Synthesis of sterically encumbered C10-arylated benzo[h]quinolines using ortho-substituted aryl boronic acids†
Marko
Weimar
and
Matthew J.
Fuchter
*
Department of Chemistry, Imperial College London, London SW7 2AZ, UK. E-mail: m.fuchter@imperial.ac.uk; Fax: +44 (0) 2075945805; Tel: +44 (0) 2075945815
First published on 5th October 2012
Abstract
The challenging coupling of 10-halobenzo[h]quinolines with ortho-substituted aryl boronic acids has been achieved using Pd(OAc)2/P(O)Ph3 as the catalytic system. High yields were obtained for diversely functionalised substrates under mild reaction conditions.
The Suzuki–Miyaura cross-coupling reaction is a highly reliable method to form aryl–aryl bonds starting from an aryl boronic acid and an aryl halide under palladium catalysis.1 It has already reached an upper level of maturity and is ubiquitously applied in synthetic chemistry, facts recently recognised by the award of a Nobel prize for one of its discoverers.2 In contrast, the highly researched field of C–H arylation is comparatively still in its infancy.3 The advantage of this methodology over standard cross-coupling reactions is the fact that it allows the coupling of (at least one) non-functionalised aromatic compounds, thus potentially saving preparative steps and chemical waste.
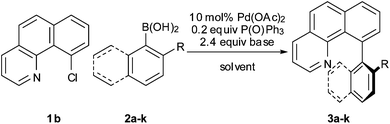
Benzo[h]quinoline represents a prevalent substrate for the development of new directed C–H arylation protocols.4 However, there is a dearth of examples of reactions using ortho-substituted arylating agents,4b,e which would afford highly sterically encumbered heterobiaryls. Through restricted rotation about the aryl–aryl bond, such bulky products would likely exist as isolatable atropoisomers and therefore be of significant interest in further conformational and stereochemical studies.5 We were surprised by this omission from C–H arylation methodology, given that in the field of conventional cross-coupling chemistry,6 especially the Suzuki–Miyaura reaction,7 the coupling of bulky substrates is an actively investigated topic. We therefore elected to study the C10-arylation of benzo[h]quinoline with bulky ortho-substituted arylating agents under both C–H arylation and more conventional coupling conditions.
Initially we screened a significant number of published procedures to examine the feasibility of arylating benzo[h]quinoline directly with ortho-substituted halobenzenes. In all cases we found that, in the case of such substrates, C–H arylation chemistry was not successful. The only procedure we found to be preparatively viable (to prepare 3a), was palladium-catalyzed C–H arylation using mixed aryl iodonium salts as reported by Sanford and co-workers.8 Since this procedure requires prior preparation of bespoke iodonium arylating agents however, the advantages of using direct arylation as opposed to more conventional coupling conditions is less clear. Consequently, we decided to study whether the Suzuki–Miyaura reaction would stand up to these challenging substrates.
Using 10-halobenzo[h]quinoline9 substrates (1a or 1b), no cross-coupling with o-toluene boronic acid (2a) occurred under standard Suzuki–Miyaura reaction conditions (10 mol% Pd(PPh3)4, 2 equiv. CsF, dioxane–H2O, 100 °C).
We reasoned that the failure of the reaction may be due to stability of the oxidative addition product,10 shutting down catalysis. Since it has been known for some time that p-benzoquinone (BQ) can function as a useful additive to promote transmetallation and reductive elimination of organopalladium complexes,11 we decided to test the effect it would have on this Suzuki reaction. To our delight, we found that the addition of 0.5 equiv. of BQ to the reaction mixture lead to the formation of 3a in 67% isolated yield (Scheme 1). While conducting control experiments, we made the surprising observation that an old batch of Pd(PPh3)4 furnished the desired product without additional BQ. We therefore concluded that one role of BQ may be the oxidation of the PPh3 ligands to P(O)Ph3 during the course of the reaction (cf. slow aerobic degradation of Pd(PPh3)4). Moreover, we found that PPh3 suppressed the reaction: An experiment with the “old” Pd(PPh3)4 catalyst and additional PPh3 did not yield any product.
