Factors affecting T cell responses induced by fully synthetic glyco-gold-nanoparticles†
Silvia
Fallarini
a,
Tiziana
Paoletti
a,
Carolina Orsi
Battaglini
a,
Paolo
Ronchi
b,
Luigi
Lay
b,
Renato
Bonomi
c,
Satadru
Jha
c,
Fabrizio
Mancin
c,
Paolo
Scrimin
*c and
Grazia
Lombardi
*c
aDepartment of Pharmaceutical Sciences, University of “Piemonte Orientale Amedeo Avogadro”, Via Bovio 6, 28100 Novara, Italy. E-mail: lombardi@pharm.unipmn.it; Fax: +39 0321 375821; Tel: +39 0321 375824
bDepartment of Organic and Industrial Chemistry, CISI, ISTM-CNR, University of Milano, Via Venezian 21, 20133 Milano, Italy
cDepartment of Chemical Sciences and ITM-CNR, University of Padova, Via Marzolo 1, 35131 Padova, Italy. E-mail: paolo.scrimin@unipd.it; Fax: +39 049 8275829; Tel: +39 049 8275276
First published on 8th November 2012
Abstract
We have synthesized and characterized nearly monodisperse and highly pure gold nanoparticles (2 and 5 nm) coated with non-immunoactive mono- and disaccharides, modelled after the capsular polysaccharide of serogroup A of the Neisseria meningitidis bacterium. We have used them to test their ability to induce immune cell responses as a consequence of their multivalency. The results indicate that they are indeed immunoactive and that immunoactivity is strongly dependent on size, and larger, 5 nm nanoparticles perform far better than smaller, 2 nm ones. Immune response (activation of macrophages) initiates with the whole nanoparticle recognition by the surface of antigen-presenting cells, independent of the saccharide oligomerization (or charge) on the nanoparticle surface. The induction of T cell proliferation and the increase of IL-2 levels, a consequence of the expression of MHC II involved in antigen presentation, require the presence of a disaccharide on the nanoparticle, not just a monosaccharide. A possible explanation is that, at this stage, the saccharides are detached from the gold surface. These results may provide leads for designing new saccharide-based, nanoparticle-conjugate vaccines.
Introduction
Monolayer-protected gold nanoparticles (Au-NPs) share with several nanosystems (liposomes, dendrimers, for instance) the property of multivalency1,2 since they present a collection of functional groups on their periphery. It is a widely accepted paradigm that multivalent systems show enhanced binding ability against multivalent counterparts.1,3 This is the case for systems governing cell–cell, protein–protein, and protein–cell interactions, for instance.4 Multivalency of Au-NPs has been exploited for many applications5,6 spanning, inter alia, enzyme mimicry,7–9 protein sensing and recognition,10–12 and gene regulation.13 For drug or gene delivery, the ability to release their payload, once internalized into cells,14,15 in a process mediated by cytoplasmic thiol compounds is an advantage of Au-NPs over other polymeric nanosystems where the cargo is covalently, and hence very strongly, bound to the polymer. One of the major problems related to the use of Au-NPs for biomedical applications is the fact that immune cells in the bloodstream and in tissues have the ability to entrap and eliminate Au-NPs.16–19 A nanoparticle's interaction with plasma proteins (opsonins) and blood components (via hemolysis, thrombogenicity and complement activation) may influence uptake and clearance and hence potentially affect distribution and delivery to the intended target sites. The conventional way to overcome this problem is to make Au-NPs stealthy to the immune system, typically by functionalization on their surface with polyethers.20,21In addition to their primary role in pathogen clearance, blood components like the complement system also promote humoral and cell-mediated immunity. Local activation of complement by nanoparticles administered via subcutaneous and intradermal routes may indeed benefit vaccine efficacy. In this paper we address the question of the interaction of saccharide-functionalized Au-NPs in vitro with blood components like monocytes with the aim to evidence their ability to induce immune responses and the factors (size, saccharide oligomerization) influencing it. Successful systems may find applications as synthetic vaccines. The design of new vaccines requires the mimicry of key pathogen features like size, shape and surface molecule organization without, of course, the ability to cause diseases.22 For these reasons nanosystems like dendrimers and polymers have been used for this purpose. Properly functionalized gold nanoparticles23,24 appear to be suitable choices for antigens because antigen processing is facilitated if antigens are particulate and have a repetitive surface organization, which increases phagocytosis and the ability, as said above, to activate complement and recruit other molecules of the innate humoral immune system. Peptide-functionalized Au-NPs have shown the ability to enhance immune responses in cells,25,26 and gold nanoparticles bearing tumor-associated glycopeptide antigens have recently been reported as promising potential cancer vaccines.27
We have recently shown28 that Au-NPs functionalized with oligosaccharides (Glyco-Au-NPs) designed to mimic the polysaccharide capsule that coats serogroup A of the Neisseria meningitidis bacterium29 (MenA) bind very strongly to polyclonal human antibodies against MenA. At the same nominal concentration of saccharides the nanoparticles showed a more than three orders of magnitude higher binding affinity than their counterparts not bound to the gold cluster: a remarkable result, fully consistent with the multivalent nature of the system. Our data showed also that the strength of the interaction with the antibodies was not dependent on the oligomerization of the saccharide used for the passivation of the gold cluster as mono-, di- and tri-saccharide-functionalized Au-NPs performed quite similarly. In contrast, the affinity constants of nanoparticle-unbound saccharides were oligomerization-dependent with the trisaccharide performing best. This suggests that the antibody when interacting with the Au-NPs can hardly differentiate a surface made up by a mono-, di- or tri-saccharide. Synthetically this is quite relevant because a monosaccharide is much more accessible than an oligomer, even short as a trisaccharide.
