 Open Access Article
Open Access ArticleElastic CNT–polyurethane nanocomposite: synthesis, performance and assessment of fragments released during use†
Wendel
Wohlleben
*ac,
Matthias W.
Meier
b,
Sandra
Vogel
c,
Robert
Landsiedel
c,
Gerhard
Cox
a,
Sabine
Hirth
a and
Željko
Tomović
*d
aBASF SE, Material Physics, GMC, 67056 Ludwigshafen, Germany. E-mail: wendel.wohlleben@basf.com; Tel: +49 621 6095339
bBASF SE, Aerosol Technology, GCP/TP, 67056 Ludwigshafen, Germany
cBASF SE, Experimental Toxicology, GVT/TB, 67056 Ludwigshafen, Germany
dBASF Polyurethanes GmbH, GMU/UE, Elastogranstrasse 60, 49448 Lemfoerde, Germany. E-mail: zeljko.tomovic@basf.com; Tel: +49 5443 124253
First published on 21st November 2012
Abstract
Intended for use in high performance applications where electrical conductivity is required, we developed a CNT–TPU composite. Such a composite can be prepared by melt processing (extrusion) on an industrial scale. Due to the known hazard upon inhalation of CNTs, we assessed three degradation scenarios that may lead to the release of CNTs from the composite: normal use, machining and outdoor weathering. Unexpectedly, we find that the relative softness of the material actually enhances the embedding of CNTs also in its degradation fragments. A release of free CNTs was not detected under any condition using several detection methods. However, since machining may induce a high acute dose of human exposure, we assessed the cytotoxicity potential of released fragments in the in vitro model of precision-cut lung slices, and found no additional toxicity due to the presence of CNTs. At very low rates over years, weathering degrades the polymer matrix as expected for polyurethanes, thus exposing a network of entangled CNTs. In a preliminary risk assessment, we conclude that this material is safe for humans in professional and consumer use.
Introduction
Nanocomposite materials can outperform established polymers with added functionalities, but the nanofiller itself or hybrid filler–matrix fragments may modify the risk profile during the lifecycle from production to consumer use to disposal.Polyurethane (PU) is one of the most versatile polymers. By changing the type and functionality of the polyol and isocyanate precursors, the properties of PU can be easily tailored, ranging from a rigid solid to a flexible elastomer. Thermoplastic polyurethane (TPU) is an elastomeric copolymer consisting of alternating hard and soft blocks which can phase separate. Because of melt processability and physical properties tunable over a wide range, TPU has been used for a variety of applications such as fibers, cable sheathing, hoses, tubes, films, sealings, protective/functional coatings and adhesives.1 Although there are already many different grades of TPU available, further improvement of the physical properties of the material will make it suitable to an increased number of applications and will provide new market opportunities.
Nanocomposites enable enhanced functionalities by incorporating into the PU matrix a variety of fillers such as silica nanoparticles,2 clays,3 glass or carbon fibers,4,5 carbon nanotubes6 and graphene sheets.7 In particular, carbon nanotubes have been extensively studied as fillers to enhance the mechanical and electrical properties of TPU nanocomposites.8–14
Here we aim at the added functionality of conductive properties, by embedding multi-wall carbon nanotubes (CNTs) in TPU. Especially if the nanofillers are CNTs with their known hazard upon inhalation,15 the extent of polymer degradation and possible CNT release must be assessed before commercialization, to ensure that human and environmental exposures remain safe.16–18 However, no more than two dozen papers have reported experimental data on release from engineered nanofiller-composites, even fewer on CNT-composites, and none on elastic materials such as TPU. We recently performed the first in vivo assessment of degradation fragments from CNT–polymer and CNT–cement nanocomposite materials, and found no additional hazard from the nanocomposite nature of the released hybrid particles.19 This result has been confirmed by independent in vivo studies at the Danish National Research Center for the Working Environment.20,21 Among the few studies to characterize the fragments released from machining of CNT-composites, there is so far consensus that the debris is vastly dominated by composite fragments of the matrix with embedded CNTs.19,22,23 The surface of the CNT–composite fragments may be decorated by a hairy layer of CNTs protruding from the particle, but it is presently unknown if this phenomenon is universal.23 Only chemical degradation of the matrix, especially by weathering, can degrade and remove the polymer matrix, leaving behind an entangled network of CNTs.16,19,24 Specifically for polyurethane with embedded graphene oxide, the presence of the nanofiller tended to slow down photo-oxidation, but the graphene flakes clearly emerged on the weathered surface.25
In the present contribution, we report on the development of a TPU–CNT composite material and evaluate three release scenarios in comparison. We address the cytotoxic potential to lung tissue in vitro. The results allow for correlations between a comparatively soft material's elastic properties and its propensity to release nanofillers, but are not intended to be extrapolated to all other variants of polyurethanes.
