Investigations regarding the utility of prodigiosenes to treat leukemia†
Deborah A. Smithena, A. Michael Forresterbc, Dale P. Corkeryd, Graham Dellairede, Julie Colpittsf, Sherri A. McFarlandf, Jason N. Bermanbcg and Alison Thompson*a
aDepartment of Chemistry, Dalhousie University, Halifax, Nova Scotia B4P 2R6, Canada. E-mail: alison.thompson@dal.ca
bDepartment of Microbiology and Immunology, Dalhousie University, Halifax, Nova Scotia B4P 2R6, Canada
cIWK Health Centre, Halifax, Nova Scotia B3K 6R8, Canada
dDepartment of Biochemistry and Molecular Biology, Dalhousie University, Halifax, Nova Scotia B4P 2R6, Canada
eDepartment of Pathology, Dalhousie University, Halifax, Nova Scotia B4P 2R6, Canada
fDepartment of Chemistry, Acadia University, Wolfville, Nova Scotia B4P 2R6, Canada
gDepartment of Pediatrics, Dalhousie University, P.O. Box 15000, Halifax, Nova Scotia B3H 4R2, Canada
First published on 15th October 2012
Abstract
Prodigiosenes, possessing a 4-methoxypyrrolyldipyrrin skeleton, are known for their anti-cancer activity. Structural modification of the C-ring resulted in a series of prodigiosenes that displayed promising activity against leukemia cell lines during in vitro analysis against the NCI 60 cancer cell line panel. Further in vivo studies of these compounds using the zebrafish model showed persistence of anti-leukemia properties in human K562 chronic myelogenous leukemia cells.
Introduction
Prodigiosin (1) is the parent member of a family of tripyrrolic natural products, isolated from several Serratia, Streptomyces and Bacillus strains of bacteria. Prodigiosin (1) possesses potent immunosuppressive, antimicrobial and cytotoxic properties.1–3 Coley's Toxin,4 consisting of a mixture of bacterial extracts including Serratia Marcescens, contains prodigiosin (1) as well as lipopolysaccharides, and was successfully used (USA 1893–1963) as an anti-cancer treatment before its withdrawal by the FDA due to systemic toxicity.5 The anti-cancer activity displayed by prodigiosin is thought to be the result of several mechanistic modes of action: for example, it is known to transport H+/Cl− over phospholipid membranes1,6–8 and to induce copper-mediated double-strand DNA cleavage.6–12Selective cytotoxicity of prodigiosin has been noted against various human cancer cell lines, for example haematopoietic,13 colon14 and gastric15 cancer cell lines. Although there are brief descriptions in the patent literature,19–21 the use of such tripyrrolic compounds for the treatment of leukemia was not published in the journal literature until 1988,16 whereby in vitro studies showed prodigiosin to be exceptionally potent against P388 leukemia, alongside significant activity against other cancerous cell lines. Omission of the methoxy (R1) substituent in ring B, as in prodigiosene 2,16,17 resulted in greatly diminished cytotoxicity and removal of all substituents (3) led to equal or further reduced activity, relative to prodigiosene 2, in the majority of cancer cell lines examined.16
This trend in activity was studied further by D'Alessio and co-workers,18–20 who found that substitution of the C-6 methoxy group with alkoxy substituents of increasing size lead to a step-wise reduction in activity and cytotoxicity. Other studies pertaining to the treatment of leukemia focused on the elucidation of signalling pathways and modes of action involved in prodigiosin-induced apoptosis. Cell lines examined include Jurkat-T cells,27–29 HL-60 leukemia cells9 and B-cell chronic lymphocytic leukemia (B-CLL) cells.21 In the latter case it was found that prodigiosin induced apoptosis of B-CLL cells through caspase activation and was the first report showing that prodigiosin induces apoptosis in human primary cancer cells.21
Myeloid leukemias are a heterogeneous group of diseases in need of novel therapeutic agents.22,23 Acute myeloid leukemia (AML) remains fatal for 40% of patients,23 both due to refractory disease and toxicity from traditional therapeutic agents. Some experts believe that AML treatment has a reached a “plateau” in efficacy.24 AML is the most common acute leukemia in adults and accounts for nearly 20% of childhood leukemias. While pediatric clinical trials over the past decades have resulted in significant improvements to overall survival of patients, cure rates for AML have peaked in the 60% range, which is significantly inferior to the 90% childhood cure rates for acute lymphoblastic leukemia (ALL).25 The need for new anti-leukemia agents is thus pressing, particularly ones that avoid overlapping toxicities with current chemotherapies, such as AraC and anthracyclines.35,36
With this in mind, we synthesized C-ring modified prodigiosenes and saw activity against leukemia cell lines in vitro, including K562 erythroleukemia cells. K562 cells harbour the t(9;22)(q34;q11) translocation termed the Philadelphia chromosome, which results in the pathognomonic BCR-ABL fusion gene found in chronic myelogenous leukemia (CML).26 Since the resulting BCR-ABL oncoprotein can be successfully targeted by tyrosine kinase inhibitors, including the prototypic imatinib mesylate (Gleevec),27 this cell line provided a positive control with which to evaluate efficacy of our prodigiosene compounds. To validate our in vitro studies, we investigated the efficacy of our novel prodigiosenes against leukemia cell survival in vivo using a novel zebrafish xenotransplantation platform that we have recently pioneered for the study of anti-leukemia agents in a live animal model.
