Synthesis of a four-component [3]catenane using three distinct noncovalent interactions†‡
Miguel Á.
Alemán García
and
Nick
Bampos
*
Department of Chemistry, Lensfield Road, University of Cambridge, Cambridge CB2 1EW, UK. E-mail: nb10013@cam.ac.uk; Tel: +44 (0)1223 762970
First published on 11th October 2012
Abstract
A multicomponent assembly is described resulting in [2] and [3]catenanes using three flexible components and three distinct noncovalent interactions. Despite the possibility of competing side-products, only the desired assemblies are generated and characterized spectroscopically.
Nature exploits many inter(intra)molecular interactions in parallel to create functional superstructures such as DNA, ribosomes and Ca2+ ATPases.1 One of the major challenges in supramolecular chemistry is to harness several orthogonal weak interactions to generate structures that display comparable complexity and functionality to those found in biological systems. However, as the diversity of building blocks and noncovalent interactions involved in the assembly process increases, it becomes exponentially more difficult to exercise control over the final structure. Therefore, most of the systems reported to date employ one or two distinct noncovalent interactions.2
Interlocked molecular complexes such as rotaxanes and catenanes have been the focus of major synthetic efforts as they offer the opportunity of being incorporated into molecular devices or smart materials.3 While coordination bonds,4 hydrogen bonds5 or π–π stacking6 interactions have been elegantly used in the construction of catenanes and rotaxanes, there are no comparable examples to those outlined in this report in which three (or more) noncovalent interactions are combined to assemble mechanically interlocked molecules.
Our interest has been directed towards exploring heteroleptic, multicomponent architectures in which a number of noncovalent interactions are exploited to generate a heteroleptic assembly, in the absence of competing side-products. As part of this study, we report the synthesis and structural characterization of a new class of multicomponent [2] and [3]catenanes (1 and 2 respectively) utilizing up to four building blocks and three orthogonal interactions (Fig. 1).
![Schematic representation (MM+ modeling) of the [2] and [3]catenane (1 and 2 respectively) in which each of the components is colored for clarity (3 in red, 4 in blue and 5 in purple).](/image/article/2013/OB/c2ob26587g/c2ob26587g-f1.gif) | ||
| Fig. 1 Schematic representation (MM+ modeling) of the [2] and [3]catenane (1 and 2 respectively) in which each of the components is colored for clarity (3 in red, 4 in blue and 5 in purple). | ||
The components of our assembly exhibit significant flexibility which comes with a ‘cost’ of organizational energy, but which also allows sampling of the various possible conformations that ultimately lead to the desired structure. This flexibility may very well explain why more than one crown ether threaded onto the heteroleptic macrocycle described below. It is also noteworthy that due to the cooperative effect of the pyridine–metal porphyrin interaction, it was possible to direct the formation of a system that would otherwise be difficult to assemble.
A distinctive feature of the [n]catenanes (where n = 2 or 3, Fig. 1) is the structure of the metallomacrocyclic ring, which consists of two components connected via two distinct noncovalent interactions: Ru–pyridine and heteroleptic Fe(II) (or Zn(II)) terpyridine. This is to the best of our knowledge the first example of clean formation of heteroleptic terpyridine complexes using Fe(II) or Zn(II)7 – in both cases the desired complex was the exclusive product.
The components of the [n]catenanes were synthesised according to Scheme 1 (see ESI†), whereby the naphthalenediimide (NDI) thread 3 was synthesized from the commercially available 1,4,5,8-naphthalenetetracarboxylic dianhydride8 in an overall yield of 13%. The crown ether macrocycle 4 was prepared using the methodology previously described by Stoddart et al.,9 while the porphyrin–terpyridyl component of the assembly 5 was obtained in 40% yield from porphyrin 810 (either as the ruthenium or zinc metallated analogues) and compound 7 in the presence of K2CO3.
