New routes to Cu(I)/Cu nanocatalysts for the multicomponent click synthesis of 1,2,3-triazoles†
Pavel
Abdulkin
a,
Yanina
Moglie
b,
Benjamin R.
Knappett
a,
David A.
Jefferson
a,
Miguel
Yus
b,
Francisco
Alonso
*b and
Andrew E. H.
Wheatley
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: aehw2@cam.ac.uk; Fax: +44 (0)1223 36362; Tel: +44 (0)1223 763966
bDepartamento de Química Orgánica, Facultad de Ciencias and Instituto de Síntesis Orgánica (ISO), Universidad de Alicante, Apdo. 99, 03080 Alicante, Spain. E-mail: falonso@ua.es; Fax: +34 96 5903549; Tel: +34 96 5903400 ext. 2406
First published on 5th November 2012
Abstract
An array of copper and copper–zinc based nanoparticles (NPs) have been fabricated employing a variety of polymeric capping agents. Analysis by TEM, XRPD and XPS suggests that by manipulating reagent, reductant and solvent conditions it is possible to achieve materials that are mono-/narrow disperse with mean particle sizes in the ≤10 nm regime. Oxidative stability in air is achieved for monometallic NPs using poly(methyl methacrylate) (PMMA) anti-agglomerant in conjunction with a variety of reducing conditions. In contrast, those encapsulated by either poly(1-vinylpyrrolidin-2-one) (PVP) or poly(4-vinylpyridine) (PVPy) rapidly show Cu2O formation, with all data suggesting progressive oxidation from Cu to Cu@Cu2O core–shell structure and finally Cu2O. Bimetallic copper–zinc systems, reveal metal segregation and the formation of Cu2O and ZnO. Catalysts have been screened in the synthesis of 1,2,3-triazoles through multicomponent azide–alkyne 1,3-dipolar cycloaddition. Whereas PMMA- and PVPy-coating results in reduced catalytic activity, those protected by PVP are highly active, with quantitative triazole syntheses achieved at room temperature and with catalyst loadings of 0.03 mol% metal for Cu and CuZn systems prepared using NaH2PO2, N2H4 or NaBH4 reductants.
Introduction
Copper has been used as a catalyst in organic synthesis at the molecular level for many decades.1–4 However, whereas the use of nanocatalysts offers enhancements in terms of more economic and sustainable processes where homogeneously catalyzed reactions presently suffer from major drawbacks,5–9 the susceptibility of nanoparticulate copper to oxidation10,11 has previously limited applications. In spite of this, processing costs and the search for new combinations of beneficial properties have led to continued interest in the replacement of established noble metal nanocatalysts with those of relatively inexpensive metals.12 In this vein, nanoparticulate copper has recently been deployed in various chemical syntheses, this having been enabled by the development of routes to the preparation of the copper-based nanomaterials Cu(0),13–16 CuO and Cu2O.17,18 As a result, copper nanoparticles (Cu NPs) have found many applications in, amongst other reactions, the reduction of functional groups,19 the formation of carbon–carbon and carbon–nitrogen bonds,20 and in so-called “click” chemistry.21,22 Our own groups have been especially concerned with the development of novel Cu NP catalysts and their application in click chemistry in particular. In this context, we have previously shown that Cu NPs could be formed either from CuCl2·2H2O or anhydrous CuCl2 and elemental lithium in the presence of catalytic 4,4′-di-tert-butylbiphenyl (DTBB) in THF at room temperature.13,19 These Cu NPs effectively catalysed the 1,3-dipolar cycloaddition of organic azides and terminal alkynes in remarkably short reaction times (10–120 min).13,23 Notwithstanding the superior catalytic activity observed when we compared these systems with other commercially available copper sources, the Cu NPs underwent dissolution under reaction conditions (Et3N, THF, 65 °C) and could not be reused. In order to overcome this problem, we introduced a catalyst consisting of oxidized copper nanoparticles immobilized on activated carbon. These could be readily prepared under mild conditions and proved extremely versatile catalysts for the multicomponent Huisgen 1,3-dipolar cycloaddition in water. Not only organic halides, but some other azide precursors, such as aryldiazonium salts, anilines, and epoxides were shown to be appropriate substrates in this process.24–27 Ullmann-type amination is another process in which we have been interested. In this context, we have recently fabricated Cu NPs capped by PVP40 [poly(1-vinylpyrrolidin-2-one), Mav = 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000] that enabled the efficient synthesis of 4-phenoxypyridine using multimode microwave heating in a microfluidic device.28 Kinetic studies have also sought to understand the processes at work during heterogeneously catalyzed reactions and, in particular, the differing affinities of reagents and products for the Cu NP surface.29 This work acted as precursor to the synthesis of bimetallic CuZn NPs that achieved enhanced yields for the same transformation under more facile reaction conditions.30 These bimetallic systems have recently been deployed in etherification chemistry with the presence of zinc used to explain the resistance of the system to copper oxide formation.31
000] that enabled the efficient synthesis of 4-phenoxypyridine using multimode microwave heating in a microfluidic device.28 Kinetic studies have also sought to understand the processes at work during heterogeneously catalyzed reactions and, in particular, the differing affinities of reagents and products for the Cu NP surface.29 This work acted as precursor to the synthesis of bimetallic CuZn NPs that achieved enhanced yields for the same transformation under more facile reaction conditions.30 These bimetallic systems have recently been deployed in etherification chemistry with the presence of zinc used to explain the resistance of the system to copper oxide formation.31
Whereas the investigation of nanostructured copper-based bimetallic systems has yielded efficient catalysts for industrially relevant processes,32,33 bimetallic CuM NPs34,35 have previously been found to deactivate on account of metal segregation and selective oxidation.36 In the case of monometallic Cu NPs, the prevailing structure-type noted has hitherto been Cu(core)@CuO/Cu2O(shell),37 with efforts to control the formation of an oxide shell predicated on the use of capping agents that inhibit molecular access to the particle surface.38–40 More recently, it has been shown that the use of polymeric capping agents may enhance the oxidative stability of NPs whilst permitting substrate access and efficient catalysis.41 Moreover, the range of polymers available for stabilizing highly reactive main group nanomaterials has also very recently been extended through the use of poly(methyl methacrylate) (PMMA) to protect Mg nanocomposites.42 The response of Cu-based nanomaterials to this capping agent is, so far as we are aware, yet to be reported.