Materials
Materials
The specific thermoplastic polyurethane (TPU) is synthesized from polytetrahydrofuran with a chain length of 1000 g mol−1 and 1,4-butanediol as diol components, and MDI (4,4′-diphenylmethane diisocyanate) as an isocyanate component, resulting in polyether soft segments. It is industrially available as Elastollan® 1185 A10 (BASF product) and was used as granules.13 The material has a melt-volume flow rate (MVR) of 33 cm3 per 10 min, and a density of 1.12 g cm−3. The industrially available multi-wall carbon nanotubes (CNTs), Nanocyl™ NC7000, were delivered by Nanocyl S.A. (Sambreville, Belgium),14 as a powder. These CNTs are produced via a catalytic carbon vapour deposition (CCVD) process. The tubes have an average diameter of 10 nm, and a length between 0.1 and 10 μm (Fig. 8e). These dimensions are supported by the specific surface of 300 m2 g−1, and by the pore size distribution derived from Hg intrusion (data not shown). The overall carbon purity is above 90% containing up to 10 wt% of metal oxide impurities that remain from the catalyst. The surface-chemistry as detected by XPS is 99% carbon and less than 1% of oxygen and aluminium each.TPU–CNT composite preparation, injection molding and extrusion
3 wt% of CNTs were mixed with TPU using a Leistritz ZSE MAXX 27 twin-screw extruder at 215 °C, a screw speed of 300 rpm, a throughput of 30 kg h−1, and obtained a composite strand cut into granules. Subsequently the granules had been used for injection molding and extrusion of 2 mm and 1.5 mm thick plates, respectively. The TPU material was dried at 90 °C for at least 3 hours before each processing step. Injection molding was performed on an Engel ES 330/80 HL (Engel, Germany), at an injection velocity of 15 mm s−1, a melt temperature of 220 °C and a mold temperature of 40 °C. Extrusion was performed on an Arenz 30 mm extruder (Arenz GmbH, Germany), a melt temperature of 185 °C and a screw speed of 25 rpm.Methods
Electrical and mechanical properties
The volume resistivity was measured according to ISO 3915 (4-point method). The hardness was measured according to DIN 53505 and elongation at break according to DIN 53504.Machining/sanding
Strong mechanical shear forces occur during drilling, sawing and sanding.18 Here we chose sanding because we estimated the chance was highest for release of powders with fine structures, as demonstrated by very similar setups on nanopigment coatings26–28 and CNT–epoxy composites.23 Our setup has been described in detail before.19 In short, background particle concentrations are reduced to 250 particles per cm3 by an air-tight housing flushed with filtered air (Fig. SI_1, ESI†). The 10 cm diameter sample (2 mm thickness) rotates against sanding paper (specification KK114F with a grain size P320 from VSM, Hannover, Germany, abrasive material is aluminum oxide nominally) at 2000 rpm, at a relative velocity of 6.5 m s−1. The total weight pressing on the sanding paper had to be reduced to 0.25 kg in the present study, because the soft TPU would otherwise arrest the rotation. Airborne fragments are aspirated onto a membrane filter and fragments fallen from the sample holder are collected for further investigations. The concentration and size distribution of the aerosol is measured by a Scanning Mobility Particle Sizer (SMPS) consisting of a dynamic mobility analyzer TSI 3071A and a condensation particle counter TSI 3022, or by the condensation particle counter alone.Taber Abraser degradation
The Taber Abraser™ test is an established method of the coatings industry to quantify wear resistance and is described in many national and international standards (e.g. DIN 53754:1977, DIN 68861-2:1981, ISO 5470-1:1999 and ASTM D 4060-95:2007). The rotating plate in the test rig was replaced by a Taber Abraser 352G equipped with a CS-0 wheel and a Taber S-42 sandpaper strip mounted to it, with 1 kg load, and the same sample collection. In contrast to the sanding experiments, the sandpaper now rolls over the samples, at a relative velocity of 0.294 m s−1.Weathering
UV radiation causes the release of pigments known as chalking. Standardized tests are established for coatings, and we primarily adhere to ISO 3892-2:2006 (with apparatus Suntest™ XLS+, standard-black temperature 65 °C), where only UV irradiation (111 W m−2 (300–400 nm)) without simulated rain is performed. We can thus ensure that degradation products are not blown or washed away, but can be safely detected and characterized. We exposed the nanocomposite and the reference testing plates in parallel for 4 (and 8) weeks, equivalent to 9 (and 18) months in Europe at approximately 50° northern latitude (acceleration factor 8).For comparison, also wet weathering was performed, using again standardized equipment (ISO 4892-2 (Verf. A)): humidity cycle (102 min dry + 18 min rain), at an average relative humidity of 50 ± 10%. The wet weathering used lower intensity (60 W m−2 (300–400 nm)), but was kept longer (1243 h), so as to simulate the same 9 months treatment, at the same standard-black temperature of 65 °C.
Photoelectron spectroscopy (XPS)
Photoelectron spectroscopy (XPS) was performed with a Phi XPS 5500 system with 300 W monochromatic Al-K alpha radiation, pass energy for surveys 117 eV (measurement time of 45 min), and detailed spectra at 23.5 eV (measurement time of 6 min). Evaluation performed by CasaXPS 2.3.15, based on the Phi standard-sensitivity factors, with Shirley background subtraction and peak shape fits as the sum of 90% Gaussian and 10% Lorentzian. Information depth is limited to the upper 10 nm of the material. We pressed the powders gently with a cover slide and measured on three locations (each integrating 0.5 mm2). The results were spatially homogeneous with the deviations not exceeding 1 at% (atom percent).Representative SEM images of entire collected powder and of air-collected filters
The SEM measurements were performed with a JEOL JSM-7500 TFE SEM at an accelerating voltage of 5 kV. The sample surfaces were sputtered with an approximately 15 nm thin Pt-layer prior to the SEM imaging in order to prevent charging of the surfaces due to the electron beam. Both gold membrane filter and silver membrane filter were employed, allowing for an increased air flow of 30 L min−1 in order to enhance sampling efficiency. Samples were collected for up to 10 minutes, and were searched for free CNTs under the SEM. Representative images of the observed airborne structures were acquired with the above SEM parameters, and selected spots were identified chemically by Energy Dispersive X-ray Spectroscopy (EDXS) at 10 kV.Laser diffraction
The size distribution of the abraded material collected on the filter was measured by laser diffraction in a Malvern Master Sizer 2000S. Particles were dispersed in water + K30 (SDS-mix) and subject to ultrasonication (protocol on page 2 of the ESI†) in order to disrupt agglomerates.Analytical ultracentrifugation (AUC)
Analytical ultracentrifugation (AUC) quantifies the amount and the diameter of dispersed nanofillers and composite fragments independently of each other (0.5–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nm diameter). Here we use interference optics (Beckman model ‘XLI proteome lab’) and the raw data are fitted by the free-ware software SedFit. The mass concentrations read directly from the interference fringe shift without further conversion. This allows the independent quantification of particulate components in the sub-100 nm range.29 The complete protocol used for sample preparation and detection is presented in detail with positive and negative controls in the ESI and Fig. SI_2.†
000 nm diameter). Here we use interference optics (Beckman model ‘XLI proteome lab’) and the raw data are fitted by the free-ware software SedFit. The mass concentrations read directly from the interference fringe shift without further conversion. This allows the independent quantification of particulate components in the sub-100 nm range.29 The complete protocol used for sample preparation and detection is presented in detail with positive and negative controls in the ESI and Fig. SI_2.†
In vitro toxicological testing method
000 μg mL−1). For cytotoxicity assays, 0.1% Triton X-100 (in DMEM/F-12, Sigma-Aldrich, Germany) was used as a technical control and DMEM/F-12 alone as a negative control. ZnO (OECD reference material NM110) was used as a positive control and the identical CNT type NC7000 as in the TPU composite was used for comparison. The uncoated ZnO NM110 has a specific surface of 12 m2 g−1, a distribution of primary particle diameters from 20 nm to 250 nm and has been characterized in detail by the prospect project.31 The ZnO and CNT nanomaterials were analysed in separate experiments and dispersed by the same ultrasonication protocol, but with 0.5% BSA in the stock solution. Their dispersed concentration after substance preparation was measured by AUC and the dilution factors were adjusted to this dispersed concentration. Agglomerated bundles accounted for less than 10% of the solid content; they settled quickly and were discarded.
