Type-II nanorod heterostructure formation through one-step cation exchange†
Meng-Yu
Chen
and
Yung-Jung
Hsu
*
Department of Materials Science and Engineering, National Chiao Tung University, Hsinchu, Taiwan 30010, Republic of China. E-mail: yhsu@cc.nctu.edu.tw; Fax: +886 5724727; Tel: +886 5712121 ext. 55317
First published on 5th November 2012
Abstract
A novel one-step cation exchange approach has been developed to prepare ZnO-decorated ZnSe nanorods (ZnSe–ZnO NRs), a prototype type-II semiconductor nanoheterostructure. Because of the staggered band offset which promoted effective charge separation, the as-synthesized ZnSe–ZnO NRs exhibited remarkable photocatalytic activities under visible light illumination, demonstrating their promising potentials in relevant photoconversion applications.
Introduction
Chemical transformation from one structure to another via exchange of atoms can be utilized to obtain materials with desirable chemical properties. Recent developments have enabled the production of semiconductor nanocrystals which preserve the morphology and crystallinity of the starting materials by applying the cation exchange concept.1 Since the pioneering work by Alivisatos and co-workers,2 cation exchange reaction has been widely employed to prepare chalcogenide semiconductor nanocrystals, especially those with structural and compositional diversities. Through carefully controlling the reaction conditions of cation exchange, various types of semiconductor nanoheterostructures including alloy nanocrystals,3 doped nanocrystals,4 core–shell structures,5 and segmented structures6 can be readily obtained.Owing to the inherently high degree of complexity, nanoheterostructures have exhibited superior synergistic properties which are difficult to acquire from their individual constituents. Particularly, great progress has been made in creating type-II semiconductor nanoheterostructures where the relative band alignment of the constituents may promote effective charge separation to favor diverse photoconversion applications.7 For instance, ZnO nanowires showed 17-fold increase in photocurrent generation once they were coated with a thin layer of ZnS. This increase is due to the more negative conduction band potential of ZnS, which ensures a quick electron transfer from ZnS to ZnO and thus the successful collection of electrons.7c Besides, upon the decoration of Ag2O nanoparticles, TiO2 nanobelts displayed significantly enhanced photocatalytic activities under both UV and visible light illumination. This enhancement derives from the staggered band offset between Ag2O and TiO2, which inhibits charge recombination to capture more charge carriers for photocatalysis utilization.7d Until now, miscellaneous kinds of semiconductor nanoheterostructures with type-II band offset have been proposed and fabricated to further the advancement of photoconversion technology.
In this work, we reported a novel one-step cation exchange method for preparation of ZnO-decorated ZnSe nanorods (denoted as ZnSe–ZnO NRs), a model system of type-II semiconductor nanoheterostructures. In the typical procedure, ZnSe NRs were obtained by conducting cation exchange reaction on Ag2Se NRs with excess Zn2+ ions in the presence of tributylphosphine at 50 °C. With extra feeding of a trace amount of H2O in the cation exchange reaction, conversion to ZnSe NRs was accompanied by the deposition of ZnO nanocrystals, resulting in the formation of ZnSe–ZnO NRs. By modulating the volume percentage of H2O added, the density of ZnO nanocrystals deposited on ZnSe NRs can be readily controlled. For ZnSe–ZnO NRs, the surface-attached ZnO can serve as an effective electron scavenger for ZnSe due to its lower conduction band potential (−0.10 V vs. NHE) than that of ZnSe (−0.61 V vs. NHE).8 Consequently, the photoexcited electrons of ZnSe NRs would preferentially transfer to ZnO nanocrystals, leaving photogenerated holes at the ZnSe domain to achieve charge separation. Time-resolved photoluminescence (PL) measurements were conducted to explore the interfacial charge carrier dynamics for the as-prepared ZnSe–ZnO NRs. By probing the emission lifetime of ZnSe, the electron transfer event between ZnSe and ZnO for ZnSe–ZnO NRs can be quantitatively analyzed. The fate of the separated charge carriers of ZnSe–ZnO NRs was further examined with a photocatalytic process. It was found that ZnSe–ZnO NRs surpassed pristine ZnSe NRs, N-doped P-25 TiO2, and commercial ZnSe powders in photodegradation of rhodamine B, conceivably resulting from the pronounced charge separation that occurred at the interface of ZnSe/ZnO.
