The enzymes of β-lactam biosynthesis
Refaat B. Hamed*ab, J. Ruben Gomez-Castellanosa, Luc Henrya, Christian Ducho†a, Michael A. McDonougha and Christopher J. Schofield*a
aUniversity of Oxford, Department of Chemistry, Chemistry Research Laboratory, Mansfield Road, Oxford, OX1 3TA, United Kingdom. E-mail: refaat.hamed@chem.ox.ac.uk; christopher.schofield@chem.ox.ac.uk; Fax: +441865275674; Tel: +441865275625
bDepartment of Pharmacognosy, Faculty of Pharmacy, Assiut University, 71256, Egypt (on leave)
First published on 7th November 2012
Abstract
Covering: up to May 2012
The β-lactam antibiotics and related β-lactamase inhibitors are amongst the most important small molecules in clinical use. Most, but not all, β-lactams including penicillins, cephalosporins, and clavulanic acid are produced via fermentation or via modification of fermented intermediates, with important exceptions being the carbapenems and aztreonam. The desire for more efficient routes to existing antibiotics and for access to new and synthetically challenging ones stimulates continued interest in β-lactam biosynthesis. We review knowledge of the pathways leading to β-lactam antibiotics focusing on the mechanisms, structures and biocatalytic applications of the enzymes involved.
 Refaat B. Hamed | Refaat Hamed got his BSc (in Pharmaceutical Sciences) and MSc (in Natural Products Chemistry) degrees in 1999 and 2004, respectively, from Assiut University, Egypt. In 2010, he finished his D.Phil. in the Chemistry Department, University of Oxford, under Prof. Chris Schofield's supervision. He then continued there as a postdoctoral research fellow. In 2011, he was appointed as a lecturer in Assiut University. His D.Phil. and postdoctoral studies targeted the mechanisms, structures and biocatalytic applications of β-lactam biosynthesis enzymes. Engineering enzymes and biosynthetic pathways redesign for the production of unnatural products of pharmaceutical interest is a main objective for Refaat's research. |
 J. Ruben Gomez-Castellanos | J. Rubén Gómez Castellanos is a doctoral candidate in the Chemical Biology programme of the Department of Chemistry, University of Oxford, under the supervision of Prof. Chris Schofield. He holds a BSc in Pharmaceutical and Biological Chemistry from La Salle University in Mexico City (2005) and an MSc in Drug Discovery from The School of Pharmacy – University of London (with distinction, 2006). He has previous experience in clinical research working for Eli Lilly and Co. in Mexico City, and is currently working as a Clinical Research Associate II at Pharm-Olam, Intl., an international contract research organisation with offices in Mexico. |
 Luc Henry | Luc Henry was born in Switzerland and studied biological chemistry in Lausanne (Switzerland), Uppsala (Sweden) and Oxford (UK), earning a M. Sc. in biological chemistry from the Ecole Polytechnique Fédérale de Lausanne (EPFL) in 2007. He then spent four years as a DPhil student in the laboratory of Prof. Christopher J. Schofield at the University of Oxford, working on the enzymology of thienamycin biosynthesis. He recently completed his doctoral studies and has returned to Lausanne to carry out postdoctoral work with Prof Kai Johnsson. His interests lie in the development of chemical tools to study biological systems. |
 Christian Ducho | Christian Ducho was born in 1976 and studied Chemistry at the University of Hamburg, Germany, from 1996 to 2001. He completed his Ph.D. in Organic Chemistry in the group of Chris Meier at the University of Hamburg in 2005. He then moved to the University of Oxford, UK, for postdoctoral research (biosynthesis of carbapenem antibiotics) in the group of Christopher Schofield from 2005 to 2007. In 2007, he commenced his independent academic career in Germany as an Assistant Professor (‘Juniorprofessor’) at the Georg-August-University Göttingen. In 2011, he moved to the University of Paderborn, Germany, as Associate Professor of Organic Chemistry. |
 Michael A. McDonough | Michael McDonough received his PhD in Biophysics, with a focus in X-ray crystallography, from the University of Connecticut in 2000 where he made significant contributions to the fundamental understanding of mechanisms by which beta-lactam antibiotic targets bind to their substrates. He then spent two years at the University of Copenhagen honing his structural skills at the Centre for Crystallographic Studies before settling in Oxford in 2003 where he has since been instrumental in the efforts to structurally characterise enzymes involved in clavulanic acid and carbapenem biosynthesis. |
 Christopher J. Schofield | Chris Schofield is currently Head of the Organic Chemistry, and a Fellow of Hertford College, University of Oxford. His current research interests include antibiotic biosynthesis and resistance, regulation of gene expression by oxygen, and epigenetics with a common theme being enzymes that catalyse chemically interesting reactions of biomedicinal importance. |
1 Introduction
The family of β-lactam antibiotics (BLAs, Fig. 1) is the most important class of clinically used antibiotics with more than half of the global antibiotic market share.1,2 However, over the past two decades, research on BLAs by most major pharmaceutical companies has decreased substantially compared to the half-century following their clinical introduction in the 1940s. The same trend is true for other classes of established antibiotics. The reasons for this decrease in activity are probably primarily commercial with the size of potential revenue being insufficient to justify the high cost of antibiotic development.3 However, the increasing problems of antibiotic resistance4 and Gram-negative infections5 coupled to a critical shortage of new antibiotics6 are now stimulating renewed interest in antibiotic development, both in the public and private sectors.4,7 Given their track record of efficacy and safety, it may be that the timing is appropriate for a resurgence of interest in BLAs.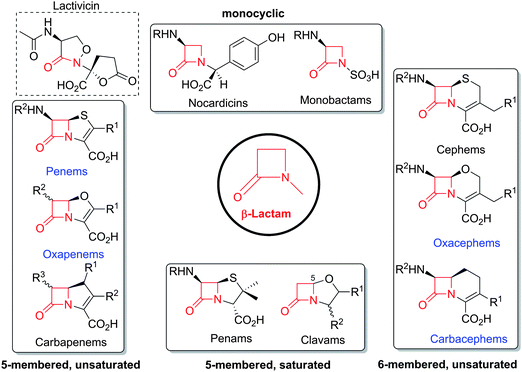 | ||
| Fig. 1 Major subfamilies of β-lactams. The names of synthetic β-lactam antibiotic (BLA) subfamilies are in blue. Lactivicin (in the dashed box) is a naturally-occurring γ-lactam antibiotic, which, like BLAs, inhibits penicillin binding proteins (PBPs). Clavulanic acid (a clavam with the (5R)-stereochemistry) is a serine β-lactamase inhibitor; clavams (with the (5S)-stereochemistry) have antibacterial and antifungal activities (the latter not via PBP inhibition). | ||
The targets of BLAs are transpeptidase enzymes which are involved in bacterial cell wall biosynthesis.8 The transpeptidases, or penicillin binding proteins (PBPs),9 catalyse peptidoglycan cross-linking (Fig. 2) and are likely amenable to modern medicinal approaches that would complement the vast structure–activity relationship data sets acquired in the period leading up to the 1990s. Such studies will benefit from advances in molecular and structural understandings of cell-wall biosynthesis and the enzymes involved (for reviews, see ref. 10, 11). However, a problem with some BLAs subfamilies is that their commercially viable synthesis remains challenging, because of the high degree of functionalisation and the enantioenriched and reactive nature of the core bicyclic ring-structures. Indeed, the issue of production costs is more of an issue with antibiotics than many other classes of pharmaceuticals. Another issue with BLAs is the need for specialised production facilities due to allergy concerns.
