various sodium chloride concentrations at 37 °C. Panel E: plot of CD intensity at 322 nm versus sodium chloride concentration.](/image/article/2013/OB/c2ob26320c/c2ob26320c-f5.gif) | ||
| Fig. 5 Panel A: plot of CD intensity at 322 nm versus [peptide]/[duplex] for the titration of duplex U4A-L4T with peptides HyS-10, HyE-10, and RY-12 at 37 °C. The proposed stoichiometric binding ratios are as indicated, with binding <2:1 considered to be predominantly 1:1. Panel B: plot of CD at 322 nm versus [peptide]/[duplex] for PyMK-10 and PyQK-10. Panel C: plot of CD at 322 nm versus [RY-12]/[duplex] at incubation temperatures of 25 °C, 31 °C and 37 °C. Panel D: plot of CD intensity at 322 nm versus [peptide]/[duplex] at various sodium chloride concentrations at 37 °C. Panel E: plot of CD intensity at 322 nm versus sodium chloride concentration. | ||
Three DNA–peptide incubation temperatures were investigated in CD studies with the dodecapeptide RY-12 (Fig. iii(a)–(f), ESI†). In the plot of Δθ versus [peptide]/[DNA] (Fig. 5C), it is evident that the change in ellipticity induced by peptide RY-12 measured at 327 nm increases steadily as the incubation temperature is lowered from 37 °C to 25 °C, indicating that the induced conformational change in the DNA minor groove is more pronounced at lower temperatures. These CD results are again in accord with the quantitative footprinting data. Recall that at 25 °C, peptide RY-12 engages in a circuit type of allosteric interaction network (marked DNA conformational changes for conveying allosteric communications), whereas at 31 °C and 37 °C the allosteric networks for this peptide switch to partial-circuit type (less DNA conformational change to power allosteric interactions).
To assess the effect of ionic strength on peptide–DNA interactions, we monitored the effect of sodium chloride on the binding of a decapeptide incorporating both XHypRK and Py motifs, Met-Hyp-Arg-Lys-(Py)4-Lys-Arg-NH2 (HyM-10), to the U4A-L4T duplex using circular dichroism. A significant, monotonic decrease in ellipticity at 320 nm was observed as the concentration of NaCl was increased (Fig. 5D), indicating a near-linear relationship (Fig. 5E). (Note again the hints of small plateaux round about the molar ratios of 1.0 and 2.0.) The obvious interpretation is that the conformation of the DNA minor groove in the peptide complex is significantly changed upon increasing the salt concentration, supporting the view that ionic interactions play an important role in the sequence-selective binding of peptides incorporating the XHypRK and Py motifs.
With some understanding of the interaction network(s) underlying cooperative sequence-selective binding of XP(Hyp)RK peptides to DNA, we went on to investigate their energetic basis. Isothermal titration calorimetry (ITC) furnishes a direct means to study the thermodynamics of binding of peptides like the five described here. For purposes of comparison, another seven peptides previously reported were included, and the chosen DNA substrate was the oligonucleotide duplex U4A-L4T (Table 3). The extra peptides included four more decapeptides, HyH-10, PyHK-10, HyM-10 and HyQ-10, and three more dodecapeptides: RPyH-12, PyHyp-12, and RHyp-12 (structures in Table 3). Representative ITC results for peptides HyS-10, HyE-10, PyMK-10 and PyQK-10 are shown in Fig. 6A; other ITC data are presented in Table 3 and in Fig. iv (ESI†). The relative concentrations of peptide and DNA duplex in each titration had to be adjusted carefully in order to produce satisfactory plots of integrated heat versus peptide/DNA molar ratio.