Results and discussion
Most structure–activity relationship (SAR) studies concerning prodigiosenes have focused on the A-ring (3),28–31 with some success in retaining the anti-proliferative behaviour of the parent natural product. Our work concerns structural modification of the C-ring within the prodigiosene skeleton (see 3, Fig. 1), an area that was previously under-developed. We hereby report the use of pendant alkyl ester groups i.e., those lacking a carbonyl group directly adjacent to the pyrrole ring. In addition, we varied the position of the functional group on the C-ring and examined the effect of di-ester functionality.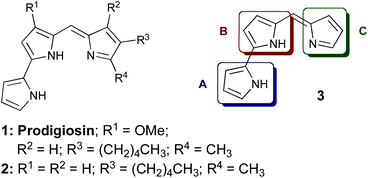 | ||
| Fig. 1 Prodigiosin (1), analogue 2 and prodigiosene core structure 3. | ||
Synthesis of C-ring modified prodigiosenes generally begins with a Knorr-type pyrrole (4, Scheme 1) bearing the desired functionality in the 4-position. It is this heterocycle that becomes the C-ring (3) of the final product. Our initial synthetic route32 involved Suzuki cross-coupling of triflate-activated dipyrrins with N-Boc pyrrole-2-boronic acid.24,25 This strategy facilitated the introduction of various C-ring pyrroles, most notably those stabilized by a deactivating carbonyl group directly adjacent to the pyrrole ring.32 However, this method was less successful for C-ring pyrroles lacking a conjugated carbonyl moiety, prompting the development of a modified protocol to include the formation of bromo-substituted dipyrrins, as a more robust alternative to the triflate analogue.33 With the exception of this modification, the syntheses followed the same pathway to that used originally,32 with the first steps being hydrogenolysis and decarboxylation of Knorr-type pyrroles 4a and 4b (Scheme 1) followed by formylation of the resulting α-free pyrroles using trimethylorthoformate (TMOF).34
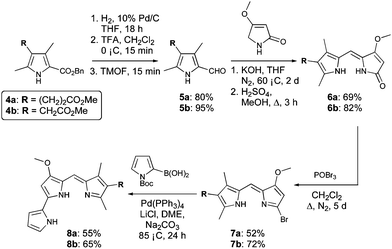 | ||
| Scheme 1 Synthesis of prodigiosenes 8a and 8b. | ||
Condensation of the 2-formyl pyrroles (5a and 5b) with 4-methoxy-3-pyrrolin-2-one,35–37 under basic conditions, followed by re-esterification of the resulting carboxylic acids in acidic methanol, afforded dipyrrinones 6a and 6b, respectively, in good yields. Reaction of 6a and 6b with phosphorous oxybromide proceeded over 5 days: the extended reaction time gave optimum conversion to the desired 2-bromodipyrrins (7a and 7b), as did the use of excess POBr3 (4 equiv.) added in two portions during the course of the reaction. Subsequent Suzuki coupling was successful for the bromide derivatives 7a and 7b, giving the target prodigiosenes 8a and 8b in yields of 55 and 65%, respectively.
Synthesis of 8a and 8b demonstrated the significance of the improved methodology and allowed us to design more challenging target compounds that also featured alkanoate substituents. Variation of the position of the C-ring substituent and incorporation of multiple substituents were of interest to further probe the SAR.