We determined the cell membrane integrity by measuring the released lactate dehydrogenase in the supernatant (LDH Cytotoxicity Detection Kit, Roche, Germany) after a centrifugation at 300g for 5 min. Triton X-100 lysed PCLuS served as a LDH positive control (100% LDH release). Results were calculated as percentage of the total LDH content (PC = 100%) or maximum viability (NC = 100%). The LDH standard was added to test preparations of ZnO, CNT, TPU and TPU + CNT and no interference with the LDH assay was detected.
The protein content was measured via the BCA Protein Assay Kit (Pierce, Rockford, IL, USA) after lysis of the PCLuS with 500 μL Triton X-100 solution in PBS. A dilution series of known BSA concentrations calibrate the protein content results. Again, we tested for interferences between the test materials and the BCA assay and found none.
Performance of the TPU + CNT
The melt processing on a larger scale that is relevant for industrial applications, has produced a TPU composite containing 3 wt% CNTs that was macroscopically homogeneous across tens of meters. The micro-morphology of injection molding and extruded plates was studied by TEM, which showed well dispersed and homogenously distributed CNTs (Fig. 1). The quality of dispersion is a prerequisite to maintain the excellent mechanical properties of TPU. We found that the hardness of TPU containing 3% CNTs was only slightly increased to 88 Shore A, while elongation at break is still as high as 560%. | ||
| Fig. 1 TEM images of (a) granules, (b) extruded plates and (c) injection molding plates, all TPU containing 3% CNTs (sample depth 1 mm). | ||
It has been recently shown on a smaller scale that the volume resistivity of TPU can drop below 104 Ω cm, starting at 2 wt% addition of Nanocyl™ 7000 industrial grade material.10 For the TPU + CNT composite of the present contribution the CNT content was increased above this threshold and the resulting volume resistivity, after injection molding and extrusion was as low as 3.5 × 102 and 1.5 × 101 Ω cm, respectively.
The performance after extrusion or injection molding enables applications such as conveyor belts, rollers, conductive foils, electromagnetic shielding of electrical equipment and electronic assemblies and electrically heatable parts.
Results from release scenarios
Machining/sanding
A significant aerosol concentration is generated by sanding either the pure matrix (TPU) or the nanocomposite (TPU + CNT). The number concentration determined by a condensation particle counter (CPC) as around 6000–8000 P cm−3 is clearly above clean-air background, and comparable to the environmental background. Over time, this process is not stable but collects fragments on the sanding paper and then generates bursts of air-borne dust every few minutes (Fig. 2), which reach 25000 P cm−3 for the TPU + CNT composite, but up to 40000 P cm−3 for the TPU reference. Note that due to burst events, a size classification in the aerosol is not reliable, because the apparatus (SMPS) is scanning over 5 minutes, and also reduced scan times do not resolve the issue due to the unpredictability of the delay between bursts. The number concentrations are not significantly different between the reference material and the nanocomposite. This result is in line with earlier findings on nanocomposites, including CNT-composites.18,19,21,26–28,32
 | ||
| Fig. 2 Aerosol concentrations over time during sanding. Reference TPU (triangles, red); TPU + CNT (diamonds, blue); measured by CPC (condensation particle counter) with time resolution, but without size discrimination, integrating the size range 10 nm–3 μm. | ||
A closer investigation has been conducted with collected dusts (homogenized over the entire abrasion duration) to assess its size distribution, morphology and chemical composition to derive an upper limit on any possible release of CNTs. The powder from the filter and collected material was dispersed as described in the ESI† to disrupt agglomerates or CNTs loosely adsorbed to fragments. We checked independently that this procedure indeed disperses pure CNTs of the same type and that free CNTs can indeed be detected and quantified in the presence of an excess of TPU powder (see ESI†). The size distribution from the sanding fragments (Fig. 3) is dominated by large fragments with 10–100 μm diameter. Some wt% of the fragments are small enough to enter the aerosol in the inhalable size range, but not in the respirable range. In the range below 150 nm, where free CNTs would appear, around 0.2 wt% of the total particulate content can be detected, which is just above the limit of detection of AUC. However, the same content in this range is found for the reference material without CNTs (Table 1).