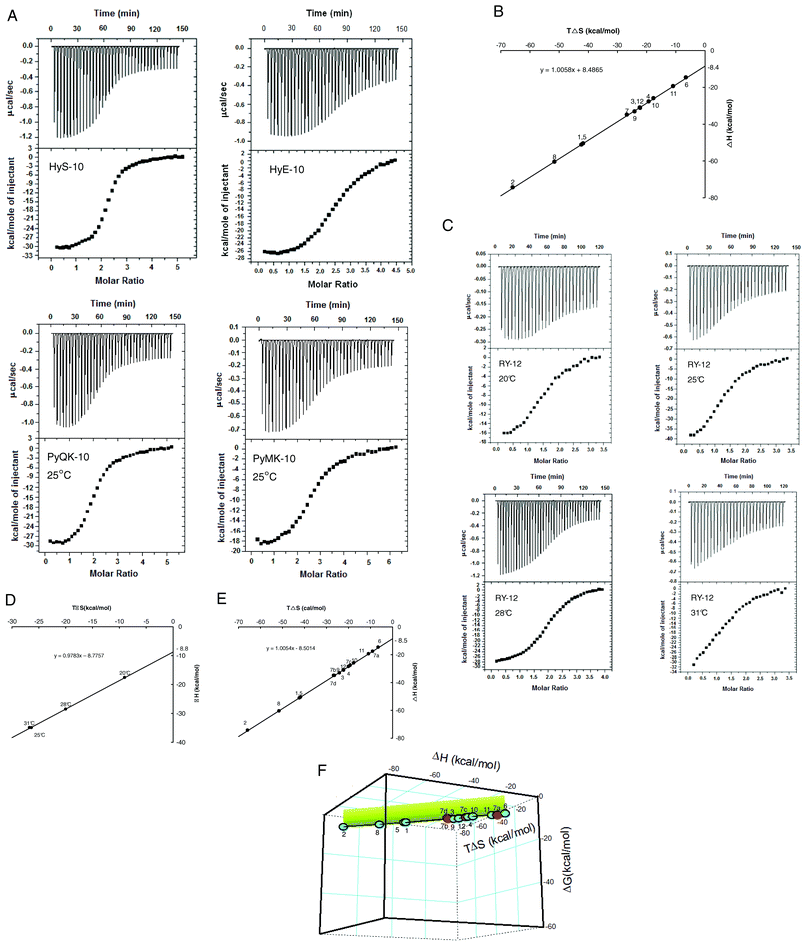 | ||
| Fig. 6 (A) ITC curves for titration of decapeptides HyS-10, HyE-10, PyMK-10 and PyQK-10 into the U4A-L4T duplex at 25 °C. For each experiment the top panel represents the raw heat of binding generated by successive additions of peptide, and in the bottom panel the integrated heat is plotted versus peptide/DNA molar ratio. Data acquisition and analysis were performed using nonlinear least-squares fitting algorithm software (Microcal Origin 7.1). (B) Plot of enthalpy versus entropy (TΔS) from ITC of twelve peptides added to the U4A-L4T duplex at 25 °C (298 K). (C) ITC curves for the titration of peptide RY-12 into the U4A-L4T duplex at various incubation temperatures (legend as in (A)). (D) Plot of enthalpy versus entropy (TΔS) from ITC of peptide RY-12 added to the U4A-L4T duplex at different temperatures. (E) Combined enthalpy–entropy plot from titrating all twelve peptides into the U4A-L4T duplex at 25 °C (with peptide RY-12 at various temperatures: 7a, 20 °C; 7b, 25 °C; 7c, 28 °C; 7d, 31 °C). (F) Three-dimensional plot of ΔH, TΔS and ΔG from titration of all twelve peptides into the U4A-L4T duplex. The short vertical projection plane (in green) corresponds to ΔG magnitudes sustained around −8.5 kcal mol−1. The figure represents a view of the ‘cube’ from below. | ||
The ITC plots for all twelve peptides versus U4A-L4T reveal a peptide/DNA binding stoichiometry near 2:1, suggesting that the peptides become bound to the model duplex in dimeric fashion, neatly in agreement with the footprinting and circular dichroism results. Moreover, all twelve peptides showed predominantly exothermic behavior in the binding reaction: ΔH was found to vary between −14.7 kcal mol−1 and −74.4 kcal mol−1, with most peptides yielding ΔH values greater than −26 kcal mol−1 measured at 25 °C (Table 3). Compared to the moderate magnitude of ΔH reported in the pioneering studies of Breslauer28 and his colleagues on the binding of netropsin, distamycin, ethidium and daunorubicin to duplex poly[d(A-T)]·[d(A-T)], where ΔH lies within the range −8.9 to −18.5 kcal mol−1, the ITC results in this study suggest that binding of the XP(Hyp)RK and KPXR peptides to DNA is overwhelmingly enthalpy-driven. However, the opposing entropy effects for all of the XP(Hyp)RK and KPXR peptides are strikingly unfavorable: TΔS falls within the range −6.5 to −65.9 kcal mol−1. In consequence, apparent net enthalpy–entropy compensation (EEC) effects (Fig. 6B) sustain a binding free energy change ΔG between −8.2 and −9.1 kcal mol−1 for all twelve peptide–DNA interactions. The linearity of ΔH versus TΔS is impressive (Fig. 6B): slope = 1.0061; y-intercept = −8.5 kcal mol−1. It originates from unusually high values of ΔH and ΔS that compensate each other. But by contrast, the individual relationship between ΔH and ΔG, and that between ΔG and TΔS, is non-linear and unpredictable (Fig. v, ESI†).
