Structural stabilization of DNA-templated nanostructures: crosslinking with 2,5-bis(2-thienyl)pyrrole monomers†
Wen
Chen
and
Gary B.
Schuster
*
Georgia Institute of Technology, Atlanta, Georgia, USA. E-mail: Schuster@gatech.edu
First published on 12th October 2012
Abstract
Ordered 2,5-bis(2-thienyl)pyrrole (SNS) zipper arrays are formed by hybridization of complementary DNA oligomers each containing covalently bound SNS monomers. Upon oxidation with HRP/H2O2, these SNS arrays are converted to oligomers having specific lengths and conformation. As a consequence of this reaction the two SNS-containing strands are permanently crosslinked.
DNA is a powerful building block for the programmed “bottom-up” assembly of the supermolecular scaffolds needed for the construction of nanoscale materials and devices.1–4 The properties of these devices can be controlled by various schemes that have been developed to modify the DNA.5–8 For example, metal ions (e.g., Ag+) electrostatically bound to the DNA phosphate groups can be reduced to form clusters that serve as nucleation sites for the formation of a metallic wire along the length of DNA.9–11 Similarly, DNA has been used as a scaffold to form porphyrin arrays that permit electronic tuning of the resulting nanostructure.12 And the special advantage of sequence programmability enabled by a DNA scaffold is demonstrated by the creation of molecular photonic wires with linearly arranged, unique chromophores.13 We are pursuing an approach to the preparation of nanowires using DNA oligomers modified by the covalent attachment of monomers for conducting polymers. The monomers are converted to conjoined DNA-conducting polymers electrochemically or chemically by treatment with horseradish peroxidase and hydrogen peroxide (HRP/H2O2).14–17 For example, DNA oligomers modified with 2,5-bis(2-thienyl)pyrrole (SNS) monomers that are covalently-linked to cytosines are converted to conducting polymers of specific length upon treatment with HRP/H2O2.18 Similarly, a DNA-programmed modular assembly approach has been used to prepare cyclic arrays of SNS polymers.19
Despite substantial progress, limitations remain in the use of modified DNA for the construction of nanostructures. Duplex DNA is usually considered to be a relatively rigid one-dimensional rod with a persistence length of ca. 50 nm.20 In reality, DNA is a flexible, dynamic substance undergoing structural and conformational changes that occur on timescales ranging from picoseconds to seconds.21 Moreover, the structural integrity of DNA is highly dependent on its environment. The stability of duplex DNA in an aqueous solution depends on pH, temperature and the ionic strength. Outside of a relatively narrow range of acceptable solution parameters, DNA denatures and its utility as a scaffold deteriorates. It is therefore crucial to develop methods for stabilization of the DNA scaffold of nano-assemblies. Several attempts have been made to “fix” or stabilize the structure of a DNA assembly through structural means (crossover, staple strands), by chemical modification22,23 or by encapsulating the assembly in a solid state matrix.24
We recently reported the use of bond formation between SNS monomers to permanently link single-stranded regions of cyclic DNA arrays.19 Here we describe the use of SNS monomers to crosslink duplex DNA oligomers so that they remain intact under conditions that would denature normal DNA. In these constructs, the SNS monomers of modified DNA are arrayed in the major groove in an ordered zipper-like arrangement (see Fig. 1). The reaction of a duplex comprised of complementary oligomers of modified DNA with HRP/H2O2 results in conversion of the SNS monomer array to a linear polymer closing the zipper and crosslinking the duplex. Physical and spectroscopic data indicate that the crosslinked assemblies retain an overall B-form DNA structure and that their stability is greatly enhanced.