 | ||
| Fig. 3 Size distribution of fragments from machining/sanding (TPU, black; TPU + CNT, red). Laser diffraction data, regarded as reliable for sizes above 1 μm. Sizes below 10 μm down to 1 nm were independently quantified by analytical ultracentrifugation, see the text and Table 1. | ||
| Sanding | Normal use | |||
|---|---|---|---|---|
| Size range below 10 μm | In the size range of free CNTs (<150 nm) | Size range below 10 μm | In the size range of free CNTs (<150 nm) | |
| TPU | 0.24 ± 0.05 wt% | 0.2 ± 0.05 wt% | 0.24 ± 0.05 wt% | 0.24 ± 0.05 wt% |
| TPU + CNT | 0.52 ± 0.1 wt% | 0.16 ± 0.05 wt% | 0.34 ± 0.07 wt% | 0.2 ± 0.05 wt% |
The colloidal size range is confirmed by SEM of the powder. We observe agglomerations of polymer fragments, where the smallest structural sizes are around 20 μm (Fig. 4). The surface is smooth down to the level of 100 nm. Tubular protrusions that would indicate CNTs emerging from the polymer are not observed. Neither can we observe any free CNTs on the SEM scans, although the magnification chosen would allow seeing them (Fig. 8e).
 | ||
| Fig. 4 Morphology of fragments from the entire collected powders from machining/sanding. (a) and (b) TPU + CNT; (c) and (d) TPU. Protrusions of CNTs from the fragments are not observed. Compare Fig. SI_3a–e† with SEM scans of directly aerosol-sampling filters. | ||
Since the entire collected debris is dominated by the large polymer fragments, we performed further sanding experiments in order to collect aerosol samples onto filters for electron microscopy. We expected that the larger polymer fragments have a very low probability to reach the filters via the aerosol, so that this procedure, in line with the earlier work,22,33,34 would increase the probability to detect free CNTs, if they are generated. The background atmosphere for these air filter experiments was kept exceptionally clean at 10 P cm−3 so that contaminations can be excluded. When the abrasion machines were switched on, the background increased minimally up to 30 P cm−3, confirming that the electrical motor is well encapsulated and does not contaminate the air filters. The filter surfaces were searched for 1.5 days in the electron microscope, but no free CNTs were detected. Representative scans are shown in Fig SI_3a–e.†
In SEM of course the statistics are limited, and AUC is not chemically sensitive, so that we performed a third attempt to detect free CNTs, by their specific surface chemistry (Fig. 5).
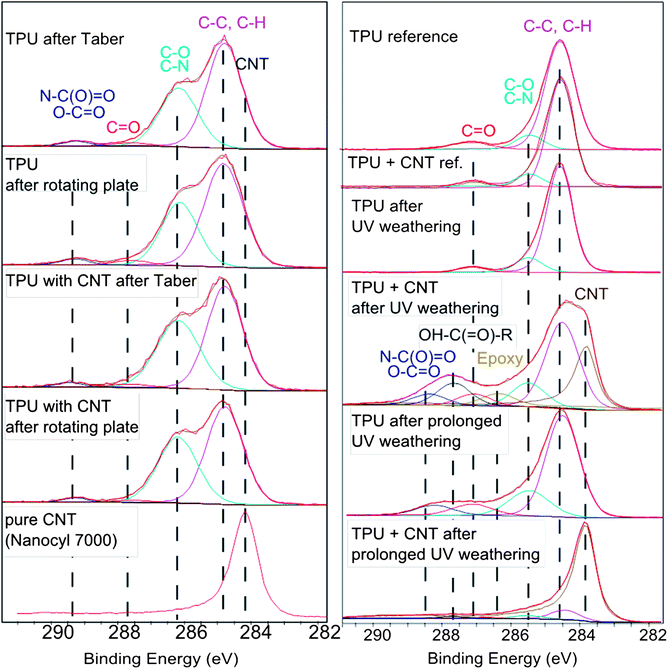 | ||
| Fig. 5 Surface chemistry in the upper 10 nm after degradation. Left column: fragments collected after mechanical treatment by Taber Abraser or by sanding (rotating plate) by X-ray photoelectron spectroscopy (XPS). The CNT positive control is shown as the bottom panel. The C(1s) detailed spectra of fragments exhibit no signal from low binding-energy-photoelectrons that are characteristic for CNTs regardless of the mechanical degradation scenario. Right column: the nanocomposite surface after weathering. When the samples were exposed to UV irradiation only, matching 9 months equivalent weathering, a shoulder appears that is characteristic for naked CNTs and cannot be explained by any other functional group. The remaining material after prolonged weathering is dominated by CNTs. Additionally, a severe degradation of the TPU matrix is observed, visible by the drastic change in the C(1s) line shape. Carboxylic groups and epoxy are observed in significant concentrations. See the ESI† for results on the XPS quantification of other relevant elements. | ||
Also the detailed evaluation of the C(1s) photoelectrons does not observe the characteristic signal of CNTs, which are hence absent in the top 10 nm of the fragments, and are instead embedded within the polymer. The C(1s) line shape which reflects the chemical bonds is identical for nanocomposite debris of TPU that contains CNTs and for the reference polymer without CNTs. The characteristic shoulder of CNT photoelectrons (Fig. 5 bottom line) can be excluded from the line shape visually, and a curve fit results in 0.5% contribution of CNTs from the reference (where they are not present) and for the nanocomposite alike.
To summarize the sanding results, there are no positive indications of released CNTs from either SEM, AUC or XPS. The size-selective quantification results by AUC (Table 1) demonstrate that a content of free CNT exceeding 0.1 wt% of the fragments would have been detected. We compare the limiting measurement uncertainty to the total content of 3 wt% in the virgin sample, and conclude that at least 97 wt% of the CNTs remain embedded in TPU and are not released. The actual value could be 100%, but cannot be better quantified with current methods.