Much controversy and interest has surrounded the EEC phenomenon,29 with various theoretical explanations adumbrated, yet the physical basis of EEC has not been convincingly elucidated. The overwhelmingly large magnitudes of ΔH and ΔS measured by ITC for DNA–peptide interaction in this study seem to offer a unique advantage for investigating the energetic basis of the compensation.
According to Williams and co-workers,30 the binding free energy ΔG can be parsed according to the following equation:
| ΔG = ΔGt+r + ΔGr + ΔGh + ΣΔGp + ΔGconf. + ΔGvdw | (1) |
where ΔGt+r refers to the unfavorable cost of association when a ligand binds to a receptor, losing its translational (t) and rotational (r) entropy considerably, relative to the receptor. ΔGr refers to the energy cost of n internal rotors. The term ΔGh represents the benefit of the net hydrophobic effect and ΣΔGp expresses the benefit from the formation of hydrogen bonding and ionic interactions. ΔGconf. is the total conformational strain energy generated upon binding, and ΔGvdw is the van der Waals energy difference between the free and bound states.
Eqn (1) was applied to the present peptide–receptor interaction data. Circular dichroism studies of the DNA–small molecule interaction, in this work as well as others, show that marked conformational changes in the DNA minor groove occur upon peptide binding. Thus, the free energy components ΔGt+r and ΔGr must include the total translational and rotational entropy of both the DNA duplex and the ligand. The strong negative entropy changes seen with all ten peptides upon binding to DNA suggest that there are significant losses in the rotational and translational motion of the DNA duplex as well as the peptide. Multiple intrastrand and interstrand bidentate interactions between the peptide moieties and DNA bases revealed by our footprinting experiments (Table 1, Fig. 1 and 3), together with ionic interactions between the positively charged Arg and Lys side chains and DNA phosphates as evidenced by CD studies (Fig. 5E), are consistent with considerable loss in translational and rotational entropy of the peptide and DNA duplex upon binding, as will be seen.
Since the net binding free energy ΔG of the twelve peptides titrated against the U4A-L4T duplex lies rather constant within the range −8.2 to −9.1 kcal mol−1, the average value of ΔG for the peptides can be taken as being around −8.5 kcal mol−1. Indeed, in a plot of ΔH vs. TΔS, the enthalpy–entropy compensation ratio ke (slope of the straight line) for the twelve peptides containing the XP(Hyp)RK and Py motifs approaches unity (slope = 1.006), with the y-axis intercept equal to the average ΔG value: −8.53 kcal mol−1 (Fig. 6B). Put mathematically,
| ΔH = keTΔS − 8.5 |
When ke approaches unity,
| ΔH = −8.5 + TΔS | (2) |
Eqn (2) well describes the strict linear dependence of ΔH on TΔS for the twelve peptide–DNA interactions studied and hence underlines the enthalpy–entropy compensation phenomenon. The almost exact compensation of energy between the favorable negative ΔH and unfavorable negative ΔS energy components is what sustains the almost constant value of net binding free energy ΔG. According to eqn (2), maintenance of a negative ΔG of acceptable magnitude seems to be crucial for DNA–peptide binding; any decrease of ΔG (less negative) would require a lowering of ΔH (less negative) and a concomitant change in ΔS (more negative)—both unfavorable for the binding process.
