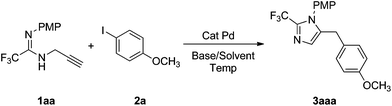
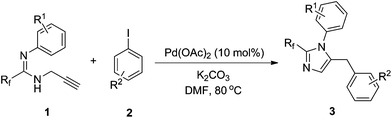
Pd-catalyzed coupling reaction of fluorinated propargyl amidines with aryl iodides†
Shan
Li
a,
Yafen
Yuan
a,
Yajun
Li
ab,
Zhengke
Li
a,
Lisi
Zhang
a and
Yongming
Wu
*a
aKey Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: ymwu@sioc.ac.cn; Fax: (+86)-021-54925190
bSchool of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, 1037 Luoyu Road, Wuhan, Hubei 430074, China
First published on 16th October 2012
Abstract
Catalyzed by ligand free Pd(OAc)2, 2,5-disubstituted imidazole was prepared in good yield by the reaction of fluorinated propargyl amidines with iodoarene. Mechanistic studies indicated that this transformation occurs through a nitropalladation–reductive elimination pathway.
Imidazoles as five-membered N-heterocyclic compounds occur in a number of natural products, in particular alkaloids.1 Owing to their biological properties, many of these are important ingredients for antibacterial, antiviral, antitumor, anticardiovascular, and antiinflammatory agents.2,3 Recently, imidazoles with fluorinated groups have received significant attention based on the fact that the physicochemical properties of organic compounds can be greatly affected by introducing fluorinated groups.4,5 With CF3I and CF3SiMe3 as the fluorinating reagents, direct trifluoromethylation has become an important approach to access these compounds.6 However, this method suffers from the use of expensive or hazardous reagents, harsh reaction conditions, and poor yield. Alternatively, imidazoles bearing fluorinated groups can be prepared from fluorinated building blocks. For example, treatment of fluorocarboxylic acid or fluorinated 1,3-dicarbonyl compounds with 1,2-diaminobenzene successfully formed 2-fluoroalkyl benzimidazoles.7 Multi-substituted imidazoles are also accessible by applying other fluorinated building blocks.8 Fluorinated propargyl amidines presented some unique properties in our work.10b Considering the reaction pattern, propargyl amidine was suitable for the preparation 2,5-disubstituted imidazoles. Herein, we disclose the synthesis of 2-fluoroalkyl-5-benzyl imidazoles from fluorinated propargyl amidines and aryl iodides using ligand free palladium acetate as the catalytic source.
In our initial study, 2-trifluoromethyl N-(p-methoxyphenyl-N-propargyl amidine 1aa was treated with p-methoxyl iodobenzene 2a (1.2 equiv.) in the presence of K2CO3 (1.5 equiv.) and Pd(OAc)2 (10 mol%) in CH3CN at room temperature. The desired product, 5-benzyl imidazole 3aaa was obtained in 26% yield. In an effort to optimize the reaction, reaction conditions were investigated by varying the temperature, base, solvent and catalyst. The outcome, however, was not promising (Table 1 in ESI†). In these reactions, 5-benzyl imidazole as the only product was obtained in low yield, no byproduct such as 5-methyl imidazole or 3-(p-methoxphenyl) prop-2-ynyl amidine was detected. The presence of the fluoroalkyl group increases the acidity of the hydrogen atom on the amidinoyl group, which leads to deprotonation under strong basic conditions, and the increased amount of propargyl amidine anion will promote the reaction. By using a 1.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.7:1.2 molar ratio of 1aa:2a:K2CO3, an improved yield of 76% was obtained when the reaction was carried out in CH3CN at 80 °C, in the presence of 10 mol% Pd(OAc)2 (Table 1, entry 1). A similar result was obtained when K3PO4 was used as the base (Table 1, entry 2). The yield of 5-benzyl imidazole 3aaa was increased to 84% when DMF was chosen as the solvent (Table 1, entry 5). The concentration of propargyl amidine was found to affect the yield of 3aaa, a lower yield was obtained with a higher concentration of 1aa (Table 1, entries 12, 13).