Experimental section
Chemicals
All chemicals were of analytical grade and used without further purification.Preparation of Se NRs
The synthesis of Se NRs was conducted in a carboxymethyl cellulose (CMC)-stabilized chemical reduction process.9 Briefly, CMC aqueous solution (Mw = 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da, 9.0 mL, 4.0 wt%) and NaOH aqueous solution (1.0 mL, 10 M), and SeO2 powder (1.0 mmol) were mixed and stirred at room temperature until the solution became transparent. Subsequently, NaBH4 solution (1.0 mL, 1.0 M) was added to carry out the reduction reaction. After stirring at 25 °C for 2 h, brown suspending solids (Se NRs) were produced. The product was collected, washed, and dried at 60 °C in vacuum for later use.
000 Da, 9.0 mL, 4.0 wt%) and NaOH aqueous solution (1.0 mL, 10 M), and SeO2 powder (1.0 mmol) were mixed and stirred at room temperature until the solution became transparent. Subsequently, NaBH4 solution (1.0 mL, 1.0 M) was added to carry out the reduction reaction. After stirring at 25 °C for 2 h, brown suspending solids (Se NRs) were produced. The product was collected, washed, and dried at 60 °C in vacuum for later use.
Transformation of Se NRs into Ag2Se NRs
Se NRs of a given amount (0.5 mmol) were re-dispersed in deionized water (5 mL) serving as the growth template for Ag2Se. The above suspension was then mixed with AgNO3 aqueous solution (1.25 mmol, 5 mL) under vigorous stirring at 25 °C. The color of the mixed solution was changed to black in 2 h, indicating the complete transformation from Se to Ag2Se. The product (Ag2Se NRs) was collected, washed, and dried at 60 °C in vacuum for later use.Cation exchange reaction for ZnSe–ZnO NRs growth
Ag2Se NRs of a given amount (0.05 mmol) were re-dispersed in methanol solution (10 mL) which contained poly(N-vinylpyrrolidone) (denoted as PVP, Mw = 10000 Da, 1.0 g) and Zn(NO3)2 (1.0 mmol). Note that PVP was used as the stabilizer to prevent NRs from agglomeration during the cation exchange process. 0.2 mL of tributylphosphine (denoted as TBP) was then injected into the above suspension to proceed with cation exchange reaction. After stirring at 60 °C for 2 h, the color of the reaction solution turned to light-brown, indicating the complete transformation from Ag2Se to ZnSe. The product (pristine ZnSe NRs) was collected, washed, and dried at 60 °C in vacuum for later characterization. In this work, an extra H2O feeding (0.8, 1.2, or 1.6 vol%) along with TBP injection into the reaction solution was also employed, which results in the formation of ZnSe–ZnO NRs. For comparison purpose, pure ZnO nanocrystals were also prepared by reacting TBP (0.2 mL) with H2O (1.6 vol%) in methanol (10 mL) at 60 °C.