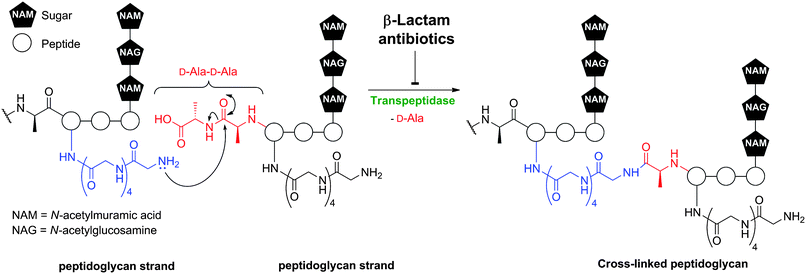 | ||
| Fig. 2 Transpeptidase-catalysed reactions during bacterial cell wall biosynthesis are inhibited by β-lactam antibiotics. Formation of the mature peptidoglycan matrix involves transpeptidase-catalysed cross-linking of linear polysaccharide chains by short peptides. The reaction is exemplified in outline for Staphylococcus aureus. | ||
With the exception of aztreonam and the carbapenems, clinically used β-lactams are produced either by isolation of fermented compounds (e.g. clavulanic acid) or by synthetic or enzymatic modification of fermented compounds (e.g. many penicillins and all such cephalosporins). For this reason, β-lactam biosynthesis has been a focal point for the application of new methodologies in molecular biology with a view to optimising production procedures. Presently, there is interest in optimising sustainable routes to pharmaceuticals. It would seem likely that if there is resurgence in interest in BLAs research, manipulation of their biosynthesis pathways might be an important tool for the efficient and sustainable production of new antibiotics.
Following the pioneering demonstration that β-lactams are elastase inhibitors,12 β-lactams are also being explored in other non-antibiotic therapeutic applications.13 β-Lactams have been used to inhibit various proteases (including HIV,14 cytomegalovirus15 and rhomboid intramembrane16 proteases), to act as cholesterol absorption inhibitors17 (ezetimibe is used clinically18,19), as vehicles for anticancer drug delivery,13 as anticancer drugs,20–22 and in the regulation of the excitatory neurotransmitter glutamate.23 The clinical success and potential of some of these compounds is stimulating continued interest in developing efficient routes to β-lactams. Aside from improving practical routes to existing antibiotics and providing access to new β-lactam structures, a further justification for work on BLA biosynthesis is that many of the reactions (and enzymes that catalyse them) are of basic (bio)chemical interest. Such enzymes include the synthetases involved in peptide and β-lactam formation and, of particular interest to our group, the oxygen-utilizing enzymes that catalyse the formation of the bicyclic ring structure (e.g. in penicillins and clavams) and other modifications. Studies on these enzymes have not only increased our understanding of how nature synthesises molecules that are synthetically inaccessible but has also impacted on fields as far varying as plant hormone signalling, collagen biosynthesis, DNA repair, epigenetics and human oxygen-sensing (for reviews, see ref. 24–28).
Here we review mechanistic and structural studies on β-lactam biosynthesis focusing on the enzymes involved and in particular on recent work (for previous reviews, see ref. 29–34). In several cases we also highlight the potential for protein engineering of β-lactam biosynthesis enzymes, both for the purpose of pathway engineering and more generally for the biocatalytic production of useful compounds.
2 Naturally-occurring β-lactams
BLAs isolated from natural origin may be classified into the penicillin, cephalosporin, carbapenem/carbapenam, clavam, and monocyclic β-lactam subfamilies (Fig. 1). All clinically used BLAs likely work by inhibition of PBPs via formation of relatively stable acyl-enzyme complexes (Fig. 2–4). PBP-inhibition results in impaired peptidoglycan biosynthesis and consequent cytolysis. It is notable that whilst many naturally-occurring BLAs which inhibit PBPs have been identified, only one naturally-occurring non β-lactam compound, lactivicin (Fig. 1), that targets PBPs, has been isolated.35–37 It is proposed that the apparently special ability of BLAs to inhibit PBPs (and possibly other enzymes which employ nucleophilic serine/threonine/cysteine residues), results from a combination of (i) efficient binding as mediated by appropriate functionalisation and stereochemistry, (ii) possible mimicry of the “tetrahedral” transition state occurring during amide hydrolysis (the Tipper–Strominger hypothesis38), (iii) irreversible reaction with the nucleophilic serine at the active site of PBPs, in part because of the highly strained nature of the β-lactam ring, and (iv) formation of an acyl-enzyme complex that is stable with respect to hydrolysis/nucleophilic attack.8,38–40 In the case of lactivicin, a γ-lactam, cycloserine derivative, the unusual fused spirocyclic ring system is proposed to enable irreversible reaction with PBPs.37,41,42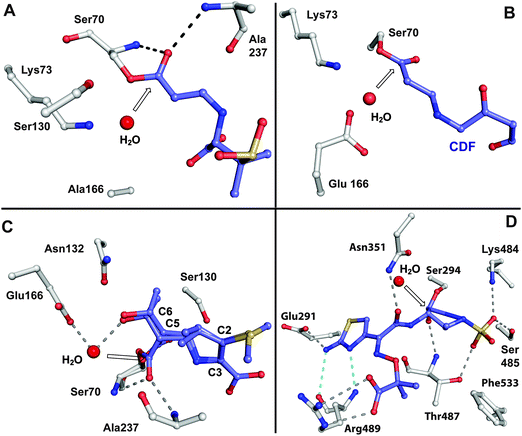 | ||
| Fig. 3 Views from crystal structures showing examples of relatively stable acyl-enzyme complexes formed by reaction of β-lactams with PBPs and β-lactamases. A: View from a structure obtained by reaction of a serine β-lactamase SHV-1 E166A with sulbactam (PDB 2A3U).679–681 Glu166, which is involved in hydrolysis of the acyl-enzyme intermediate, was substituted for alanine to enable trapping of the complex; B: View from a complex resulting from reaction of the Mycobacterium tuberculosis β-lactamase BlaC with clavulanic acid (PDB 3CG5). Following nucleophilic attack by Ser70 and β-lactam ring opening, the complex decarboxylates to generate the shown adduct with the clavulanate-derived fragment (CDF,682 which is also shown in blue in Fig. 4iii); C: View from a structure of a serine β-lactamase (SHV-1)/meropenem acyl-enzyme complex (PDB 2ZD8). Two conformations were observed, one with the carbonyl of the acyl-enzyme complex in the oxyanion hole (formed by NH amides of Ser70 and Ala237), and another with the carbonyl directed away from the oxyanion hole. Only part of the C-2 substituent of meropenem is shown; D: View from a PBP3 (Pseudomonas aeruginosa)/aztreonam acyl-enzyme complex (PDB 3PBS).683 The white arrows point to the carbonyl carbon of the (hydrolysed) β-lactam. Structures of sulbactam and aztreonam are given in Fig. 5, while those of clavulanic acid and meropenem are in Fig. 6 and Fig. 46, respectively. | ||
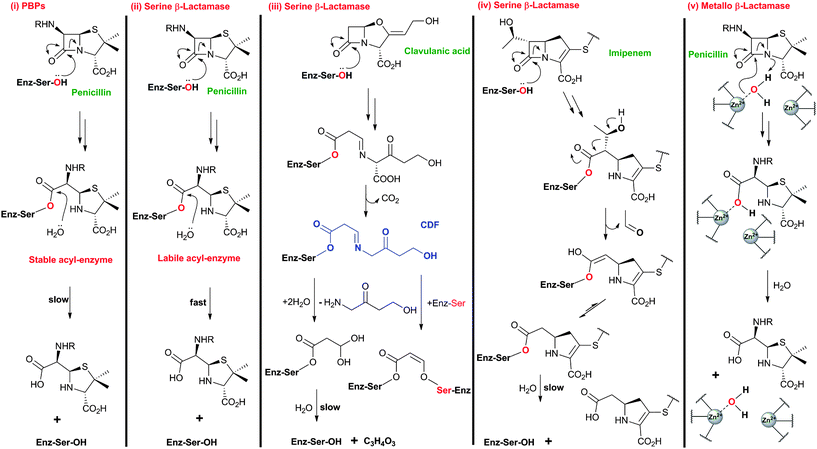 | ||