Pyrroles 4c and 4d (Scheme 2) were both derived from methyl-4,6-dioxoheptanoate (12, Scheme 2),34 itself synthesised from tert-butyl acetoacetate (9). Indeed, reaction of tert-butyl acetoacetate with magnesium methoxide afforded the Grignard reagent 10,38 and subsequent reaction of 10 with 3-methoxycarbonyl propionyl chloride, followed by thermolysis of the resulting Boc-protected product (11) in the presence of catalytic p-tosic acid gave 12 in a 39% yield over the three steps. From here, the 5-methyl and 5-ethyl ethanoate diketones 13a and 13b were synthesized by reaction of 12 with methyl iodide or ethyl bromoacetate, respectively.34 The two diketones were subsequently employed in separate Knorr-type reactions to generate the pyrroles 4c and 4d. Hydrogenolysis of the benzyl esters followed by decarboxylation and formylation gave the 2-formyl pyrroles 5c and 5d (Scheme 3).
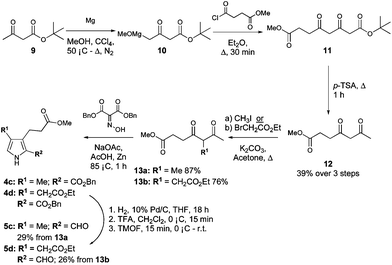 | ||
| Scheme 2 Synthesis of 3,4-disubstituted pyrroles. | ||
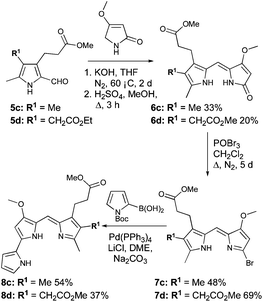 | ||
| Scheme 3 Synthesis of 8c and 8d. | ||
Condensation of 5c and 5d with 4-methoxy-3-pyrrolin-2-one,35–37 followed by re-establishment of the esters gave 6c and 6d (Scheme 3). In the latter case (6d), this resulted in two terminal methyl ester groups, as opposed to the precursor (5d), which possessed both ethyl and methyl ester groups. Bromination of 6c and 6d followed by palladium-catalyzed cross-coupling of the resulting bromodipyrrins 7c and 7d with N-Boc pyrrole-2-boronic acid completed the syntheses of 8c and 8d, respectively (Scheme 3).
We were interested to learn whether the prodigiosenes 8a–d had the ability to induce copper-mediated DNA cleavage, akin to prodigiosin (1). Compound 8a was selected as representative of the group and subjected to agarose gel electrophoresis with supercoiled plasmid DNA in the presence of Cu(OAc)2.11 Following incubation at 37 °C for two hours (8a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu 1:1), the concentration required to effect 50% DNA cleavage (EC50) was calculated based on its fit to a non-linear sigmoidal dose–response curve. Significant DNA cleavage was observed from 8a (EC50 = 14.1 μM), indicating that C-ring modified prodigiosenes maintain the cleavage ability of prodigiosin (1, EC50 = 9.2 μM).
:Cu 1:1), the concentration required to effect 50% DNA cleavage (EC50) was calculated based on its fit to a non-linear sigmoidal dose–response curve. Significant DNA cleavage was observed from 8a (EC50 = 14.1 μM), indicating that C-ring modified prodigiosenes maintain the cleavage ability of prodigiosin (1, EC50 = 9.2 μM).
All four novel prodigiosenes 8a–d were selected for screening by the National Cancer Institute (NCI) against their standard panel of 60 human cell lines, derived from nine cancer cell types. The averaged results of these in vitro screens are shown in Table 1, alongside those previously determined for prodigiosin (1).
| Entry | Compound | Log10 mean GI50 | Log10 mean TGI | Log10 mean LC50 |
|---|---|---|---|---|
| 1 | 1 | −7.85 | −5.68 | −6.65 |
| 2 | 8a | −6.24 | −5.44 | −4.59 |
| 3 | 8b | −6.70 | −5.71 | −4.81 |
| 4 | 8c | −6.27 | −5.27 | −4.52 |
| 5 | 8d | −5.45 | −4.82 | −4.29 |
With the exception of 8d (entry 5) all compounds showed sufficient anti-cancer activity to warrant further study. Prodigiosene 8b (entry 3) was selected by the NCI for additional toxicity studies and it was found that this compound caused no deaths in mice at 50 mg kg−1versus acute system toxicity at 4 mg kg−1 for prodigiosin (1). Of further interest, during the original 60-line screen prodigiosenes 8a–d displayed enhanced activity against leukemia cell lines compared to lines representing the other eight types of cancer included in the NCI panel (Fig. 2). Indeed, compounds 8a–c uniformly effected cell kill against the leukemia cell lines: note the negative growth percentages across the leukemia cell lines versus both negative and positive growth of cells across all of the other cancer types. This preferential activity was not observed for any of the previous prodigiosenes that have been synthesized in our group and evaluated in the NCI 60-line screen.