PL lifetime measurement
Time-resolved PL spectra were measured using a home-built single photon counting system (Horiba Jobin Yvon) which delivers an instrument response function down to 25 ps FWHM. A GaN diode laser (λ = 375 nm) was used as the excitation source. The signals collected at the excitonic emission of ZnSe (λem = 460 nm) were dispersed with a grating spectrometer, detected by a high-speed photomultiplier tube, and then correlated using a single photon counting card. Upon laser excitation, the photoexcited electrons of ZnSe were injected into ZnO due to the higher conduction band potential of ZnSe (−0.61V vs. NHE) than that of ZnO (−0.10V vs. NHE), which leads to a significant quenching on the emission of ZnSe. By comparing the emission decay profiles between ZnSe and ZnSe–ZnO, the electron transfer from ZnSe to ZnO can be quantitatively deduced. The emission decay data were analyzed and fitted with a biexponential kinetics model which generates two lifetime values, τ1 and τ2, and the corresponding amplitudes, A1 and A2. All the fitting results are summarized in Table 1. The near-unity value of goodness (χ2) of the fitting indicated a good fit to the experimental data. It should be noted that the wavelength of excitation laser is too long to excite ZnO, with which the emission decay of ZnSe at 460 nm was not interfered by ZnO and thus can be precisely interpreted.| A 1 | τ 1 (ns) | A 2 | τ 2 (ns) | χ 2 | k et (s−1) | |
|---|---|---|---|---|---|---|
| Pristine ZnSe | 237.8 | 14.36 | 737.8 | 0.80 | 1.035 | — |
| ZnSe–ZnO-1 | 211.3 | 11.38 | 861.7 | 0.73 | 1.095 | 1.20 × 108 |
| ZnSe–ZnO-2 | 225.0 | 12.81 | 840.2 | 0.76 | 1.084 | 0.66 × 108 |
| ZnSe–ZnO-3 | 228.5 | 13.22 | 814.9 | 0.78 | 1.074 | 0.32 × 108 |
Photocatalytic performance measurement
The photocatalytic performance of the sample was evaluated by the photodegradation of rhodamine B (denoted as RhB) under visible light illumination. A quartz tube with a capacity of 30 mL was used as the photoreactor vessel. The optical system used for photodegradation consisted of a xenon lamp (500 W, with a light intensity of 175 mW cm−2) and a bandpass filter (with the bandwidth of 400–700 nm) that allowed the irradiation in the visible range. Seven kinds of samples including pristine ZnSe NRs, pure ZnO nanocrystals, three ZnSe–ZnO NRs, N-doped P-25 TiO2,10 and commercial ZnSe powders were compared in the photodegradation of RhB. Typically, a fixed amount of sample (3 mg) was added into RhB aqueous solution (15 mL, 1.0 × 10−5 M) in the photoreactor vessel. Prior to irradiation, the suspension was aerated and stirred in the dark for 30 min to reach the adsorption equilibrium between RhB and the sample. At certain time intervals of irradiation, 1.5 mL of the reaction solution was withdrawn and centrifuged to remove sample particles. The filtrates were analyzed with a UV–visible spectrophotometer to measure the concentration variation of RhB through recording the corresponding absorbance of the characteristic peak at 554 nm.Characterizations
The morphology and dimensions of the samples were examined with a field-emission scanning electron microscope (SEM, JEOL, JSM-6500F) and a high-resolution transmission electron microscope (HRTEM, JEOL, JEM-3000) operated at 300 kV. The crystallographic structure of the samples was investigated with X-ray diffraction (XRD, MAC Science, MXP18). The elemental analysis of NRs was conducted with the energy dispersive X-ray spectrometry (EDS), the accessory of SEM (JSM-6500F) and TEM (JEM-3000). For steady-state PL spectroscopy, a Kimmon IK3001R-G equipped with a He–Cd laser (720 W) was used. UV–visible absorption spectra were obtained using a Hitachi U-3900H spectrophotometer at room temperature. The Brunauer–Emmett–Teller (BET) specific surface area of the sample was estimated from the N2 adsorption–desorption analysis.Results and discussion