| Fig. 4 Comparison of outline reaction mechanisms of different β-lactams with PBPs and β-lactamases. (i) β-Lactams bind to the transpeptidase domain of PBPs to form relatively stable acyl-enzyme complexes; (ii) β-Lactams react with serine β-lactamases to form acyl-enzyme complexes labile to hydrolysis; (iii) Clavulanic acid, and other β-lactam-based serine β-lactamase inhibitors, react with serine β-lactamases to form stable acyl-enzyme complexes;684 (iv) Thienamycin derivatives (e.g. imipenem) react with some serine β-lactamases to form a stable acyl-enzyme complex;56,685 (v) β-Lactams react with zinc-dependent metallo-β-lactamases forming a complex that is labile to hydrolysis (aztreonam is an exception48). Note that the reaction mechanisms are more complex than shown here and variations on the shown outline mechanisms occur. The water molecule shared between ZnII ions likely exists as a hydroxide ion, which can act as the nucleophile during metallo-β-lactamase catalysis.686,687 | ||
Resistance to BLAs was identified very soon after their first clinical use in the 1940s,43 and is now known to be mediated via processes including44 (i) mutation of PBPs to block inhibitor binding,45 (ii) ejection of β-lactams by efflux pumps,46 (iii) reduced permeability to β-lactams,47 and (iv) the production of β-lactamases that catalyse β-lactam ring hydrolysis.48–50 The largest family of identified β-lactamases likely evolved from PBPs and similarly employ a serine-residue as a nucleophilic catalyst.51–53 Metallo-β-lactamases,54 although not yet as clinically relevant as their serine-relative, employ one or two zinc ions to activate a “hydrolytic” water molecule and are an increasing threat because of their ability to hydrolyse almost all types of BLAs (a notable exception being aztreonam54,55). The BLAs show different degrees of stability with respect to β-lactamases, with the cephalosporins and, particularly, the carbapenems showing, in general, increased stability compared to the penicillins (like β-lactamase inhibitors, carbapenems can undergo fragmentation on reaction with serine β-lactamases, Fig. 3 and 4).56
Notably, clinically used serine β-lactamase inhibitors are themselves β-lactams, e.g. clavulanic acid (naturally-occurring), sulbactam and tazobactam (Fig. 5). These inhibitors react with the β-lactamase active site serine to form an acyl-enzyme complex, but the initially formed complex fragments to form an ester that is unusually stable with respect to hydrolysis. Because these compounds (Fig. 5) are insufficiently potent as antibiotics, they are used in combination with a BLA (e.g. amoxicillin and clavulanic acid are combined as Augmentin®). As with the antibacterial activity of BLAs, one current objective for β-lactamase inhibitor development is to increase the spectrum of activity. The currently used β-lactamase inhibitors57,58 only efficiently inhibit Class A of the three (A, C and D) classes of serine β-lactamases and do not inhibit the metallo-β-lactamases (Class B). AM-112,59 an oxapenem, and NXL-104 (Avibactam),60–62 a synthetic bridged γ-lactam (Fig. 5), have both been developed as broad spectrum β-lactamase inhibitors; Avibactam is presently in clinical trials.
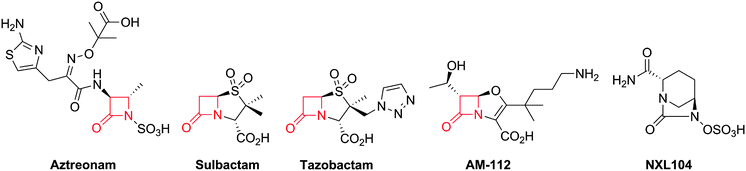 | ||
| Fig. 5 Structures of aztreonam and some β-lactamase inhibitors. Note that some of the inhibitors (e.g. sulbactam, tazobactam and AM-112) also show antibacterial activity. Clavulanic acid (Fig. 6), sulbactam688 and tazobactam689 are used clinically in combination with β-lactam antibiotics to protect the latter against the effects of β-lactamases. AM-11259 and NXL104 (Avibactam)60–62 are examples of broad spectrum serine β-lactamase inhibitors. | ||
3 Overview of β-lactam biosynthesis
An important step in the development of clinically useful penicillins was the discovery that different penicillins could be produced by modification of growth media and strains. The combination of corn-steep liquor as a growth medium, a high yielding strain of Penicillium chrysogenum and use of aerated deep fermentation had a historically important role in the development of penicillins. It was found that supplementing the fermentation medium with aromatic acids could result in the production of specific penicillins, e.g. addition of phenoxyacetic acid results in formation of penicillin V, which has a 6-phenoxyacetamido-side chain (Fig. 6), as the major product.63 Penicillin V was developed for oral clinical use because of its increased resistance to acid-catalysed hydrolysis in the stomach compared to penicillin G (the presence of the electron-withdrawing phenoxy group hinders attack of the side chain carbonyl-oxygen on the β-lactam carbonyl).64 The development of procedures to produce penicillins with neutral hydrophobic side chains (via supplementing the fermentation medium with appropriate precursors)63,65 was important because it enables the purification of penicillins from the fermentation medium by extraction. This work was a pioneering example of metabolic engineering. However, the type of penicillins that could be produced by this technique was limited. A crucial further advancement was the development of procedures for the production of 6-aminopenicillanic acid (6-APA)66 from extracted hydrophobic penicillins (e.g. penicillin G). This procedure is catalysed by penicillin acylases67 (Section 4.10). The 6-APA can then be acylated to give “semi-synthetic” penicillins with diverse side chains – many have found clinical applications, e.g. piperacillin, amoxicillin, and ampicillin (Fig. 7A).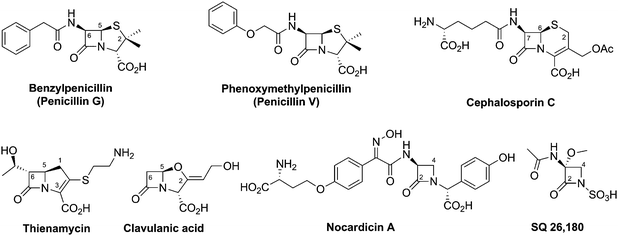 | ||
| Fig. 6 Prototypical examples of different subfamilies of naturally-occurring β-lactam antibiotics. | ||
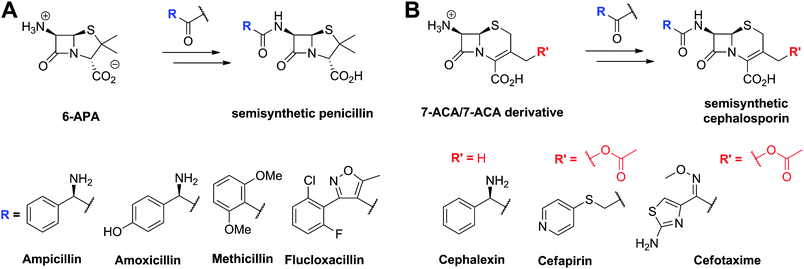 | ||
| Fig. 7 Examples of semisynthetic penicillins (A) and cephalosporins (B) derived from 6-aminopenicillanic acid (6-APA) and 7-aminocephalosporanic acid (7-ACA)/7-ACA derivative, respectively. | ||