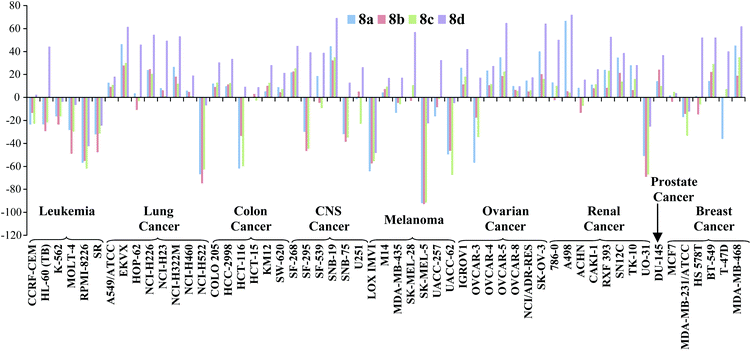 | ||
| Fig. 2 In vitro activity of prodigiosenes 8a–d, at 10 μM concentration, against 60 human cancer cell lines representing 9 different cancer types; negative growth % represents cell death; http://dtp.cancer.gov. | ||
We explored this new-found activity against leukemia cells in vivo and decided to employ the zebrafish model, an emmerging robust model for vertebrate blood development and leukemia.39 We chose to use mutant casper fish40 which lack all pigmentation and are thus ideal for tracking fluorescently-labelled cells in vivo during real-time analysis. Micro-injection41 (Fig. 3) of CM-DiI-labelled K562 human leukemia cells (∼50 cells) directly into the yolk sac of casper embryos at 48 hours of life, allowed successful xenotransplantation to be determined via the presence of a fluorescent mass at the injection site 24 hours post injection (hpi).
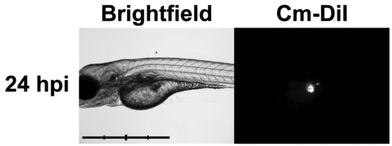 | ||
| Fig. 3 Baseline proliferation (24 hpi) of xenotransplanted K562 leukemia cells in zebrafish embryo. Brightfield (left) and 555 nm fluorescent (right) images of casper zebrafish embryos following successful injection of K562 human leukemia cells at 48 hours of life. Scale bar = 1 mm, intervals = 250 μm. (Abbreviations: hpi = hours post-injection.) | ||
We then sought to determine whether the prodigiosenes 8a–d could inhibit growth of the leukemia cells: injected embryos were divided into six groups; two controls and one each with addition of a prodigiosene (8) to the water. Prodigiosenes were resuspended in DMSO and the optimal dose (50% of maximum tolerated dose, MTD) of each compound was determined by conducting toxicity curves using 48-hour zebrafish embryos treated for 72 hours. Prodigiosenes 8c (2 μM) and 8d (3 μM) were better tolerated than 8a (0.2 μM) and 8b (0.2 μM). When the MTD was exceeded with 8a and 8b, the fish died; with 8c the embryos exhibited reduced activity, but were still alive; and with 8d, the embryos seemed unaffected by increasing concentrations, even above the MTD value that was selected as higher concentrations exhibited incomplete dissolution and started to undesirably stain the embryos a brown-red colour. At the MTD concentrations, all prodigiosenes were water-soluble: no precipitation was observed for any compound. Engrafted embryos were followed using live cell microscopy and living embryos were imaged every 24 hours to monitor cell numbers and migration (Fig. 4). We observed that K562 cells engrafted in vehicle and drug-treated fish, and either exhibited proliferation and extensive migration throughout the fish (i.e. proliferative), a single stable mass of cells (i.e. cytostatic), or visible regression of tumour mass (i.e. cytotoxic) over time. We classified the embryos into these three categories, via scoring.