A complex cascade of events is required for inducing immune responses. These events include adaptive events that take place on the surface of antigen-presenting cells (APCs), which display foreign antigens coupled with MHC to a T cell receptor complex for effective T cell priming and B/T cell activation. APCs collaborate also with the innate system by cytokines, which steers the differentiation of T helper (Th) cells into Th1 or Th2 subsets.30 Accordingly, in addressing the ability of our Glyco-Au-NPs to elicit immune cell responses, we have evaluated: (a) the activation of APCs; (b) the induction of effector functions in the same cells; and (c) the ability to stimulate T cells. The role of the size and of the oligomerization of the saccharide present on the surface of the nanoparticles in the different processes studied has been addressed by considering Glyco-Au-NPs of two different sizes (2 and 5 nm core diameter) passivated with thiols functionalized with a mono- and a disaccharide. Liposome-, polymer- or dendrimer-based vaccines are typically of a size of at least 10 nm.22 We used Glyco-Au-NPs with a maximum overall diameter of ca. 9 nm corresponding to a gold core of 5 nm because a 5.5 nm diameter of the gold core is considered as the threshold limit to allow renal clearance in gold-based nanoparticles31 and, hence, to avoid accumulation of the metal in the organism: a critical issue for possible in vivo applications. Thus our size limit was the result of considering both renal clearance and maximization of APC interactions for possible future vaccine development. In spite of the fact that the two sizes studied are not much different, the results indicate that the behavior of the Glyco-Au-NP is quite diverse. Our selected pathogen has been the Neisseria meningitidis bacterium to compare our results with the previous ones28 concerning polyclonal antibody affinities but also for the high relevance of this bacterium that causes the highest incidence of epidemics in Saharan and sub-Saharan countries of Africa (the so-called “African meningitis belt”).32 Furthermore, bacterial meningitis remains a global problem worldwide, accounting for an estimated annual number of 1.2 million cases with at least 5–10% case fatality in industrialized countries and 20% in the developing world.33 It is thus not surprising that the scientific interest in new approaches for effective vaccine development remains very high.34–36
Results and discussion
Synthesis and characterization of Au nanoparticles
We have prepared following our protocol,37 which is a modification adapted for water-soluble Au-NPs of the two-phase Brust synthesis38 (Fig. 1), 2 and 5 nm nanoparticles‡ functionalized with monomeric and dimeric saccharides29 (Glyco-Au-NP 1–2 and 3–4, respectively). Since incorporation of a triethylenglycol (TEG) unit in the monolayer provides to Au-NPs water solubility, prevents nanoparticle aggregation that occurs at the ionic strength of the experiments with cells, and non-specific interactions with biomolecules,20 TEG-Au-NPs 5 and 6 were used as Au-NP controls in all experiments, while saccharides 8 and 9 were used as monovalent comparison molecules. Glyco-Au-NP 7 is equivalent to Glyco-Au-NP 4 as for dimension and saccharide composition. It differs from 4 for the presence of 5% of fluorescein as a fluorescent tag and was prepared to monitor cell internalization of the nanoparticles. In disaccharide 9 and that present in gold nanoparticles 3 and 4, the native phosphodiester bridge connecting the glycosidic units of MenA has been substituted with a phosphonate29 as a hydrolytically more stable moiety to prevent early degradation with loss of the Glyco-Au-NPs properties. The Au-NPs were analyzed, for their size, by transmission electron microscopy (TEM) and dynamic light scattering (DLS) (see Table 1 for relevant physical properties) and, for the purity of the monolayer, by 1H-NMR and thermogravimetry (see ESI†). While TEM provides the size of the gold core, DLS gives the hydrodynamic diameter of the nanoparticle (gold core and surrounding monolayer). DLS is particularly sensitive to the presence of aggregated nanoparticles because of the strong increase of scattered intensity with size. All our samples showed no evidence of aggregation, thus suggesting that the passivation with the saccharides makes them very soluble and not prone to interacting reciprocally. The thiol/particle ratio that gives information on the multivalency of the Glyco-Au-NPs has been obtained from the thermogravimetric analysis and size data as reported.41 Glyco-Au-NPs with a monolayer composed of a monosaccharide (1 and 2) are neutral, while those composed of a disaccharide (3 and 4) are anionic, as proven by the respective Z potentials (Table 1).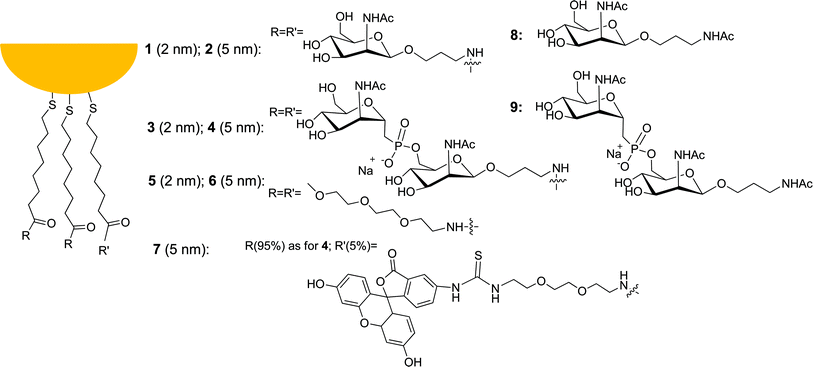 | ||
| Fig. 1 Gold nanoparticles and thiolated saccharides described in this work. | ||
| Au-NP | Size, nm | Thiols/particlec | Z potential, mV | |
|---|---|---|---|---|
| TEMa | DLSb | |||
| a Diameter of the gold core. b Hydrodynamic diameter of the entire nanoparticle. c Mean value estimated from the thermogravimetric analysis and the size of the gold core (see ref. 39 for details). | ||||
| 1 | 1.8 ± 0.5 | 6.2 ± 1.8 | 80 | +3 ± 4 |
| 2 | 4.9 ± 0.9 | 9.3 ± 2.2 | 480 | +4 ± 4 |
| 3 | 1.8 ± 0.4 | 6.5 ± 2.0 | 70 | −29 ± 3 |
| 4 | 5.0 ± 0.8 | 9.6 ± 2.1 | 450 | −30 ± 4 |
| 5 | 1.9 ± 0.4 | n.d. | n.d. | n.d. |
| 6 | 4.8 ± 1.0 | n.d. | n.d. | n.d. |
Macrophage activation
Preliminarily, we ruled out the possibility that immune cell responses were due to the presence of endotoxin contaminations, such as lypopolysaccharide (LPS), introduced into the system through chemicals, materials, or equipment. Limulus amebocyte lysate endochrome assays were performed in four different batches of each compound and the concentration of LPS was measured. LPS concentrations were always below the value of 0.01 ng ml−1, sufficient to stimulate immune cell responses,42 suggesting that the compounds under testing were LPS-free and biologically evaluable (data not shown). In addition, to exclude the possibility that the compounds are cytotoxic, cell viability was evaluated by a Trypan blue exclusion test after 24 h cell exposure to each compound. All compounds showed no toxicity at all concentrations tested (data not shown).Macrophages are potent immune cells that provide an immediate response against pathogens and may act as APCs to activate adaptive immune responses. Exposure to pathogens can induce a rapid activation of macrophages, which stop proliferation and acquire effector cell functions.43
We, thus, analyzed in vitro the effects of nanoparticles 1–6 and saccharides 8 and 9 on macrophage proliferation using RAW 264.7 cells, a widely used cellular model that accurately replicates APC biology, such as antigen uptake, processing, and presentation.44 Cells were rendered quiescent prior to each assay by 18 h serum deprivation. After 24 h exposure to increasing concentrations (1 × 10−2 to 1 × 102 μM)§ of each compound, cell proliferation was significantly inhibited (−55 ± 1% over compound-untreated cells as controls) only by 5 nm Glyco-Au-NPs 2 and 4 at a 1 × 102 μM concentration (Fig. 2). To demonstrate that the inhibition of macrophage proliferation is specific and not related to a reduction in cell viability, the Trypan blue exclusion test was performed at the end of cell exposure to each compound. The results indicate that the observed decrease in cell proliferation is not due to cytotoxicity as cell viability remains >95%. Apoptosis and necrosis were also analyzed under epifluorescence microscopy in cells treated with 1 × 102 μM Glyco-Au-NPs 1–4 for 24 h. Cells treated with the non-conjugated saccharides 8 and 9 or the TEG-functionalized Au-NPs 5 and 6 devoid of the saccharides were used as a comparison. Actinomicyn D, a cytotoxic inducer of apoptosis,45 was used as a positive control. None of the tested Glyco-Au-NPs significantly affected cell death, indicating that the inhibition of cell proliferation occurs through a specific blockage of the cell cycle progression (ESI, Fig. S6†).