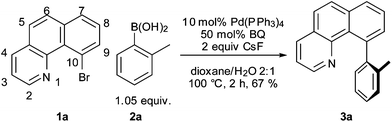 | ||
| Scheme 1 Suzuki–Miyaura cross-coupling reaction with BQ. | ||
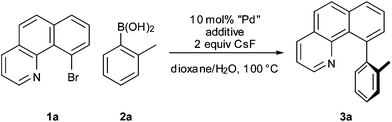
In light of these results, we turned our attention to phosphine-free palladium precatalysts and found that a combination of Pd(OAc)2 and P(O)Ph3 led to a fast reaction with complete consumption of 1a within 10 min at 100 °C (Table 1, entry 2). Denmark and co-workers have previously reported the beneficial effect of P(O)Ph3 in cross-coupling reactions of electron-rich arylsilanolates.12 At this temperature, comparable results were obtained when a Pd(0) or a Pd(II) source was used (Table 1, entries 3 and 4). Only the heterogeneous catalyst Pd black afforded an incomplete conversion even after 18 h (Table 1, entry 5). Interestingly, with a Pd(0) source (Pd(dba)2) as the catalyst, the addition of P(O)Ph3 was not necessary for the reaction to occur at 100 °C (Table 1, entry 7) whereas with a Pd(II) source (Pd(OAc)2) only traces of 3a were formed in the absence of P(O)Ph3 (Table 1, entry 6). Upon lowering the reaction temperature however, we observed that a quantitative yield of 3a was afforded even at ambient temperature when a Pd(II) precatalyst and P(O)Ph3 was used (Table 2, entry 1), whereas the use of a Pd(0) source (Pd(dba)2) was far less effective (Table 2, entry 2). This result highlights the preparative usefulness of a Pd(II) source combined with P(O)Ph3. Furthermore, to our surprise chlorobenzo[h]-quinoline 1b gave a much faster reaction time than with bromo compound 1a. Indeed, the coupling of substrate 1b proceeded quantitatively within 0.5 or 2 h, respectively, with CsF or Na2CO3 as the base (Table 2, entries 3 and 4), while for 1a stirring overnight was necessary to reach full conversion.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Additive (equiv.) | Time (h) | Conversionb (%) |
a Reactions were performed on a 0.2 mmol scale with 1.05 equiv. 2a in 1.5 ml dioxane–H2O 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
b Determined by NMR based on the ratio of 1a and 3a.
c Isolated yield. :1.
b Determined by NMR based on the ratio of 1a and 3a.
c Isolated yield.
|
||||
| 1 | Pd(PPh3)4 | BQ (0.5) | 2 | 67c |
| 2 | Pd(OAc)2 | P(O)Ph3 (0.2) | 10 min | 86 |
| 3 | PdCl2 | P(O)Ph3 (0.2) | 0.5 | 98 |
| 4 | Pd(dba)2 | P(O)Ph3 (0.2) | 0.5 | 83 |
| 5 | Pd black | P(O)Ph3 (0.2) | 18 | 64 |
| 6 | Pd(OAc)2 | — | 1 | 1 |
| 7 | Pd(dba)2 | — | 0.5 | 86 |
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | X | Catalyst | Base | Solvent | Time (h) | Yieldc (%) |
|
a Reactions were performed on a 0.1 mmol scale with 1.05 equiv. 2a in 0.75 ml dioxane–H2O 2:1 or 0.5 ml dioxane or 0.5 ml MeOH.
b For more results, see ESI.†
c Isolated yield.
d 1 mol% Pd(OAc)2.