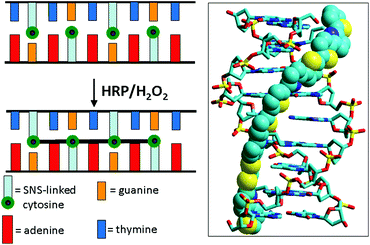 | ||
| Fig. 1 Illustration of DNA-templated crosslinking polymerization of SNS monomers covalently linked to cytosines on each of the two strands of a DNA duplex. The image on the right is the simulated 3D structure of (SNS)4 formed from the reaction of 5′-AGTXAGTXC-3′/3′-TXAGTXAGG-5′ in which X represents the SNS-linked cytosines. | ||
Results
The DNA oligomers used in this study are shown in Fig. 2 where X represents a cytosine modified with an SNS monomer linked covalently through a trimethylene group (CH2)3 at its N4 position. DNA(1,2) is an unmodified duplex oligomer used in control experiments. DNA(3,2) is similar to DNA(1,2) except that a single cytosine in the center of DNA(3) is replaced by X. DNA(3,4) has two SNS modified cytosines, one on each strand, separated by a single A/T base-pair in the duplex. Similarly, duplex oligomers DNA(3,5), DNA(6,7) and DNA(8,9) contain three, four and six SNS monomers, respectively, each separated by an A/T base pair. | ||
| Fig. 2 Structures of the DNA oligomers used in this work. Xn represents the number of SNS monomers linked to the DNA duplex oligomer. Tm is the observed melting temperature of the duplex monitored at 260 nm where the absorption of the DNA bases dominates the spectrum. | ||
Melting temperature
Evidence showing the assembly of the modified oligomers into duplex structures comes from analysis of their melting temperatures (Tm) monitored at 260 nm in 10 mM sodium citrate buffer solution containing 200 mM of NaCl at pH 4.5. Comparison of DNA(1,2) with DNA(3,2) and DNA(3,4), each comprised of 21 base pairs, shows that each replacement of a cytosine with an X results in a ca. 4 °C reduction in melting temperature, see Fig. 2. Similarly, DNA(6,7) and DNA(8,9), which are comprised of 24 and 27 base pairs, respectively, show reversible melts at ca. 63 °C. Clearly, the SNS-containing oligomers form stable duplex structures, but replacement of C by X decreases the stability of the duplex (presumably by perturbation of hydrogen bonding, see ESI Fig. S-1†), but even with six SNS monomers the duplex is stable in buffer solution well above room temperature.Absorption spectroscopy
The UV-Vis spectra of the modified DNA duplexes, Fig. 3, show a characteristic peak at ca. 260 nm that is typical of the DNA nucleobases and a weaker absorption between 300 nm and 380 nm that is characteristic of the SNS group (see ESI Fig. S-2,† the UV-Vis spectrum of SNS).18 The absorption spectrum of the SNS monomer in citrate buffer solution (4–12 μM) that also contains duplex DNA (1.5 μM) exhibits a maximum at 332 ± 2 nm (when deconvoluted from the DNA absorption, see ESI Fig. S-3†). The inset in Fig. 3 is an expansion of the deconvoluted SNS portion of the absorption spectra of the modified duplex oligomers. The absorption of DNA(3,2), which contains one SNS monomer, shows a peak at 348 nm. Thus the covalent linkage of the SNS group positioning it in the major groove results in a red shift of its absorption maximum of 16 nm compared with that of the SNS monomer in buffer solution. This shift is due at least in part to the more hydrophobic environment of the DNA major groove.25,26 As the number of SNS units positioned in the major groove increases from one to six, the SNS absorption maximum shifts to shorter wavelength. In DNA(3,4) for which there are two SNS monomers, the absorption maximum shifts from 348 nm to 341 nm. Similarly for DNA(3,5), which has three SNS monomers, the absorption maximum is observed at 338 nm. As the number of SNS groups is increased further, there is a concomitant increase in the observed blue shift. This shift in absorption maximum is attributed to the π-stacking of the SNS groups in the DNA major groove resulting in the formation of weakly associated H-aggregate-like assemblies.27,28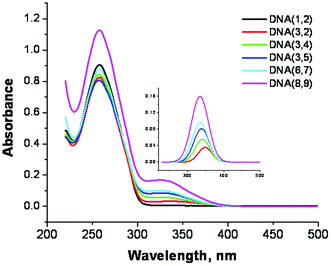 | ||
| Fig. 3 UV-Vis spectra of SNS linked DNA duplexes in buffer solution. The number of SNS monomers = 0–6. Inset: The spectra of the SNS groups mathematically deconvoluted from the DNA absorption bands. | ||
The structure of the SNS monomers arrayed in the major groove was further assessed by monitoring the duplex melting at 320 nm where the primary absorbing species are the SNS groups. The results (Fig. 4) reveal a temperature dependent hyperchromicity that is ascribed to the unstacking of the weakly associated SNS monomers when the duplex denatures. The Tm observed at 320 nm is the same as is observed when monitoring the absorption of the DNA bases at 260 nm indicating that the same structural change is being measured.