Single-crystalline Se NRs with a trigonal crystallographic structure were first prepared using a CMC-stabilized chemical reduction method.9 By reacting with AgNO3 at room temperature, direct insertion of Ag+ into Se lattices can be achieved for Se NRs, resulting in the complete conversion to orthorhombic Ag2Se NRs (Fig. S1,† ESI). Further replacement of Ag+ of Ag2Se NRs with Zn2+ was performed by conducting cation exchange reaction in the presence of TBP at an elevated temperature of 50 °C. Note that the conversion from Ag2Se to ZnSe is thermodynamically unfavorable unless a complexation ligand is employed at the elevated reaction temperature.1 Specifically, the complexation of TBP with Ag+ of Ag2Se may facilitate the replacement of Ag+ by Zn2+ at the anionic framework of NRs, which is conducive to the successful transformation of NRs into ZnSe. As shown in Fig. 1a, the resultant wurtzite ZnSe NRs inherited the dimensions of the initial Se, which had a typical diameter of 40–60 nm and length up to 1 μm. Interestingly, with extra feeding of a trace amount of H2O (0.8 vol%) in the reaction solution, conversion to ZnSe NRs was accompanied by the deposition of spherical nanocrystals. As displayed in Fig. 1b, these nanocrystals were of 80–100 nm in size and dispersed exclusively on the surface of ZnSe NRs. The corresponding XRD (Fig. 1e) and TEM-EDS analyses (Fig. 2b) confirm the composition of these nanocrystals as wurtzite ZnO. The density of ZnO nanocrystals deposited on the ZnSe NR surface can be further controlled by modulating the volume percentage of H2O added. Fig. 1c and d display the gradual increase in ZnO nanocrystal density for ZnSe–ZnO NRs, which was achieved by introducing H2O of increasing volume. The samples obtained with H2O volume percentage of 0.8, 1.2, and 1.6 vol% were respectively denoted as ZnSe–ZnO-1, ZnSe–ZnO-2, and ZnSe–ZnO-3. SEM-EDS analysis shows that the weight percentage of ZnO was 62.0, 70.9, and 76.5% for ZnSe–ZnO-1, ZnSe–ZnO-2 and ZnSe–ZnO-3, respectively.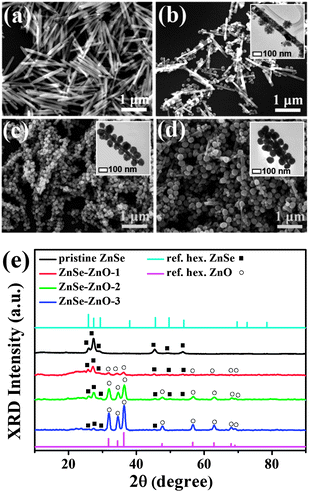 | ||
| Fig. 1 SEM images of (a) pristine ZnSe and (b), (c), (d) ZnSe–ZnO NRs prepared with H2O volume percentage of 0.8, 1.2, and 1.6 vol%, respectively. Insets show the representative TEM images. The corresponding XRD patterns are shown in (e). | ||
 | ||
| Fig. 2 (a) TEM image, (b) TEM-EDS analysis, and (c) HRTEM image of ZnSe–ZnO NRs. In (c), the ZnSe/ZnO interface is highlighted by a dashed line. | ||
Fig. 2a shows the typical TEM image of the as-obtained ZnSe–ZnO NRs. Evidently, ZnO-free ZnSe NRs or free-standing ZnO nanocrystals were rarely observed in the products, demonstrating the advantage of the current approach to obtain semiconductor nanoheterostructures with high structural integrity. In Fig. 2c, an HRTEM image taken at the interface of NR and nanocrystal region clearly reveals two distinct sets of lattice fringes, which can be respectively assigned to wurtzite ZnSe and wurtzite ZnO crystals. Moreover, the substantial interface of ZnSe/ZnO was existent in ZnSe–ZnO NRs. This interface was essentially important because it ensured the successful electron transfer from ZnSe to ZnO upon light irradiation and thereby the achievement of charge separation.