 | ||
| Fig. 2 Concentration–response curves of the tested compounds on macrophage proliferation (RAW 264.7 cells) expressed as the percentage over compound-untreated cells (CTRL). Compound identification number as in Fig. 1. The data represent means ± SEM of five experiments run in triplicate. *P < 0.05; **P < 0.01 versus compound-untreated cells (CTRL). | ||
This result suggests that the conjugation to Au-NPs confers to the saccharides the ability to activate macrophages and that this property is dependent on the Au-NP size, with 5 nm nanoparticles giving comparable results to those obtained when the polysaccharide bacterium capsule (MenA) is used as a natural antigen. Moreover, there is little difference in the activity of 5 nm Glyco-Au-NPs, regardless of whether they are functionalized with a monomeric or a dimeric saccharide (compare 2vs.4). Thus, macrophage activation is dependent on size, but very little on oligomerization (or charge) of the saccharide on the Au-NP surface.
Since the blockage of macrophage cycle progression correlates with a cell phenotype differentiation from macrophage- to dendritic-like cells, we then studied the effects of the nanoparticles on RAW 264.7 cell morphology. As shown in Fig. 3, native cells (CTRL) had a macrophage-like morphology with small nuclei and finely granulated cytoplasm. Cells appeared elongated with two short cytoplasmic processes. A morphological transformation of RAW 264.7 cells from macrophage-like cells into dendritic-like cells was clearly observed after cell treatment with Glyco-Au-NPs (1 × 102 μM). Dendritic morphology was characterized by multiple prominent cytoplasmic processes. Glyco-Au-NP-treated cells acquired larger nuclei, prominent nucleoli, and increased cytoplasmic granularities. This morphological transformation is particularly evident in cells treated with 5 nm Glyco-Au-NPs, as the percentage of dendritic-like cells was almost twice that observed with the smaller (2 nm) Glyco-Au-NPs (Table 2).
 | ||
| Fig. 3 Glyco-Au-NPs induced macrophage phenotype differentiation. The pictures show the effects of the tested compounds (1 × 102 μM, in the case of the nanoparticles it refers to the organic component) on macrophage (RAW 264.7 cells) morphology after 24 h exposure (scale bar = 30 nm). The inset for 4 represents a 2.5-fold magnification. | ||
| Sample | Dendritic-like cells, % | MHC II expression, % |
|---|---|---|
| CTRL | 6 ± 0.5 | 5.3 |
| MenA | 73 ± 3 | 29.8 |
| 1 | 37 ± 2 | 18.4 |
| 2 | 70 ± 2 | 35.5 |
| 3 | 32 ± 1.5 | 9.0 |
| 4 | 68 ± 2 | 29.3 |
| 5 | 6 ± 0.8 | 5.9 |
| 6 | 4 ± 0.3 | 6.8 |
| 8 | 10 ± 1.1 | 5.7 |
| 9 | 9 ± 1.5 | 6.8 |
At the same concentration (1 × 102 μM) Glyco-Au-NPs elicited similar responses to that observed with MenA. No significant effects were observed in the cells treated either with the non-conjugated saccharides 8 and 9 or the TEG-functionalized Au-NPs 5 and 6. This result indicates that the functionalization, and not just the Au-NPs per se, is at the origin of the observed effects.
The blockage of macrophage proliferation, together with the acquisition of a dendritic-like phenotype, clearly indicates that the Glyco-Au-NPs are immunoactive.
The next step was to investigate whether Glyco-Au-NPs activate effector functions in the same cells. Fig. 4 and 5 show the effects induced by each compound on the production of typical pro-inflammatory mediators (e.g. ROS and TNF-α). The data demonstrate that, after processing Glyco-Au-NPs 1–4 as antigenic molecules, macrophages become active APCs (although to different extents) that attract responder cells to the site of the inflammation and start adaptive immune cell responses.
 | ||
| Fig. 4 Effects of the different compounds on intracellular reactive oxygen species (ROS) production: (a) monosaccharide-functionalized Glyco-Au-NPs 1 and 2 and unbound monosaccharide 8; (b) disaccharide-functionalized Glyco-Au-NPs 3 and 4 and unbound disaccharide 9. The percentage of ROS was measured by incubating the cells with the fluorescent probe DCFH-DA. RAW 264.7 cells were seeded (0.5 × 105 cells per well) in 24-well plates and treated with increasing concentrations (10−2 to 102 μM) of each compound for 1 h. TEG-functionalized Au-NPs 5 and 6 were used as Au-NP controls while compound-untreated cells (CTRL) as negative controls. *P < 0.05; **P < 0.01 vs. CTRL. | ||
 | ||
| Fig. 5 Effects of the different compounds on production of pro-inflammatory mediator tumor necrosis factor (TNF)-α: (a) monosaccharide-functionalized Glyco-Au-NPs 1 and 2 and unbound monosaccharide 8; (b) disaccharide-functionalized Glyco-Au-NPs 3 and 4 and unbound disaccharide 9. RAW 264.7 cells were seeded at a density of 0.6 × 105 cells per well in 24-well plates and treated with increasing concentrations (10−2 to 102 μM) of each compound for 24 h. TEG-functionalized Au-NPs 5 and 6 were used as Au-NP controls while compound-untreated cells (CTRL) as negative controls. *P < 0.05; **P < 0.01 vs. CTRL. | ||
The membrane expression of class II major histocompatibility complex molecules (MHC II), which represents the first marker of a mature APC, was then examined by flow cytometry in RAW 264.7 cells treated with the different systems (1 × 102 μM). No change in the expression levels of MHC II was observed in cells treated with non-conjugated saccharides 8 and 9 or TEG-functionalized Au-NPs 5 and 6, while a 3- and a 6-fold increase in the MHC II expression was measured in cells treated with 2 or 5 nm Glyco-Au-NPs, respectively (Table 2 and Fig. S7†). It is noteworthy that 5 nm Glyco-Au-NPs exert identical (or even larger) MHC II expression levels to MenA CPS. Once again, nanoparticles are differentiated by their size and not by the presence of a monomeric or dimeric saccharide (or charge) on their surface.