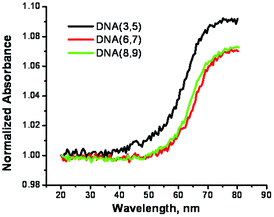 | ||
| Fig. 4 Melting profiles for the duplexes formed from SNS-containing DNA recorded at 320 nm in buffer solution. | ||
Circular dichroism spectroscopy
The configuration of the linked SNS groups in the DNA major groove was investigated by means of circular dichroism (CD) spectroscopy. Fig. 5a shows that the CD spectrum of DNA(1,2), which has no attached SNS groups, has a negative peak at 241 nm and a positive peak at 278 nm that are characteristic of B-form DNA.29 The CD spectrum of DNA(3,2), one SNS group, and DNA(3,4), two SNS groups, are essentially indistinguishable from that of DNA(1,2). Apparently, the SNS monomers in the DNA major groove do not significantly affect the overall B-form structure. Moreover, there is no detectable induced circular dichroism (ICD) of the SNS monomer in the CD spectrum of DNA(3,2) or DNA(3,4). The CD spectra of DNA oligomers that contain three, four and six SNS groups show the CD features typical of B-form DNA and a negative ICD peak in the region of the SNS absorption at ca. 340 nm (see Fig. 5b). The ICD is attributed to the ordered assembly of SNS monomers in the major groove of the B-form right-handed helix, which appears to require at least three SNS groups. The temperature dependence of the CD spectrum for DNA(8,9), shown in Fig. 5c, supports an origin for the ICD with the helical structure of the duplex DNA. The ICD band in the SNS absorption region gradually decreases as the temperature is increased from 20 to 80 °C with an apparent transition at ca. 60 °C corresponding to the Tm of this oligomer. Analogous temperature dependent changes are observed in the ICD spectra of DNA(3,5) and DNA(6,7) (see ESI Fig. S-4†).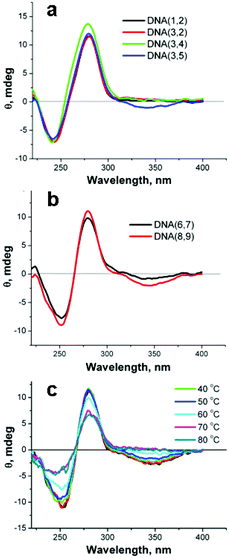 | ||
| Fig. 5 a,b – CD spectra of DNA duplexes formed from single strands each containing 0–6 covalently linked SNS monomers. c – The temperature dependence of the CD spectrum of DNA(8,9). | ||
Reaction with HRP/H2O2
We have previously reported that the reaction of SNS-modified DNA oligomers with HRP/H2O2 results in efficient polymerization of the monomers.18 This reaction is characterized by the unique optical absorption spectrum of polySNS that displays the characteristic features of a conducting polymer in the leuco, polaron or bipolaron oxidation state. In previously studied examples of this reaction the SNS monomers were linked to one strand of a DNA module that was used to form simple duplexes or more elaborate cyclic structures. In the current case, both strands of a duplex are modified with covalently linked SNS monomers. In this circumstance, bond formation between the SNS monomers results in crosslinking of the duplex, which will prevent its denaturation into single strands. The progress of the crosslinking reaction was monitored by UV-Vis spectroscopy, melting temperature and by denaturing polyacrylamide gel electrophoresis (PAGE).The reaction of DNA(1,2) with HRP/H2O2 and a catalytic amount of ABTS,18,19 as expected, results in no meaningful change to its absorption spectrum because it does not contain an SNS monomer. In contrast, the addition of HRP/H2O2 to an aqueous buffer solution of duplex DNA(3,2) results in a rapid decrease of the SNS absorption band at 340 nm accompanied by the appearance of new absorption bands with apparent maxima at 460 and 730 nm with an apparent isosbestic point at ca. 390 nm (see Fig. 6a). DNA(3,2) contains a single SNS