In the present study, we describe recent work aimed at systematically extending the chemical reduction methods that we have previously used to fabricate a range of Cu-based nanoparticulates.41 Efforts focus on understanding the influence of (i) different steric capping agents, (ii) the choice of metal substrate and reducing agent, (iii) the presence or not of water during the synthesis, and (iv) reagent concentrations and reaction temperature. We report on whether the resulting Cu-based NPs undergo the formation of an oxide coat and on the long-term resistance of these materials to oxidation upon air exposure. Applications in the multicomponent click synthesis of model 1,2,3-triazoles are tested (Scheme 1), with an array of Cu-based nanomaterials screened for activity, selectivity and longevity under different conditions.
 | ||
| Scheme 1 Representative multicomponent synthesis of 1,2,3-triazoles. | ||
Results and discussion
Catalyst preparation and structure
Initial particle preparations sought to extend our former work on PVP-capped Cu28,41 and CuZn30 systems using the general route outlined in Scheme 2. The effects of varying polymer chain length were probed, with monometallic Cu fabricated in the presence both of PVP40 and PVP29 (average Mw = 40000 and 29000, respectively) by reducing CuSO4·5H2O with sodium hypophosphite hydrate, NaH2PO2·H2O, before modulating the pH with aqueous NaOH. The resulting narrow-disperse particles showed mean particle sizes of 7.23 ± 2.81 and 5.19 ± 1.02 nm, respectively (Table 1, samples 1 and 3, see also Fig. 1 for the latter). The fabrication of Cu using PVP29 made monometallic work consistent with that previously reported for PVP29-coated CuZn systems (also see below). However, for both samples 1 and 3, XRPD analysis demonstrated lines clearly corresponding to oxide formation.
 | ||
| Scheme 2 Representative conditions for Cu and CuZn NP synthesis. | ||
| Sample | Cu reagent | Polymer | Solvent–reductant | Mean size (nm) |
|---|---|---|---|---|
|
a av. Mw = 40000.
b Ethylene glycol.
c av. Mw = 29000.
d Heat to reflux.
e av. Mw = 60000.
f av. Mw = 120000.
|
||||
| 1 | CuSO4·5H2O | PVP40a | EGb–NaH2PO2·H2O | 7.23 ± 2.81 |
| 2 | CuSO4 | PVP40 | EG–NaH2PO2 | 7.03 ± 1.91 |
| 3 | CuSO4·5H2O | PVP29c | EG–NaH2PO2·H2O | 5.19 ± 1.02 |
| 4 | CuSO4 | PVP29 | EG–NaH2PO2 | 5.02 ± 0.99 |
| 5 | CuSO4 | PVP29 | EtOHd–NaBH4 | 4.42 ± 0.99 |
| 6 | CuSO4 | PVP29 | EtOH–NaBH4 | 8.91 ± 3.41 |
| 7 | Cu(OAc)2 | PVP29 | EtOH–NaBH4 | 2.50 ± 0.54 |
| 8 | Cu(OAc)2 | PVP29 | EtOHd–NaBH4 | 3.19 ± 0.86 |
| 9 | Cu(OAc)2 | PVP29 | THF–N2H4 | 4.14 ± 0.77 |
| 10 | CuCl2 | PVP29 | EtOHd–NaBH4 | 3.35 ± 0.92 |
| 11 | CuCl2 | PVP29 | THF–N2H4 | 5.02 ± 0.86 |
| 12 | CuSO4 | PVPy60e | EG–NaH2PO2 | 2.58 ± 0.60 |
| 13 | Cu(OAc)2 | PMMA120f | THFd–NaBH4 (in EtOH) | 2.42 ± 0.48 |
| 14 | Cu(OAc)2 | PMMA120 | 1:1 THF:EtOHd–NaBH4 |
4.34 ± 0.68 |
| 15 | Cu(OAc)2 | PMMA120 | THF–NaBH4 (in EtOH) | 2.48 ± 0.40 |
| 16 | Cu(OAc)2 | PMMA120 | THFd–N2H4 | 8.03 ± 1.34 |
| 17 | Cu(OAc)2 | PMMA120 | THF–N2H4 | 2.97 ± 0.78 |
| 18 | CuCl2 | PMMA120 | THFd–NaBH4 (in EtOH) | 3.01 ± 0.34 |
| 19 | CuCl2 | PMMA120 | 1:1 THF:EtOHd–NaBH4 |
3.96 ± 0.56 |
| 20 | CuCl2 | PMMA120 | THFd–N2H4 | 8.94 ± 1.45 |
| 21 | CuSO4 | PMMA120 | THFd–NaBH4 (in EtOH) | 3.54 ± 0.74 |
 | ||
| Fig. 1 Representative HRTEM images and particle size distributions for Cu catalyst samples 3 (top) and 4 (bottom) at ×300k magnification. Samples prepared by the NaH2PO2 reduction of CuSO4 in the presence of PVP29 under hydrous and anhydrous conditions, respectively. | ||
Subsequent work sought to replicate the synthesis of samples 1 and 3 in the absence of water in the expectation of achieving greater control over particle size distribution and/or stability, with solvents being distilled and degassed with argon immediately prior to use and NaOH being added as a solution in the reaction solvent. CuSO4 was reduced using two eq. NaH2PO2 in the presence of 10 eq. PVP following the synthetic procedure described in Scheme 2 (samples 2 and 4, see Fig. 1 for the latter). Similar particle size distributions were observed and XRPD revealed that any enhancement in oxidative stability was modest, with Cu, Cu2O and CuO all being observed (see ESI†). This led to the conclusion that the elimination of water from the synthesis had not resulted in particles with a long-term resistance to oxidation. Overall, particles synthesized under strictly anhydrous conditions closely resembled those prepared in the presence of water in terms of both morphology and mean particle size and distribution.