![Anti-leukemia activity of prodigiosenes 8a–din vivo against K562 human leukemia cells injected into casper zebrafish. Embryos injected at 48 hours of life with K562 human leukaemia cells. K562 cell numbers and migration were monitored through live cell microscopy every 24 hours up to 96 hpi. Brightfield (left panels in each pair) and 555 nm fluorescent (right panels in each pair) images of casper zebrafish embryo at indicated time-points. Embryo displayed in side profile, anterior to the left. (A–F) Embryos were treated with (A) 0.3% DMSO (negative control); (B) 20 μM IM (positive control);41 (C) 0.2 μM 8a; (D) 0.2 μM 8b; (E) 2 μM 8c; and (F) 3 μM 8d. Drugs were added directly to water at 50% maximum tolerated dose [MTD]. Treatment was initiated when embryos were 24 hpi, and incubation in chemical continued until end of assessment. Asterisks indicate autofluoresence of prodigiosenes in the zebrafish liver [*] and swim bladder [**]; white arrows indicate leukaemia cells; IM = imatinib mesylate; hpi = hours post-injection.](/image/article/2013/OB/c2ob26535d/c2ob26535d-f4.gif) | ||
| Fig. 4 Anti-leukemia activity of prodigiosenes 8a–din vivo against K562 human leukemia cells injected into casper zebrafish. Embryos injected at 48 hours of life with K562 human leukaemia cells. K562 cell numbers and migration were monitored through live cell microscopy every 24 hours up to 96 hpi. Brightfield (left panels in each pair) and 555 nm fluorescent (right panels in each pair) images of casper zebrafish embryo at indicated time-points. Embryo displayed in side profile, anterior to the left. (A–F) Embryos were treated with (A) 0.3% DMSO (negative control); (B) 20 μM IM (positive control);41 (C) 0.2 μM 8a; (D) 0.2 μM 8b; (E) 2 μM 8c; and (F) 3 μM 8d. Drugs were added directly to water at 50% maximum tolerated dose [MTD]. Treatment was initiated when embryos were 24 hpi, and incubation in chemical continued until end of assessment. Asterisks indicate autofluoresence of prodigiosenes in the zebrafish liver [*] and swim bladder [**]; white arrows indicate leukaemia cells; IM = imatinib mesylate; hpi = hours post-injection. | ||
Embryos treated with 0.3% DMSO served as a negative control, showing abundant K562 cell proliferation and entry of cells into circulation. Embryos treated with 20 μM imatinib mesylate (IM), a targeted inhibitor of the BCR-ABL1 oncoprotein in K562 cells, served as a positive control, demonstrating no cell proliferation or migration.41 By comparison, prodigiosenes (8a–d) inhibited K562 cell activity to a similar or greater degree than IM, preventing proliferation of the leukemia cells (Fig. 5). The effects of prodigiosenes appeared to be graduated: 8a (n = 7/13) was predominantly cytostatic, 8b (n = 6/14) and 8d (n = 5/14) were moderately cytotoxic. Activity of compound 8c (n = 8/11) was of particular note, being strongly cytotoxic. Migration of K562 cells was sometimes observed in embryos treated with prodigiosenes, independent of apparent cytostatic activity of the compound. This indicates that prodigiosenes are not likely to affect cell migration. Migration of K562 cells in fish with clear cytotoxic effects and tumour regression were rarely seen.
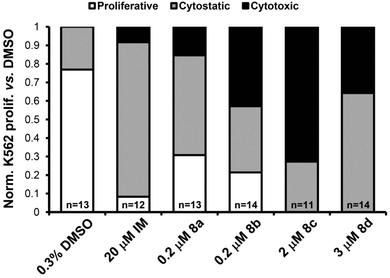 | ||
| Fig. 5 Bar graph summarizing in vivo activity of prodigiosenes 8a–d against human K562 leukemia cell proliferation in zebrafish. The effects of treatments on K562 activity were scored relative to DMSO control, then normalized to a percentage value of the total number of embryos of that treatment group. White bar = “proliferative”; grey bar = “cytostatic”; black bar = “cytotoxic”; n = number of embryos used for analysis. | ||
Conclusions
Four previously inaccessible prodigiosenes bearing pendant alkyl ester groups on the C-ring were synthesised. These compounds showed particular activity for leukemia cells in vitro that was explored in vivo using the zebrafish model. Initial results indicate that all compounds demonstrate anti-proliferation and, in some cases, selective induction of cell-death in K562 leukemia cells. Our work also extends the use of zebrafish xenografts to determine the in vivo sensitivity of human cancer cells to novel drugs and suggests that tailored prodigiosenes may represent novel therapeutic agents for leukemia with improved anti-cancer potency and reduced toxicity.Experimental section
General procedure for the synthesis of 2-formyl pyrroles (5)
(a) Triethylamine (∼10 drops) was added to a solution of the 2-benzyl ester pyrrole (4, 4 mmol, 1 eq.) in ethanol (95%) or THF (65 mL) and the solution was degassed with nitrogen for 10 minutes. 