Since the dodecapeptide RY-12 displayed notable differences in binding preference at lower temperatures, an attempt was made to perturb the enthalpy–entropy compensation by varying the incubation temperature (Fig. 6C). Accordingly, peptide RY-12 was titrated against duplex U4A-L4T at 20, 25, 28, and 31 °C (unstable calorimetric readings were recorded at 37 °C). Strikingly, the ΔH and TΔS values measured for peptide RY-12 over this temperature range were still found to be linearly related, with a slope of 0.98. Although the temperature variation undoubtedly produces significant deviations in ΔH and TΔS individually, the ΔG values still lie within the range −8.2 to −8.6 kcal mol−1 (Table 3). Plotting enthalpy versus entropy for all twelve peptides binding to the model duplex, including the data for peptide RY-12 at different temperatures (Fig. 6E), still gives a straight line of slope 1.005, so evidently the global value of ΔG does remain remarkably constant. To underline the EEC phenomenon in the DNA–peptide interaction, it is instructive to plot a 3-dimensional figure of ΔH, TΔS and ΔG. The result is a near-straight line suspended in thermodynamic space, nicely illustrating the linear enthalpy–entropy-free energy relationship (Fig. 6F). The conclusion is that stringent constraints act in nature to preserve EEC so as to maintain favorable ΔG within a constant or narrow range for successful binding of ligands to DNA.
On the other hand, we know that temperature affects the nature of allosteric interaction networks on the pBR322 fragments because of the quantitative footprinting experiments (Fig. 3), which also establish that binding of peptides to the d(AAAA)-d(TTTT) and d(AAA)-d(TTT) sites on the pBR322 fragments occurs with significant positive cooperativity (Table 1). These DNA–peptide interaction results are consistent with Cooper and Dryden's suggestion31 that positive cooperativity in ligand binding will induce a ‘stiffening’ or tightening effect in the protein/receptor, initiated by the loss of many internal vibrational degrees of freedom. Williams and co-workers30,32 concluded that highly exothermic binding is associated with positive cooperativity, allied to improved bonding within the receptor in its drug-bound state, which would account for the very unfavorable negative entropy change. As predicted by Williams30 positive cooperativity and negative cooperativity can coexist in the same system. Our previous studies show that DNA binding of the peptide PyPro-1218 does involve concomitant positively and negatively cooperative site binding, although positive cooperativity is always predominant for peptides incorporating the XPRK motifs. If positive cooperativity is associated with improved bonding within the DNA duplex upon peptide binding, it is most likely related to the existence of multiple intrastrand monodentate interactions and interstrand bidentate interactions between the peptide moieties and the DNA bases. Doubtless ionic forces between the positively charged Arg and Lys side chains of the peptides and DNA phosphates also restrict the conformational mobility of the DNA duplex and thereby help to account for the dramatic decrease in total entropy. According to the law of conservation of energy, any loss in entropic energy must transform to exothermic energy, which in turn causes an increase in enthalpy. On the other hand, high exothermic energy promotes allosteric adjustment of DNA conformation within the binding loci by means of hydrogen bonding and ionic interactions between the DNA duplex and peptide, augmenting the favorable energy components ΔGh, ΣΔGp, ΔGconf., and ΔGvdw, and facilitating positively cooperative binding of approaching peptide molecules. This is fully consistent with the proposal of Williams30 that highly exothermic binding is associated with significant positive cooperativity, and is verified by our discovery of positively cooperative peptide binding to multiple DNA recognition sites by DNase I footprinting experiments in this study (Tables 1 and 2) as well as previously.17,18 This communication of cooperativity between multiple binding loci forms the basis of the DNA–peptide interaction networks proposed here.