The need to combat penicillin resistance provoked a search for new BLAs, which resulted in the isolation of cephalosporin C68 from Acremonium chrysogenum (previously known as Cephalosporium acremonium). Cephalosporin C (Fig. 6), like other cephalosporins, shows resistance to some serine β-lactamases (Class A). Procedures were developed for the production of 7-aminocephalosporanic acid (7-ACA, Fig. 7), though these were hindered by the fact that, in contrast to penicillins, it is not yet possible to (at least efficiently) produce cephalosporins with hydrophobic side chains by the simple addition of hydrophobic acetic acid derivatives to the wildtype cephalosporin-producing fermentations (see Section 4.11). One successful approach to address this problem has been the development of chemical methods for the ring-expansion of penicillins with hydrophobic side chains to give cephalosporins.69 Enzymic or chemical deacylation is then used to hydrolyse the side chain at the 7-position of the resultant cephalosporins (Section 4.10, where other approaches to producing 7-ACA are also described). Compared to the penicillins, the cephalosporins have an additional site for modification, i.e. at the C-3′ position. Esterases have been identified that efficiently catalyse the hydrolysis of the C-3′ ester of cephalosporin C to give an allylic alcohol which can be subsequently modified, so enabling the production of many cephalosporin derivatives. Thousands of cephalosporin derivatives, modified at C-7 and/or C-3′, have been produced by semi-synthesis with many achieving clinical utility (Fig. 7B).70,71
The enzymes used in the production of 6-APA, 7-ACA (Section 4.10) and C-3′ hydroxymethyl-cephalosporins are highly efficient on industrial scale. Because these enzymes are not part of the BLA biosynthesis pathways, they are not the subject of this review (see ref. 1, 2, 72 for reviews), but it should be noted that some are structurally and mechanistically related to enzymes involved in BLA biosynthesis (Section 4.5). For example, the penicillin acylases, used in the commercial production of 6-APA from hydrophobic penicillins, and isopenicillin N acyl-transferase, which is involved in the biosynthesis of hydrophobic penicillins, are both members of the N-terminal nucleophilic (Ntn) hydrolase family.73,74
In the late 1970s, new strategies in which supersensitive strains of bacteria, some with β-lactamase inhibitory activities, were used to screen massive number of samples containing soil micro-organisms for BLAs.75,76 The new screens coupled to advances in chromatography led to the discovery of three new subfamilies of BLAs: the clavams, monobactams and carbapenems (Fig. 6).
Clavulanic acid (Fig. 6) was the first member of the clavam subfamily of BLAs to be isolated (from Streptomyces clavuligerus), and it was found to be a potent inhibitor of Class A β-lactamases (Fig. 3 and 4) but a relatively weak antibiotic.77 The clavams are distinguished from the penicillins by replacement of the sulfur atom of the thiazolidine ring with an oxygen atom to give an oxazolidine ring.
Nocardicin A (Fig. 6) was the first naturally-occurring monocyclic β-lactam to be isolated (from Nocardia uniformis subsp. Tsuyamanensis).78 Nocardicin A has weak to moderate activity against Gram-negative and Gram-positive bacteria, and some resistance to β-lactamases; to date the nocardicins have not found clinical application because of their relatively poor antibacterial properties.79 The development of monobactams, with the aim of increasing their binding affinity to PBPs, resulted in the discovery of aztreonam (Fig. 5) which is effective against Gram-negative bacteria and unusually resistant to hydrolysis by β-lactamases.79
Thienamycin (Fig. 6), named for its novel β-thioenamine group,80 was the first of the clinically important carbapenem antibiotics to be isolated (from Streptomyces cattleya).81,82 The carbapenems are distinguished from the penicillins by replacement of the sulfur atom in the five-membered ring with a methylene group, and by desaturation between C-2 and C-3. Thienamycin has been described as one of the most potent broad-spectrum antibacterial agents isolated from natural sources;83 it has good activity against many Gram-positive and Gram-negative bacteria, and inhibits some serine β-lactamases. Despite its activity, the chemical and metabolic instability of thienamycin have prevented its clinical use. Instead, stable derivatives of thienamycin, e.g. ertapenem, meropenem and doripenem, have been developed and are now widely used as antibiotics (Section 6.1).84
An important difference between the penicillin and cephalosporin development stories and that of the clavams, monobactams, and carbapenems is that in the latter three cases the semisynthetic approach has been much less productive. Whilst some clavulanic acid derivatives have been prepared by modifications of the C-2 allylic alcohol,85–88 modifications at the C-6 position have been limited by the lack of a functional group at this position and inefficiency of synthetic methodologies for C-6 modification. This is a pity because it is known that clavulanic acid derivatives functionalised at C-6 (e.g. with the hydroxyethyl group of carbapenems) have a useful spectrum of β-lactamase inhibitory activity,89 as do the related synthetic oxapenems (e.g. AM-112, Fig. 5).59 The situation with fermented carbapenems has been similarly frustrating. Despite extensive efforts, it was not possible to increase fermentation titres of carbapenems to useful large-scale levels. As a consequence, total synthesis methods were developed to produce clinically used carbapenems. The development of these methods enabling the production of densely functionalised and relatively labile carbapenems is one of the triumphs of the 20th century synthetic chemistry. Whilst the routes are remarkably efficient (for reviews, see ref. 80, 90), the available synthetic methodology limits the range of accessible compounds, with one particular limitation being the reliance on 4-acetoxy-3-[1-(tert-butyldimethylsilyloxy)ethyl]azetidin-2-one (AOSA, Section 6.1) as a synthetic intermediate.90–92 The problem of efficient and inexpensive biotechnological production of modified clavams and carbapenems has stimulated efforts to understand and manipulate their biosynthesis pathways through exploiting the techniques of molecular and structural biology. As with the related pioneering studies on penicillin and cephalosporin biosynthesis, this work may have come too late to have a major impact in the first golden age of BLAs, but it is possible that it may help enable production of new and existing antibiotics by efficient and sustainable methods.