The success of the current one-step cation exchange route to ZnSe–ZnO NRs was achieved by utilizing H2O as the alkalization reagent. A plausible mechanism for the formation of ZnSe–ZnO NRs was proposed as follows. At the initial stage of reaction, TBP captured Ag+ ions of Ag2Se NRs to form intermediate complexes, which allows the mutual diffusion of Ag+ (outbound) and Zn2+ (inbound) at the anionic framework of NRs. Because of the relatively small volume change during the transformation process,11 ZnSe NRs which preserved the morphology and single crystallinity of the starting Ag2Se formed. When a trace amount of H2O was present, additional reaction associated with TBP alkalization and OH− liberation could occur. As reported in the literature,12 when added to a solution containing alcohol, water and formaldehyde, TBP undergoes alkalization to dissociate into phosphonium cations and OH− ions. Since the increase in the pH value for the current TBP methanolic solution upon the H2O addition was apparent (from pH = 5.25 for solution without H2O to pH = 6.40 for solution with 0.8 vol% H2O), we believed that alkalization of TBP would take place to generate a considerable amount of OH−. On the other hand, during the cation exchange process, PVP was added as the stabilizer to prevent NRs from agglomerating. The PVP-stabilized ZnSe NRs may exhibit capability of Zn2+ adsorption by virtue of the effective coordination between PVP and Zn2+.13 These surface-adsorbed Zn2+ ions then reacted with the TBP-derived OH− to form ZnO through proper dehydration. As a result, ZnO nanocrystals were grown and deposited on the surface of ZnSe NRs. The mechanism of the H2O-induced TBP alkalization described above can be further verified by considering the effect of the increased H2O volume percentage on the ZnO growth of ZnSe–ZnO NRs. As revealed in Fig. 1, the increase in H2O volume percentage caused the formation of ZnSe–ZnO NRs with high ZnO density. This consequence was mainly a result of the more OH− ions generated in TBP methanolic solution with larger H2O volume percentage, which is corroborated by the corresponding increase in solution pH. As more and more OH− ions were involved, formation of ZnO may be facilitated to grow ZnO with an increased amount. Accordingly, ZnSe–ZnO NRs with high ZnO nanocrystal density were obtained upon raising the H2O volume percentage.
Owing to the band alignment between ZnSe and ZnO, the present ZnS–ZnSe NRs exhibited pronounced charge separation upon light irradiation, which can be quantitatively characterized with time-resolved PL measurement and photocatalysis experiment. Fig. 3a represents the time-resolved PL spectra of pristine ZnSe NRs and the three ZnSe–ZnO NR samples. These spectra were fitted with a biexponential function to yield a slow (τ1) and a fast (τ2) decay component, which are relatively assigned to radiative recombination and nonradiative relaxation pathways. The fitting results are summarized in Table 1. For ZnSe–ZnO NRs, emission lifetimes of both components were shorter than those of pristine ZnSe, implying the emergence of an additional nonradiative pathway from the electronic interaction between ZnSe and ZnO. This supposition can be verified by the steady-state PL depression observed for ZnS–ZnO NRs (Fig. S2†). If electron transfer from ZnSe to ZnO was the predominant process dictating the emission quenching of ZnSe for ZnSe–ZnO NRs, we can estimate the electron-transfer rate constant (ket) by the expression ket(ZnSe → ZnO) = (1/τ2)(ZnSe–ZnO) – (1/τ2)(ZnSe).14 Using the emission lifetime data in Table 1, we obtained ket values as 1.20 × 108, 0.66 × 108, and 0.32 × 108 s−1 for ZnSe–ZnO-1, ZnSe–ZnO-2, and ZnSe–ZnO-3, respectively. An obvious decreasing trend in the ket value was noticed for ZnSe–ZnO NRs with increasing ZnO nanocrystal density. This observation suggests that charge separation of ZnSe–ZnO NRs turned decreasingly conspicuous as ZnO nanocrystals density increased. Note that when put in contact with semiconductor nanocrystals, electron scavenger with an excess amount exerts a detrimental influence on the overall charge separation efficiency.15 This adverse effect is due to the consumption of the separated charge carriers in the later-emerging electron–hole recombination process, which reduces the extent of electron scavenging to lead to the less effective charge separation.