Initial attempts at using reducing agents other than hypophosphite for Cu systems failed to afford NPs that were observable by HRTEM. This was independent of Cu(II) sources tested but was particularly pronounced for any synthetic schemes involving water. However, by imposing anhydrous conditions (vide supra) and dissolving NaBH4 and NaOH in either ethylene glycol or EtOH it was possible to obtain a black/brown nanoparticle dispersion amenable to HRTEM analysis. In this way anhydrous CuSO4 was reduced using 2 eq. NaBH4 (an effective eight-fold excess of reducing agent) in the presence of 6 eq. PVP29 (samples 5 and 6). Analysis revealed that the use of elevated temperature gave a smaller mean particle size (sample 5) and that sample 6 was composed of NPs that were both significantly larger and had a greater than hoped for size dispersion. Mean particle size was lowered most effectively by varying Cu(II) source, with both Cu(OAc)2 and CuCl2 yielding narrow disperse NPs using NaBH4 (samples 7, 8 and 10). However, for both of these substrates, the alternative use of hydrazine as reductant gave a slight increase in mean particle size (samples 9 and 11). In each case, TEM images revealed lattice spacings attributable to metallic Cu, with XRPD on samples 7–10 showing progressive oxidation (see ESI†). In particular, sample 7 provided interesting new insights into the process by which particle oxidation had occurred (Fig. 2). Cu NPs have previously been shown to decompose to Cu(core)@CuO/Cu2O(shell) structures.37 Fringe analysis suggests that this process was not uniform in the present work. Hence, for example, the analysis of sample 7 suggests the co-existence of Cu, Cu2O and Cu@Cu2O NPs.
 | ||
| Fig. 2 HRTEM analysis of sample 7 suggests the presence of Cu (top), Cu@Cu2O (centre) and Cu2O (bottom) particles. | ||
Remaining with anhydrous preparations, the use of more aggressive anti-agglomerant was investigated whilst employing conditions otherwise similar to those used to synthesize sample 4.
A reduction in mean particle size along with a proportionately slightly raised particle size distribution was noted when using PVPy60 [poly(4-vinylpyridine), average Mw = 60000] capping agent (Table 1), with Cu NPs of 2.58 ± 0.60 nm seen (sample 12, cf. 5.02 ± 0.99 nm for sample 4).
We next sought to extend the idea that variations in Cu(II) reagent and reductant influence both mean particle size and dispersion, whilst recognizing that PVP(y) capping agents were unlikely to yield particles with extensive oxidative stability. We therefore sought to develop a remarkable recent study in which Mg nanocomposites were successfully passivated with respect to oxidation.42 In a typical experiment a Cu(II) solution was pretreated with PMMA120 [poly(methyl methacrylate); average Mw = 120000] before being stirred at 40 °C under Ar. The addition of reducing agent resulted in a colour change to dark brown/black. Having allowed the reaction to complete (1.5–2.5 h), purification was achieved by precipitating the sample using ethanol (samples 13–21; Table 1). The reduction of CuSO4, Cu(OAc)2 and CuCl2 was investigated using N2H4 and NaBH4 reductants. In almost all cases, mono- or narrow-disperse sub-5 nm particles were obtained. The presence of a higher proportion of EtOH favoured agglomeration upon NaBH4 reduction (cf. samples 13, 14 and also 18, 19). Reduction by N2H2 at ambient temperature afforded similarly small NPs (sample 17; Fig. 3), though the use of elevated temperatures led to narrowly dispersed, but significantly agglomerated particles (samples 16, 20). All samples fabricated in the presence of PMMA120 demonstrated excellent oxidative stability, undergoing no visible changes according to XRPD over a period of 2 weeks in the open laboratory (Fig. 3).
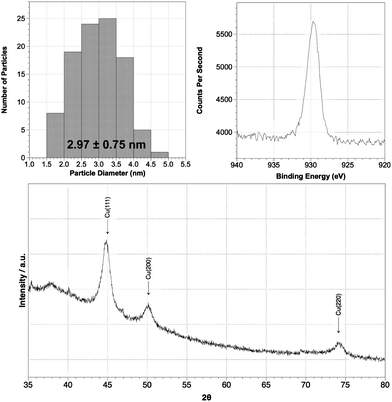 | ||
| Fig. 3 Analytical data for a representative PMMA120-coated sample of Cu NPs. Sample 17 was prepared using Cu(OAc)2 and N2H4 under anhydrous conditions at room temperature (Table 1). HRTEM reveals monodisperse behaviour (top right) while XPS shows oxidative stability by analysis of the Cu 2p3/2 region (top right). This is retained after 2 weeks according to XRPD (bottom). | ||
Moving to the preparation of bimetallic systems by the introduction of ZnCl2 into the synthesis, EDS revealed the presence of Kα1 emission lines corresponding to Cu and Zn for systems 22–24 (Scheme 2 and Table 2), with the ratio of peak intensities varying in nominal accordance with reaction stoichiometry when EDS was conducted with a broad beam-width (see ESI†). However, narrow beam EDS plainly revealed that these bimetallic samples were, in fact, comprised of Cu-rich and Zn-rich phases, arguing against individually bimetallic nanoparticles and consistent with the known ability of such systems to undergo dynamic segregation of the metals.36