|
||||||
| 1 | Br | Pd(OAc)2 | CsF | Dioxane–H2O 2:1 |
18 | 98 |
| 2 | Br | Pd(dba)2 | CsF | Dioxane–H2O 2:1 |
18 | 52 |
| 3 | Cl | Pd(OAc)2 | CsF | Dioxane–H2O 2:1 |
0.5 | 100 |
| 4 | Cl | Pd(OAc)2 | Na2CO3 | Dioxane–H2O 2:1 |
2 | 99 |
| 5 | Cl | Pd(OAc)2 | Na2CO3 | Dioxane | 18 | 92 |
| 6 | Cl | Pd(OAc)2 | Na2CO3 | MeOH | 5 min | 100 |
| 7 | Cl | Pd(OAc)2d |
Na2CO3 | MeOH | 0.25 | 84 |
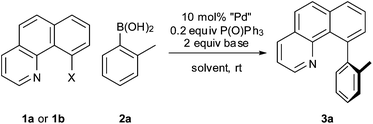
In terms of suitable solvents, we found that under non-aqueous conditions (for example anhydrous dioxane) the reaction took up to 18 h to go to completion (Table 2, entry 5), whereas it was fastest in MeOH where the reaction was complete after about 5 min (Table 2, entry 6). Pleasingly, when the catalyst loading was reduced to 1 mol%, the reaction was still suitably fast, although the product yield dropped slightly (Table 2, entry 7). While we surveyed other phosphine oxide additives, for example P(O)Cy3 or XPhos(O), we found these to be less effective than P(O)Ph3. Additional optimisation conditions can be found in Table S1, ESI.†
Next, the scope of the coupling reaction was examined (Table 3). A variety of ortho-substituents were tolerated. In general, electron donating (Me, OMe) and electron withdrawing substituents (CF3 and Cl) all furnished a high product yield (Table 3, entries 1 to 4). To our surprise, 2-bromobenzeneboronic acid (Table 3, entry 5) was also a suitable substrate, giving the corresponding product in 99% yield; the ortho-bromo group proving inert to the reaction conditions. Additional substituents on the aromatic ring did not generally alter the reactivity of the boronic acids (compare Table 3, entry 1 with entry 7) unless the 6-position was occupied in addition to the 2-position. For example, 2,6-dimethylbenzeneboronic acid (Table 3, entry 8) only underwent coupling when the reaction was performed at elevated temperature. Indeed, more sterically encumbered substrates reduced the reaction rate and, depending on the solvent used, led to a side reaction where 1a or 1b were converted to the 10-methoxy derivative (in methanol) or the 10-hydroxy derivative (in dioxane–H2O), respectively. Pleasingly, this side reaction could be avoided simply by switching to THF as solvent. While 10 mol% catalyst was arbitrarily selected from our optimisation studies, we were delighted to observe that analogous results were obtained using 5 mol% (Table 3, entries 4 and 12). The robustness of our method could be demonstrated when the reaction of 2-methoxy-1-naphthylboronic acid with 1b was conducted on gram scale affording the coupling product in 92% isolated yield (Table 3, entry 11).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Boronic acid | Solvent | Base | T (°C) | Time (h) | Yieldc (%) |
|
a Reactions were performed on a 0.1 mmol scale with 1.2 equiv. boronic acid in 0.75 ml dioxane–H2O 2:1 or 0.5 ml THF.
b For more results, see ESI.†
c Isolated yield.
d Isolated yield using 5 mol% Pd(OAc)2.
e On gram scale (4.2 mmol 1b).
|
||||||
| 1 | 2a | Dioxane–H2O 2:1 |
CsF | rt | 0.5 | 100 |
| 2 |
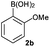
|
Dioxane–H2O 2:1 |
CsF | rt | 18 | 74 |
| 3 |
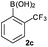
|
Dioxane–H2O 2:1 |
CsF | rt | 18 | 100 |
| 4 |
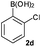
|
Dioxane–H2O 2:1 |
CsF | rt | 10 min | 93 (96d) |
| 5 |
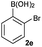
|
Dioxane–H2O 2:1 |
CsF | rt | 2 min | 99 |
| 6 |
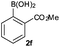
|
THF | Na2CO3 | 60 | 18 | 51 |
| 7 |
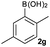
|
Dioxane–H2O 2:1 |
CsF | rt | 0.5 | 100 |
| 8 |
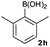
|
THF | CsF | 60 | 18 | 84 |
| 9 |
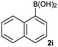
|
Dioxane–H2O 2:1 |
Na2CO3 | rt | 18 | 85 |
| 10 |
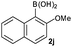
|
THF | CsF | 60 | 2 | 88 |
| 11e | 2j | THF | CsF | 60 | 2 | 92 |
| 12 |
|
THF | CsF | 60 | 18 | 72 (84d) |
In conclusion, we have developed a highly robust and versatile method to prepare bulky C10-arylated benzo[h]quinoline products. We envisage these products will be of significant interest in the study of dynamic conformation chemistry and as stereochemical switches. In the developed method, the use of P(O)Ph3 as an additive enabled the formation of a number of sterically encumbered, functionalised products. It is worth reiterating that use of chlorobenzo[h]quinoline (1b) lead to shorter reaction times than the bromo compound (1a), a fact that is counterintuitive for the known reactivity profile of oxidative addition of a Pd(0) species into the carbon–halide bond.13 Future studies will be aimed at elucidating the mechanistic nature of this interesting transformation and its use in target based synthesis.
We would like to thank the Leverhulme Trust (grant F/07058/BG) for funding this work.