Here we review knowledge of the pathways leading to the BLAs with an emphasis on recent work on the biosynthesis enzymes and reactions of chemical interest.
4 Penicillin and cephalosporin biosynthesis pathways
4.1 Introduction – overview of the pathways
To date, the formation of hydrophobic penicillins (Fig. 8) has only been reported in fungi, e.g. P. chrysogenum. In contrast, the hydrophilic cephalosporins are produced by both fungi (e.g. A. chrysogenum), and bacteria. The cephamycins have been isolated from Gram-positive bacteria, e.g. Streptomyces sp., while the cephabacins and related structures (Fig. 8) have been isolated from Gram-negative bacteria, e.g. Flavobacterium sp.,93–95Xanthomonas lactamgena96–99 and Lysobacter lactamgenus.98,99 The Gram-positive actinomycetes are probably able to produce the most diverse array of β-lactam containing structures, including not only the penicillins, cephalosporins and cephamycins but also carbapenems, clavams, and nocardicins.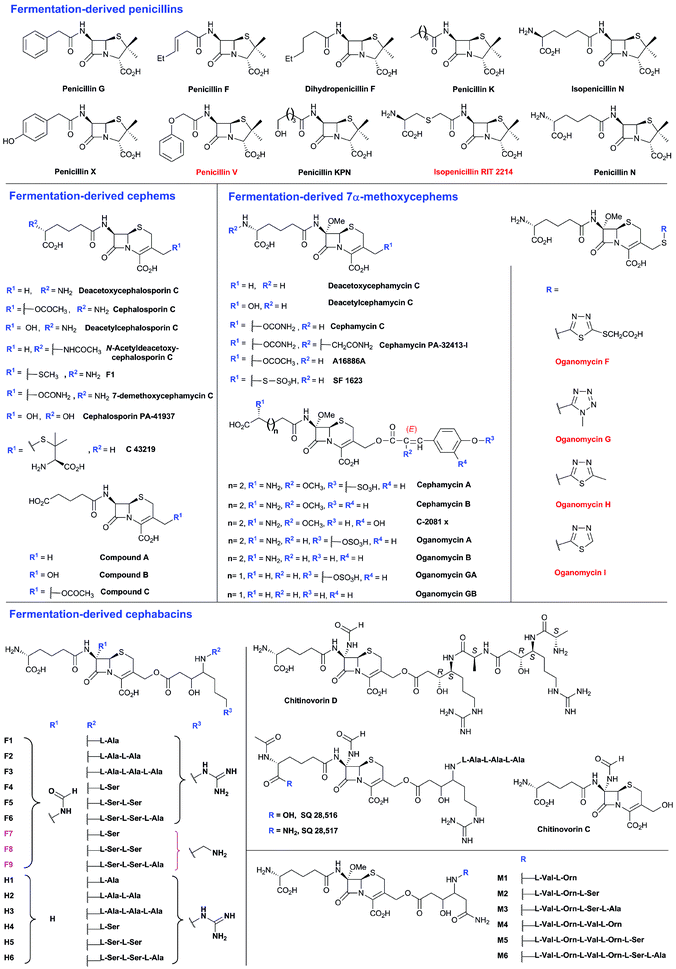 | ||
| Fig. 8 Penicillins and cephalosporins isolated from natural sources. Compounds in red are obtained as the result of specific precursor addition to the fermentation medium: phenoxyacetic acid for penicillin V,63L-S-carboxymethylcysteine for RIT 2214690 and aromatic thiols for the oganomycins (F-I).113 Most of the shown penicillins (except penicillin N and KPN) are produced by Penicillium chrysogenum and/or P. notatum in varying ratios depending on the strain and culture conditions. Penicillin F, a major product of P. notatum, is designated so to identify it as the penicillin discovered by Fleming.691 Penicillin G is a major product of P. chrysogenum grown on corn-steep liquor. Penicillin KPN is produced by strains of the genus Paecillomyces.692 Cephalosporins with α-ketoglutaryl side chain (e.g. compounds A, B and C)693 were detected in culture broth of some strains belonging to the genus Acremonium, and are predicted to be produced by an oxidase-mediated deamination of the C-7 D-amino-adipate side chain.367 The 3′-thiomethylcephems F-1111 and C-43219112 were isolated from culture broth of mutants of Acremonium chrysogenum, while SF-1623110 was isolated from culture broth of Streptomyces chartreusis. Cephalosporin PA-41937 was isolated from the PA-41937 Streptomyces strain.694 Cephamycin PA-32413-I was isolated from the PA-32413 S. clavuligerus strain.695 7α-Methoxycephems with cinnamoyl-side chains at C-3′ were isolated from Streptomyces spp.320,345,696,697 The cephabacins are produced by some Gram-negative bacteria e.g. Flavobacterium sp. (SQ 28516/7, chitinovorin C D−1),93–95Xanthomonas lactamgena (M1–6, F4–9, H4–6)96–99 and Lysobacter lactamgenus (F1–3 and H1–3).98,99 | ||
The biosynthetic routes leading to the core nuclei of penicillins and cephalosporins (Fig. 9) have been extensively investigated (for prior reviews, see ref. 1, 30, 31, 100–103). The committed step in these routes is the assembly of the tripeptide LLD-ACV from L-α-aminoadipic acid, L-cysteine and L-valine (Fig. 9).104LLD-ACV biosynthesis is catalysed by the multidomain, multimodular, non-ribosomal peptide synthetase ACVS – which in addition to catalysing formation of two peptide bonds, also catalyses inversion of the C-α stereochemistry of L-valine to the D-valine residue in LLD-ACV105 (Fig. 9). The first β-lactam to be formed, isopenicillin N, is then produced from LLD-ACV in a remarkable oxidative cyclisation reaction catalysed by the FeII-dependent oxidase isopenicillin N synthase31,106 (Fig. 9). In some fungi, e.g. P. chrysogenum, acyl-transferases act upon isopenicillin N to give various acyl-penicillin derivatives, e.g. penicillin G (Fig. 8). In some fungi and bacteria, e.g. Streptomyces spp, the side chain epimerisation of isopenicillin N to penicillin N, catalysed by isopenicillin N epimerase, enables branching of the penicillins/cephalosporins pathway (Fig. 9). Penicillin N transformation into cephalosporin C proceeds in 3 steps (i) expansion of the 5-membered thiazolidine ring of penicillin N into the 6-membered dihydrothiazine ring of deacetoxycephalosporin C (DAOC), as catalysed by the 2-oxoglutarate (2OG) dependent oxygenase deacetoxycephalosporin C synthase (DAOCS); (ii) hydroxylation of DAOC to give deacetylcephalosporin C (DAC), as catalysed by DAOC hydroxylase; and (iii) acetylation of DAC to give cephalosporin C, as catalysed by DAC acetyltransferase (Fig. 9).