| Sample | Compositiona (mol%) | Cu reagent | Polymer | Solvent–reductant | Mean size (nm) |
|---|---|---|---|---|---|
| a By reaction stoichiometry. | |||||
| 3 | 100 Cu | CuSO4·5H2O | PVP29 | EG–NaH2PO2·H2O | 5.19 ± 1.02 |
| 22 | 25:75 CuZn |
CuSO4·5H2O | PVP29 | EG–NaH2PO2·H2O | 6.11 ± 1.66 |
| 23 | 50:50 CuZn |
CuSO4·5H2O | PVP29 | EG–NaH2PO2·H2O | 4.66 ± 0.69 |
| 24 | 75:25 CuZn |
CuSO4·5H2O | PVP29 | EG–NaH2PO2·H2O | 4.55 ± 1.09 |
| 4 | 100 Cu | CuSO4 | PVP29 | EG–NaH2PO2 | 5.02 ± 0.99 |
| 25 | 25:75 CuZn |
CuSO4 | PVP29 | EG–NaH2PO2 | 4.97 ± 0.96 |
| 26 | 50:50 CuZn |
CuSO4 | PVP29 | EG–NaH2PO2 | 4.88 ± 1.07 |
| 27 | 75:25 CuZn |
CuSO4 | PVP29 | EG–NaH2PO2 | 5.10 ± 1.38 |
| 12 | 100 Cu | CuSO4 | PVPy60 | EG–NaH2PO2 | 2.58 ± 0.60 |
| 28 | 25:75 CuZn |
CuSO4 | PVPy60 | EG–NaH2PO2 | 4.21 ± 1.57 |
| 29 | 50:50 CuZn |
CuSO4 | PVPy60 | EG–NaH2PO2 | 3.95 ± 2.06 |
| 30 | 75:25 CuZn |
CuSO4 | PVPy60 | EG–NaH2PO2 | 2.91 ± 1.36 |
In line with EDS observations, the XRPD analysis of sample 23 revealed both Cu2O and ZnO. These last data are supported by the diffraction patterns obtained for this sample by the FFT of representative HRTEM images. Lattice spacings observed in different particles corresponded most closely to either Cu2O or ZnO. Taken together, these observations suggest the formation of segregated regions of either oxide. ICP-OES (see ESI†) demonstrated that the reaction stoichiometry was well reflected in the product composition for Cu rich syntheses, with samples 24, 27, 30 revealing 70.6–78.0 mol% Cu. However, attempts to incorporate equimolar Cu and Zn (samples 23, 26, 29) resulted in an excess of the latter element (27.1–42.9 mol% Cu). Attempts to fabricate Zn-rich NPs led to the observation of significant levels of zinc, in particular when PVP capping agent was used.
Adjusting the ratios of CuSO4 and ZnCl2 in anhydrous preparations whilst maintaining a constant total metal content:polymer:reducing agent ratio yielded CuZn NPs (samples 25–27) that were similar in nature to samples 22–24 – that is, revealing the same tendency for metal segregation and ZnO formation (see ESI†).
HRTEM analysis revealed particles with essentially equivalent size distributions of 5.02 ± 0.99, 4.97 ± 0.96, 4.88 ± 1.07, and 5.10 ± 1.38 nm for PVP-capped samples 4, 25–27. In contrast, PVPy-capped samples 12, 28–30 increased in mean particle size from 2.58 ± 0.60 to 4.21 ± 1.57 nm as a function of Zn contribution to reaction stoichiometry. However, an analysis of particle size histograms showed that for reactions containing more Zn an increasingly bimodal particle size distribution resulted. This was most clear for sample 28 (Fig. 4), for which EDS detected that regions dominated by smaller particles (ca. 3 nm wide) were Cu-rich and regions dominated by larger particles (ca. 7 nm wide) were Zn-rich. The view that the latter in fact constitute ZnO was reinforced by the observation of lattice spacings corresponding to the oxide. Overall, these data suggest that in each of samples 28–30, Cu-based NPs accounted for the smaller particles observed and were relatively unchanged in each case, while larger ZnO agglomerates had come to dominate in Zn-rich reactions.
 | ||
| Fig. 4 Representative FFTs of ×800k magnification TEM images of individual NPs (left: average d = 2.47 Å, Cu2O (111); right: average d = 2.81 Å, ZnO (100)) and the size distribution for CuZn catalyst 28 (prepared from CuSO4–ZnCl2 with dry NaH2PO2) suggest that the metals have segregated. | ||
Catalyst screening
The multicomponent 1,3-dipolar cycloaddition of terminal alkynes and azides (generated in situ from alkyl halides and NaN3) was chosen as a route for catalyst screening (Scheme 3 and Table 3), utilizing aliquots of as-prepared catalyst MeOH dispersions of known concentration. The reaction with activated benzyl (Bn) bromide 1 was found to be significantly promoted by dispersions of both mono- and bimetallic catalysts without the need for high temperatures and using substantially lower loadings than have previously proved effective for such transformations (0.03 mol% metal vs. 1–10 mol% previously).43–51 Accordingly, Cu (samples 1 and 2; entries 1 and 2 in Table 3) as well as 50:50 CuZn (samples 23 and 26; entries 22 and 25), and 75:25 CuZn (sample 24; entry 23) gave yields of 3 that were good-to-quantitative even at room temperature. However, attempts to generate 25:75 CuZn catalysts led to NPs with a very low Cu content (see ESI† and vide supra), and this was reflected in the poor activity of catalyst 25 (entry 24).