monomer and the absorption features that result from its reaction with HRP/H2O2 are essentially identical with those reported for similar SNS radical cations (SNS˙+).18,30 Evidently, the normally rapid dimerization of SNS˙+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31 is inhibited when it is embedded in the DNA major groove and additional stabilization of the radical cation may result from its charge neutralization by the fixed anionic phosphate groups of the DNA backbone.32 Similarly, the addition of HRP/H2O2 to DNA(3,4), which contains two covalently linked SNS monomers, one on each strand, results in a prominent new absorption band at 820 nm and two weaker absorption bands at 420 nm and 465 nm (see Fig. 6b). These spectral changes indicate that oxidation of duplex DNA(3,4) results in the formation of the dimeric (SNS–SNS)˙+ by covalent bond formation between the two SNS groups linked to complementary strands. This reaction causes chemical crosslinking of the DNA. The reaction of the other duplex DNA oligomers examined in this work with HRP/H2O2 results in spectroscopic changes characteristic of the formation of (SNS)n˙+ oligomers where n = 3–6 (see ESI Fig. S-5†). As n increases, the absorption features of the (SNS)n˙+ oligomers consistently shift to the red demonstrating the formation of additional SNS-to-SNS bonds.
31 is inhibited when it is embedded in the DNA major groove and additional stabilization of the radical cation may result from its charge neutralization by the fixed anionic phosphate groups of the DNA backbone.32 Similarly, the addition of HRP/H2O2 to DNA(3,4), which contains two covalently linked SNS monomers, one on each strand, results in a prominent new absorption band at 820 nm and two weaker absorption bands at 420 nm and 465 nm (see Fig. 6b). These spectral changes indicate that oxidation of duplex DNA(3,4) results in the formation of the dimeric (SNS–SNS)˙+ by covalent bond formation between the two SNS groups linked to complementary strands. This reaction causes chemical crosslinking of the DNA. The reaction of the other duplex DNA oligomers examined in this work with HRP/H2O2 results in spectroscopic changes characteristic of the formation of (SNS)n˙+ oligomers where n = 3–6 (see ESI Fig. S-5†). As n increases, the absorption features of the (SNS)n˙+ oligomers consistently shift to the red demonstrating the formation of additional SNS-to-SNS bonds.
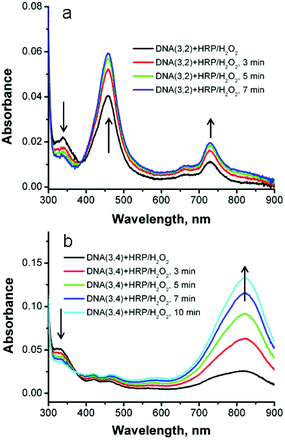 | ||
| Fig. 6 (a) UV-Vis spectra of SNS-containing duplex DNA(3,2) at various times after the addition of HRP/H2O2. (b). DNA(3,4). | ||
The melting behavior of the duplex oligomers after reaction with HRP/H2O2 was examined to assess the increase in stability that results from crosslinking. The results of reaction of DNA(8,9) are shown in Fig. 7a. Before reaction this duplex has a Tm = 63.6 °C and exhibits a hyperchromicity on melting of 0.16 absorbance units. After reaction with HRP/H2O2 the crosslinked duplex has a Tm = 75 °C and the hyperchromicity is reduced by about half to 0.08. The 12 °C increase in Tm shows that the stability of the duplex has been increased by crosslinking. The reduction in hyperchromicity suggests that only the uncrosslinked “tails” of the duplex are capable of denaturation. In support of this conclusion, the Tm of DNA(3,2), which has only one SNS modification and thus cannot be crosslinked, is hardly changed by reaction with HRP/H2O2, (see ESI Fig. S-6†).