 | ||
| Fig. 3 (a) Time-resolved PL spectra for pristine ZnSe and ZnSe–ZnO NRs. (b) Results of RhB photodegradation for seven relevant samples. | ||
To further examine the fate of the separated charge carriers for the present ZnSe–ZnO NRs, we performed photocatalysis experiment by using RhB as the test pollutant. Fig. 3b compares the RhB photodegradation results of seven relevant samples, from which several points can be observed. First, pure ZnO nanocrystals showed a small but not negligible photocatalytic activity under visible light illumination, ascribable to the self-photosensitized effect between RhB molecules and ZnO.16 Second, all the three ZnSe–ZnO NRs performed better toward RhB photodegradation than pristine ZnSe NRs did, which can be accounted for by the effective charge separation that took place at the interface of ZnSe/ZnO. This demonstration again addresses the advantage of type-II semiconductor nanoheterostructures for photoconversion applications. Third, as compared to the relevant commercial products such as N-doped P-25 TiO2 and ZnSe powders, the three ZnSe–ZnO NR samples exhibited superior photocatalytic performance under visible light illumination, demonstrating their potential as an efficient visible light-responsive photocatalyst. Lastly, ZnSe–ZnO NRs showed depressed photocatalytic efficiency as the ZnO nanocrystal density increased. This outcome was attributed to the consumption of charge carriers observed for ZnSe–ZnO-2 and ZnSe–ZnO-3 samples as demonstrated in the time-resolved PL investigation. To quantitatively compare the photocatalytic performance of different ZnSe–ZnO samples, the apparent rate constant of RhB photodegradation (kRhB) was calculated using the pseudo-first-order approximation.10 Using the result of Fig. 3b, we obtained kRhB values of 0.042, 0.033, and 0.014 min−1 for ZnSe–ZnO-1, ZnSe–ZnO-2, and ZnSe–ZnO-3, respectively. Evidently, the trend in kRhB variation for ZnSe–ZnO NRs with increasing ZnO nanocrystal density corresponded well with that in ket change. As illustrated in Fig. 4, the correspondence between kRhB and ket can be realized by the causal relation between electron transfer and hole generation. As ZnO nanocrystal density increased, the separated electrons at ZnO became abundant such that electron–hole recombination across the ZnSe/ZnO interface was encouraged, simultaneously leaving delocalized holes of a decreased amount in ZnSe. The reduction in the number of photogenerated holes further led to the depression in the resulting photocatalytic performance. Note that in the case of the present ZnSe–ZnO NRs, photogenerated holes were the exclusive oxidation species for RhB degradation, with which the correlation between charge carrier dynamics and photocatalytic performance (Fig. 4) can be validated. RhB has an oxidation potential of 0.95 V vs. NHE, which makes it a good hole acceptor for ZnSe–ZnO NRs.17 The direct hole-oxidation path for the current RhB photodegradation can be further verified from the controlled experiments. When tert-butyl alcohol, an effective ˙OH radical scavenger,18 was added to RhB solution, no rate change of photodegradation was observed for ZnSe–ZnO NRs. In contrast, if methanol, a typical hole scavenger,19 with a commensurate amount was introduced, significant suppression in RhB photodegradation resulted. These trials reveal that direct hole-oxidation was the primary path for RhB photodegradation in the presence of ZnSe–ZnO NRs.
 | ||
| Fig. 4 Correlations of ket and kRhB for ZnSe–ZnO NRs. Inset shows the relative band structure of ZnSe–ZnO. | ||
It might be argued that the observed difference in photocatalytic activity among the three ZnSe–ZnO samples resulted from the variation in the surface area. The BET characterization shows that the surface areas of different ZnSe–ZnO NRs were comparable, which are 19.4, 22.5, and 23.7 cm2 g−1 for ZnSe–ZnO-1, ZnSe–ZnO-2, and ZnSe–ZnO-3, respectively. This result signifies that the variation in photocatalytic performance for different ZnSe–ZnO NRs is mainly related to the interfacial charge carrier dynamics rather than the surface area effect. To further demonstrate the remarkable photocatalytic properties for ZnSe–ZnO NRs, we investigated the stability of the samples by examining their microstructures after used in RhB photodegradation. As shown in Fig. S3,† no appreciable change in the XRD pattern was found upon photocatalysis operation, revealing the considerably high stability for the present ZnSe–ZnO NRs.
Conclusions
In conclusion, a novel one-step cation exchange method has been developed to prepare ZnSe–ZnO NRs which showed a prominent charge separation property. The success of the synthetic approach relied on the utilization of H2O as the alkalization reagent, which enables the deposition of ZnO nanocrystals along with the growth of ZnSe NRs in the cation exchange reaction. The current cation exchange approach can be extended to obtain other semiconductor nanoheterostructures with type-II band offset such as CdO-decorated CdSe NRs (Fig. S4†).Acknowledgements
This work was financially supported by the National Science Council of Republic of China (Taiwan) under grants NSC-100-2113-M-009-004 and NSC-101-3113-P-009-005.Notes and references
- G. D. Moon, S. Ko, Y. Min, J. Zeng, Y. Xia and U. Jeong, Nano Today, 2011, 6, 186–203 CrossRef CAS.