 | ||
| Scheme 3 Conditions for the multicomponent 1,3-dipolar cycloaddition of terminal alkynes and in situ generated azides. | ||
| Entry | Catalyst | T (°C) | t (h) | 3 (%) | 5 (%) |
|---|---|---|---|---|---|
| a Determined from the GLC peak area, isolated yields in parentheses. b 19% alkyne homocoupling also observed. c Ar atmosphere. | |||||
| 1 | 1 | rt | 7 | 94 | — |
| 2 | 2 | rt | 10 | 81b | |
| 3 | 4 | rt | 7 | 60 | — |
| 4 | 4 | rt | 24 | 80 | — |
| 5 | 4 | rtc | 7 | 46 | — |
| 6 | 4 | rtc | 24 | 48 | — |
| 7 | 7 | rt | 7 | 56 | — |
| 8 | 7 | rt | 24 | 100 (98) | — |
| 9 | 7 | rtc | 24 | 94 | — |
| 10 | 9 | rt | 24 | 100 (98) | — |
| 11 | 9 | rtc | 24 | 96 | — |
| 12 | 9 | rt | 18 | — | 0 |
| 13 | 9 | Reflux | 6 | — | 100 (98) |
| 14 | 12 | rt | 7 | 16 | — |
| 15 | 12 | Reflux | 2 | 100 | — |
| 16 | 15 | rt | 24 | 0 | — |
| 17 | 15 | Reflux | 24 | 100 | — |
| 18 | 15 | rtc | 24 | 0 | — |
| 19 | 17 | rt | 24 | 0 | — |
| 20 | 17 | Reflux | 24 | 60 | — |
| 21 | 17 | rtc | 24 | 0 | — |
| 22 | 23 | rt | 7 | 98 | — |
| 23 | 24 | rt | 7 | 91 | — |
| 24 | 25 | rt | 7 | 19 | — |
| 25 | 26 | rt | 7 | 100 (99) | — |
| 26 | 26 | rt | 16 | — | 10 |
| 27 | 26 | Reflux | 6 | — | 100 (98) |
| 28 | 28 | rt | 24 | 14 | — |
| 29 | 28 | Reflux | 7 | 100 | — |
| 30 | 29 | rt | 7 | 3 | — |
| 31 | 29 | Reflux | 2 | 100 | — |
| 32 | 30 | rt | 7 | 5 | — |
| 33 | 30 | Reflux | 2 | 100 | — |
Consistent with PVP-coated NPs demonstrating Cu(I) oxide formation (vide supra) there is a difference observed between catalyst reactivities under atmospheres of air or Ar. Hence, sample 4 gave rather lower yields of 3 when used under Ar (compare entries 5 and 6 with entries 3 and 4). The generally lower efficiency of this catalyst under either atmosphere may be interpreted in terms of the inclusion of sulfur in the NP preparation, though it is of interest to note that CuZn systems 24 and 26 resisted comparable deactivation. The possibility of sulfur inhibiting the action of Cu NPs was overcome in catalyst samples 7 and 9, which were sourced from Cu(OAc)2 (entries 7–11). These catalysts revealed enhanced yields of 3 after 24 h at room temperature under air (entries 8 and 10) and, in fact, returned only small losses of yield under Ar (entries 9 and 11). The temperature responses of the most successful catalyst systems (samples 9 and 26) were further studied in 1,2,3-triazole synthesis using the less reactive, non-activated alkyl halide n-nonyl iodide (Scheme 3), with the yield of 5 found to be quantitative under reflux conditions (entries 12, 13, 26 and 27). As with PVP-coated catalysts, PVPy-coated systems revealed oxide formation (vide supra). However, inferior reactivity was noted at room temperature for both Cu (catalyst sample 12) and CuZn (samples 28–30) based systems coated with the more aggressive anti-agglomerant. Hence, while each of these samples gave quantitative click conversions under reflux conditions, room temperature reactions revealed yields in the range 3–16% (entries 14, 15, 28–33). Structural studies showed that PMMA-coated Cu NPs resist oxidation even after sustained air exposure. Both PMMA-coated samples 15 and 17 were found to be completely inactive with respect to triazole synthesis at room temperature under either air or Ar atmospheres (entries 16, 18, 19 and 21) but active when heated to reflux in an aerobic environment (entries 17 and 20). Taken together with the catalytic data for PVPy-capped samples these data suggest that catalytic inactivity cannot be attributed to NP resistance to Cu(I) formation alone, but that reagent access to the catalyst surface is also significant.
During the screening of Cu based nanocatalysts in the click synthesis of 1,2,3-triazoles a remarkable observation was made concerning the ability of air-exposed catalysts to promote the alkynylation of triazoles (Scheme 4). To the best of our knowledge, the synthesis of 5-alkynyl 1,2,3-triazoles52 following a similar approach has been accomplished only by Porco's group.53 In that report, organic azides and terminal alkynes reacted in a system comprised of Cu(CH3CN)4PF6, N,N,N′-trimethylethylenediamine as ligand, Hünig's base, molecular oxygen, and 4-methylmorpholine N-oxide as co-oxidant in dichloromethane. Presently, both Cu (sample 1) and 50:50 CuZn NPs (sample 23) yielded not only triazole 3 upon treatment with BnBr, NaN3 and PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH, but also C-alkynylated heterocycle 6 and BnN37 (Table 4, entries 1 and 3). The selectivity towards 6 was found to be considerably enhanced by either using catalyst sample 2 (entry 2) which, unlike catalyst 1, was fabricated under anhydrous conditions, or by omitting the 12 h pre-treatment but conducting the reaction in an aerobic atmosphere for sample 1 (entry 4). Conversely, bimetallic catalyst 23 was found to be inactive towards alkynylation (entry 5) except when air exposure was extended (entry 3), suggesting that the preferential oxidation of Zn rendered the need for an air pre-treatment more important. Failure to expose pure Cu nanocatalyst to air either before or during reaction resulted in the enhanced formation of 3 and a lower level of alkynylation (compare entries 2 and 6), with the subsequent addition of phenylacetylene 2 to the reaction in entry 6 yielding even more 3.
CH, but also C-alkynylated heterocycle 6 and BnN37 (Table 4, entries 1 and 3). The selectivity towards 6 was found to be considerably enhanced by either using catalyst sample 2 (entry 2) which, unlike catalyst 1, was fabricated under anhydrous conditions, or by omitting the 12 h pre-treatment but conducting the reaction in an aerobic atmosphere for sample 1 (entry 4). Conversely, bimetallic catalyst 23 was found to be inactive towards alkynylation (entry 5) except when air exposure was extended (entry 3), suggesting that the preferential oxidation of Zn rendered the need for an air pre-treatment more important. Failure to expose pure Cu nanocatalyst to air either before or during reaction resulted in the enhanced formation of 3 and a lower level of alkynylation (compare entries 2 and 6), with the subsequent addition of phenylacetylene 2 to the reaction in entry 6 yielding even more 3.