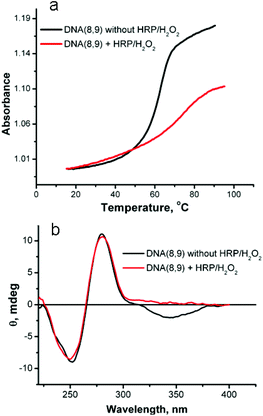 | ||
| Fig. 7 (a) Melting profile of DNA(8,9) before and after reaction with HRP/H2O2. (b) Circular dichroism spectra of DNA(8,9) before and after reaction with HRP/H2O2. | ||
Fig. 7b shows the affect of reaction with HRP/H2O2 on the CD spectrum of DNA(8,9). This duplex oligomer contains six SNS monomers – three on each strand. As noted above, the CD spectrum of DNA(8,9) before reaction shows two bands that are typical of B-form DNA and an ICD band for the stacked SNS monomers. After reaction with HRP/H2O2 the SNS monomer band disappears (the primary absorption band for (SNS)6˙+ is in the near IR region and not recorded in the CD spectrum) but the two DNA bands are essentially unaffected by the reaction. This result shows that conversion of the SNS monomers to a polymer does not measurably distort the B-form DNA scaffold. Similar results are obtained for the other SNS-modified duplexes studied in this work (see ESI Fig. S-7†).
The DNA-templated crosslinking reaction with HRP/H2O2 was analyzed by denaturing PAGE. Under the conditions of this experiment normal duplex DNA is denatured and the two strands migrate separately. Fig. 8a shows the results of denaturing PAGE for DNA(3,2) and DNA(3,4) where DNA(3) has been labeled with 32P at its 5′-ends for visualization by autoradiography. Lane 1 shows the position of DNA(3) on the gel. Lane 2 shows that the reaction of DNA(3,2) with HRP/H2O2 does not change the position of DNA(3). With only one SNS monomer DNA(3,2) cannot be crosslinked. Lane 3 of Fig. 8a shows the results of reaction of DNA(3,4) with HRP/H2O2. In this case there are two SNS monomers, one linked to each strand of the duplex, and reaction with HRP/H2O2 results in crosslinking as evidenced by the slower moving band. Fig. 8b shows the results of the denaturing PAGE experiment for DNA(6,7) and DNA(8,9) where DNA(6) and DNA(8) have been radiolabeled. In these cases each duplex strand has two or three SNS monomers arrayed in a zipper-like fashion in the major groove and the crosslinking is essentially 100% efficient.