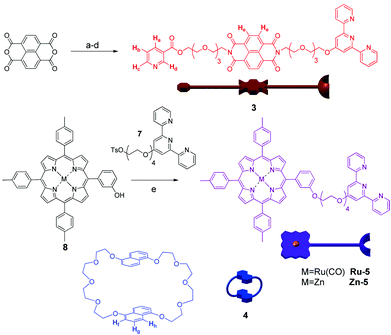 | ||
Scheme 1 Synthesis of the components 3 and 5: (a) 2-(2-(2-(2-aminoethoxy)ethoxy)ethoxy)ethanol, DMF, μw 5 min, 89%; (b) HOBt, DCC, nicotinic acid/THF–CH2Cl2 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 12 h, 34%; (c) Ag2O, TsCl/CH2Cl2, RT, 70%; (d) K2CO3/HO-terpy, acetone, 70 °C, 61%; (e) DMF, 70 °C, 40%. Chemical structure and schematic representation of 4 is also shown. :1, 12 h, 34%; (c) Ag2O, TsCl/CH2Cl2, RT, 70%; (d) K2CO3/HO-terpy, acetone, 70 °C, 61%; (e) DMF, 70 °C, 40%. Chemical structure and schematic representation of 4 is also shown. | ||
The initial target structure ([2]catenane, 1) was assembled using a “capping” approach (Scheme 2) in which the donor–acceptor interaction between the electron deficient NDI (3) and the electron rich crown ether (4) was enhanced by lowering the temperature.11 Once the pseudo rotaxane was formed at 195 K, the two ends were capped with a single molecular component Ru-5 possessing two distinct recognition units. At one end, the ruthenium porphyrin interacts with the pyridyl group of the pseudo rotaxane, while addition of Fe(II) salt connects the two terpyridyl ends of the assembly to form the heteroleptic [2]catenane 1. Since the formation of 1 was not quantitative when one equivalent of crown ether 4 was used, higher concentrations of 4 were required to drive the equilibrium towards the exclusive formation of the [2]catenane. Spectroscopic characterization indicated formation of the unexpected [3]catenane 2 in addition to the predicted [2]catenane 1 (as the major product). To prepare the [3]catenane (2) a one-pot stepwise synthesis was adopted (Scheme 2a) in which the first step involved the addition of a saturated solution of 4 to a solution containing the NDI 3 in CD2Cl2. A characteristic pale red color was observed (Fig. S1†) indicating that the charge transfer complex had formed (which is in equilibrium with the free components at room temperature).11 The solution was then cooled to 195 K to shift the equilibrium in favor of the donor–acceptor complex, and one equivalent of ruthenium terpyridyl Ru-5 was added to bind and block the pyridyl end of the NDI 3. Finally one equivalent of Fe(BF4)2 (in CD3OD) was added to coordinate the two terpyridyl ends of the heteroleptic complex. The [2]catenane 1 was generated in solution using the same sequence of additions, except that in the first step one equivalent of the crown ether 4 was added.
![(a) Graphical representation of the one-pot assembly of the [2] and [3]catenanes, 1 and 2 respectively. In this sequence of additions the crown 4 is added to the NDI 3 in the first step, and depending on the stoichiometry of the components, it is possible to bias the equilibrium (at low temperature) towards the [2] and [3]pseudorotaxane. Addition of 5 leads to coordination of the pyridyl end of 3 to the metalloporphyrin, after which Fe(BF4)2 (or Zn(TfO)2) coordinates the terpyridyl ends of the complex, leading to 1 and 2. (b) Pre-formation of the heteroleptic complex 6 from a 1 : 1 mixture of 3 and Ru-5 suggests that cooperative effects of the assembly do not allow any of the crown 4 to thread onto the metallomacrocyclic ring at room temperature.](/image/article/2013/OB/c2ob26587g/c2ob26587g-s2.gif) | ||
| Scheme 2 (a) Graphical representation of the one-pot assembly of the [2] and [3]catenanes, 1 and 2 respectively. In this sequence of additions the crown 4 is added to the NDI 3 in the first step, and depending on the stoichiometry of the components, it is possible to bias the equilibrium (at low temperature) towards the [2] and [3]pseudorotaxane. Addition of 5 leads to coordination of the pyridyl end of 3 to the metalloporphyrin, after which Fe(BF4)2 (or Zn(TfO)2) coordinates the terpyridyl ends of the complex, leading to 1 and 2. (b) Pre-formation of the heteroleptic complex 6 from a 1:1 mixture of 3 and Ru-5 suggests that cooperative effects of the assembly do not allow any of the crown 4 to thread onto the metallomacrocyclic ring at room temperature. | ||
The porphyrin–pyridine interaction was crucial in the first step of the heteroleptic assembly, as confirmed by the addition of one equivalent of Fe(BF4)2 to a one-to-one mixture of the NDI (3) and Ru-5 (Scheme 2b). In the absence of any cooperativity one might expect a statistical 1:2:1 mixture (3·3, 3·5/5·3, 5·5) of Fe(II)-terpyridyl complexes, however 1H NMR and HR-MS proved that the heteroleptic complex 6 was the exclusive product. The heteroleptic dimer was also observed when the same experiment was carried out using the weaker coordinating zinc metallated analogue of 5.