 | ||
| Scheme 4 Domino multicomponent 1,2,3-triazole synthesis/triazole alkynylation. | ||
:50 CuZn) in multicomponent 1,2,3-triazole synthesis/triazole alkynylation to give 6
| Entry | Sample | Conditions | T (°C) | t (h) | 3 (%) | 6 (%) | 7 (%) | 8 (%) |
|---|---|---|---|---|---|---|---|---|
| a Yield of 3 after subsequent addition of 1 mmol 2 (1 mmol = 1 eq. wrt BnBr) given in parentheses. b Use 2 eq. 2. | ||||||||
| 1 | 1 | Catalyst stirred in air for 12 h prior to reaction in air | rt | 24 | 34 | 32 | 34 | — |
| 2 | 2 | rt | 24 | 13 | 77 | 10 | — | |
| 3 | 23 | rt | 24 | 39 | 33 | 26 | — | |
| 4 | 1 | Reaction in air | rt | 24 | 10 | 81 | 9 | — |
| 5 | 23 | rt | 24 | 40 | — | — | — | |
| 6 | 2 | Reaction under Ar | rt | 16 | 63 (77)a | 23 | 14 | — |
| 7 | 2 | Reaction in airb | rt | 24 | — | — | — | — |
| 8 | 2 | Catalyst stirred in air for 2 h prior to reaction in airb | rt | 24 | 32 | 57 | — | 3 |
| 9 | 2 | Reflux | 24 | 32 | 57 | — | 3 | |
| 10 | 2 | Catalyst stirred in air for 1 h prior to reaction in air | Reflux | 24 | 88 | 10 | — | 2 |
| 11 | 2 | Catalyst stirred in air for 1 h prior to reaction in airb | Reflux | 24 | 72 | 16 | — | — |
| 12 | 1 | Reaction in airb | rt | 24 | 29 | 58 | — | — |
| 13 | 1 | Reflux | 24 | 79 | 5 | — | — | |
The employment of 2 eq. 2 at the outset of a triazole synthesis reaction failed to have a positive effect on the yield of alkynylated product at either room temperature or reflux and instead led to enhanced yields of 3 (entries 7–9 and 11–13). It was also generally clear that alkynylation was deleteriously affected at higher temperature (e.g. compare entries 12 and 13). Taken together, these data suggest that the alkynylation of triazoles is best promoted by catalysts rich in Cu2O, but that catalysts that are excessively exposed to oxygen or that have a lower copper content (e.g. bimetallic catalysts) give less alkynylation. The suggestion is that the formation of 6 is strongly dependent on the level of Cu oxidation manifest and that it is inhibited by the presence of less active CuO (e.g. sample 1 after an extended pre-treatment in air) or by the resistance of Cu to oxidation by the sacrificial formation of ZnO (e.g. sample 23). This last point is of particular interest, with Zn having a clear effect on catalytic activity in spite of evidence (vide supra) for metal segregation.
In seeking to better understand whether the alkynylation process occurred in tandem with triazole formation, catalyst sample 2 was treated with a mixture of 2 and 3 under various conditions. In agreement with Porco's observations,53 the failure to note any detectable reaction argues strongly against C–H activation in pre-formed 3 facilitating alkynylation by 2. Similarly, the treatment of a pre-formed mixture of 3, 6 and 7 with phenylacetylene yielded no 6 and only a small amount of 8. Lastly, the failure of a mixture of 1, NaN3 and 8 to undergo detectable reaction in the presence of catalyst 2 suggested that 6 is not formed by the click reaction of diyne 8.
Conclusion
In summary, the fabrication of a variety of Cu-based NPs in the 3–9 nm regime has been allowed by the use different reducing agents (NaH2PO2(·H2O), N2H4, NaBH4) and polymeric capping agents. The exclusion of water from NP synthesis was found to have a limited beneficial effect on stability of the resulting NPs. Electron microscopy suggested that samples incorporating either PVP or PVPy anti-agglomerants rapidly underwent oxidation to give Cu@Cu2O NPs alongside unoxidized Cu NPs, and also the segregation and preferential oxidation of Zn for bimetallic systems. Meanwhile, the use of PMMA capping agent gave oxidatively stable Cu(0) NPs as evidenced by XPS and XRPD.A selection of as-prepared NPs were screened in the multicomponent 1,3-dipolar synthesis of 1,2,3-triazoles using in situ generated organic azides. PVPy- and PMMA-coated NPs were found to be inactive at room temperature, suggesting that these polymers represent a barrier against substrate access to the catalyst surface. However, NPs coated with PVP were found to be highly active, producing quantitative yields of triazoles from benzyl bromide at room temperature and from the less reactive n-nonyl iodide at reflux. Unprecedentedly low catalyst loadings could be used and alkynylation of the triazole C-5 position was also recorded under conditions that promoted limited catalyst oxidation.
Studies will now seek to develop a more detailed understanding of specific triazole syntheses using the most promising catalysts identified. Efforts will focus on extending the range of azide precursors, with anilines, aryldiazonium salts and epoxides all being tested, and of alkynes, including aliphatic alkynes. Zinc-containing catalysts will be studied with a view to developing systems that are active for both terminal and internal alkynes.54 Moreover, the presence of Zn clearly influences the resistance of Cu to oxidation and also plays a role in controlling the formation of alkynylated triazoles. In order to develop catalyst reusability, we are also initiating studies that marry azide–alkyne 1,3-dipolar cycloaddition catalysis with our own recent work on the immobilization of Cu-based NPs.31
Experimental
General synthetic and analytical details
Chemicals, including hydrazine (1 M in THF), were purchased from Sigma-Aldrich (reagents: analytical grade; solvents: HPLC grade) and used without further purification. Aqueous solutions were prepared using deionized water (Millipore, specific resistivity ≥18.2 MΩ cm). Ethanol was distilled immediately before use. Prior to synthesis, flasks were flushed and all solutions were degassed with argon.Catalyst preparation
Catalyst characterization
Samples analyzed on an image aberration corrected FEI Titan 80–300 operated at 300 kV providing an information limit of 0.08 nm in TEM mode were prepared by dropping a suspension of the NPs in MeOH onto carbon-coated Ni grids (Quantifoil). Scanning transmission electron microscopy (STEM) was performed using a high-angle annular dark-field (HAADF) detector with a nominal spot size of 0.14 nm. For spectroscopy, a nominal spot size of ∼0.5 nm was used in STEM mode with a Gatan Tridiem image filter for electron energy loss spectroscopy (EELS) and an EDAX S-UTW EDS detector.