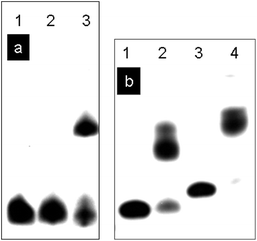 | ||
| Fig. 8 (a) Denaturing PAGE analysis of DNA oligomers after reaction with HRP/H2O2. Lanes 1–3, DNA(3) labelled with 32P. Lane 1, DNA(3); lane 2, DNA(3,2) + HRP/H2O2; lane 3, DNA(3,4) + HRP/H2O2. (b) Denaturing PAGE analysis of DNA oligomers after the reaction with HRP/H2O2. Lane 1–2, DNA(6) labelled with 32P. Lane 3–4, DNA(8) labelled with 32P. Lane 1, DNA(6); lane 2, DNA(6,7) + HRP/H2O2; lane 3, DNA(8); lane 4, DNA(8,9) + HRP/H2O2. | ||
Significantly, ESI mass spectrometric analysis of DNA(3) and DNA(5) before reaction of the duplex with HRP/H2O2 reveals that they have the expected molecular weight of 6663 and 7006 amu, respectively. The complete oligomerization of DNA(3,5) will result in conversion of its three covalently linked SNS monomers to an (SNS)3 trimer crosslinking DNA(3,5). With the formation of two new SNS–SNS bonds, the molecular weight of crosslinked DNA(3,5) should be reduced by 4 amu. Mass spectrometric analysis of the crosslinked DNA(3,5) resulting from trimer formation (SNS)3 confirms this expectation. After treatment with HRP/H2O2, the molecular weight of DNA(3,5) is found to be 13665 amu, which is 4 amu less than the sum of the molecular weight of DNA(3) and DNA(5) (see ESI Fig. S-8†). This mass spectrometric result confirms the essentially complete conversion of SNS monomer to a trimer (SNS)3 in which all monomers have reacted thus crosslinking DNA(3) and DNA(5). In addition to the peak of 13665 amu, the mass spectrum of DNA(3,5) shows additional higher m/z peaks at intervals of 16 amu corresponding to addition of oxygen atoms to the crosslinked trimer. The formation of these oxygen adducts may occur by nucleophilic attack by water during the HRP/H2O2 reaction, oxidation by air during sample purification or by oxidation during the ESI mass analysis. Nonetheless, the mass spectroscopic analysis clearly demonstrates that the (SNS)3 trimer is formed in this reaction.
Conclusions
DNA is a powerful building block for the programmed “bottom-up” assembly of the supermolecular scaffolds needed for the construction of nanoscale materials and devices. In this work we demonstrate that SNS zipper arrays are formed by hybridization of complementary DNA oligomers each containing covalently bound SNS monomers. UV-Vis absorption and circular dichroism spectroscopic studies demonstrate that the SNS monomers are in the DNA major groove in a face-to-face H-aggregate-like chiral structure induced by the right-handed helical B-form DNA. Upon oxidation with HRP/H2O2, these SNS arrays are converted to oligomers having specific lengths and conformation. As a consequence of this reaction the two SNS-containing strands are permanently crosslinked. These findings provide a clear demonstration of a facile pathway from the DNA-directed orderly array of monomers to a robust nanostructure resistant to denaturation. These materials may be useful for building stable nanostructures that find application in chemical biology.Acknowledgements
This work is supported by the Vasser Woolley Foundation, for which we are grateful.Notes and references
- A. Heckel and M. Famulok, Biochimie, 2008, 90, 1096–1107 CrossRef CAS.
- X. D. Liu, H. Y. Diao and N. Nishi, Chem. Soc. Rev., 2008, 37, 2745–2757 RSC.
- N. C. Seeman, Nature, 2003, 421, 427–431 CrossRef.
- N. Badi and J. F. Lutz, Chem. Soc. Rev., 2009, 38, 3383–3390 RSC.
- L. Zhu, P. S. Lukeman, J. W. Canary and N. C. Seeman, J. Am. Chem. Soc., 2003, 125, 10178–10179 CrossRef CAS.
- T. Nguyen, A. Brewer and E. Stulz, Angew. Chem., Int. Ed., 2009, 48, 1974–1977 CrossRef CAS.
- I. Bouamaied, L. A. Fendt, D. Haussinger, M. Wiesner, S. Thoni, N. Amiot and E. Stulz, Nucleosides, Nucleotides Nucleic Acids, 2007, 26, 1533–1538 CAS.
- J. B. Ravnsbaek, M. F. Jacobsen, C. B. Rosen, N. V. Voigt and K. V. Gothelf, Angew. Chem., Int. Ed., 2011, 50, 10851–10854 CrossRef CAS.