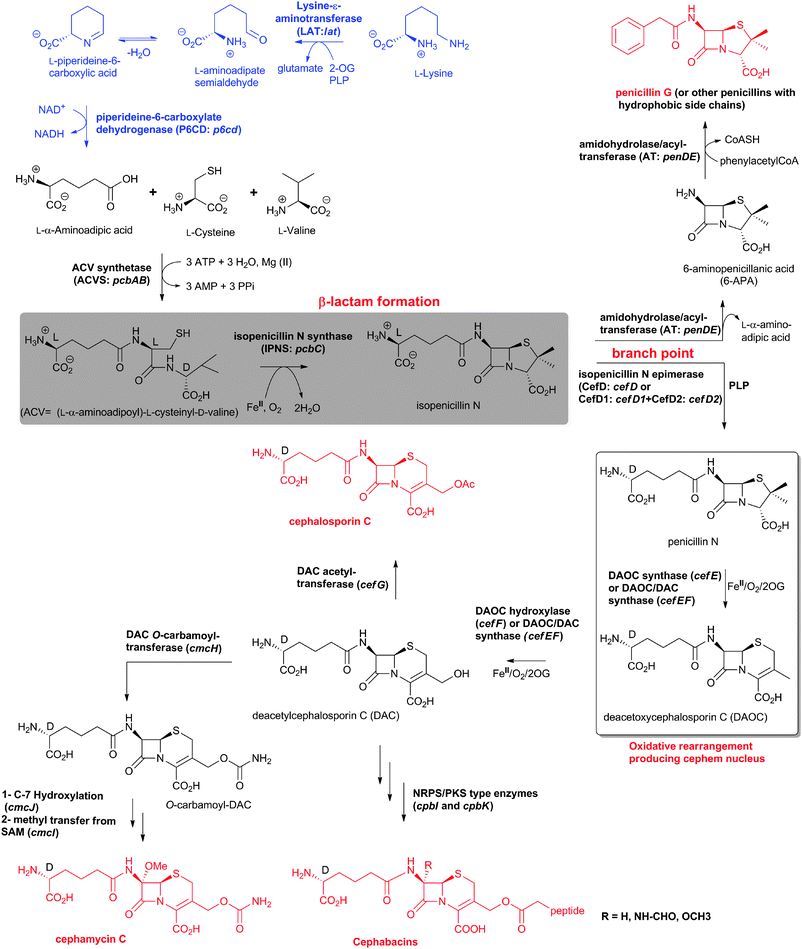 | ||
| Fig. 9 Biosynthetic pathways leading to penicillins and cephalosporins in fungi and bacteria. Whilst bacterial cephamycin-producers have a specific pathway to form L-α-aminoadipic acid (L-AAA) from lysine (shown in blue), L-AAA is an intermediate of lysine biosynthesis in penicillin-producing fungi. The isopenicillin N epimerase in cephalosporin-producing bacteria (i.e. CefD) is pyridoxal phosphate (PLP)-dependent; an alternative isopenicillin N epimerisation system (comprising CefD1 and CefD2) operates in fungi.248,253 In prokaryotes, the DAOC synthase and DAOC hydroxylase reactions are carried out by separate enzymes (encoded by cefE and cefF, respectively), whereas in fungi, a bifunctional enzyme (encoded by cefEF) catalyses both reactions. CmcJ was previously not considered as belonging to the stereotypical 2OG oxygenase superfamily;109 however, bioinformatic analyses suggest that it is likely a 2OG-dependent hydroxylase.330 Acronyms for the genes encoding for enzymes are in parentheses (see Fig. 10, Table 1). Selected cofactors and co-substrates are shown. A non-ribosomal peptide synthetase (NRPS)/polyketide synthase (PKS) hybrid system (CpbI/CpbK) is proposed to be involved in the assembly of the C-3′ side chain of the cephabacins.114,115 | ||
The cephamycins are 7α-methoxy-cephem derivatives with different side chains at C-3′ (for reviews, see ref. 30, 107). Cephamycin C biosynthesis gene cluster (Fig. 10, Table 1) contains specific genes related to L-α-aminoadipic acid formation, and genes encoding for enzymes involved in DAC modification.30,107 Late steps in cephamycin C biosynthesis (Fig. 9) involve carbamoylation of the C-3′-hydroxyl group, as catalysed by the O-carbamoyltransferase CmcH,108 and 7α-methoxylation, as catalysed by CmcJ and CmcI (Fig. 9).109 The side chain at the 3′-position of cephamycins and oganomycins (Fig. 8) can be acetyl (e.g. A-16884A), carbamoyl (e.g. cephamycin C), as well as esters of p-coumaric acid derivatives (e.g. cephamycin A and B, oganomycins). Derivatives of 3-thiomethylcephems (Fig. 8) have also been reported in the culture broth of S. chartreusis (e.g. SF-1623110), mutants of A. chrysogenum (e.g. F-1111 and C-43219112), and after specific precursor (e.g. heterocyclic thiols) addition to the culture broth of S. oganonensis (e.g. oganomycins F-I113).