Normal use/Taber Abraser
Also with the tenfold lower relative velocity of the Taber Abraser, a significant amount of the material is removed from the polymer disk and most of it falls onto the bottom of the chamber. The aerosol concentration remains at the background level for the TPU reference (below 250 P cm−3); for the TPU + CNT, two of three replicates show a peak at 30 nm diameter between 200 and 900 P cm−3 (Fig. 6). An attempt to collect these particles on filters for structural identification remained unsuccessful due to the vanishingly low total amounts. We can hence not exclude that this peak consists of CNTs or CNT–polymer-fragments, but we can provide upper limits on the number concentrations. Only one experiment (Fig. 6b, green line) showed an airborne concentration of several hundred nanometers. We reproduced the experiments with identical equipment in a chamber with the ultra-low background of 10 P cm−3, and found that the size peak between 20 nm and 100 nm is reproduced. It can now be quantified with total concentrations of 100 P cm−3 above the background, in accordance with the small peak observed in the previous experiments (Fig. 6b, blue and magenta lines). The green line is hence an occasional burst event, similar to the observations with high-speed sanding (Fig. 2). | ||
| Fig. 6 Aerosol size distribution during normal use/Taber Abraser, measured by SMPS in three replicates. (a) TPU; (b) TPU + CNT. The clean air background is at 250 P cm−3. | ||
Additionally, the entire powder after the normal use testing was collected from the test chamber floor and characterized for its size distribution, morphology and surface chemistry, in order to identify any possible presence of released CNTs. The powder was dispersed with probe sonication (150 W, 50%) and dispersant (SDS) to disrupt agglomerates or CNTs loosely adsorbed to fragments (see ESI†). The fragments with 10–200 μm diameter (Fig. 7) are slightly larger than from the high-speed sanding. A tail towards diameters between 1 μm and 10 μm is reflected also by the AUC determination of the solid content below 10 μm (Table 1), but the decisive range of free CNTs (below 150 nm) is identical for the TPU reference and for the TPU + CNT nanocomposite, both around 0.2 wt% absolute concentration in a 50 g L−1 suspension. SEM images confirm the agglomerates of polymer fragments, where the smallest structural sizes are around 20 μm (Fig. 8). The surface is smooth without tubular protrusions.
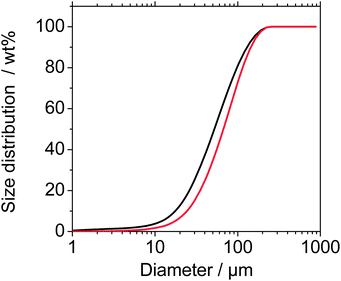 | ||
| Fig. 7 Size distribution of fragments from normal use/Taber Abraser (TPU, black; TPU + CNT, red). Fraunhofer diffraction data, regarded as reliable for sizes above 1 μm. Sizes below 10 μm down to 1 nm were independently quantified by analytical ultracentrifugation, see the text and Table 1. | ||
 | ||
| Fig. 8 Morphology and size of fragments from normal use/Taber Abraser. (a) and (b) TPU + CNT; (c) and (d) TPU. (e) CNT positive control on the same scale. The scans are dominated by polymeric particles, occasionally a fiber shape, which is at least one order of magnitude larger and longer than CNTs, and hence is identified as polymer fiber from the increased temperature during the testing. | ||
The chemical identification by the C(1s) photoelectrons (Fig. 5) shows that the upper 10 nm of the powder (where released CNTs would appear, loosely agglomerated) do not contain CNTs above the limit of detection. The exposed surface is polymer, not nanofiller. The full XPS scans reveal, however, that up to 4 at% of the fragments from normal use testing are Si (Fig. SI_4†), which is observed for both TPU and TPU + CNT from the Taber testing and not from the sanding assay.
Weathering
The TPU + CNT nanocomposite material follows the previously observed behaviour of an organic matrix with inorganic (nano)fillers: under weathering the matrix degrades, but not the (nano)fillers, such that their chemical signature becomes dominant on the outermost sample layer.19,24 The surface after dry weathering clearly shows the morphology of CNTs (Fig. 9) in a progressive way from 0 to 9 to 18 months equivalent irradiation. The tubular structures are confirmed as naked CNTs by their surface chemistry with the same approach via the C(1s) photoelectrons as above (Fig. 5). | ||
| Fig. 9 SEM surface images of the reference material TPU ((a–c) without CNTs), and the injection-molded TPU + CNT ((d–f), 3% CNTs). The virgin materials are on top (a and d), followed by 9 months (b and e) and 18 months (c–f) equivalent weathering with UV light only. As a positive control, CNTs on the same magnification are shown in Fig. 8e. | ||
A similar progression with the time-integral of UV intensity was observed for the UV degradation of a PU nanocomposite with 2 wt% graphene oxide, first emerging nanofillers on the weathered surface after 11 months-equivalent.25 However, that material was significantly different: a water-based aliphatic polyester urethane resin applied as a thin film.
In a further investigation we simulated outdoor weathering, but kept the UV dose constant. We then find that 72 ± 3% of the top 10 nm of the weathered sample are CNTs (from XPS, Fig. SI_5†), which is a factor of 3 faster removal of the polymer matrix than without humidity. The SEM scans confirm the interpretation that the polymer matrix has receded stronger, but the CNTs remain (Fig. SI_6†). A difference between samples prepared by injection molding or by extrusion has not been observed (Fig. SI_6a and c†).
The exposed network of entangled CNTs does not necessarily allow the release of the fillers known as ‘chalking’ for paints. Run-off from weathered TPU surfaces was simulated here by simply immersing the weathered sample in a few mL of H2O + SDS. Both indoor (UV) and outdoor (UV + humidity) weathered samples were tested. We observed no spontaneous change of the color or colloidal content of the water (by AUC) – hence no spontaneous release. However, since no methods are validated to assess the actual release of CNTs after weathering of CNT-composites, we do not further investigate this issue in the present contribution.