- D. H. Son, S. M. Hughes, Y. Yin and A. P. Alivisatos, Science, 2004, 306, 1009–1012 CrossRef CAS.
- (a) A. M. Smith and S. Nie, J. Am. Chem. Soc., 2011, 133, 24–26 CrossRef CAS; (b) L. De Trizio, M. Prato, A. Genovese, A. Casu, M. Povia, R. Simonutti, M. J. P. Alcocer, C. D'Andrea, F. Tassone and L. Manna, Chem. Mater., 2012, 24, 2400–2406 CrossRef CAS; (c) Y. Yu, J. Zhang, X. Wu, W. Zhao and B. Zhang, Angew. Chem., Int. Ed., 2012, 51, 897–900 CrossRef CAS; (d) X. Wu, Y. Yu, Y. Liu, Y. Xu, C. Liu and B. Zhang, Angew. Chem., Int. Ed., 2012, 51, 3211–3215 CrossRef CAS; (e) E. Dilena, D. Dorfs, C. George, K. Miszta, M. Povia, A. Genovese, A. Casu, M. Prato and L. Manna, J. Mater. Chem., 2012, 22, 13023–13031 RSC.
- (a) A. K. Samal and T. Pradeep, J. Phys. Chem. C, 2010, 114, 5871–5878 CrossRef CAS; (b) J. Zhang, S. Liu, J. Yu and M. Jaroniec, J. Mater. Chem., 2011, 21, 14655–14662 RSC.
- (a) P. H. C. Camargo, Y. H. Lee, U. Jeong, Z. Zou and Y. Xia, Langmuir, 2007, 23, 2985–2992 CrossRef CAS; (b) J. M. Pietryga, D. J. Werder, D. J. Williams, J. L. Casson, R. D. Schaller, V. I. Klimov and J. A. Hollingsworth, J. Am. Chem. Soc., 2008, 130, 4879–4885 CrossRef CAS; (c) Y. F. Zhu, D. H. Fan and W. Z. Shen, J. Phys. Chem. C, 2008, 112, 10402–10406 CrossRef CAS; (d) J. Park and S.-W. Kim, J. Mater. Chem., 2011, 21, 3745–3750 RSC; (e) B. Mukherjee, A. Peterson and V. Subramanian, Chem. Commun., 2012, 48, 2415–2417 RSC; (f) C. Dong, A. Korinek, B. Blasiak, B. Tomanek and F. C. J. M. van Veggel, Chem. Mater., 2012, 24, 1297–1305 CrossRef CAS; (g) M. Casavola, M. A. van Huis, S. Bals, K. Lambert, Z. Hens and D. Vanmaekelbergh, Chem. Mater., 2012, 24, 294–302 CrossRef CAS; (h) H. Li, R. Brescia, R. Krahne, G. Bertoni, M. J. P. Alcocer, C. D'Andrea, F. Scotognella, F. Tassone, M. Zanella, M. D. Giorgi and L. Manna, ACS Nano, 2012, 6, 1637–1647 CrossRef CAS; (i) K. A. Abel, P. A. FitzGerald, T.-Y. Wang, T. Z. Regier, M. Raudsepp, S. P. Ringer, G. G. Warr and F. C. J. M. van Veggel, J. Phys. Chem. C, 2012, 116, 3968–3978 CrossRef CAS; (j) S. Panigrahi and D. Basak, RSC Adv., 2012, 2, 11963–11968 RSC.