A control experiment was undertaken in order to establish whether it was possible to observe threading of crown ether 4 after the formation of the heteroleptic macrocycle 6. When a saturated solution of the crown (4) was added to the preassembled complex, there was no indication of threading of the crown over a period of six months (Scheme 2b). This finding is rather surprising since previously reported ruthenium(II) porphyrin assemblies were shown to be in thermodynamic equilibrium.12 The absence of such an equilibrium indicates that the heteroleptic terpyridine complex and the Ru-5–pyridine bond act as a “molecular clamp”.
The stepwise construction of the [3]catenane was monitored and characterized by 1H NMR spectroscopy (Fig. 2). The free crown 4 (Fig. 2a) and NDI 3 (Fig. 2b) are shown for comparison, and their relative chemical shifts do not undergo significant temperature dependent changes. Addition of 4 to a solution of NDI 3 resulted in the appearance of diagnostic resonances indicative of the formation of the donor–acceptor complex at room temperature (Fig. 2c). For example, the resonance of the NDI unit broadened and shifted from 8.62 ppm to 8.50 ppm. Likewise the resonances of the crown 4 were recorded as three broad peaks at 7.65, 7.10 and 6.44 ppm as a result of fast exchange on the NMR timescale. At low temperature (Fig. 2d) any exchange broadening is suppressed as the equilibrium slows down sufficiently on the NMR timescale to allow identification of sharp peaks attributed to the bound crown and NDI, with the excess crown peaks (4) matching those in Fig. 2a. Addition of one equivalent of Ru-5 led to an ‘open’ heteroleptic assembly by virtue of the ruthenium porphyrin bound pyridyl resonances of 3 (Fig. S3†). As the pyridyl protons Hc and Hd experience the diamagnetic ring current of the porphyrin ring, a significant change in their chemical shift was observed for Hc (Δδ −7.11 ppm) and for Hd (Δδ −7.10 ppm), while Ha and Hb experience less dramatic movement of peaks (Δδ 1.58 and Δδ −2.05 ppm respectively), as they are further from the distance dependent effects of the porphyrin ring current. When one equivalent of Fe(BF4)2 was then added at 195 K, the cyclic assembly was finally ‘locked’.