:0.7:1.2 molar ratio of 1aa:2a:K2CO3, an improved yield of 76% was obtained when the reaction was carried out in CH3CN at 80 °C, in the presence of 10 mol% Pd(OAc)2 (Table 1, entry 1). A similar result was obtained when K3PO4 was used as the base (Table 1, entry 2). The yield of 5-benzyl imidazole 3aaa was increased to 84% when DMF was chosen as the solvent (Table 1, entry 5). The concentration of propargyl amidine was found to affect the yield of 3aaa, a lower yield was obtained with a higher concentration of 1aa (Table 1, entries 12, 13).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Base | Temp/°C | Yield/%a |
|
a Isolated yield.
b Calculated by 19F NMR.
c The molar ratio of 1aa:2a = 1:0.8.
d The molar ratio of 1aa:2a = 1:1.
|
|||||
| 1 | Pd(OAc)2 | CH3CN | K2CO3 | 80 | 76 |
| 2 | Pd(OAc)2 | CH3CN | K3PO4 | 80 | 71 |
| 3 | Pd(OAc)2 | CH3CN | Na2CO3 | 80 | 65b |
| 4 | Pd(OAc)2 | CH3CN | Py | 80 | 58b |
| 5 | Pd(OAc)2 | DMF | K2CO3 | 80 | 84 |
| 6 | Pd(OAc)2 | DMF | K3PO4 | 80 | 75 |
| 7 | Pd(OAc)2 | DMF | Py | 80 | 46b |
| 8 | Pd(OAc)2 | DMF | K2CO3 | 60 | 70 |
| 9 | Pd(PPh3)2Cl2 | CH3CN | K2CO3 | 80 | 68 |
| 10 | Pd(PPh3)2Cl2 | DMF | K2CO3 | 80 | 77 |
| 11 | Pd(PPh3)2Cl2 | DMF | K3PO4 | 80 | 62 |
| 12 | Pd(OAc)2 | DMF | K2CO3 | 80 | 74c |
| 13 | Pd(OAc)2 | DMF | K2CO3 | 80 | 55b,d |
The scope of the palladium-catalyzed coupling of fluorinated propargyl amidines with aryl iodides by this procedure is summarized in Table 2. To our delight, this reaction shows good compatibility towards aryl iodides containing many functional groups, such as ester and acetyl groups (Table 2, entries 5, 6). Iodoarenes with both electron-donating and electron-withdrawing groups gave moderate to good yields of the corresponding products (Table 2, entries 1–7). Iodoarenes with an electron-withdrawing group give a lower yield (Table 2, entries 5–7). Steric hindrance of iodoarene imposed by ortho-substitution has little impact on the yield except for the substrate with ortho-fluorine (Table 2, entries 8, 9, entry 10). Steric and electronic effects of propargyl amidines were also negligible for the coupling reaction (Table 2, entries 11–13). Substrates with –CF2Br are too unstable under the reaction condition to afford any product (Table 1, entry 14), while good yield was obtained from substrates with –CF2H (Table 1, entry 15).
|
|
||||
|---|---|---|---|---|
| Entry | Rf | R1 | R2 | Yield/%a |
| a Isolated yield. | ||||
| 1 | –CF3 | p-OCH3 | p-OCH3 | 3aaa/84 |
| 2 | –CF3 | p-OCH3 | H | 3aab/80 |
| 3 | –CF3 | p-OCH3 | p-Cl | 3aac/85 |
| 4 | –CF3 | p-OCH3 | m-CH3 | 3aad/74 |
| 5 | –CF3 | p-OCH3 | p-COOEt | 3aae/61 |
| 6 | –CF3 | p-OCH3 | p-COCH3 | 3aaf/68 |
| 7 | –CF3 | p-OCH3 | p-CF3 | 3aag/79 |
| 8 | –CF3 | p-OCH3 | o-OCH3 | 3aah/81 |
| 9 | –CF3 | p-OCH3 | o-CF3 | 3aai/75 |
| 10 | –CF3 | p-OCH3 | o-F | 3aaj/42 |
| 11 | –CF3 | p-Cl | p-OCH3 | 3aba/72 |
| 12 | –CF3 | o-OCH3 | p-OCH3 | 3aca/86 |
| 13 | –CF3 | Naphth | p-OCH3 | 3ada/80 |
| 14 | –CF2Br | p-OCH3 | p-OCH3 | — |
| 15 | –CF2H | H | p-OCH3 | 3cea/77 |
This transformation may proceed through two pathways.9 In path I, the C–C triple bond in the propargyl amidine coordinates to the σ-aryl palladium complex. A nitrogen atom attacks the activated C–C triple bond and the propargyl amidine accepts the attack of the nitrogen atom to afford a σ-aryl-σ-(5-imidazolymethyl) palladium intermediate, which then undergoes reductive elimination to form 5-benzyl imidazole (path I). Alternatively, 3-aryl prop-2-yne amidine would be produced first from the σ-aryl palladium complex with a terminal alkyne, and then it undergoes 5-exo-dig cyclization to give the desired product (path II).