Cellular uptake of 5 nm Glyco-Au-NPs 7 by RAW 264.7 cells was next determined by immunofluorescence and confocal microscopy analysis. As shown in Fig. 6, fluorescein-functionalized Glyco-Au-NPs 7 are internalized (green signal) by APC with no absorption onto the cell surface (red signal). The overall data confirm that the Glyco-Au-NPs enter the cells and start expressing molecules (MHC II) involved in antigen presentation.
 | ||
| Fig. 6 Laser confocal fluorescence microscopy showing the intracellular localization of 5 nm fluorescent Glyco-Au-NPs 7 (green) after cell treatment (3 h, panel a). In (b) Alexa Fluor 647-conjugate anti-CD14 monoclonal antibodies (red staining) were used as surface cellular markers. Images are representative of six different cell preparations with similar results. | ||
Glyco-Au-NPs induce T cell responses
To better explore the potentiality of these Glyco-Au-NPs as leads for new vaccine design, their ability to induce T cell responses was determined in human homologous monocytes/T cell co-cultures.40 The generated T cell responses are essential for developing a functional T cell memory following vaccination. Thus, human monocytes were treated with increasing concentrations (1 × 10−2 to 1 × 102 μM) of each compound, and, after six days of cell co-cultures, T cell proliferation (Fig. 7 and S8†) and IL-2 levels (Table 3 and Fig. S9†) were determined. Non-conjugated saccharides 8 and 9, TEG-functionalized Au-NPs 5 and 6, as well as Glyco-Au-NPs 1 and 2 (i.e. the nanoparticles functionalized with the monosaccharide), failed to induce significant T cell proliferation at all concentrations tested, while both Glyco-Au-NPs 3 and 4 were effective. Glyco-Au-NP 4 was more potent than 3; its maximal effect was a 13-fold increase at 1 × 101 μM in comparison with controls (native cells). Consistently, the IL-2 levels in the cell culture medium (Table 3) significantly increased after Glyco-Au-NP 4 cell treatment (almost a 12-fold increase at 1 × 101 μM vs. controls). The overall data clearly demonstrate that Glyco-Au-NPs 4 are the most effective nanosystems in activating T cell responses. | ||
| Fig. 7 Effects of the different compounds on T cell proliferation: (a) monosaccharide-functionalized Glyco-Au-NPs 1 and 2 and unbound monosaccharide 8; (b) disaccharide-functionalized Glyco-Au-NPs 3 and 4 and unbound disaccharide 9. T-cell proliferation was analyzed by FACS, and expressed as mean percentages ± SEM of proliferated T cells. Cells treated with 1 mg ml−1 phytohaemagglutinin (PHA), a widely used mitogen able to trigger cell division in T-lymphocytes, were considered as positive controls, while TEG-functionalized Au-NPs 5 and 6 were used as Au-NPs controls, and compound-untreated cells (CTRL) as negative controls. *P < 0.05; **P < 0.01 vs. CTRL. | ||
The picture that emerges from our results is the following (see Fig. 8a): (i) Glyco-Au-NPs activate macrophages leading to the blockage of their cycle progression and inducing their differentiation into dendritic-like cells; (ii) they induce on macrophages the expression of MHC II involved in antigen presentation to T lymphocytes; (iii) following their internalization and degradation, the constituent saccharides are loaded onto MHC II and are transported to the APC surface for presentation; (iv) 5 nm Glyco-Au-NPs, functionalized with disaccharide, significantly induce T cell proliferation and IL-2 release. Processes (i) and (ii) require intact nanoparticles taking advantage of their multivalency and proceed via Glyco-Au-NPs surface recognition. Processes (iii) and (iv) involve very likely Glyco-Au-NP degradation into APCs, a process that implies chemical modifications of the nanoparticles.
 | ||
| Fig. 8 (a) Cartoon representation of the effect of the Glyco-Au-NPs on macrophages and consequent T cell activation (see text for description of steps i–iv); (b) monosaccharide- (circles) and disaccharide- (squares) functionalized nanoparticle effects on APC proliferation (blue) and MHC II expression (red) as a function of size; the lines were drawn for facilitating the connection of the data points; (c) monosaccharide- (circles) and disaccharide- (squares) functionalized nanoparticle effects on T cell proliferation (red) and IL-2 expression (blue) as a function of size; the lines were drawn for facilitating the connection of the data points. | ||
As in the case of the interaction with the antibodies,28 the efficiency of the nanosystems is dependent on their size (5 nm are better than 2 nm) but very little on the oligomerization (and charge) of the saccharide on the surface (the effect is very similar for mono- and di-saccharide-functionalized nanoparticles, which are neutral and anionic, respectively), see Fig. 8b. The occurrence of a surface-recognition process controlling steps (i) and (ii) requires a packed surface, a situation more easily obtained with a less curved surface such as that of the larger, 5 nm, nanoparticles as compared to the smaller, 2 nm, ones. It is interesting to point out that, in contrast, when a substrate needs to penetrate into the organic monolayer, the opposite situation occurs, i.e. smaller Au-NPs perform better than larger ones, as we have recently shown.46 Processes (iii) and (iv) require nanoparticle degradation and, consistently, disaccharide-functionalized nanoparticles are required in this case, see Fig. 8c. Likely, within the cell, the saccharide-functionalized thiols are detached from the surface and, at this stage, being monomeric or dimeric becomes relevant for T cell activation. It has been shown that the thiolic units can be displaced from the gold surface by glutathione.15,47 This mode of release utilizes the dramatic differential between extra- and intracellular glutathione levels. It is possible that this is what happens in this case, too. This is supported by the increased fluorescence intensity of internalized nanoparticles 7 (see Fig. 6), reasonably a consequence of the release of the fluorophore from the Au-NP surface.14 The fluorescence intensity of fluorescein when bound to the gold surface is very small due to its quenching14 by the gold nanoparticle core while it increases when released into the solution. In any case, T cells appear to recognize dimeric, but not monomeric saccharide-functionalized gold nanoparticles: whether this is due to differences in their intracellular processing, as we suggest, or antigenic determinants is a matter of plausible speculation and this point should be clarified in future work.