10% Pd/C (∼10% by weight of pyrrole 4) was then added and the reaction was placed under a hydrogen atmosphere, with use of a hydrogen-filled balloon, with stirring at r.t. overnight. The reaction mixture was then filtered through Celite, concentrated to dryness and the residue was dissolved in 5% aq. ammonium hydroxide (60 mL) and washed with ethyl acetate. The aqueous phase was then acidified to pH 4 (pH paper) with 5% citric acid and the product was extracted with ethyl acetate (3 × 80 mL). The organic extracts were combined and washed with brine, dried over magnesium sulfate, filtered and concentrated to give the 2-carboxy pyrrole product, which did not require further purification. (b) TFA (44 mmol, 11 eq.) was added drop-wise, over 15 minutes, to a solution of 2-carboxy pyrrole (4 mmol, 1 eq.) in freshly distilled dichloromethane (10 mL), with stirring at 0 °C under N2 for 15 minutes. Trimethylorthoformate (20 mmol, 5 eq.) was then added drop-wise, over 15 minutes, and the reaction mixture was stirred under N2 for a further 20 minutes, warming to r.t., before being poured into ice-water (50 mL) followed by slow addition of 6 M aq. sodium hydroxide (32 mmol, 8 eq.) and extraction with chloroform (3 × 50 mL). The combined organic extracts were carefully washed with 5% aq. NaHCO3 solution (2 × 80 mL), water (80 mL) and brine (80 mL), before being dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to give the crude product, which was purified using column chromatography on silica, eluting with 40–60% ethyl acetate in hexanes, to give the product (5).General procedure for the synthesis of dipyrrinones (6)
Aqueous potassium hydroxide (4 M, 20 mmol, 2 equiv.) was added drop-wise to a solution of 4-methoxy-3-pyrrolin-2-one (15.5 mmol, 1.55 equiv.) in THF (100 mL) and the resulting solution was purged with nitrogen for 15 minutes before warming to 60 °C, with stirring under nitrogen for 1 hour. The reaction mixture was then cooled slightly and the 2-formyl pyrrole (5, 10 mmol, 1 equiv.) was added slowly before re-heating to 60 °C with stirring under nitrogen for 48 hours. After this time the reaction mixture was concentrated in vacuo, the residue was dissolved in methanol and conc. HCl (12 M, 30 mmol, 3 equiv.) was added drop-wise. The solution was then left to cool in the freezer, whereby a precipitate formed and was collected by filtration, and washed with hexane to give the saponified product as a yellow solid. Re-esterification was achieved by heating a solution of the preceding carboxylic acid (10 mmol, 1 equiv.) and sulfuric acid (9 mmol, 0.9 equiv.) in methanol (750 mL) to reflux temperature for 3 hours, with stirring under nitrogen. The reaction mixture was then concentrated in vacuo and the residue was dissolved in dichloromethane (80 mL), washed with water (50 mL) and brine (50 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to give the product (6).General procedure for the synthesis of 2-bromodipyrrins (7)
Phosphoryl bromide (4 mmol, 2 eq.) was added to a solution of dipyrrinone (6, 2 mmol, 1 eq.) in anhydrous dichloromethane (60 mL) under N2 and the reaction mixture was heated to reflux temperature with stirring for 24 hours. The reaction mixture was then cooled slightly before further phosphoryl bromide (4 mmol, 2 eq.) was added. Reflux temperature was then re-established, with continued stirring under nitrogen for 72 hours or until consumption of starting material was observed using TLC. The reaction mixture was then cooled to 0 °C and saturated aq. NaHCO3 (40 mL) was added slowly with stirring. When gas evolution had ceased, the reaction mixture was diluted with water (40 mL) and thoroughly extracted with dichloromethane (4 × 50 mL). The organic fractions were combined and dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to give the crude product, which was dissolved in dichloromethane and filtered through a short pad of neutral alumina, washing with dichloromethane, to give the product (7).General procedure for the synthesis of prodigiosenes (8)
N-Boc-pyrrole-2-boronic acid (0.36 mmol, 1.2 equiv.), lithium chloride (0.9 mmol, 3 equiv.) and tetrakis(triphenylphosphine)palladium (0.03 mmol, 0.1 equiv.) were added to a solution of the preceding 2-bromodipyrrin (7, 0.3 mmol, 1 equiv.) in 1,2-dimethoxyethane (12 mL, 0.025 M) and the resulting mixture was purged with nitrogen for 15 minutes. Aqueous sodium carbonate (2 M, 1.2 mmol, 4 equiv.), previously bubbled with nitrogen, was then added drop-wise and the reaction mixture was heated to 85 °C, with stirring under nitrogen for 24 hours. After cooling to room temperature, the reaction mixture was diluted with water (30 mL) and thoroughly extracted with ethyl acetate (4 × 30 mL). The organic extracts were combined and washed with water (60 mL) and brine (60 mL), then dried over anhydrous magnesium sulfate before filtering and concentrating in vacuo to give the crude product, which was purified using column chromatography over neutral alumina, eluting with 2–40% ethyl acetate in hexanes, to give the product (8).Acknowledgements
The authors thank Drs. Donald Small and Robert Arceci (Johns Hopkins School of Medicine) for provision of the K562 cell line. A.M.F. was supported by graduate Awards from the Canadian Institutes of Health Research (CIHR) and The Canadian Cancer Society, in conjunction with The Beatrice Hunter Cancer Research Institute. D.C. was supported by a trainee award from the Beatrice Hunter Cancer Research Institute (BHCRI) with funds provided by The Canadian Breast Cancer Foundation (CBCF) – Atlantic Region. G.D. is a Canadian Institutes of Health New Investigator (CIHR) and both G.D. and J.N.B. are Senior Scientists of the BHCRI. This work was funded by research grants to: A.T. from CIHR and the Nova Scotia Health Research Foundation; G.D. from the CBCF-Atlantic; J.N.B. from the Dalhousie Clinical Research Scholar Program and S.A.M. from NSERC. Human cell-line screening was performed by the Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute (http://dtp.cancer.gov).Notes and references
- A. Fürstner, Angew. Chem., Int. Ed., 2003, 42, 3582–3603 CrossRef.