With the inductive electron-withdrawing effect of fluoroalkyl, deprotonation of amidinolyl readily takes place under basic conditions, and then undergoes intramolecular nucleophilic reaction with the activated alkyne to give the desired product. In our system, 5-benzyl imidazole was found to be the sole product. To distinguish these two pathways, we prepared N-(3-phenyl) prop-2-ynyl amidine 1aab. Under the same conditions, only a low yield (63%) of 5-benzyl imidazole 3aab was detected by 19F NMR, which was lower than from the reaction of fluorinated proparygyl amidine with iodobenzene (Table 2, entry 2). In the presence of an iodoarene, 1aab underwent a cyclization–arylation to give 5-diarymethyl imidazole 3aaba in very low yield due to large steric hindrance. Judging from these observations, it is very likely that the transformation proceeds through path I. The proposed mechanism is shown in Scheme 1. 5-Benzyl imidazole 3 is most likely generated by reductive elimination from the intermediate B, which is more stable than the σ-vinyl-σ-aryl palladium species A (Scheme 2).10
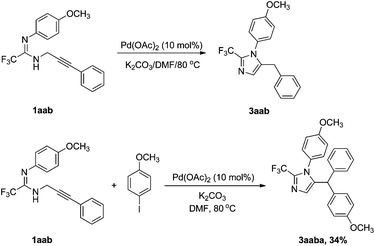 | ||
| Scheme 1 Transformations of 1aab catalyzed by Pd(OAc)2. | ||
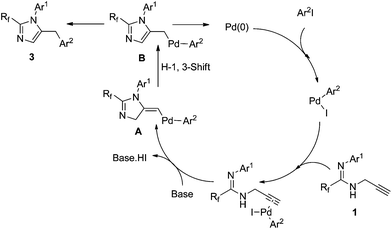 | ||
| Scheme 2 Proposed mechanism for the formation of 2,5-disubstituted imidazoles. | ||
In conclusion, a convenient protocol to generate 2,5-disubstituted imidazoles has been developed. With ligand free Pd(OAc)2 as the catalyst, 2-fluoroalkyl-5-benzyl imidazoles can be obtained in moderate to good yields from the reaction of fluorinated propargyl amidines with aryl iodides. This transformation is compatible with a wide range of functional groups. Mechanistic investigations revealed that the reaction most likely proceeds via a nitropalladation–reductive elimination pathway.
Notes and references
- (a) K. L. Rinehart, P. D. Shaw and L. S. Shield, Pure Appl. Chem., 1981, 53, 795–817 CrossRef CAS; (b) G. P. Kalemkerian and X. Ou, Cancer Chemother. Pharmacol., 1999, 43, 145–150 CrossRef CAS; (c) R. M. Jones and G. Bulaj, Conotoxins—new vistas for peptide therapeutice [J], Curr. Pharm. Des., 2000, 6, 1249–1285 CrossRef CAS; (d) M. J. Jtowle and K. A. Salvato, J. Cancer Res., 2001, 61, 1013 Search PubMed; (e) R. W. Sparidans, E. Stokvis and J. M. Jimeno, Anti-Cancer Drugs, 2001, 12, 575 CrossRef CAS; (f) C. Gajate, J. Biol. Chem., 2002, 277, 41580–41589 CrossRef CAS.