| Incubation temperature | Binding site position | Recognition sequence | K a | n H | Position of interstrand bidentate interactions | Type of interaction network |
|---|---|---|---|---|---|---|
| Details as in the footnote to Table 1. | ||||||
| 25 °C | U84-90 | 5′-AATACCG-3′ | 1.6 × 106 | 3.4 | 84–86 | Circuit |
| U107-116 | 5′-GAAAATACCG-3′ | 1.9 × 106 | 2.9 | 107–112 | ||
| L86-80 | 5′-ATTTCAC-3′ | 2.1 × 106 | 3.4 | |||
| L112-105 | 5′-ATTTTCTC-3′ | 3.3 × 106 | 2.7 | |||
| 31 °C | U106-116 | 5′-AGAAAATACCG-3′ | 2.1 × 106 | 2.8 | 106–112 | Partial-circuit |
| L112-105 | 5′-ATTTTCTC-3′ | 3.5 × 106 | 2.2 | |||
| 37 °C | U108-115 | 5′-AAAATACC-3′ | 1.9 × 106 | 1.4 | 108–112 | Partial-circuit |
| U84-90 | 5′-AATACCG-3′ | — | — | |||
| L112-105 | 5′-ATTTTCTC-3′ | 1.6 × 106 | 2.4 | |||
3. Conclusion
An important outcome of the present study stems from the ITC experiments on twelve peptides, showing that a dramatic decrease in entropy occurs upon binding to DNA, consistent with the presence of multiple monodentate and bidentate interactions, as well as ionic forces that mediate positively cooperative binding, resulting in a ‘tightening’ effect of DNA conformation. Allosteric communication between multiple DNA loci via cooperative peptide binding establishes the basis of the DNA–peptide interaction networks described here. Temperature-dependence studies with peptide RY-12 using quantitative DNase I footprinting are in accord with the results of CD experiments, namely that a lower temperature (25 °C) favors a circuit type of allosteric interaction network demanding more drastic DNA conformational changes, whereas higher temperatures (31 and 37 °C) afford only a partial-circuit type of interaction network that requires fewer DNA conformational changes. The enthalpy–entropy compensation effects associated with peptide RY-12 binding to the 13-mer DNA duplex persist through various temperatures, while the magnitude of ΔH and TΔS, as well as the type of allosteric interaction networks seen with RY-12 on the 135-mer and 158-mer DNA duplexes, are changed. The unusually high values of negative ΔH recorded for DNA–peptide binding furnish exothermic energy that facilitates conformational adjustment of DNA binding loci to promote positively cooperative binding of approaching peptide molecules. The net energy compensation between the favorable negative ΔH and unfavorable negative ΔS components sustains the value of net binding free energy ΔG within a remarkably constant range. We derive a linear equation which effectively describes the strict linear dependence of ΔH on TΔS for the twelve peptide–DNA interactions studied, and hence underlines the enthalpy–entropy compensation phenomenon: ΔH = −8.5 + TΔS.Thus it appears that the preservation of a negative ΔG value of acceptable magnitude is central to the EEC of many DNA–peptide interactions, and hence the findings illuminate the universal EEC paradox commonly manifested in biochemical reactions. In other words, stringent restraints affecting enthalpy–entropy compensation are conserved in nature to sustain a favorable ΔG value within a narrow range for the successful binding of ligands to DNA. We propose that the emergence of allosteric interaction networks featuring significant positive cooperativity in many DNA–small ligand interactions is also aimed at assuring an optimum binding free energy. These insights will foster the design of new DNA–binding small molecules for further study of DNA–peptide allosteric interactions as well as interaction networks.
4 Experimental
4.1 Chemicals and biochemicals
All the protected amino acid derivatives were purchased from Bachem California (Torrance, CA) and AnaSpec, Inc. (San Jose, CA) or synthesized in our own laboratory according to published procedures. All other analytical reagents were purchased from Acros, Tedia or Sigma. The radiolabelled nucleoside triphosphate [γ-32P]ATP was obtained from NEN Life Science Products at a specific activity of 6000 Ci mmol−1. Taq polymerase, T4 polynucleotide kinase, and DNase I were purchased from Promega. All other chemicals were analytical grade reagents, and all solutions were prepared using deionized, Millipore-filtered water.4.2 Chemical methods
Melting points were determined on a Mel-Temp apparatus (Cambridge, Mass) and are uncorrected. Optical rotations were determined on a Rudolph Autopol II instrument. Semi-preparative and analytical HPLC (Vydac reversed-phase columns, TP201; column 1, 1 × 25 cm; column 2, 0.4 × 25 cm) were performed using a Hitachi L-7100 pump equipped with a gradient elution device and a Soma S-3702 variable wavelength UV detector connected to a PC computer installed with Hitachi HPLC analytical software. Mass spectra were determined with a Finnigan/Thermo Quest MAT 95XL instrument operating in the electrospray ionization (ESI) mode in Chung-Hsing University.:1); ESIMS requires: 1316.47, found: 1316.0.