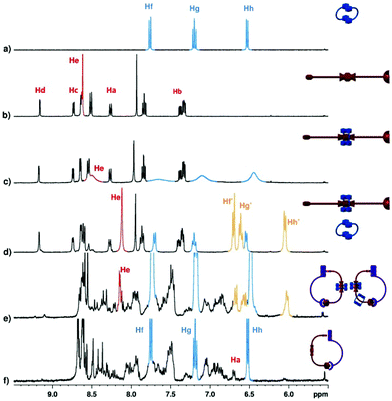 | ||
| Fig. 2 Partial 1H NMR spectra (400 MHz, CD2Cl2) of: (a) crown 4 at 298 K; (b) NDI 3 at 298 K; (c) NDI 3 in a saturated solution of crown 4 at 298 K; (d) same sample as in spectrum c, at 195 K; (e) NDI 3 in a saturated solution of crown 4, with one equivalent of Ru-5 and one equivalent of Fe(BF4)2 (added as a CD3OD solution) at room temperature; (f) pre-formation of 6 before the addition of crown 4 (at 298 K). | ||
At the final point of the stepwise construction of the assembly, two sets of peaks for the crown ether were identified at room temperature (Fig. 2e). One set corresponded to the free crown 4 (an excess was added in the first step) at 6.65, 7.23 and 7.71 ppm, which did not change significantly throughout the experiment, while a second set of peaks at 6.00, 6.54 and 6.65 ppm were coupled to each other, according to a 1H–1H COSY experiment, and attributed to those participating in the donor–acceptor interaction. A third set overlapping with those of the free crown were assigned to the fraction of crown trapped on the [3]catenane that did not directly contribute to the donor–acceptor complex. Accordingly to MM+ molecular modeling, the metallomacrocycle is flexible and large enough to accommodate two bound crown ethers. Significantly, pre-forming the heteroleptic complex (6) and then adding the crown (4) resulted in a 1H NMR spectrum with no evidence of bound crown resonances (Fig. 2f).
When the heteroleptic supramolecular metallomacrocycle is formed (in 1, 2 or 6), all signals corresponding to the pyridyl protons split into sets of signals, which by 1H NMR spectroscopy suggests that a number of different conformations of the assembly exist in solution (Fig. S5 and S6†).
Formation of the [2] and [3]catenanes 1 and 2 was also investigated by DOSY NMR spectroscopy (Fig. S7†) in which the constituent components were identified with distinct diffusion coefficients indicative of their relative sizes (in this case 1 and 2 have similar hydrodynamic radii, but the free crown 4 can be clearly distinguished from 1 and 2). Further evidence for the formation of the heteroleptic assemblies was obtained from HR-MS (Fig. 3, Fig. S8–S12†), where the heteroleptic complex 6 as well as the [2] and [3]catenanes (1 and 2) were identified. In contrast, when a mixture of the heteroleptic macrocycle 6 and the crown ether 4 (Scheme 2b) was analyzed by High Resolution Mass Spectrometry (HR-MS), only 6 and 4 were observed, confirming the structural integrity of 2 resulting from the stepwise assembly (Scheme 2a).
![HR-MS of the [3]catenane solution described in Fig. 2e.](/image/article/2013/OB/c2ob26587g/c2ob26587g-f3.gif) | ||
| Fig. 3 HR-MS of the [3]catenane solution described in Fig. 2e. | ||
Threading of crown ether macrocycles into polyethylene glycol chains (PEG's) has been reported in the literature.13 The idea of introducing tetra ethylene glycol chains into our molecular building blocks was to investigate what effect the addition of alkali cations, such as Na+ or Li+, will have on the conformation of the final structures. Furthermore, it has been reported that addition of Na/Li salts can enhance the donor–acceptor interaction between crown ether and NDI molecules – an effect referred to by Sanders et al. as ‘cation reinforcement’.14 However, due to the limited solubility of complexes 1 and 2 after the addition of Na/Li salts it was not possible to analyze the effects of alkali cations on the assemblies, but we are exploring ways to overcome these limitations to assemble more elaborate heteroleptic multi-component assemblies.
In summary, this report describes a one-pot sequential synthesis, using three building blocks and three distinct noncovalent interactions, to construct heteroleptic interlocked supramolecular metallomacrocycles 1 and 2 as the major or exclusive products. Current investigations are directed towards understanding the length dependence of the ethylene glycol chains in the formation of the assemblies, and incorporating different metalloporphyrins in order to tune one of the three interactions. We predict that the use of multiple orthogonal interactions in the construction of multicomponent assemblies, such as those described in the present work, will give access to structures of much higher complexity and pave the way towards functional systems.
Acknowledgements
We are grateful to the Consejo Nacional de Ciencia y Tecnologia (CONACYT) Mexico for funding a studentship (MAAG).References
- (a) E. Gouaux and R. MacKinnon, Science, 2005, 310, 1461 CrossRef CAS; (b) J. H. Cate, M. M. Yusupov, G. Z. Yusupova, T. N. Earnest and H. F. Noller, Science, 1999, 285, 2095 CrossRef CAS; (c) J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737 CrossRef CAS.