- (a) R. R. Wexler, W. J. Greenlee, J. D. Irvin, M. R. Goldberg, K. Prendergast, R. P. Smith and P. B. M. W. M. Timmermans, J. Med. Chem., 1996, 39, 625 CrossRef CAS; (b) F. G. Salituro, C. T. Baker, J. J. Court, D. D. Deininger, E. E. Kim, B. Li, P. M. Novak, B. G. Rao, S. Pazhanisamy, M. D. Porter, W. C. Schairer and R. D. Tung, Bioorg. Med. Chem. Lett., 1998, 8, 3637–3642 CrossRef CAS; (c) W. M. Kazmierski, F. G. Salituro, R. D. Tung and L. L. Wright, Bioorg. Med. Chem. Lett., 2000, 10, 1159–1162 CrossRef; (d) G. A. Reichard, C. Stengone, S. Paliwal, I. Mergelsberg, S. Majmundar, C. Wang, R. Tiberi, A. T. McPhail, J. J. Piwinkski and N. Y. Shih, Org. Lett., 2003, 5, 4249–4251 CrossRef CAS; (e) R. Morphy and Z. Rankovic, J. Med. Chem., 2005, 48, 6523 CrossRef CAS; (f) C. Sabourin and J. M. H. Robert, J. Enzyme Inhib. Med. Chem., 2008, 23, 659–667 CrossRef CAS; (g) Ł. Albrecht, H. Jiang, G. Dickmeiss, B. Gschwend, S. G. Hansen and K. A. Jørgensen, J. Am. Chem. Soc., 2010, 132, 9188 CrossRef.
- (a) R. Liebl, R. Handte, H. Mildenberger, K. Bauer and H. Bieringer, Ger. Offen. Pat., 3604042, 1987 Search PubMed; (b) P. Chiesi, V. Servadio and R. Razzetti, EP Pat., 301422, 1989 Search PubMed; (c) E. O. Renth, K. Schromm, R. Anderskewitz, F. Birke, A. Fuegner and H. Heuer, Pat., WO 9421616, 1994 Search PubMed; (d) A. F. Pozherskii, A. T. Soldatenkov and A. R. Katritzky, Heterocycles in Life and Society, Wiley, New York, 1997, 179–180 Search PubMed.
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef.
- (a) M. Hudlicky, Chemistry of organic Fluorine Compounds, Ellis Horwood Ltd, Chichester, 1992 Search PubMed; (b) I. Ojima, J. R. McCarthy and J. T. Welch, Biomedical Frontiers of Fluorine Chemistry, ACS Symposium Series, no. 639, Washington, 1996 Search PubMed; (c) L. Strekowski and H. Lee, J. Fluorine Chem., 2000, 104, 281 CrossRef CAS; (d) H. Kagoshima and T. Akiyama, Org. Lett., 2000, 2, 1577 CrossRef; (e) B. Crousse and D. Bonnet-Delpon, J. Org. Chem., 2000, 65, 5009 CrossRef CAS; (f) P. Jeschke, ChemBioChem, 2004, 5, 570–589 CrossRef CAS; (g) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- (a) H. Kimoto, S. Fujii and A. Louis, J. Org. Chem., 1982, 47, 2867–2872 CrossRef CAS; (b) M. Medebielle, S. Fujii and K. Kato, Tetrahedron, 2000, 56, 2655–2664 CrossRef CAS; (c) M. S. Wiehn, E. V. Vinogradova and A. Togni, J. Fluorine Chem., 2010, 131, 951–957 CrossRef CAS; (d) Y. Ye, S. H. Lee and M. S. Sanford, Org. Lett., 2011, 13, 5464–5467 CrossRef CAS; (e) C.-P. Zhang, Z.-L. Wang, Q. Y. Chen, C.-T. Zhang, Y.-C. Gu and J.-C. Xiao, Angew. Chem., Int. Ed., 2011, 50, 1896–1900 CrossRef CAS; (f) L. Chu and F. Qing, J. Am. Chem. Soc., 2012, 134, 1298–1304 CrossRef CAS.