Notes and references
- (a) A. Suzuki, J. Organomet. Chem., 1999, 576, 147 CrossRef CAS; (b) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (c) A. Suzuki, in Metal-Catalyzed Cross-Coupling Reactions, ed. F. Diederich and P. J. Stang, Wiley-VCH, Weinheim, Germany, 1998, p. 49 Search PubMed.
- X.-F. Wu, P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9047 CrossRef CAS.
- For reviews discussing C–H arylation, see: (a) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (b) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS; (c) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS; (d) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS; (e) G. P. McGlacken and L. M. Bateman, Chem. Soc. Rev., 2009, 38, 2447 RSC; (f) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS.
- (a) L. Ilies, M. Kobayashi, A. Matsumoto, N. Yoshikai and E. Nakamura, Adv. Synth. Catal., 2012, 354, 593 CrossRef CAS; (b) T. W. Lyons, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 4455 CrossRef CAS; (c) B. Li, Z.-H. Wu, Y.-F. Gu, C.-L. Sun, B.-Q. Wang and Z.-J. Shi, Angew. Chem., Int. Ed., 2011, 50, 1109 CrossRef CAS; (d) H. Miura, K. Wada, S. Hosokawa and M. Inoue, Chem.–Eur. J., 2010, 16, 4186 CrossRef CAS; (e) N. Luo and Z. Yu, Chem.–Eur. J., 2010, 16, 787 CrossRef CAS; (f) W. Jin, Z. Yu, W. He, W. Ye and W.-J. Xiao, Org. Lett., 2009, 11, 1317 CrossRef CAS; (g) M. Kim, J. Kwak and S. Chang, Angew. Chem., Int. Ed., 2009, 48, 8935 CrossRef CAS; (h) X. Zhao and Z. Yu, J. Am. Chem. Soc., 2008, 130, 8136 CrossRef CAS; (i) K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2007, 129, 11904 CrossRef CAS.
- (a) J. Clayden, S. P. Fletcher, S. J. M. Rowbottom and M. Helliwell, Org. Lett., 2009, 11, 2313 CrossRef CAS; (b) J. Clayden, S. P. Fletcher, J. J. W. McDouall and S. J. M. Rowbottom, J. Am. Chem. Soc., 2009, 131, 5331 CrossRef CAS; (c) M. S. Betson, A. Bracegirdle, J. Clayden, M. Helliwell, A. Lund, M. Pickworth, T. J. Snape and C. P. Worrall, Chem. Commun., 2007, 18, 754 RSC.
- (a) C. Valente, S. Çalimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314 CrossRef CAS; (b) G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151 RSC; (c) G. C. Fu, Acc. Chem. Res., 2008, 41, 1555 CrossRef CAS; (d) W. Su, S. Urgaonkar, P. A. McLaughlin and J. G. Verkade, J. Am. Chem. Soc., 2004, 126, 16433 CrossRef CAS.
- (a) A. Chartoire, M. Lesieur, L. Falivene, A. M. Z. Slawin, L. Cavallo, C. S. J. Cazin and S. P. Nolan, Chem.–Eur. J., 2012, 18, 4517 CrossRef CAS; (b) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461 CrossRef CAS.
- D. Kalyani, N. R. Deprez, L. V. Desai and M. S. Sanford, J. Am. Chem. Soc., 2005, 127, 7330 CrossRef CAS.
- (a) D. Kalyani, A. R. Dick, W. Q. Anani and M. S. Sanford, Tetrahedron, 2006, 62, 11483 CrossRef CAS; (b) A. R. Dick, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 2300 CrossRef CAS.
- M. Alami, C. Amatore, S. Bensalem, A. Choukchou-Brahim and A. Jutand, Eur. J. Inorg. Chem., 2001, 2675 CrossRef CAS.
- (a) C. Sköld, J. Kleimark, A. Trejos, L. R. Odell, S. O. Nilsson Lill, P.-O. Norrby and M. Larhed, Chem.–Eur. J., 2012, 18, 4714 CrossRef; (b) K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 9651 CrossRef CAS and references cited therein.
- S. E. Denmark, R. C. Smith and S. A. Tymonko, Tetrahedron, 2007, 63, 5730 CrossRef CAS.
- (a) A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176 CrossRef CAS; (b) V. V. Grushin and H. Alper, Chem. Rev., 1994, 94, 1047 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and 1H and 13C NMR spectra for all new compounds. See DOI: 10.1039/c2ob26806j |
| This journal is © The Royal Society of Chemistry 2013 |