Particle sizes were analysed using Macnification 2.0.1 by counting the diameters of 100 particles in lower magnification images,55 defining size intervals of 0.2 nm between dmin ≤ d ≤ dmax and counting the number of particles falling into these intervals. The data were then used to construct particle size distributions using DataGraph 3.0.
The detailed analysis of particle morphology was done using Digital Micrograph 3.6.5. The values of average d-spacing were obtained from Fourier transforms of high magnification images (×800k, ×1M, ×1.2M) using the expression d = D/20 where D is the diameter (nm) of rings obtained. The average d-spacing was then verified using the profile tool in Digital Micrograph counting at least 10 d-spacings. To determine the error in the value of d-spacing thus obtained, detailed TEM examination of CeO2 and Au nanoparticles was undertaken. The relationship between the diameter of the FT rings and the DV value (a measure of the objective lens focusing voltage) was established for DV values between −6 and +6 and the value of standard deviation in d-spacing was established to be 10% when compared to literature values. Where oxidation state was determined based on d-spacings obtained by TEM measurements, values within 1 standard deviation (±5%) of literature values were used.
Multicomponent synthesis of 1,2,3-triazoles
NaN3 (72 mg, 1.1 mmol), organic halide [benzyl bromide (119 μl, 1 mmol) or 1-iodononane (197 μl, 1 mmol)], and phenylacetylene (110 μl, 1 mmol or 220 μl, 2 mmol) were added to a suspension of Cu NPs or CuZn NPs in MeOH (2 ml; the metal content of which had been determined by ICP-OES prior to dilution to a known concentration) under an atmosphere of air or argon (see Tables 3 and 4). The reaction mixture was maintained at room temperature or at reflux for a specified time, with the progress being monitored by TLC. Water (30 ml) was added and the mixture extracted with EtOAc (3 × 10 ml). The organic phase was dried with anhydrous MgSO4, and the solvent removed in vacuo to give crude material. This was analyzed by GLC and did not require any further purification when high conversions into the triazoles 3, 5 and 6 were reached. Triazoles 3,23525 and 656 were characterized by comparison of their physical and spectroscopic data with those described in the literature.
Acknowledgements
We thank the the Royal Society of Chemistry (Journals Grant for International Authors), the Royal Society (International Exchanges Scheme 1212/R1) and Dr Christian Kübel of the Karlsruhe Nano Micro Facility for technical assistance with HRTEM/EDS analysis. We also acknowledge the UK EPSRC (EP/F019823/1) and Dr D. J. Morgan (Cardiff University) and Dr Peter Thüne (Technical University of Eindhoven) for XPS access. This work was generously supported by the Spanish Ministerio de Ciencia e Innovación (MICINN; CTQ2007-65218 and Consolider Ingenio 2010-CSD2007-00006), the Generalitat Valenciana (GV; PROMETEO/2009/039), and FEDER. Y. M. and B. R. K. thank the ISO of the Universidad de Alicante, the UK EPSRC and the University of Cambridge for grants.Notes and references
- D. Dollimore, Copper in Catalysis, in Handbook of Copper Compounds and Applications, ed. H. W. Richardson, 1997, Dekker, New York, pp. 231–264 Search PubMed.
- I. P. Beletskaya and A. V. Cheprakov, Coord. Chem. Rev., 2004, 248, 2337 CrossRef CAS.
- Handbook of Reagents for Organic Synthesis. Catalyst Components for Coupling Reactions, ed. G. Molander, John Wiley & Sons, Chichester, 2008, pp. 222–226 Search PubMed.
- B. H. Lipshutz and Y. Yamamoto, Chem. Rev., 2008, 108, 2793 CrossRef CAS.
- M. Kidwai, Nanoparticles in Green Catalysis, in Handbook of Green Chemistry, ed. P. T. Anastas and R. H. Crabtree, Wiley-VCH, Weinheim, 2009, vol. 2, pp. 81–92 Search PubMed.
- J. M. Campelo, D. Luna, R. Luque, J. M. Marinas and A. A. Romero, ChemSusChem, 2009, 2, 18 CrossRef CAS.
- R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481 RSC.
- V. Polshettiwar and R. S. Varma, Green Chem., 2010, 12, 743 RSC.
- N. Yan, C. Xiao and Y. Kou, Coord. Chem. Rev., 2010, 254, 1179 CrossRef CAS.
- A. Yabuki and S. Tanaka, Mater. Res. Bull., 2011, 46, 2323 CrossRef CAS.
- D. E. Diaz-Droguett, R. Espinoza and V. M. Fuenzalida, Appl. Surf. Sci., 2011, 257, 4597 CrossRef CAS.
- Catalysis Without Precious Metals, ed. R. M. Bullock, Wiley-VCH, Weinheim, 2010 Search PubMed.
- F. Alonso, Y. Moglie, G. Radivoy and M. Yus, Tetrahedron Lett., 2009, 50, 2358 CrossRef CAS.
- H. A. Orgueira, D. Fokas, Y. Isome, P. C. M. Chan and C. M. Baldino, Tetrahedron Lett., 2005, 46, 2911 CrossRef CAS.
- L. D. Pachon, J. H. van Maarseveen and G. Rothenberg, Adv. Synth. Catal., 2005, 347, 811 CrossRef.
- A. Sarkar, T. Mukherjee and S. Kapoor, J. Phys. Chem. C, 2008, 112, 3334 CAS.
- G. Molteni, C. L. Bianchi, G. Marinoni, N. Santo and A. Ponti, New J. Chem., 2006, 30, 1137 RSC.
- I. S. Park, M. S. Kwon, Y. Kim, J. S. Lee and J. Park, Org. Lett., 2008, 10, 497 CrossRef CAS.
- F. Alonso and M. Yus, Pure Appl. Chem., 2008, 80, 1005 CrossRef CAS.
- B. C. Ranu, R. Dey, T. Chatterjee and S. Ahammed, ChemSusChem, 2012, 5, 22 CrossRef CAS.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef CAS.