4.3 Polymerase chain reaction (PCR) and end-labeling of PCR products
The 158-mer DNA duplex (Watson strand 5′-32P-labeled) and 135-mer DNA duplex (Crick strand 5′-32P-labeled) were prepared by PCR amplification in a thermal cycler (ABI model 9700), as reported previously.14 The DNA concentration was determined by UV spectroscopy to lie in the range 600–800 nM.4.4 DNase I footprinting
Reactions were carried out following procedures as described in detail previously.17,184.5 Circular dichroism (CD) studies
CD spectra were measured at 37 °C with a Jasco J-815 instrument in the Institute of Chemistry, Academia Sinica. The duplex DNA was adjusted to 1.0 μM in 5 mM sodium cacodylate buffer (pH 6.5) and peptides, dissolved in the same buffer, were added to maintain final concentrations of 0.2, 1.0, 2.2, 2.4, 2.6, 2.8, 3.0, 3.2, 3.4, 4.0 and 5.0 μM. Spectra were recorded after 60 min incubation at 37 °C.4.6 Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) experiments were performed in the Institute of Chemistry, Academia Sinica using a Microcal VP-ITC (MicroCal, Northampton, MA) with a reaction cell volume of 2 mL for the oligonucleotide duplex (U4A-L4T) at 25 °C or other temperatures, as indicated in Table 3, and all samples were degassed under vacuum for 5 min. The DNA solutions of appropriate concentration in 5 mM sodium cacodylate, pH 6.5 (Table 3) were placed in the calorimeter cell. The peptide, at an appropriate concentration in 5 mM sodium cacodylate, pH 6.5 (Table 3, 273 μL), was placed in the titration syringe and injected in aliquots of 7 μL with 220 s intervals between the individual injections whilst stirring at 304 rpm, for a total of 39 injections. Data acquisition and analysis were performed using nonlinear least-squares fitting algorithm software (single site binding model, Microcal Origin 7.1 software). The Ka values were obtained from the ITC instrument by computer fit of the ITC isotherms and the corresponding ΔG values were calculated from the equation: ΔG = −RTln Ka| Entry | Peptide (mM) | DNA (mM) | ΔH (kcal mol−1) | ΔS (cal mol−1) | TΔS (kcal mol−1) | ΔG (kcal mol−1) | K a (M−1) |
|---|---|---|---|---|---|---|---|
| All ITC experiments were performed at 25 °C unless otherwise stated. The relative concentrations of peptide and DNA in each titration were adjusted in order to produce satisfactory plots of integrated heat versus peptide/DNA molar ratio. The amino acid sequences of the peptides are: Met-Hyp-Arg-Lys-(Py)4-Lys-Arg-NH2 (HyM-10), Gln-Hyp-Arg-Lys-(Py)4-Lys-Arg-NH2 (HyQ-10), Ser-Hyp-Arg-Lys-(Py)4-Lys-Arg-NH2 (HyS-10), Glu-Hyp-Arg-Lys-(Py)4-Lys-Arg-NH2 (HyE-10), His-Hyp-Arg-Lys-(Py)4-Lys-Arg-NH2 (HyH-10), His-Pro-Arg-Lys-(Py)4-Lys-Arg-NH2 (PyHK-10), Tyr-Pro-Arg-Lys-(Py)4-Lys-Arg-Pro-Tyr-NH2 (RY-12), His-Hyp-Arg-Lys-(Py)4-His-Hyp-Arg-Lys-NH2 (PyHyp-12), His-Hyp-Arg-Lys-(Py)4-Lys-Arg-Hyp-His-NH2 (RHyp-12), His-Pro-Arg-Lys-(Py)4-Lys-Arg-Pro-Tyr-NH2 (RPyY-12), Lys-Pro-Met-Lys-(Py)4-Lys-Arg-NH2 (PyMK-10), Lys-Pro-Gln-Lys-(Py)4-Lys-Arg-NH2 (PyQK-10). | |||||||
| 1 | HyM-10 (0.15) | 0.0033 | −50.5 | −140 | −41.7 | −8.8 | 2.9 × 106 |