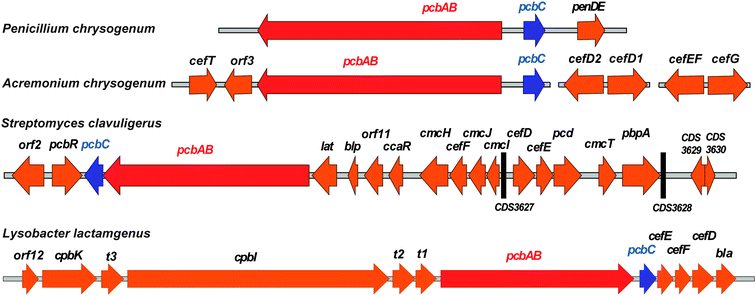 | ||
| Fig. 10 Examples of penicillin and cephalosporin biosynthesis gene clusters from fungi (Penicillium chrysogenum and Acremonium chrysogenum) and bacteria (Streptomyces clavuligerus and Lysobacter lactamgenus).266,698,699 For the role of the biosynthetic proteins encoded by the genes shown, see Table 1 and Fig. 9. The protein encoded by orf12 (L. lactamgenus) shows similarity to methyltransferases;115 however, the reported cephabacins from L. lactamgenus are not methoxylated at C-7, hence the role of orf12 is unclear. The orf2 gene (S. clavuligerus) is part of the clavulanic acid gene cluster (Fig. 28). The L. lactamgenus cluster, the only cluster reported so far for cephabacin biosynthesis, appears to be incomplete (e.g. the genes encoding for the proteins responsible for the introduction of the C-7 formylamino moiety and for L-aminoadipate biosynthesis are not yet identified115,700). | ||
| Gene | AA | (Proposed) function of encoded protein |
|---|---|---|
| pcbABa–d | 3746a | ACV Synthetase (ACVS); formation of LLD-ACV tripeptide.134–137 |
| pcbCa–d | 331a | Isopenicillin N synthase (IPNS); LLD-ACV oxidative cyclisation.106,172 |
| penDEa | 357 | Isopenicillin N acyltransferase (AT).225–228 |
| cefTb | 561 | Efflux pump protein for the export of cephalosporin C.701 |
| orf3b | Unknown. | |
| cefD2b | 383 | Isopenicillin N-CoA epimerase (CefD2).253 |
| cefD1b | 609 | Isopenicillin N-CoA synthetase (CefD1).253 |
| cefEFb | 332 | Deacetylcephalosporin C synthase/hydroxylase (DAOCS/DACS).255–258 |
| cefGb | 385 | Acetyl-coenzyme A:DAC O-acetyltransferase (CefG).337 |
| pcbRc | 551 | Protein involved in BLA resistance.702 |
| latc | 457 | L-Lysine-ε-aminotransferase (LAT).124,703 |
| blpc | 182 | Extracellular β-lactamase inhibitory protein.704 |
| orf11c | Unknown. | |
| ccaRc | 256 | Transcriptional activator (CcaR) essential for the biosynthesis of cephamycin, clavulanic acid and clavams.704 |
| cmcHc | 521 | O-Carbamoyltransferase (CmcH).108,703 |
| cefFc,d | 318 | Deacetylcephalosporin C hydroxylase (DACS).259–262 |
| cmcJc | 310 | 7α-Hydroxylase (CmcJ, 2OG oxygenase).109,330 |
| cmcIc | 236 | Methyl transferase (CmcI).109 |
| cefDc,d | 398c | Isopenicillin N epimerase (CefD); epimerisation of L- to D-α-aminoadipoyl side chain.246–248 |
| cefEc,d | 311 | Deacetylcephalosporin C synthase (DAOCS).259–262 |
| pcdc | 496 | Piperidine-6-carboxylate dehydrogenase (Pcd).125,704 |
| cmcTc | 489 | Efflux pump protein for the export of cephamycin C.125,704,705 |
| pbpAc | 409 | Protein involved in resistance against β-lactam antibiotics.704 |
| orf12d | 302 | Methyltransferase.115 |
| cpbKd/cpbId | 1056/5049 | Non-ribosomal peptide synthetase/polyketide synthase hybrid involved in biosynthesis of the C-3′-tetrapeptide of cephabacins.114,115 |
| t1d | ABC-transporter protein.115,700 | |
| t2d | ABC-transporter protein.115,700 | |
| t3d | ABC-transporter protein involved in secretion of cephabacins.700,706 | |
| blad | 385 | Protein involved in BLA resistance.115 |
The Gram-negative bacteria Flavobacterium sp.,93–95X. lactamgena96–99 and L. lactamgenus98,99 are able to further modify DAC to produce cephabacins which have a substituent at C-3′ comprising acetate and oligopeptide moieties, and may (e.g. cephabacin F group) or may not (e.g. cephabacin H group) contain 7α-formylamino98 or 7α-methoxy groups (e.g. cephabacin M group96,97) (Fig.8). The substituent at C-3′ of the cephem nucleus of cephabacins is proposed to be assembled by a non-ribosomal peptide synthetase/polyketide synthase hybrid encoded for by cpbI/cpbK.114,115
4.2 Biosynthesis of L-α-aminoadipic acid
The L-α-aminoadipoyl side chain of the tripeptide ACV is crucial for the biosynthesis of penicillins as substantiated by the observation that a mutant of P. chrysogenum blocked in the biosynthesis of L-α-aminoadipic acid only produced penicillin G when L-α-aminoadipic acid was added to the growth medium.116L-α-Aminoadipic acid is an intermediate in lysine biosynthesis in (penicillin-producing) fungi;117–119 penicillin production in P. chrysogenum is inhibited by lysine due to end-product control exerted by lysine decreasing the availability of α-aminoadipate for penicillin biosynthesis.120 Cephamycin-producing bacteria (e.g. Amycolatopsis lactamdurans121 and S. clavuligerus122) form lysine via the diaminopimelic acid pathway123 which does not involve L-α-aminoadipic acid as an intermediate. Thus, these organisms have a specific system to form L-α-aminoadipic acid from lysine via two enzyme-catalysed steps involving L-lysine-ε-aminotransferase (LAT, encoded by the lat gene)124 and piperideine-6-carboxylate dehydrogenase (P6CD, encoded by the pcd gene)125 (Fig. 9). Lysine-supplemented cultures of A. lactamdurans showed higher titres of cephamycin C,126 as did the overexpression of the lat gene from strong heterologous promoters.127 LAT employs 2OG, as an amino-group acceptor, and pyridoxal phosphate, as a cofactor, to enable deamination of lysine to give P6C.121,122,128 LAT also accepts L-ornithine and, to a lesser extent, N-acetyl-L-lysine as amino-group donors.119 P6CD converts P6C/NAD+ into L-α-aminoadipic acid/NADH.125,129 The specific activity of LAT peaks before cephamycins production and decreases as cephamycins are formed.124 P6CD does not accept pyrroline-5-carboxylate, an analogous intermediate in proline/carbapenem biosynthesis, as a substrate analogue.129 In S. clavuligerus, whilst the lat gene mutants lose the ability to produce cephamycins, the pcd disrupted mutants are still able to form 30–70% of wildtype levels, implying the existence of an alternative mechanism to convert P6C into L-α-aminoadipic acid.130–132 Cephamycin production by pcd mutants can be restored to wildtype levels either by addition of α-aminoadipic acid to the fermentation medium, or by complementation of the mutated strain with a copy of the pcd gene.1314.3 δ-(L-α-Aminoadipoyl)-L-cysteinyl-D-valine synthetase (ACVS)