Results from in vitro toxicity testing
The cytotoxicity of TPU + CNT in lung tissue was assessed in comparison to TPU alone using the in vitro method ‘Precision Cut Lung Slices’ (PCLuS).In order to be able to expose PCLuS with the TPU + CNT abrasion material a dispersion in DMEM/F12 medium with 0.05% BSA had to be prepared 24 hours before application. Thus, the PCLuS could be treated, in duplicates, submersed for 24 h with several TPU concentrations and controls in parallel. Cytotoxicity was evaluated by measuring the membrane integrity by lactate dehydrogenase (LDH) release from the slices as well as the mitochondrial activity by the WST-1 assay. Triton X-100 is the standard positive control for PCLuS assays; ZnO particles served as an additional positive control. ZnO caused severe cytotoxicity already at a concentration of 10 μg mL−1. CNT alone and TPU with and without CNT were tested at high concentrations (1000 μg mL−1 and up to 20000 μg mL−1, respectively). Protein concentration determination was assessed to compare the negative control with historic protein data. The results show a good protein amount per sample (300 μg mL−1, Fig. 10). The two cytotoxicity assays showed congruent results: LDH release was increased and mitochondrial activity was decreased for the positive controls (Triton X-100 and ZnO) but not for TPU with or without CNT or CNT alone. Likewise, this indicates no cytotoxic effects of TPU with or without CNT on lung tissue in vitro. Also for CNT alone, no cytotoxicity was detected. This is consistent with the corresponding in vivo inhalation studies, where CNT did not cause cytotoxicity (necrosis) but inflammation of the lung.35 The PCLuS model used in this study is not proficient for these inflammatory processes. The model remains one of the few possible screening methods for physiological effects, given that the mass distribution of the fragments between 20 μm and 100 μm is prohibitive for in vivo inhalation or instillation.36
 | ||
| Fig. 10 Mean cytotoxicity (LDH release plotted in black bars and mitochondrial activity plotted in grey bars) and protein content (triangles) of PCLuS treated with positive controls (Triton X-100 and ZnO) and TPU with or without CNT (mean ± MIN/MAX of 2 replicates). The mitochondrial activity (WST-1) was calculated relative to the negative control (=100%). The cell membrane integrity (LDH release) was calculated relative to the Triton X-100 positive control (=100%). Protein content was calculated from the standard curve included in the experiment. | ||
Discussion
In terms of a systematic exploration of the correlation between the material's properties and its propensity to release nanofillers, the elastic TPU nanocomposite opens a new perspective. The published literature on release from nanocomposites has investigated relatively hard materials such as those for structural parts (engineering thermoplastics19,37,38 and epoxies22,23,33,34) or even harder matrices from the coating applications.25–27,39 Some observations from hard materials are recurring also for our very soft material: (a) the mass distribution remains dominated by micron particles despite the presence of significant numbers of submicron particles. Each of these two modes is related to material-specific properties and their variations do not correlate.26,27 (b) The fragment diameters from the nanocomposite tend to be slightly larger than from the pure matrix for TPU + CNT (Fig. 5) just like for PA + SiO2 or POM + CNT or cement + CNT.19 (c) The numbers of airborne particles are within the error bars independent of the presence of nanofillers in the matrix,26,27 sometimes even lower.38,40However, the absolute values are strongly material-dependent. In mass metrics the released fragments from TPU + CNT have diameters between 10 μm and 100 μm. These sizes are an order of magnitude larger than e.g. from sanding of epoxy + CNT, where diameters range between sub-micron and 10 μm, and even smaller for drilling.22,23,34,38,41
Most strikingly, the protruding CNTs that are typically observed on the surface of sanding fragments from epoxy + CNT,23,34 are absent on the surface of sanding fragments from TPU + CNT, as demonstrated by both morphology (SEM, Fig. 4 and 8) and chemical identification (XPS, Fig. 5). We interpret that the elasticity of TPU with its 600% elongation at break allows the polymer to reflow around the CNTs during fragmentation, whereas epoxy is stronger but brittle with an elongation at break around 5% only.
What about free CNTs? The aerosol time courses of TPU and TPU + CNT are not identical and their behaviour of bursts is different. This may be due to the modified mechanical properties during sanding, to the modified dispersibility by the antistatic TPU + CNT fragments, or to the release of CNTs. In several previous aerosol studies during mechanical treatment by scratching,37 sanding,26,27 and shredding40 there was no confirmation of airborne nanofillers, based on aerosol number concentrations and TEM/SEM sampling. Drilling in general produces high absolute aerosol concentration.22,38 Especially drilling without cooling liquids can release nanofillers.33 Evidence of individual freely released fillers was found by cutting or grinding on epoxy composites with CarbonNanoFibers (CNF),41 which range between CNTs and conventional carbon fibers in terms of diameter, length and stiffness. In a systematic investigation, release phenomena were linked to a high filler content (30%) or/and the presence of CNT agglomerates as predefined failure spots.37 It was argued that SEM/TEM sampling is more sensitive than aerosol number concentrations to detect freely released CNTs.34
Given the absence of agglomerates in our TPU + CNT (Fig. 1), the absence of CNT observations on SEM/TEM aerosol samples (Fig. SI_3†) or in the total collected swarf (Fig. 4 and 8), the exclusion of CNTs below the limit of detection of XPS chemical identification (Fig. 5) and below the limit of detection by size-selective fractionation (Table 1), we have no indication of the release of free CNTs from the TPU + CNT material.
The release probability has to be reconsidered if the matrix is chemically degraded, such as by UV radiation or by temperatures above the onset of polymer decomposition. According to the weathering hypothesis of Nguyen, developed on CNT–epoxy systems, the matrix is hydrolysed from the top few hundred nanometers, leaving behind an entangled network of CNTs that act as a passivating and UV-protecting layer.24 A recent study investigated CNT–epoxy and graphene–epoxy coatings, and found a linear enhancement of durability under UV and humidity with the CNT content increasing from 0 to 2% and fourfold reduction of the coating loss.42 Our results confirm that by photodegradation of the polymer matrix the remaining CNTs form an entangled network, that does not spontaneously release into run-off waters, in contrast to particulate nanofillers such as TiO2 (ref. 43) or SiO2.24
The very reliable thermal decomposition of CNTs between 500 and 650 °C is in fact exploited for their characteristic identification in thermogravimetric analysis.44 By well-ventilated incineration at temperatures above 1000 °C, even the much thicker CNF in a composite were effectively destroyed in the combustion gases.45 Incineration hence appears to be a favorable route of disposal.