| 2 | HyQ-10 (0.15) | 0.01 | −74.4 | −221 | −65.9 | −8.6 | 1.9 × 106 |
| 3 | HyS-10 (0.15) | 0.006 | −31.2 | −74.5 | −22.2 | −9.0 | 4.2 × 106 |
| 4 | HyE-10 (0.15) | 0.007 | −27.8 | −64.6 | −19.3 | −8.5 | 1.7 × 106 |
| 5 | HyH-10 (0.05) | 0.0033 | −51.2 | −142 | −42.3 | −8.8 | 2.9 × 106 |
| 6 | PyHK-10 (0.2) | 0.0066 | −14.7 | −21.8 | −6.5 | −8.2 | 9.8 × 105 |
| 7a (20 °C) | RY-12 (0.05) | 0.0033 | −17.6 | −30.7 | −9.0 | −8.6 | 2.7 × 106 |
| 7b (25 °C) | RY-12 (0.15) | 0.01 | −35.0 | −89.7 | −26.7 | −8.2 | 1.0 × 106 |
| 7c (28 °C) | RY-12 (0.15) | 0.008 | −28.6 | −66.4 | −20.0 | −8.6 | 1.6 × 106 |
| 7d (31 °C) | RY-12 (0.05) | 0.0033 | −35.0 | −86.7 | −26.4 | −8.6 | 1.6 × 106 |
| 8 | RPyY-12 (0.05) | 0.0045 | −60.5 | −173 | −51.6 | −8.9 | 3.3 × 106 |
| 9 | PyHyp-12 (0.05) | 0.004 | −33.2 | −80.9 | −24.1 | −9.1 | 4.4 × 106 |
| 10 | RHyp-12 (0.15) | 0.005 | −26.0 | −59.1 | −17.6 | −8.4 | 1.4 × 106 |
| 11 | PyMK-10 (0.15) | 0.005 | −19.4 | −36.5 | −10.9 | −8.5 | 1.8 × 106 |
| 12 | PyQK-10 (0.15) | 0.006 | −31.0 | −74.9 | −22.3 | −8.7 | 2.2 × 106 |
Acknowledgements
This work is dedicated to the memory of Professor Dudley H. Williams. We thank professors L. S. Kan, S. T. Chen, S. H. Wu, D. K. Chang and Y. T. Tau, Academia Sinica, for generous access to ITC and CD facilities as well as helpful advice, and professors Lin Ma, A. N. Ko, A. Yeh, C. W. Ong, and F. B. Yeh for helpful discussions and encouragement. The work was supported by grant NSC97-2113-M029-005 and funds from Tunghai Christian University, the Jeng Ching Ho Foundation, and the Inpac International Corporation.References
- K. R. Fox and M. J. Waring, Methods Enzymol., 2001, 340, 412–430 CAS.
- J. B. Chaires, K. R. Fox, J. E. Herrera, M. Britt and M. J. Waring, Biochemistry, 1987, 26, 8227–8236 CrossRef CAS.
- D. L. Boger, J.-H. Chen and K. W. Saionz, J. Am. Chem. Soc., 1996, 118, 1629–1644 CrossRef CAS.
- C. Bailly and J. B. Chaires, Bioconjugate Chem., 1998, 9, 513–538 CrossRef CAS and references cited therein.
- Y. H. Chen and J. W. Lown, J. Am. Chem. Soc., 1994, 116, 6995–7005 CrossRef CAS.
- W. L. Walker, E. M. Landaw, R. E. Dickerson and D. S. Goodsell, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 5634–5639 CrossRef CAS.
- A. Blasko and T. C. Bruice, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 10018–10022 CrossRef CAS.
- R. P. L. DeClairac, B. N. Geierstanger, M. Mrksich, P. B. Dervan and D. E. Wemmer, J. Am. Chem. Soc., 1997, 119, 7909–7916 CrossRef.
- E. J. Fechter and P. B. Dervan, J. Am. Chem. Soc., 2003, 125, 8476–8485 CrossRef CAS and references cited therein.
- M. Mrksich, M. E. Parks and P. B. Dervan, J. Am. Chem. Soc., 1994, 116, 7983–7988 CrossRef CAS.
- K. L. Buchmueller, A. M. Staples, C. M. Howard, S. M. Horick, P. B. Uthe, N. Le Minh, K. K. Cox, B. Nguyen, K. A. O. Pacheco, W. D. Wilson and M. Lee, J. Am. Chem. Soc., 2005, 127, 742–750 CrossRef CAS.
- T. Brown, H. Mackay, M. Turlinton, A. Sutterfield, T. Smith, A. Sielaff, L. Westrate, C. Bruce, J. Kluza, C. O'Hare, B. Nguyen, W. D. Wilson, J. A. Hartley and M. Lee, Bioorg. Med. Chem., 2008, 16, 5266–5276 CrossRef CAS.