- M. G. Finn and V. V. Fokin, Copper-Catalyzed Azide–Alkyne Cycloaddition (CuAAC), in Catalysis Without Precious Metals, ed. R. M. Bullock, Wiley-VCH, Weinheim, 2010, pp. 235–260 Search PubMed.
- F. Alonso, Y. Moglie, G. Radivoy and M. Yus, Eur. J. Org. Chem., 2010, 1875 CrossRef CAS.
- F. Alonso, Y. Moglie, G. Radivoy and M. Yus, Adv. Synth. Catal., 2010, 352, 3208 CrossRef CAS.
- F. Alonso, Y. Moglie, G. Radivoy and M. Yus, Org. Biomol. Chem., 2011, 9, 6385 CAS.
- F. Alonso, Y. Moglie, G. Radivoy and M. Yus, J. Org. Chem., 2011, 76, 8394 CrossRef CAS.
- F. Alonso, Y. Moglie, G. Radivoy and M. Yus, Heterocycles, 2012, 84, 1033 CrossRef CAS.
- F. Benaskar, V. Engels, E. V. Rebrov, N. Patil, V. Hessel, L. A. Hulshof, J. C. Schouten and A. E. H. Wheatley, Tetrahedron Lett., 2010, 51, 248 CrossRef CAS.
- F. Benaskar, V. Engels, N. Patil, E. V. Rebrov, J. Meuldijk, V. Hessel, L. A. Hulshof, A. E. H. Wheatley and J. C. Schouten, 2012, manuscript in preparation.
- V. Engels, F. Benaskar, N. G. Patil, E. V. Rebrov, V. Hessel, L. A. Hulshof, D. A. Jefferson, S. Karwal, J. C. Schouten and A. E. H. Wheatley, Org. Process Res. Dev., 2010, 14, 644 CrossRef CAS.
- F. Benaskar, V. Engels, E. V. Rebrov, N. G. Patil, J. Meuldijk, P. C. Thüne, P. C. M. M. Magusin, B. Mezari, V. Hessel, L. A. Hulshof, E. J. M. Hensen, A. E. H. Wheatley and J. C. Schouten, Chem.–Eur. J., 2012, 18, 1800 CrossRef CAS.
- J. Greeley, A. A. Gokhale, J. Kreuser, J. A. Dumesic, H. Topsøe, N.-Y. Topsøe and M. Mavrikakis, J. Catal., 2003, 213, 63 CrossRef CAS.
- P. L. Hansen, J. B. Wagner, S. Helveg, S. R. Rostrup-Nielsen, B. S. Clausen and H. Topsøe, Science, 2002, 295, 2053 CrossRef CAS.
- R. E. Cable and R. E. Schaak, Chem. Mater., 2007, 19, 4098 CrossRef CAS.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 846 CrossRef and refs. therein.
- V. Calò, A. Nacci, A. Monopoli, E. Ieva and N. Cioffi, Org. Lett., 2005, 7, 617 CrossRef.
- P. Kanninen, C. Johans, J. Merta and K. Kontturi, J. Colloid Interface Sci., 2008, 318, 88 CrossRef CAS.
- J. G. Yang, Y. L. Zhou, T. Okamoto, R. Ichino and M. Okido, Surf. Eng., 2007, 23, 448 CrossRef CAS.
- M. Tomonari, K. Ida, H. Yamashita and T. Yonezawa, J. Nanosci. Nanotechnol., 2008, 8, 2468 CrossRef CAS.
- M. Valle-Orta, D. Diaz, P. Santiago-Jacinto, A. Vázquez-Olmos and E. Reguera, J. Phys. Chem. B, 2008, 112, 14427 CrossRef CAS.
- V. Engels, F. Benaskar, D. A. Jefferson, B. F. G. Johnson and A. E. H. Wheatley, Dalton Trans., 2010, 6496 CAS.
- K.-J. Jeon, H. R. Moon, A. M. Ruminski, B. Jiang, C. Kisielowski, R. Bardhan and J. J. Urban, Nat. Mater., 2011, 10, 286 CrossRef CAS.
- P. Appukkuttan, W. Dehaen, V. V. Fokin and E. Van der Eycken, Org. Lett., 2004, 6, 4223 CrossRef CAS.
- M. Lakshmi Kantam, V. Swarna Jaya, B. Sreedhar, M. Mohan Rao and B. M. Choudary, J. Mol. Catal. A: Chem., 2006, 256, 273 CrossRef.
- Y.-B. Zhao, Z.-Y. Yan and Y.-M. Liang, Tetrahedron Lett., 2006, 47, 1545 CrossRef CAS.
- T. Miao and L. Wang, Synthesis, 2008, 363 CAS.
- H. Sharghi, M. H. Beyzavi, A. Safavi, M. M. Doroodmand and R. Khalifeh, Adv. Synth. Catal., 2009, 351, 207 CrossRef CAS.
- D. Kumar, G. Patel and V. Buchi Reddy, Synlett, 2009, 399 CrossRef CAS.
- V. Bénéteau, A. Olmos, T. Boningari, J. Sommer and P. Pale, Tetrahedron Lett., 2010, 51, 3673 CrossRef.
- J. Yan and L. Wang, Synthesis, 2010, 447 CAS.
- T. Shamim and S. Paul, Catal. Lett., 2010, 136, 260 CrossRef CAS.
- L. Ackermann and H. K. Potukuchi, Org. Biomol. Chem., 2010, 8, 4503 CAS.
- B. Gerard, J. Ryan, A. B. Beeler and J. A. Porco Jr, Tetrahedron, 2006, 62, 6405 CrossRef CAS.
- X. Meng, X. Xu, T. Gao and B. Chen, Eur. J. Org. Chem., 2010, 5409 CrossRef CAS.
- OriginLab Origin 8.6, OriginLab, Northampton, MA, 2011 Search PubMed.
- J. Deng, Y.-M. Wu and Q.-Y. Chen, Synthesis, 2005, 2730 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of nanoparticle synthesis and characterization and triazole syntheses. See DOI: 10.1039/c2nr32570e |
| This journal is © The Royal Society of Chemistry 2013 |