Conclusions
In conclusion, we have demonstrated that the conjugation of small size, fully synthetic, non-immunogenic saccharides to Au-NPs confers upon them the ability to induce immune cell responses. The importance of multivalency to increase the selectivity and the strength of binding but also to induce the immunoactivity is clearly evidenced by the ineffectiveness of non-conjugated saccharides, which bind to MenA antibodies but are not immunoactive. Our data show how a complex interplay of features is required with saccharide-functionalized gold nanoparticles for eliciting the complex cascade of events that induce immune responses which eventually may result in immunization. On one hand, for maximizing the cell interaction, size is a key requisite, in full agreement with the reported observation that large size nanoparticles perform better in interacting with cell surfaces48 and mediating cell responses.49 On the other hand, for T cell-mediated responses a structured saccharide (in our case a disaccharide) is required. A plausible explanation is that, at this stage, the nanosystem has been in part degraded and the covering functional thiols have been detached from the gold core for presentation by MHC II. The need to comply with both requisites makes 5 nm Glyco-Au-NPs 4 functionalized with a disaccharide the best of the studied nanosystems for inducing immune responses in cells.The present results may provide significant leads for the design of nanoparticles for developing nanoparticle-based vaccines. The next obvious step will be to test Glyco-Au-NPs in vivo to determine their ability to induce immunization,22 a feasible development in view of the claimed non-toxicity of gold nanoparticles of the size discussed here.50–53
Experimental section
Synthesis of the saccharides
Synthesis of the non-conjugated saccharides 8 and 9. The non-conjugated saccharides 8 and 9 were synthesized from their corresponding 1-O-aminopropyl glycoside precursors as follows.Monosaccharide 8: the aminopropyl glycoside (29 mg, 0.104 mmol) was dissolved in dry pyridine (1.0 ml) under Ar atmosphere, and acetic anhydride (0.1 ml, 1.05 mmol); thereafter, a catalytic amount of N,N-dimethylamino pyridine was added. The reaction mixture was stirred at room temperature for 3 h, and then concentrated in vacuum. The crude residue was dissolved in dry MeOH (3.0 ml) under Ar atmosphere, and then a 0.1 M solution of MeONa in dry MeOH (0.2 ml) was added dropwise. The reaction mixture was stirred at room temperature for 1 h, then neutralised with Dowex 50W X8 resin (H+ form), filtered, and concentrated in vacuum. Flash chromatography (CHCl3/MeOH 8/2) afforded compound 8 (26 mg, 78%).
Disaccharide 9: the aminopropyl glycoside (20 mg, 0.034 mmol) was treated with acetic anhydride (0.038 ml, 0.34 mmol) and N,N-dimethylamino pyridine, as described for 8. The crude residue was purified by flash chromatography (CHCl3/MeOH from 9/1 to 7/3) providing the fully O- and N-acetylated derivative (22 mg, 77%). This compound was dissolved in dry MeOH (1.0 ml) under Ar atmosphere, and then a 0.1 M solution of MeONa in dry MeOH (0.026 ml) was added. After stirring at room temperature for 3 h, the reaction mixture was neutralised with aq. HCl 5% and the solution concentrated in vacuum. The residue was dissolved in H2O, first eluted through a column filled with Dowex 50W X8 resin (H+ form), and then through a column filled with the same resin in Na+ form. The eluate was lyophilized to afford 9 (15 mg, 91%).
Nanoparticle synthesis
The monomeric and dimeric saccharides used for nanoparticle passivation as well as Glyco-Au-NPs 1–4 were synthesized as previously described.27,28 The triethyleneglycol (TEG)-functionalized Au-NPs (TEG-Au-NPs) 5 and 6 were synthesized as reported.36Fluorescein-tagged GlycoNPs 7: the nanoparticles used for cell internalization studies were prepared following the same procedure, but using a mixture of two thiols, the disaccharide-functionalized thiol and the fluorescein-tagged thiol. The scheme for the preparation of the Fluorescein-tagged thiol (iv, see ESI) is reported in the ESI.† To a solution of TOABr (94 mg, 0.171 mmol) in 68 ml toluene, an aqueous solution of HAuCl4·H2O (27 mg, 0.0685 mmol) in 1.0 ml Milli-Q water was added and shaken vigorously until the aqueous layer became transparent. The reddish-orange organic layer was carefully separated from the aqueous solution and transferred in a flask with a magnetic bar. Dioctylamine (21 mg, 0.0856 mmol) was added into the solution under vigorous stirring at room temperature for 3 h. A solution of NaBH4 (26 mg, 0.685 mmol) in 1.0 ml Milli-Q H2O was added drop by drop to the organic layer under vigorous stirring (1 h). The color of the solution immediately changed to purple. The organic layer was again carefully separated and transferred into a flask with a magnetic bar. The freshly prepared mixture of disaccharide-functionalized thiol (12 mg, 0.0162 mmol) and iv (0.70 mg, 0.0010 mmol) in DMF (1 ml) was added and allowed to stir until the toluene layer became colorless. A red precipitate was obtained after centrifugation followed by decantation of the organic layer. After washing with Et2O (3 × 30 ml) and EtOAc (2 × 30 ml) the solid residue was dissolved with the minimum amount of water. On slow addition of THF a red material precipitated from the aqueous layer and the precipitate was collected after centrifugation followed by removal of solvents. The same procedure was repeated to afford pure 7 (17 mg).
Nanoparticle characterization
TEM images were recorded on a copper grid using a Jeol 300PX instrument. TGA analyses were recorded by exploring a 30–800 °C interval using a temperature ramp of 20 °C min−1 under nitrogen using a SDT 2960 model TA instrument. Dynamic light scattering (DLS) and Z potential measurements were carried out with a Malvern Zetasizer Nano S instrument thermostatted at 25 °C, using 0.1 mg ml−1 solutions of nanoparticles in water. NMR spectra were recorded on a Bruker AC-300 or AC-250 spectrometer. Diffusion-ordered 1H NMR spectra were recorded using the low eddy currents distortion bipolar gradients (LEDbp) sequence, which allows differentiation of molecules based on their diffusion coefficients. This provides not only an unequivocal confirmation that the thiols are bound to the Au-NPs surface, but also a way to assess the purity of the samples. Further determinations of the organic content of each preparation were performed both by TGA and by analyzing the soluble material using 1H-NMR after treatment with I2. Experimental results are in the ESI.†Competitive ELISA
Ninety-six-well flat-bottomed plates were incubated overnight at 4 °C with a mixture of Men-A CPS (1 mg ml−1) and methylated human serum albumin (mHSA; 1 mg ml−1) (Men-A CPS and mHSA were generous gifts from Novartis Farma SpA, Siena, Italy). A solution of 5% fetal bovine serum (FBS) in phosphate buffered saline (PBS) supplemented with Brij-35 (0.1%) and sodium azide (0.05%) was added to each well to block unspecific binding sites. The plates were incubated overnight at 4 °C with a solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :400) of mouse anti-MenA, used as reference serum. This serum was obtained from mice vaccinated with an anti-meningococcal A + C + W135 + Y polysaccharide vaccine. When non-conjugated saccharides 8 and 9 or Glyco-Au-NPs 1–6 were tested, they were added to each well just before the addition of the reference serum. The plates were then incubated with alkaline phosphatase-conjugate anti-mouse, stained with p-nitrophenyl phosphate, and the absorbance was measured at 405 nm with an Ultramark microplate reader. Experiments were repeated in triplicate. Control experiments to verify unspecific binding were performed by coating the ELISA plates with colominic acid from Escherichia coli.