- E. Braun, Y. Eichen, U. Sivan and G. Ben-Yoseph, Nature, 1998, 391, 775–778 CrossRef CAS.
- C. T. Wirges, J. Timper, M. Fischler, A. S. Sologubenko, J. Mayer, U. Simon and T. Carell, Angew. Chem., Int. Ed., 2009, 48, 219–223 CrossRef CAS.
- M. Fischler, U. Simon, H. Nir, Y. Eichen, G. A. Burley, J. Gierlich, P. M. E. Gramlich and T. Carell, Small, 2007, 3, 1049–1055 CrossRef CAS.
- L. A. Fendt, I. Bouamaied, S. Thoni, N. Amiot and E. Stulz, J. Am. Chem. Soc., 2007, 129, 15319–15329 CrossRef CAS.
- M. Heilemann, R. Kasper, P. Tinnefeld and M. Sauer, J. Am. Chem. Soc., 2006, 128, 16864–16875 CrossRef CAS.
- B. Datta, G. B. Schuster, A. McCook, S. C. Harvey and K. Zakrzewska, J. Am. Chem. Soc., 2006, 128, 14428–14429 CrossRef CAS.
- B. Datta and G. B. Schuster, J. Am. Chem. Soc., 2008, 130, 2965–2973 CrossRef CAS.
- S. Srinivasan and G. B. Schuster, Org. Lett., 2008, 10, 3657–3660 CrossRef CAS.
- W. Chen, M. Josowicz, B. Datta, G. B. Schuster and J. Janata, Electrochem. Solid-State Lett., 2008, 11, E11–E14 CrossRef CAS.
- W. Chen, G. Guler, E. Kuruvilla, G. B. Schuster, H. C. Chiu and E. Riedo, Macromolecules, 2010, 43, 4032–4040 CrossRef CAS.
- W. Chen and G. B. Schuster, J. Am. Chem. Soc., 2012, 134, 840–843 CrossRef CAS.
- S. Geggier and A. Vologodskii, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 15421–15426 CrossRef CAS.
- A. Perez, F. J. Luque and M. Orozco, Acc. Chem. Res., 2012, 45, 196–205 CrossRef CAS.
- P. Kočalka, A. H. El-Sagheer and T. Brown, ChemBioChem, 2008, 9, 1280–1285 CrossRef.
- H. Xiong and F. Seela, Bioconjugate Chem., 2012, 23, 1230–1243 CrossRef CAS.
- E. Auyeung, R. J. Macfarlane, C. H. J. Choi, J. I. Cutler and C. A. Mirkin, Adv. Mater., 2012, 5181–5186 CrossRef CAS.
- S. K. Kim and B. Nordén, FEBS Lett., 1993, 315, 61–64 CrossRef CAS.
- T. Biver, Appl. Spectrosc. Rev., 2012, 47, 272–325 CrossRef CAS.
- H. Asanuma, K. Shirasuka, T. Takarada, H. Kashida and M. Komiyama, J. Am. Chem. Soc., 2003, 125, 2217–2223 CrossRef CAS.
- J. A. Jung, S. H. Lee, B. Jin, Y. Sohn and S. K. Kim, J. Phys. Chem. B, 2010, 114, 7641–7648 CrossRef CAS.
- D. M. Gray, S. H. Hung and K. H. Johnson, Methods Enzymol., 1995, 246, 19–34 CAS.
- V. M. Domingo, C. Aleman, E. Brillas and L. Julia, J. Org. Chem., 2001, 66, 4058–4061 CrossRef CAS.
- R. E. Niziurski-Mann, C. Scordilis-Kelley, T. L. Liu, M. P. Cava and R. T. Carlin, J. Am. Chem. Soc., 1993, 115, 887–891 CrossRef CAS.
- J. Heinze, B. A. Frontana-Uribe and S. Ludwigs, Chem. Rev., 2010, 110, 4724–4771 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ob26716k |
| This journal is © The Royal Society of Chemistry 2013 |