- (a) E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72 CrossRef CAS; (b) A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2012, 41, 19 RSC; (c) R. S. Forgan, J. P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434 CrossRef CAS; (d) J.-F. Ayme, J. E. Beves, D. A. Leigh, R. T. McBurney, K. Rissanen and D. Schultz, Nat. Chem., 2012, 4, 15 CrossRef CAS; (e) V. E. Campbell, X. D. Hatten, N. Delsuc, B. Kauffmann, I. Huc and J. R. Nitschke, Nat. Chem., 2010, 2, 684 CrossRef CAS; (f) K. Mahata, M. L. Saha and M. Schmittel, J. Am. Chem. Soc., 2010, 132, 15933 CrossRef CAS; (g) N. Christinat, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2008, 47, 1848 CrossRef CAS; (h) C. D. Pentecost, K. S. Chichak, A. J. Peters, G. W. V. Cave, S. J. Cantrill and J. F. Stoddart, Angew. Chem., Int. Ed., 2007, 46, 218 CrossRef CAS.
- M. Schmittel and K. Mahata, Angew. Chem., Int. Ed., 2008, 47, 5284 CrossRef CAS.
- (a) J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed. Engl., 2011, 50, 9260 CrossRef CAS; (b) D. J. Mercer and S. J. Loeb, Dalton Trans., 2011, 40, 6385 RSC.
- (a) C. A. Hunter, J. Am. Chem. Soc., 1992, 114, 5303 CrossRef CAS; (b) A. G. Johnston, D. A. Leigh, L. Nezhat, J. P. Smart and M. D. Deegan, Angew. Chem., Int. Ed., 1995, 34, 1212 CrossRef CAS; (c) A. G. Johnston, D. A. Leigh, A. Murphy, J. P. Smart and M. D. Deegan, J. Am. Chem. Soc., 1996, 118, 10662 CrossRef CAS.
- (a) S. J. Loeb, Chem. Soc. Rev., 2007, 36, 226 RSC; (b) D. J. Mercer and S. J. Loeb, Dalton Trans., 2011, 40, 6385 RSC; (c) J. F. Stoddart and H. R. Tseng, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4797 CrossRef CAS.
- (a) C. Mugemana, P. Guillet, S. Hoeppener, U. S. Schubert, C. A. Fustin and J. F. Gohy, Chem. Commun., 2010, 46, 1296 RSC; (b) E. C. Constable, Chem. Soc. Rev., 2007, 36, 246 RSC.
- K. M. Mullen, K. D. Johnstone, M. Webb, N. Bampos, J. K. M. Sanders and M. J. Gunter, Org. Biomol. Chem., 2008, 6, 278 CAS.
- C. J. Bruns, S. Basu and J. F. Stoddart, Tetrahedron Lett., 2010, 51, 983 CrossRef CAS.
- K. M. Mullen, K. D. Johnstone, D. Nath, N. Bampos, J. K. M. Sanders and M. J. Gunter, Org. Biomol. Chem., 2009, 7, 293 CAS.
- I. R. Fernando, S. G. Bairu, G. Ramakrishna and G. Mezei, New. J. Chem., 2010, 34, 2097 RSC.
- M. J. Gunter, N. Bampos, K. D. Johnstone and J. K. M. Sanders, New J. Chem., 2001, 25, 166–173 RSC.
- Y. F. Ng, J. C. Meillon, T. Ryan, A. P. Dominey, A. P. Davis and J. K. M. Sanders, Angew. Chem., Int. Ed., 2001, 40, 1757 CrossRef CAS.
- S. I. Pascu, T. Jarrosson, C. Naumann, S. Otto, G. Kaiser and J. K. M. Sanders, New J. Chem., 2005, 29, 80 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic and experimental procedures are described in addition to spectroscopic data. See DOI: 10.1039/c2ob26587g |
| ‡ This manuscript is dedicated to Prof. Maxwell J. Gunter in recognition of his guidance, support and friendship. |
| This journal is © The Royal Society of Chemistry 2013 |