- R. A. Manderville, Curr. Med. Chem.: Anti-Cancer Agents, 2001, 1, 195–218 CrossRef CAS.
- B. Montaner and R. Pérez-Tomás, Curr. Cancer Drug Targets, 2003, 3, 57–65 CrossRef CAS.
- G. Rook, Nature, 1992, 357, 545 CrossRef CAS.
- R. Péréz-Tomas, B. Montaner, E. Llagostera and V. Soto-Cerrato, Biochem. Pharmacol., 2003, 66, 1447–1452 CrossRef.
- A. Fürstner and E. J. Grabowski, ChemBioChem, 2001, 2, 706–709 CrossRef.
- M. S. Melvin, M. W. Calcutt, R. E. Noftlet and R. A. Manderville, Chem. Res. Toxicol., 2002, 15, 742–748 CrossRef CAS.
- M. S. Melvin, D. C. Ferguson, N. Lindquist and R. A. Manderville, J. Org. Chem., 1999, 64, 6861–6869 CrossRef CAS.
- M. S. Melvin, J. T. Tomlinson, G. Park, C. S. Day, G. R. Saluta, G. L. Kucera and R. A. Manderville, Chem. Res. Toxicol., 2002, 15, 734–741 CrossRef CAS.
- M. S. Melvin, J. T. Tomlinson, G. R. Saluta, G. L. Kucera, N. Lindquist and R. A. Manderville, J. Am. Chem. Soc., 2000, 122, 6333–6334 CrossRef CAS.
- M. S. Melvin, K. E. Wooton, C. C. Rich, G. R. Saluta, G. L. Kucera, N. Lindquist and R. A. Manderville, J. Inorg. Biochem., 2001, 87, 129–135 CrossRef CAS.
- G. Park, J. T. Tomlinson, M. S. Melvin, M. W. Wright, C. S. Day and R. A. Manderville, Org. Lett., 2003, 5, 113–116 CrossRef CAS.
- B. Montaner, S. Navarro, M. Pique, M. Vilaseca, M. Martinell, E. Giralt, J. Gil and R. Perez-Tomas, Br. J. Pharmacol., 2000, 131, 585–593 CrossRef CAS.
- B. Montaner and R. Perez-Tomas, Life Sci., 2001, 68, 2025–2036 CrossRef CAS.
- C. Diaz-Ruiz, B. Montaner and R. Perez-Tomas, Histol. Histopathol., 2001, 16, 415–421 CAS.
- D. L. Boger and M. Patel, J. Org. Chem., 1988, 53, 1405–1415 CrossRef CAS.
- W. R. Hearn, M. K. Elson, R. H. Williams and J. Medina-Castro, J. Org. Chem., 1970, 35, 142–146 CrossRef CAS.
- R. D'Alessio, A. Bargiotti, O. Carlini, F. Colotta, M. Ferrari, P. Gnocchi, A. M. Isetta, N. Mongelli, P. Motta, A. Rossi, M. Rossi, M. Tibolla and E. Vanotti, J. Med. Chem., 2000, 43, 2557–2565 CrossRef CAS.
- R. D'Alessio and A. Rossi, Synlett, 1996, 513–514 CrossRef CAS.
- V. Rizzo, A. Morelli, V. Pinciroli, D. Sciangula and R. D'Alessio, J. Pharm. Sci., 1999, 88, 73–78 CrossRef CAS.