- M. Munde, M. A. Ismail, R. Arafa, P. Peixoto, C. J. Collar, l. Yang, L. Hu, M. H. David-Cordonnier, A. Lansiaux, C. Bailly, D. W. Boykin and W. D. Wilson, J. Am. Chem. Soc., 2007, 129, 13732–13743 CrossRef CAS.
- J.-C. Chang, C.-H. Yang, P.-J. Chou, W.-H. Yang, I.-C. Chou, C.-T. Lu, P.-H. Lin, R. C.-W. Hou, K.-C. G. Jeng, C.-C. Cheng and L. Sheh, Bioorg. Med. Chem., 2004, 12, 53–61 CrossRef CAS.
- C.-H. Yang, W. F. Chen, M.-C. Jong, B.-J. Jong, J.-C. Chang, M. J. Waring, L. Ma and L. Sheh, J. Am. Chem. Soc., 2004, 126, 8104–8105 CrossRef CAS.
- C. H. Yang, K. C. G. Jeng, W. H. Yang, Y. L. Chen, C. C. Hung, J. W. Lin, S. T. Chen, S. Richardson, C. R. H. Martin, M. J. Waring and L. Sheh, ChemBioChem, 2006, 7, 1187–1196 CrossRef CAS.
- K. L. Kao, J. C. T. Huang, C. K. Yang, K. C. G. Jeng, J. C. Chang, W. C. Yao, S. C. Hsien, M. J. Waring, M. H. Chen, L. Ma and L. Sheh, Bioorg. Med. Chem., 2010, 18, 366–376 CrossRef CAS; J. T. B. Huang, R. C. K. Yang, W. K. Hung, M. J. Waring and L. Sheh, in Molecular Recognition, ed. J. A. McEvoy, Nova Science Publishers Inc., New York, 2011. ISBN 978-1-61122-734-5, internet open access available Search PubMed.
- J. T. B. Huang, Y. C. Chen, J. C. Chang, K. C. G. Jeng, K. L. Kao, C. K. Yang, L. S. Kan, M. T. Wey, M. J. Waring, C. S. Chen, W. J. Chien and L. Sheh, Bioorg. Med. Chem., 2010, 18, 2575–2585 CrossRef CAS.
- M. E. A. Churchill and M. Suzuki, EMBO J., 1989, 8, 4189–4195 CAS.
- M. Suzuki, Nature, 1990, 344, 562–565 CrossRef CAS.
- D. E. Koshland Jr. and K. Hamadani, J. Biol. Chem., 2002, 277, 46481–46844 Search PubMed and references cited therein.
- W. E. Calvano, et al. , Nature, 2005, 437, 1032–1037 CrossRef.
- R. B. Russell and P. Aloy, Nat. Chem. Biol., 2008, 4, 666–673 CrossRef CAS.
- M. Ciubotaru, L. M. Ptaszek, G. A. Baker, S. N. Baker, F. V. Bright and D. G. Schatz, J. Biol. Chem., 2003, 278, 5584–5596 CrossRef CAS.
- A. A. Ouameur and H.-A. Tajmir-Riahi, J. Biol. Chem., 2004, 279, 42041–42054 CrossRef CAS.
- I. Tato, S. Zunzunegui, F. de la Cruz and E. Cabezon, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8156–8161 CrossRef CAS.
- N. M. Luscombe, R. A. Laskowski and J. M. Thornton, Nucleic Acids Res., 2001, 29, 2860–2874 CrossRef CAS.
- K. J. Breslauer, D. P. Rometa, W. Y. Chou, R. Ferrante, J. Curry, D. Zaunczkowski, J. G. Snyder and L. A. Marky, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 8922–8926 CrossRef CAS.
- E. B. Starikov and B. Norden, J. Phys. Chem. B, 2007, 111, 14431–14435 CrossRef CAS and references quoted therein.
- D. H. Williams, E. Stephens, D. P. O'Brien and M. Zhou, Angew. Chem., Int. Ed., 2004, 43, 6596–6616 CrossRef CAS.
- A. Cooper and D. T. F. Dryden, Eur. Biophys. J., 1984, 11, 103–109 CrossRef CAS.
- H. Shiozawa, B. C. S. Chia, N. L. Davies, R. Zerella and D. H. Williams, J. Am. Chem. Soc., 2002, 124, 3914–3919 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c2ob26320c |
| ‡ R. C. K. Y. and J. T. B. H. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2013 |