:400) of mouse anti-MenA, used as reference serum. This serum was obtained from mice vaccinated with an anti-meningococcal A + C + W135 + Y polysaccharide vaccine. When non-conjugated saccharides 8 and 9 or Glyco-Au-NPs 1–6 were tested, they were added to each well just before the addition of the reference serum. The plates were then incubated with alkaline phosphatase-conjugate anti-mouse, stained with p-nitrophenyl phosphate, and the absorbance was measured at 405 nm with an Ultramark microplate reader. Experiments were repeated in triplicate. Control experiments to verify unspecific binding were performed by coating the ELISA plates with colominic acid from Escherichia coli.
Cell cultures
The mouse monocyte/macrophage cell line RAW 264.7 was cultured in high glucose Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% heat-inactivated FBS, 2 mM L-glutamine, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin in a 37 °C incubator with 5% CO2. Cells were grown in 75 cm2 flasks and sub-cultured by scraping when they reached 90% confluence with a 1:5 or 1:10 ratio in fresh medium. Peripheral blood mononuclear cells (PBMC) were isolated by gradient centrifugation onto Ficoll-Hystopaque of venous blood obtained from healthy volunteers after their informed consent.37 PBMC were suspended in RPMI-1640 medium containing 10% heat inactivated FBS, 100 μg ml−1 kanamycin, 1 mM sodium pyruvate, 2 mM L-glutamine, 1% MEM amino acid solution and 0.01 mM 2-mercaptoethanol for immediate use. Human monocytes were isolated from heparinised venous blood of healthy volunteers, and their purity was assessed with the pan-leukocyte anti-CD45 (HLE-1) and the anti-CD14 (Leu-M3) monoclonal Abs, as previously described.37 Cells were used on the day of isolation under endotoxin-free conditions. Before each experiment, the viable cell count was assessed (always >95%) by Trypan blue staining and cells were seeded, in complete fresh medium, at different densities 24 h prior to compound treatments. Compound-untreated cells were considered as negative controls, while cells treated with Men-A (1 × 102 μg ml−1) were considered as positive controls. Monocytes and PBMC used for co-culture experiments were always isolated from the same healthy donor.
Cell proliferation
Proliferation was analyzed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, as previously described.38 Briefly, RAW 264.7 cells were seeded (0.5 × 105 cells per well) in 24-well culture plates and treated with increasing concentrations (1 × 10−2 to 1 × 102 μM) of compounds 1–8 for 24 h at 37 °C in a 5% CO2 humidified incubator. Absorbance was measured at 570–630 nm using an Ultramark microplate reader. The percentage of cell proliferation was calculated as (x × 100)/y, where x and y are the absorbance read in compound-treated and compound-untreated cells, respectively.Annexin V and propidium iodide assays
The extent of apoptosis and necrosis was determined by using annexin V-FITC and propidium iodide (PI) staining, respectively. RAW 264.7 cells were seeded (1 × 104 cells per well) in 24-well culture plates and treated with compounds 1–8 (1 × 102 μM) for 24 h at 37 °C in a 5% CO2 humidified incubator. After treatment with test compounds, culture media were removed, cells washed twice with annexin binding buffer (10 mM Hepes sodium salt, 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, pH 7.4), and incubated in binding buffer containing annexin V-FITC (1 μg ml−1) and PI (20 μg ml−1) for 15 min at 37 °C in the dark. After two washes with binding buffer, cells were analyzed under epifluorescence microscopy. For each sample, apoptotic and necrotic cells were counted in six random high-power fields. The relative apoptotic and necrotic cells were expressed as the percentage of the total cells in each sample. Actinomycin D (0.5 mg ml−1) was included as a positive control of apoptosis.Macrophage morphology assessment
RAW 264.7 cells were seeded (1 × 105 cells per well) in 6-well culture plates and treated with compounds 1–8 (1 × 102 μg ml−1) for 24 h at 37 °C in a 5% CO2 humidified incubator. Following incubation, cells were washed once with PBS and analyzed under optical microscope. Six different fields were observed for control cells (CTRL; compound-un-treated cells), as well as for cells treated with different compounds (1–6 and 8, 9), and used for counting cells with normal or dendritic-like morphology. Cells with dendritic-like morphology in the view field were calculated as the percentage of all cells observed.Production of reactive oxygen species
Intracellular reactive oxygen species production was measured by incubating the cells with the fluorescent probe 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA), as previously described.54 RAW 264.7 cells were seeded (0.5 × 105 cells per well) in 24-well plates and treated with increasing concentrations (10−2 to 102 μM) of each compound for 1 h. Accumulation of dichlorofluorescein was measured as an increase in the fluorescence at 525 nm, when the samples were excited at 488 nm using a microplate spectrofluorometer.Production of TNF-α
Compounds were tested for their effects on the production of pro-inflammatory mediators, such as tumor necrosis factor (TNF)-α, as previously described.54 RAW 264.7 cells were seeded at a density of 0.6 × 105 cells per well in 24-well plates and treated with increasing concentrations (10−2 to 102 μM) of each compound for 24 h. At the end of each experiment, supernatants were collected and stored until measurements.The amounts of TNF-α in cell culture medium were assayed using enzyme-linked immunosorbent assay (ELISA). The concentrations of the cytokine in the samples were determined by extrapolation from specific reference standard curves.