- C. Campas, M. Dalmau, B. Montaner, M. Barragan, B. Bellosillo, D. Colomer, G. Pons, R. Perez-Tomas and J. Gil, Leukemia, 2003, 17, 746–750 CrossRef CAS.
- G. Marcucci, T. Haferlach and H. Doehner, J. Clin. Oncol., 2011, 29, 475–486 CrossRef CAS.
- G. J. Roboz, Hematology Am. Soc. Hematol. Educ. Program, 2011, 2011, 43–50 CrossRef.
- B. Lowenberg, T. Pabst, E. Vellenga, P. W. van, H. C. Schouten, C. Graux, A. Ferrant, P. Sonneveld, B. J. Biemond, A. Gratwohl, G. G. E. de, L. F. Verdonck, M. R. Schaafsma, M. Gregor, M. Theobald, U. Schanz, J. Maertens and G. J. Ossenkoppele, N. Engl. J. Med., 2011, 364, 1027–1036 CrossRef.
- W. G. Woods, Pediatr. Blood Cancer, 2006, 46, 565–569 Search PubMed.
- R. P. Gale and E. Canaani, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 5648–5652 CrossRef CAS.
- T. P. Hughes, A. Hochhaus, S. Branford, M. C. Muller, J. S. Kaeda, L. Foroni, B. J. Druker, F. Guilhot, R. A. Larson, S. G. O'Brien, M. S. Rudoltz, M. Mone, E. Wehrle, V. Modur, J. M. Goldman and J. P. Radich, Blood, 2010, 116, 3758–3765 CrossRef CAS.
- C. M. Baldino, J. Parr, C. J. Wilson, S.-C. Ng, D. Yohannes and H. H. Wasserman, Bioorg. Med. Chem. Lett., 2006, 16, 701–704 CrossRef CAS.
- K. Daïri, S. Tripathy, G. Attardo and J.-F. Lavallée, Tetrahedron Lett., 2006, 47, 2605–2606 CrossRef.
- H. H. Wasserman, A. K. Petersen, M. Xia and J. Wang, Tetrahedron Lett., 1999, 40, 7587–7589 CrossRef CAS.
- H. H. Wasserman, M. Xia, J. Wang, A. K. Petersen, M. Jorgensen, P. Power and P. Parr, Tetrahedron, 2004, 60, 7419–7425 CrossRef CAS.
- J. Regourd, A. Al-Sheikh Ali and A. Thompson, J. Med. Chem., 2007, 50, 1528–1536 CrossRef CAS.
- M. I. Uddin, S. Thirumalairajan, S. M. Crawford, T. S. Cameron and A. Thompson, Synlett, 2010, 2561–2564 CAS.
- T. D. Lash, U. N. Mani, E. A. Lyons, P. Thientanavanich and M. A. Jones, J. Org. Chem., 1999, 64, 478–487 CrossRef CAS.
- L. Duc, J. F. McGarrity, T. Meul and A. Warm, Synthesis, 1992, 391–394 CrossRef CAS.
- R. C. F. Jones and A. D. Bates, Tetrahedron Lett., 1986, 27, 5285–5288 CrossRef CAS.
- K. S. Kochhar, H. J. Carson, K. A. Clouser, J. W. Elling, L. A. Gramens, J. L. Parry, H. L. Sherman, K. Braat and H. W. Pinnick, Tetrahedron Lett., 1984, 25, 1871–1874 CrossRef CAS.
- A. R. Battersby, C. J. R. Fookes, M. J. Meegan, E. McDonald and H. K. W. Wurziger, J. Chem. Soc., Perkin Trans. 1, 1981, 2786–2799 RSC.
- J. F. Amatruda and L. I. Zon, Dev. Biol., 1999, 216, 1–15 CrossRef CAS.
- R. M. White, A. Sessa, C. Burke, T. Bowman, J. LeBlanc, C. Ceol, C. Bourque, M. Dovey, W. Goessling, C. E. Burns and L. I. Zon, Cell Stem Cell, 2008, 2, 183–189 CrossRef CAS.
- D. P. Corkery, G. Dellaire and J. N. Berman, Br. J. Haematol., 2011, 153, 786–789 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General Experimental; Zebrafish Husbandry, Embryo Collection and Embryo Staging; Xenotransplantation of Human Leukemia Cells Into Zebrafish Embryos, Dissociation, and Immunofluorescence; Experimental Procedures and Data; NMR spectra for all novel compounds; and HPLC traces for all prodigiosenes. See DOI: 10.1039/c2ob26535d |
| This journal is © The Royal Society of Chemistry 2013 |