Taken together, these results suggest the following indicators for the probability of protruding or released nanofillers under mechanical stress:
• brittle matrix, i.e. low elongation at break
• presence of agglomerates after production
• thick, stiff and long fillers (CNF vs. CNT)
• chemical degradation of the polymer matrix
All the above essentially prevent the polymer reflow at and around the nanofiller–polymer interface during fragmentation. None of the above applies to as-produced TPU + CNT. The softer material is in summary less vulnerable to CNT release, and additionally produces larger fragments that reduce the respirable fraction of the total debris mass.
Conclusions
The present results apply for the specific material based on polyether soft segment polyurethane and cannot necessarily be extrapolated to other TPUs or all other polyurethanes (e.g. TPU based on polyester soft segments or different hardness, partially crosslinked elastomers, highly crosslinked duromers, foams).For the specific thermoplastic polyurethane nanocomposite tested (polyether soft segments) that contained 3 wt% CNTs, we found that the relative softness and high elongation at break contribute to large average fragment sizes after mechanical degradation. The absolute numbers of aerosol during high-speed sanding are clearly above clean-air background, and comparable to the environmental background. Importantly, the numbers are not significantly different between the reference material and the nanocomposite. In mass metrics, only larger fragments with 10–200 μm diameters are generated. Free CNTs are not observed: not by morphology, not by quantitative surface chemistry, not in the quantitative size distribution. We conclude from our limits of detection that at least 97 wt% of the CNTs remain embedded in TPU and are not released. Protrusions of CNTs on the polymer fragments were not observed either in morphology or in surface chemistry. Under normal use, the absolute concentrations of respirable dust are around clean-air background level and thus far below normal environmental concentrations. The in vitro tests of fragments generated by sanding showed that TPU with compounded CNTs is not cytotoxic to lung tissue. This finding is supported by in vivo and in vitro studies on comparable materials.19–21 After simulated weathering the TPU + CNT nanocomposite material follows the previously observed behaviour of an organic matrix with inorganic (nano)fillers: the matrix degrades, and the more persistent CNTs remain as an entangled network. On the methodical side, we have demonstrated XPS line shape analysis as a highly selective method to quantify the presence of free or protruding CNTs.
In a preliminary risk assessment, we conclude that the human risk of workers and consumers by the known critical route of inhalation is very low, since the airborne dust (a) does not contain free CNTs above the detection limit of state-of-the-art complementary methods, (b) is too large to even reach the alveoli,36 and since (c) in vitro assays found no cytotoxicity potential.
This study is the first to provide quantitative limits of the amounts of released CNTs, and the methods are directly applicable to other nanocomposites. In contrast to other nanocomposites studied for their lifecycle release so far, the high elongation at break of TPU supports the tendency of the CNTs to remain embedded during mechanical treatments.
Acknowledgements
We thank Alexandra Aumann, Klaus Vilsmeier, Steffen Donde, Annemarie Hess, Arkadius Boron, Ulrich Flörchinger, Klaus Schill, Dennis Schomaeker for excellent laboratory support.Notes and references
- D. Randall and S. Lee, The Polyurethane Book, John Wiley & Sons, New York, 2002 Search PubMed.
- Z. S. Petrovic, Y. J. Cho, I. Javni, S. Magonov, N. Yerina, D. W. Schaefer, J. Ilavsky and A. Waddon, Polymer, 2004, 45, 4285 Search PubMed.
- Y. Qian, C. I. Lindsay, C. Macosko and A. Stein, ACS Appl. Mater. Interfaces, 2011, 3, 3709 CrossRef CAS.
- J. Jancar, Polym. Compos., 2000, 21, 369 Search PubMed.
- M. S. Sanchez-Adsuar, A. Linares-Solano, D. Cazorla-Amoros and L. Ibarra-Rueda, J. Appl. Polym. Sci., 2003, 90, 2676 Search PubMed.
- H. Koerner, G. Price, N. A. Pearce, M. Alexander and R. A. Vaia, Nat. Mater., 2004, 3, 115 CrossRef CAS.
- H. Kim, Y. Miura and C. W. Macosko, Chem. Mater., 2010, 22, 3441 CrossRef CAS.
- U. Khan, P. May, A. O'Neill, J. J. Vilatela, A. H. Windle and J. N. Coleman, Small, 2011, 7, 1579 CrossRef CAS.
- R. Zhang, A. Dowden, H. Deng, M. Baxendale and T. Peijs, Compos. Sci. Technol., 2009, 69, 1499 CrossRef CAS.
- P. Potschke, L. Haussler, S. Pegel, R. Steinberger and G. Scholz, KGK, Kautsch. Gummi Kunstst., 2007, 60, 432 Search PubMed.
- W. Chen and X. M. Tao, Macromol. Rapid Commun., 2005, 26, 1763 CrossRef CAS.
- H. Koerner, W. D. Liu, M. Alexander, P. Mirau, H. Dowty and R. A. Vaia, Polymer, 2005, 46, 4405 CrossRef CAS.
- http://www.polyurethanes.basf.de/pu/Elastollan/Elastollan_Materialeigenschaften, accessed April 2012.
- http://www.nanocyl.com/en/Products-Solutions/Products/Nanocyl-NC-7000-Thin-Multiwall-Carbon-Nanotubes, accessed April 2012.
- R. Landsiedel, L. Ma-Hock, A. Kroll, D. Hahn, J. Schnekenburger, K. Wiench and W. Wohlleben, Adv. Mater., 2010, 22, 2601 CrossRef CAS.
- E. Petersen, L. Zhang, N. T. Mattison, D. M. O'Carroll, A. J. Whelton, N. Uddin, T. L. Nguyen, Q. Huang, T. B. Henry, R. D. Holbrook and K. L. Chen, Environ. Sci. Technol., 2011, 45, 9837–9856 Search PubMed.
- B. Nowack, J. F. Ranville, S. Diamond, J. A. Galego-Urrea, C. Metcalfe, J. Rose, N. Horne, A. A. Koelmans and S. J. Klaine, Environ. Toxicol. Chem., 2012, 31, 50 CrossRef CAS.