- (a) R. D. Robinson, B. Sadtler, D. O. Demchenko, C. K. Erdonmez, L.-W. Wang and A. Paul Alivisatos, Science, 2007, 317, 355–358 CrossRef CAS; (b) B. Sadtler, D. O. Demchenko, H. Zheng, S. M. Hughes, M. G. Merkle, Ul. Dahmen, L.-W. Wang and A. Paul Alivisatos, J. Am. Chem. Soc., 2009, 131, 5285–5293 CrossRef CAS; (c) J. M. Luther, H. Zheng, B. Sadtler and A. Paul Alivisatos, J. Am. Chem. Soc., 2009, 131, 16851–16857 CrossRef CAS; (d) B. Zhang, Y. Jung, H.-S. Chung, L. V. Vugt and R. Agarwal, Nano Lett., 2010, 10, 149–155 CrossRef CAS; (e) A. K. Samal and T. Pradeep, Nanoscale, 2011, 3, 4840–4847 RSC; (f) K. Miszta, D. Dorfs, A. Genovese, M. R. Kim and L. Manna, ACS Nano, 2011, 5, 7176–7183 CrossRef CAS; (g) A. Som and T. Pradeep, Nanoscale, 2012, 4, 4537–4543 RSC.
- (a) Z. Wang, B. Huang, Y. Dai, X. Qin, X. Zhang, P. Wang, H. Liu and J. Yu, J. Phys. Chem. C, 2009, 113, 4612–4617 CrossRef CAS; (b) J. Hensel, G. Wang, Y. Li and J. Z. Zhang, Nano Lett., 2010, 10, 478–483 CrossRef CAS; (c) A. Bera and D. Basak, ACS Appl. Mater. Interfaces, 2010, 2, 408–412 CrossRef CAS; (d) W. Zhou, H. Liu, J. Wang, D. Liu, G. Du and J. Cui, ACS Appl. Mater. Interfaces, 2010, 2, 2385–2392 CrossRef CAS; (e) Y. Shemesh, J. E. Macdonald, G. Menagen and U. Banin, Angew. Chem., Int. Ed., 2011, 50, 1185–1189 CrossRef CAS; (f) S. Cho, J.-W. Jang, J. Kim, J. S. Lee, W. Choi and K.-H. Lee, Langmuir, 2011, 27(27), 10243–10250 CrossRef CAS.
- J. Chung, J. Myoung, J. Oh and S. Lim, J. Phys. Chem. Solids, 2012, 73, 535–539 CrossRef CAS.
- Y.-D. Chiou and Y.-J. Hsu, Appl. Catal., B, 2011, 105, 211–219 CrossRef CAS.
- T.-T. Yang, W.-T. Chen, Y.-J. Hsu, K.-H. Wei, T.-Y. Lin and T.-W. Lin, J. Phys. Chem. C, 2010, 114, 11414–11420 CAS.
- G. D. Moon, S. Ko, Y. Xia and U. Jeong, ACS Nano, 2010, 4, 2307–2319 CrossRef CAS.
- W. J. Vullo, J. Org. Chem., 1968, 33, 3665–3667 CrossRef CAS.
- J. Zhang, H. Liu, Z. Wang and N. Ming, J. Cryst. Growth, 2008, 310, 2848–2853 CrossRef CAS.
- I. V. Lightcap and P. V. Kamat, J. Am. Chem. Soc., 2012, 134, 7109–7116 CrossRef CAS.
- (a) Y.-C. Pu, Y.-C. Chen and Y.-J. Hsu, Appl. Catal., B, 2010, 97, 389–397 CrossRef CAS; (b) Y.-C. Chen, Y.-C. Pu and Y.-J. Hsu, J. Phys. Chem. C, 2012, 116, 2967–2975 CrossRef CAS.
- J. Guo, J. Li, A. Yin, K. Fan and W. Dai, Chin. J. Chem., 2010, 28, 2144–2150 CrossRef CAS.
- Z. Xiong, L. L. Zhang, J. Ma and X. S. Zhao, Chem. Commun., 2010, 46, 6099–6101 RSC.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2010, 26, 3894–3901 CrossRef CAS.
- J. Zhuang, W. Dai, Q. Tian, Z. Li, L. Xie, J. Wang and P. Liu, Langmuir, 2010, 26, 9686–9694 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SEM images, XRD patterns and steady-state PL spectra of relevant samples. See DOI: 10.1039/c2nr32879h |
| This journal is © The Royal Society of Chemistry 2013 |