- Some example reactions: (a) M. S. Hashtroudi, S. S. Nia, H. Asadollahi and S. Balalaie, Indian J. Heterocycl. Chem., 2000, 9, 307–308 CAS; (b) G. Navarrete-Vazquez, R. Cedillo, A. Hernandez-Campos, L. Yepez, F. Hernandez-Luis, J. Valdez, R. Morales, R. Cortes, M. Hernandez and R. Castillo, Bioorg. Med. Chem. Lett., 2001, 11, 187–190 CrossRef CAS; (c) N. V. Gabriel, R. V. Maria de Monserrat, Y. M. Lilian, M. Victor, G. Lucia, H. C. Alicia, C. Rafael and H. L. Francisco, Eur. J. Med. Chem., 2006, 41, 135–141 CrossRef; (d) F.-L. Ge, Z.-X. Wang, W. Wang, W.-C. Lu and J. Hao, Tetrahedron Lett., 2007, 48, 3251–3254 CrossRef CAS; (e) P. Chen, C. G. Caldwell, R. J. Mathvink, B. Leiting, F. Marsilio, R. A. Patel, J. K. Wu, H. He, K. A. Lyons, N. A. Nancy and A. E. Weber, Bioorg. Med. Chem. Lett., 2007, 17, 5853–5857 CrossRef CAS; (f) V. I. Filyakova, N. S. Boltacheva, D. V. Sevenard and V. N. Charushinam, Russ. Chem. Bull., 2010, 59, 1791–1795 CrossRef CAS; (g) H. M. Alkahtani, A. Y. Abbs and S. Wang, Bioorg. Med. Chem. Lett., 2012, 22, 1317–1321 CrossRef CAS.
- (a) S. Fujii, Y. Maki, H. Kimoto and L. A. Cohen, J. Fluorine Chem., 1986, 32, 329–343 CrossRef CAS; (b) Y. Kamitori, J. Heterocycl. Chem., 2001, 38, 773–776 CrossRef CAS; (c) H. Kim, J.-T. Lee and C. Lee, Bull. Korean Chem. Soc., 2000, 21, 345–347 CAS; (d) G. Duan, N. Zhu and V. W. Yam, Chem.–Eur. J., 2010, 16, 13199–13209 CrossRef CAS.
- (a) B. M. Nilsson and U. Hecksell, J. Heterocycl. Chem., 1989, 26, 269 CrossRef CAS; (b) P. Wipf, L. T. Rahman and S. R. Rector, J. Org. Chem., 1998, 63, 7132 CrossRef CAS; (c) J. Freeman and E. W. Huber, J. Heterocycl. Chem., 1990, 27, 343 CrossRef; (d) D. C. Clark and D. A. Travis, Bioorg. Med. Chem., 2001, 9, 2857 CrossRef CAS; (e) A. Arcadi, S. Cacchi, L. Cascia, G. Fabrizi and F. Marinelli, Org. Lett., 2001, 3, 2501 CrossRef CAS; (f) P. Wipf, Y. Aoyama and T. E. Benedum, Org. Lett., 2004, 20, 3593 CrossRef; (g) A. Bacchi, M. Costa, B. Gabriele, G. Pelizzi and G. Salerno, J. Org. Chem., 2002, 67, 4450 CrossRef CAS; (h) E. Merkul and T. J. J. Müller, Chem. Commun., 2006, 4817 RSC; (i) E. M. Beccalli, E. Borsini, G. Broggini, G. Palmisano and S. Sottocornola, J. Org. Chem., 2008, 73, 4746 CrossRef CAS.
- (a) J. P. Weyrauch, A. S. K. Hashmi, A. Schuster, T. Hengst, S. Schetter, A. Littmann, M. Rudoiph, M. Hamzic, J. Visus, F. Rominger, W. Frey and J. W. Bats, Chem.–Eur. J., 2010, 16, 956–963 CAS; (b) S. Li, Z.-K. Li, Y.-F. Yuan, D.-J. Peng, Y.-J. Li, L.-S. Zhang and Y.-M. Wu, Org. Lett., 2012, 14, 1130–1133 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General procedure for 2,5-disubstituted imidazole synthesis; Characterization data and NMR spectra for compounds 3aaa–3cea, 3aaba. See DOI: 10.1039/c2ob26377g |
| This journal is © The Royal Society of Chemistry 2013 |