- T. A. J. Kuhlbusch, C. Asbach, H. Fissan, D. Göhler and M. Stintz, Part. Fibre Toxicol., 2011 DOI:10.1186/1743-8977-8-22.
- W. Wohlleben, S. Brill, M. W. Meier, M. Mertler, G. Cox, S. Hirth, B. von Vacano, V. Strauss, S. Treumann, K. Wiench, L. Ma-Hock and R. Landsiedel, Small, 2011, 7, 2384 Search PubMed.
- A. Saber, N. Jacobsen, A. Mortensen, J. Szarek, P. Jackson, A. Madsen, K. Jensen, I. Koponen, G. Brunborg, K. Gutzkow, U. Vogel and H. Wallin, Part. Fibre Toxicol., 2012, 9, 4 Search PubMed.
- A. Saber, I. K. Koponen, K. A. Jensen, N. R. Jacobsen, L. Mikkelsen, P. Moeller, S. Loft, U. Vogel and H. Wallin, Nanotoxicology, 2012, 6, 776–788 Search PubMed.
- D. Bello, B. L. Wardle, N. Yamamoto, R. G. deVilloria, E. J. Garcia, A. J. Hart, K. Ahn, M. J. Ellenbecker and M. Hallock, J. Nanopart. Res., 2009, 11, 231 Search PubMed.
- L. G. Cena and T. M. Peters, J. Occup. Environ. Hyg., 2011, 8, 86 Search PubMed.
- T. L. Nguyen, B. Pellegrin, C. Bernard, X. Gu, J. M. Gorham, P. Stutzman, D. Stanley, A. Shapiro, E. Byrd, R. Hettenhouser and J. Chin, J. Phys.: Conf. Ser., 2011, 304, 012060 Search PubMed.
- C. Bernard, T. L. Nguyen, B. Pellegrin, R. D. Holbrook, M. Zhao and J. Chin, J. Phys.: Conf. Ser., 2011, 304, 012063 Search PubMed.
- D. Göhler, M. Stintz, L. Hillemann and M. Vorbau, Ann. Occup. Hyg., 2010, 54, 615 Search PubMed.
- I. K. Koponen, K. A. Jensen and T. Schneider, J. Exposure Sci. Environ. Epidemiol., 2011, 21, 408 Search PubMed.
- I. K. Koponen, K. A. Jensen and T. Schneider, J. Phys.: Conf. Ser., 2009, 151, 012048 Search PubMed.
- K. L. Planken and H. Cölfen, Nanoscale, 2010, 2, 1849 RSC.
- J. S. Taurozzi, V. A. Hackley and M. R. Wiesner, Nanotoxicology, 2010, 5, 711 Search PubMed.
- C. Singh, S. Friedrichs, N. Gibson, K. A. Jensen, M. Levin, R. Birkedal, G. Pojana, W. Wohlleben, S. Schulte, K. Wiench, D. Marshall, K. Hund-Rinke, W. Kördel and C. L. Klein, NM-Series of Representative Manufactured Nanomaterials: Zinc Oxide NM-110, NM-111, NM-112, NM-113. Characterisation and Test Item Preparation, 2011 Search PubMed.
- M. Vorbau, L. Hillemann and M. Stintz, J. Aerosol Sci., 2009, 40, 209 CrossRef CAS.
- D. Bello, B. L. Wardle, J. Zhang, N. Yamamoto, C. Santeufemio, M. Hallock and M. A. Virji, Int. J. Occup. Environ. Health, 2010, 16, 434 Search PubMed.
- L. Schlagenhauf, B. Chu, J. Buha, F. A. Nüesch and J. Wang, Environ. Sci. Technol., 2012, 46, 7366 Search PubMed.
- L. Ma-Hock, S. Treumann, V. Strauss, S. Brill, F. Luizi, M. Mertler, K. Wiench, A. O. Gamer, B. van Ravenzwaay and R. Landsiedel, Toxicol. Sci., 2009, 112, 468 CrossRef CAS.
- G. Oberdorster, E. Oberdorster and J. Oberdorster, Environ. Health Perspect., 2005, 113, 823 CrossRef CAS.
- L. Golanski, A. Guiot, M. Pras, M. Malarde and F. Tardif, J. Nanopart. Res., 2012, 14, 1.
- S. Sachse, F. Silva, H. Zhu, A. Irfan, A. Leszczynska, K. Pielichowski, V. Ermini, M. Blazquez, O. Kuzmenko and J. Njuguna, J. Nanomater., 2012, 189386 Search PubMed.
- L. Golanski, A. Gaborieau, A. Guiot, G. Uzu, J. Chatenet and F. Tardif, J. Phys.: Conf. Ser., 2011, 304, 012062 Search PubMed.
- P. C. Raynor, J. I. Cebula, J. S. Spangenberger, B. A. Olson, J. M. Dasch and J. B. D'Arcy, J. Occup. Environ. Hyg., 2012, 9, 1 Search PubMed.
- M. Methner, C. Crawford and C. Geraci, J. Occup. Environ. Hyg., 2012, 9, 308 Search PubMed.
- R. Asmatulu, G. A. Mahmud, C. Hille and H. E. Misak, Prog. Org. Coat., 2011, 72, 553 Search PubMed.
- R. Kaegi, A. Ulrich, B. Sinnet, R. Vonbank, A. Wichser, S. Zuleeg, H. Simmler, S. Brunner, H. Vonmont, M. Burkhardt and M. Boller, Environ. Pollut., 2008, 156, 233 CrossRef CAS.
- E. Mansfield, A. Kar and S. Hooker, Anal. Bioanal. Chem., 2010, 396, 1071 Search PubMed.
- N. Uddin and M. R. Nyden, Characterization of nanoparticle release from polymer nanocomposites due to fire, conference proceedings Nanotech2011, pp. 526–532 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Method to quantify the content of CNTs, figures of the degradation simulation setups, of contamination identification after testing, of supportive weathering results, of additional in vitro results. See DOI: 10.1039/c2nr32711b |
| This journal is © The Royal Society of Chemistry 2013 |