Glyco-Au-NP uptake by macrophages
The cellular uptake of 5 nm Glyco-Au-NPs was assessed analyzing intracellular localization of FITC-labeled 5 nm Glyco-Au-NPs (14) by confocal scanning laser microscopy. RAW 264.7 cells (1 × 104 cells per well) were allowed to adhere overnight on sterile cover slips in 24-well culture plates and pulsed (3 h) with (1 × 102 μg ml−1) FITC-labeled 5 nm Glyco-Au-NPs in complete medium. The cover slips were washed twice with ice-cold PBS and cells fixed with paraformaldehyde 3% for 15 min at 4 °C. After two additional PBS washings, cell membranes on the cover slips were labelled with ALexa Fluor 647-conjugated anti-mouse CD14 monoclonal Ab at room temperature for 1 h in the dark. After one wash with PBS, the cover slips were mounted on glass slides by SlowFade® reagent. Cover slips were maintained in the dark overnight at 4 °C and analyzed by a confocal scanning laser microscope (Leica TCS P2). Green fluorescence was observed with a 505–550 nm band-pass emission filter under 488 nm laser illumination and red fluorescence was observed with a 560 nm long-pass emission filter under 543 nm laser illuminations.Flow cytometry analysis
The acquisition of effector functions by macrophages was assessed analyzing the levels of major histocompatibility complex class II (MHC II) surface expression. RAW 264.7 cells were seeded (1 × 105 cells per well) in 6-well culture plates and treated with compounds 1–8 (1 × 102 μM) for 24 h at 37 °C in a 5% CO2 humidified incubator. Following incubation, cells were washed once with PBS, resuspended in 100 μl PBS and incubated with Alexa Fluor 647-conjugated anti-mouse I-A/I-E monoclonal Ab for 15 min at room temperature in the dark. Cells were then washed twice with PBS and analyzed by flow cytometry. Emitted fluorescence was analyzed by using FACSDiva software.T cell activation assays
Monocytes (1 × 105 cells per well), used as APCs, were pulsed with increasing concentrations (1 × 10−2 to 1 × 102 μM) of each compound 1–8 for 24 h. Responder PBMC (1 × 106 cells per ml) were labeled with 0.25 μM carboxyfluorescein succinimidyl ester (CFSE) in serum-free PBS for 30 min at 37 °C. Filtered FBS was then added to stop the reaction, and cells were washed several times with RPMI-1640. 5 × 104 CFSE-labeled responder T cells were co-cultured with antigen-pulsed or unpulsed homologous monocytes for six days at 37 °C, 5% CO2 in a humidified incubator. Phytohemagglutinin (PHA) (1 μg ml−1) was used as a positive control of PBMC proliferation. After six days of co-culture, cells were harvested, washed three times with warmed PBS, resuspended in fresh PBS, and analyzed by flow cytometry. Cell proliferation was measured as reduction of CFSE fluorescence intensity due to cell division by using FACSDiva software.Measurement of IL-2 production
Monocytes (1 × 105 cells per well), used as APCs, were pulsed with increasing concentrations (1 × 10−2 to 1 × 102 μM) of compounds 1–8 for 24 h. Responder PBMC (5 × 104 cells per well) were co-cultured with antigen-pulsed or unpulsed homologous monocytes for six days at 37 °C, 5% CO2 in a humidified incubator. PHA (1 μg ml−1) was added in positive control wells. After six days of co-culture, the supernatants were collected and stored at −20 °C until assays. The levels of IL-2 were measured by ELISA using a mouse anti-human IL-2 monoclonal Ab and quantified with a recombinant IL-2 standard. Absorbance was measured at 450 nm using an Ultramark microplate reader, and the concentration of IL-2 in each sample was determined by extrapolation from reference standard curves. The detection limit of this method was 4 pg ml−1.Statistical analysis
Results are expressed as means ± SEM of at least five different experiments run in triplicate. Statistical significance was evaluated by Student's t-test for unpaired varieties. Differences were considered statistically significant when p < 0.05. The software Origin version 6.0 (Microcal Software) was used as a nonlinear regression model for analysis of the concentration–response data to obtain the 50% inhibitory concentration value (IC50).Acknowledgements
The authors thank Novartis Farma SpA (Siena, Italy) for providing MenA CPS and mouse anti-MenA polyclonal Ab and Dr Antonella Vallario for flow cytometry measurements. Financial support was provided by MIUR-Italy (PRIN 2008 contract 2008CZ3NP3) and “Comune di Milano” (contract 55/2008).Notes and references
- M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2755 CrossRef CAS.
- N. Jayaraman, Chem. Soc. Rev., 2009, 38, 3463 RSC.
- C. W. Cairo, J. E. Gestwicki, M. Kanai and L. L. Kiessling, J. Am. Chem. Soc., 2002, 124, 1615 CrossRef CAS.
- L. L. Kiessling, J. E. Gestwicki and L. E. Strong, Curr. Opin. Chem. Biol., 2000, 4, 696 CrossRef CAS.
- O. Martinez-Avila, L. M. Bedoya, M. Marradi, C. Clavel, J. Alcami and S. Penades, ChemBioChem, 2009, 10, 1806 CrossRef CAS.
- O. Martinez-Avila, K. Hijazi, M. Marradi, C. Clavel, C. Campion, C. Kelly and S. Penades, Chem.–Eur. J., 2009, 15, 9874 CrossRef CAS.
- F. Manea, F. B. Houillon, L. Pasquato and P. Scrimin, Angew. Chem., Int. Ed., 2004, 43, 6165 CrossRef CAS.
- P. Pengo, S. Polizzi, L. Pasquato and P. Scrimin, J. Am. Chem. Soc., 2005, 127, 1616 CrossRef CAS.
- P. Pengo, L. Baltzer, L. Pasquato and P. Scrimin, Angew. Chem., Int. Ed., 2007, 46, 400 CrossRef CAS.
- J. M. Nam, C. S. Thaxton and C. A. Mirkin, Science, 2003, 301, 1884 CrossRef CAS.
- C. Guarise, L. Pasquato, V. De Filippis and P. Scrimin, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3978 CrossRef CAS.
- D. Aili, M. Mager, D. Roche and M. M. Stevens, Nano Lett., 2011, 11, 1401 CrossRef CAS.
- N. L. Rosi, D. A. Giljohann, C. S. Thaxton, A. K. R. Lytton-Jean, M. S. Han and C. A. Mirkin, Science, 2006, 312, 1027 CrossRef CAS.
- B. Kim, G. Han, B. J. Toley, C. K. Kim, V. M. Rotello and N. S. Forbes, Nat. Nanotechnol., 2010, 5, 465 CrossRef CAS.
- P. S. Ghosh, C. K. Kim, G. Han, N. S. Forbes and V. M. Rotello, ACS Nano, 2008, 2, 2213 CrossRef CAS.
- M. A. Dobrovolskaia, P. Aggarwal, J. B. Hall and S. E. McNeil, Mol. Pharm., 2008, 5, 487 CrossRef CAS.
- M. A. Dobrovolskaia, D. R. Germolec and J. L. Weaver, Nat. Nanotechnol., 2009, 4, 411 CrossRef CAS.
- M. A. Dobrovolskaia and S. E. McNeil, Nat. Nanotechnol., 2007, 2, 469 CrossRef CAS.
- M. A. Dobrovolskaia, A. K. Patri, J. W. Zheng, J. D. Clogston, N. Ayub, P. Aggarwal, B. W. Neun, J. B. Hall and S. E. McNeil, Nanomedicine-Nanotechnology Biology and Medicine, 2009, 5, 106 CrossRef CAS.
- A. S. Karakoti, S. Das, S. Thevuthasan and S. Seal, Angew. Chem., Int. Ed., 2011, 50, 1980 CrossRef CAS.
- J. V. Jokerst, T. Lobovkina, R. N. Zare and S. S. Gambhir, Nanomedicine, 2011, 6, 715 CrossRef CAS.
- M. F. Bachmann and G. T. Jennings, Nat. Rev. Immunol., 2010, 10, 787 CrossRef CAS.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS.
- D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 3280 CrossRef CAS.
- W. H. Cheung, V. S. F. Chan, H. W. Pang, M. K. Wong, Z. H. Guo, P. K. H. Tam, C. M. Che, C. L. Lin and W. Y. Yu, Bioconjugate Chem., 2009, 20, 24 CrossRef CAS.
- N. G. Bastus, E. Sanchez-Tillo, S. Pujals, C. Farrera, C. Lopez, E. Giralt, A. Celada, J. Lloberas and V. Puntes, ACS Nano, 2009, 3, 1335 CrossRef CAS.
- R. P. Brinas, A. Sundgren, P. Sahoo, S. M. Morey, K. Rittenhouse-Olson, G. E. Wilding, W. Deng and J. J. Barchi, Bioconjugate Chem., 2012, 23, 1524 CrossRef.
- F. Manea, C. Bindoli, S. Fallarini, G. Lombardi, L. Polito, L. Lay, R. Bonomi, F. Mancin and P. Scrimin, Adv. Mater., 2008, 20, 4348 CrossRef CAS.
- M. I. Torres-Sanchez, C. Zaccaria, B. Buzzi, G. Miglio, G. Lombardi, L. Polito, G. Russo and L. Lay, Chem.–Eur. J., 2007, 13, 6623 CrossRef CAS.
- P. J. Delves and I. M. Roitt, N. Engl. J. Med., 2000, 343, 37 CrossRef CAS.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. I. Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165 CrossRef CAS.
- L. H. Harrison, C. L. Trotter and M. E. Ramsay, Vaccine, 2009, 27, B51 CrossRef.
- L. K. K. Tan, G. M. Carlone and R. Borrow, N. Engl. J. Med., 2010, 362, 1511 CrossRef CAS.
- R. D. Astronomo and D. R. Burton, Nat. Rev. Drug Discovery, 2010, 9, 308 CrossRef CAS.
- M. M. Giuliani, J. Adu-Bobie, M. Comanducci, B. Arico, S. Savino, L. Santini, B. Brunelli, S. Bambini, A. Biolchi, B. Capecchi, E. Cartocci, L. Ciucchi, F. Di Marcello, F. Ferlicca, B. Galli, E. Luzzi, V. Masignani, D. Serruto, D. Veggi, M. Contorni, M. Morandi, A. Bartalesi, V. Cinotti, D. Mannucci, F. Titta, E. Ovidit, J. A. Welsch, D. Granoff, R. Rappuoli and M. Pizza, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10834 CrossRef CAS.
- P. Gardner, N. Engl. J. Med., 2006, 355, 1466 CrossRef CAS.
- F. Manea, C. Bindoli, S. Polizzi, L. Lay and P. Scrimin, Langmuir, 2008, 24, 4120 CrossRef CAS.
- M. Brust, M. Walker, D. Bethell, D. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801 RSC.
- S. Fallarini, T. Paoletti, L. Panza and G. Lombardi, Biochem. Pharmacol., 2008, 76, 738 CrossRef CAS.
- G. Miglio, F. Varsaldi and G. Lombardi, Biochem. Biophys. Res. Commun., 2005, 338, 1875 CrossRef CAS.
- M. J. Hostetler, J. E. Wingate, C. J. Zhong, J. E. Harris, R. W. Vachet, M. R. Clark, J. D. Londono, S. J. Green, J. J. Stokes, G. D. Wignall, G. L. Glish, M. D. Porter, N. D. Evans and R. W. Murray, Langmuir, 1998, 14, 17 CrossRef CAS.
- H. Vallhov, J. Qin, S. M. Johansson, N. Ahlborg, M. A. Muhammed, A. Scheynius and S. Gabrielsson, Nano Lett., 2006, 6, 1682 CrossRef CAS.
- D. O. Adams and T. A. Hamilton, Annu. Rev. Immunol., 1984, 2, 283 CrossRef CAS.
- S. F. G. van Helden, F. N. van Leeuwen and C. G. Figdor, Immunol. Lett., 2008, 117, 191 CrossRef CAS.
- J. Kleeff, M. Kornmann, H. Sawhney and M. Korc, Int. J. Cancer, 2000, 86, 399 CrossRef CAS.
- M. Lucarini, P. Franchi, G. F. Pedulli, C. Gentilini, S. Polizzi, P. Pengo, P. Scrimin and L. Pasquato, J. Am. Chem. Soc., 2005, 127, 16384 CrossRef CAS . See also: M. Lucarini, P. Franchi, G. F. Pedulli, P. Pengo, P. Scrimin and L. Pasquato, J. Am. Chem. Soc., 2004, 126, 9326 CrossRef.
- R. Hong, G. Han, J. M. Fernandez, B. J. Kim, N. S. Forbes and V. M. Rotello, J. Am. Chem. Soc., 2006, 128, 1078 CrossRef CAS.
- P. Decuzzi and M. Ferrari, Biomaterials, 2007, 28, 2915 CrossRef CAS.
- W. Jiang, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Nat. Nanotechnol., 2008, 3, 145 CrossRef CAS.
- C. J. Murphy, A. M. Gole, J. W. Stone, P. N. Sisco, A. M. Alkilany, E. C. Goldsmith and S. C. Baxter, Acc. Chem. Res., 2008, 41, 1721 CrossRef CAS.
- E. E. Connor, J. Mwamuka, A. Gole, C. J. Murphy and M. D. Wyatt, Small, 2005, 1, 325 CrossRef CAS.
- N. Khlebtsov and L. Dykman, Chem. Soc. Rev., 2011, 40, 1647 RSC.
- L. M. Browning, K. J. Lee, T. Huang, P. D. Nallathamby, J. E. Lowman and X.-H. N. Xu, Nanoscale, 2009, 1, 138 RSC.
- T. Paoletti, S. Fallarini, F. Gugliesi, A. Minassi, G. Appendino and G. Lombardi, Eur. J. Pharmacol., 2009, 620, 120 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S10 mentioned in the text related to the syntheses of nanosystems, nanoparticle characterization, and biological tests. See DOI: 10.1039/c2nr32338a |
| ‡ For the sake of clarity we refer to the gold core to identify the size of the nanoparticles throughout the paper. |
| § The concentrations indicated in the case of the nanoparticles refer to the organic component. The Au-NP concentration can be derived by dividing these numbers by the number of ligands present on the monolayer as reported in Table 1. |
| This journal is © The Royal Society of Chemistry 2013 |