The ATP-dependent ACVS reaction is common to the biosynthesis of all penicillins and hence all cephems (Fig. 9). ACVS is a monomeric133 multidomain nonribosomal peptide synthetase (NRPS) with a molecular mass of 404–426 kDa (depending on the source organism);134–137 for general reviews on NRPSs, see ref. 138–142. ACVS was the first fungal NRPS to be identified. At least under some conditions, LLD-ACV formation is proposed to be the rate-limiting step143,144 in the penicillin/cephalosporin biosynthesis.The ACVS encoding gene was designated as pcbAB (Fig. 10) because 2 genetic loci were initially thought to be responsible for ACV synthesis;145,146 however, genetic evidence indicates that a single gene encodes all the activities needed to form ACV.147–149 Comparison of the predicted amino acid sequence of ACVSs to those of NRPSs with biochemically-characterised domains150 reveals that ACVS possesses three modules (and probably at least ten catalytic domains). These modules contain different types of domains: (A) adenylation domain, (T) thiolation/pantothenylation/peptidyl carrier domain (which has a covalently attached 4′-phosphopantetheine prosthetic group), (C) condensation/peptide-bond formation domain, (E) epimerisation domain, and (TE) thioesterase domain. The domains within the two N-terminal modules of ACVS are arranged as (C/A/T) units. The C-terminal module of ACVS contains the (E) and (TE) domains (Fig. 11B).151,152
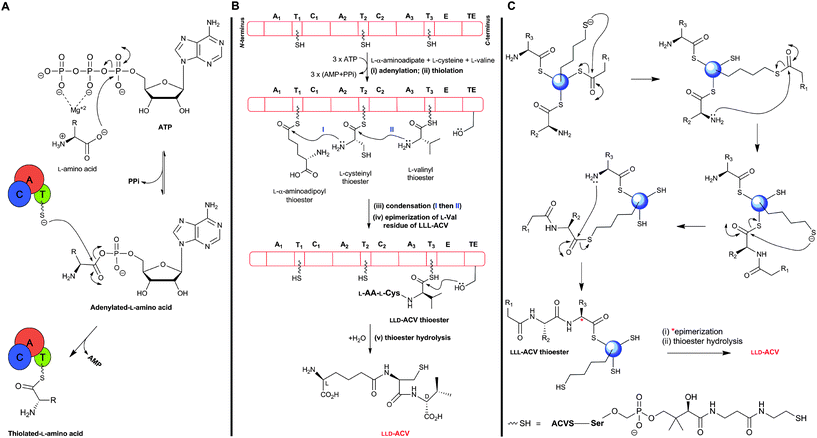 | ||
| Fig. 11 Outline potential mechanism for δ-(L-α-aminoadipoyl)-L-cysteinyl-D-valine synthetase (ACVS). (A) Amino acid substrates are activated by adenylation followed by transfer to the thiol of the 4′-phosphopantetheinyl cofactor(s); (B) Proposed domain organisation of ACVS outlining the multiple carrier thiotemplate mechanism. A1–A3: adenylation domains, T1–T3: thiolation domains, C1 and C2: condensation domains, E: epimerisation domain, and TE: thioesterase domain. Each amino acid is recognized and activated by its cognate adenylation domain (A), and attached as a thioester to the 4′-phosphopantetheinyl cofactor at the corresponding thiolation domain (T). Peptide bond formation is catalysed by the condensation domain (C). The L-valine residue of the tripeptide LLL-ACV is isomerised, at the α-carbon, by the epimerisation domain (E) followed by release of the final LLD-ACV by the thioesterase domain (TE); (C) Proposed role of the 4′-phosphopantetheinyl moiety as a “swinging arm” in peptide bond formation. | ||
Unlike (normal) ribosomal peptide synthesis, NRPS can accommodate modified residues, as exemplified in the case of ACV/ACVS by the presence of an L-α-aminoadipate and a D-residue. The mechanism of ACVS catalysis (Fig. 11), like those of other NRPSs, is proposed to operate via a thiotemplate mechanism involving the recognition and activation of the substrate (L-amino acid) carboxylate group via adenylation (in (A) domain), followed by thiolation (in (T) domain) where transfer of the activated amino acid is coordinated by the 4′-phosphopantetheine cofactor (which acts as a “swinging arm”) to the condensation domain (C) for peptide bond formation. There is evidence that epimerisation of L-valine to the D-configuration, catalysed by the (E) domain, takes place at the tripeptide LLL-ACV level.153 Finally, release of the enzyme-bound LLD-ACV occurs through the action of the (TE) domain.105,151
Thiol-blocking agents (e.g. N-ethylmaleimide and iodoacetamide) inhibit ACVS activity revealing the importance of thiol groups in ACVS catalysis.136,154 The conversion of the apo-form of ACVS into its active form, via transfer of a phosphopantetheinyl moiety from coenzyme A to a conserved serine residue in the three (T) domains of ACVS, is catalysed by a 4′-phosphopantetheinyl transferase (PPTase). A gene encoding for a specific PPTase in Aspergillus nidulans has been reported to be essential for penicillin biosynthesis.155,156
Studies on the mechanism of the ACVS-catalysed L-valine epimerisation reveal that D-valine is not a substrate for ACVS and does not significantly induce ATP/PPi exchange.154,157,158 To date, enzyme-bound thioester intermediates have not been detected in ACVS-catalysis. However, substrate analogue studies in which cysteine was replaced by L-O-methylserine led to isolation of both L-O-methylserinyl-L-valine and L-O-methylserinyl-D-valine dipeptides, implying that epimerisation occurs after formation of (at least one) peptide bond.159,160 Modification of the (TE) domain of A. nidulans ACVS (by substituting Ser3599 in the conserved GXSXG motif for alanine, aiming to block the final hydrolytic step and trap an enzyme-bound intermediate) led to a ∼95% decrease in penicillin production.153 The purified ACVS S3599A variant showed a ∼50% reduction in peptide formation rate, with LLL-ACV being the major product implying that (i) the (TE) domain activity is linked to the (E) domain, (ii) Ser3599 is not essential for peptide release but likely involved in selecting the isomer to be released,153 and (iii) the intermediacy of LLL-ACV in penicillin biosynthesis. The epimerisation step possibly takes place at the enzyme-bound thioester level of LLL-ACV105,137,161 (similarly to that in actinomycin biosynthesis162). As the epimerisation step is likely to be reversible,137,141,161 a selection mechanism for LLD-ACV hydrolysis over LLL-ACV is proposed (note that LLL-ACV is not a substrate for the next enzyme in the pathway, i.e. IPNS). Mutagenesis studies on P. chrysogenum ACVS163 revealed that residues E3371, H3373, R3375 and E3376 are essential for the epimerase activity. Overexpression of the sequence encoding the (TE) domain of ACVS complemented mutants lacking the (TE) domain suggested that a “stand-alone” (TE) domain can recognize and interact with the other ACVS domains.163
Wildtype ACVS has been found to accept a variety of substrate analogues,105e.g. ACVS has been reported to substitute L-S-carboxyethylcysteine for L-α-aminoadipic acid, DL-homocysteine, L-S-methylcysteine, L-cystathionine, allylglycine and vinylglycine for L-cysteine (demonstrating that the thiol group of cysteine is not essential for peptide formation), and L-allo-isoleucine, L-α-aminobutyrate, L-norvaline, L-allylglycine or L-leucine for L-valine.154,158,164–166 These findings suggest that the biosynthesis of unnatural peptides for use as IPNS-substrates might be possible via the use of ACVS engineered variants.
Structural information on ACVS is of interest not only to reveal how an unusually small NRPS operates, but also because it may guide the rational redesign of ACVS to produce unnatural peptides. Progress has been made with structural studies on other NRPSs (e.g. the X-ray structure of a termination module (four domains) in Bacillus subtilis,167–169 (T/C) didomain structure in tyrocidine synthetase, and (T/TE) in enterobactin synthetase170,171), suggesting that ACVS may be amenable to crystallographic analyses.
4.4 Isopenicillin N synthase
The IPNS-catalysed reaction is exceptional in that a densely functionalised bicyclic ring is formed in a single four-electron oxidation step from a simple tripeptide (Fig. 9).106,172 Only the reduced form of LLD-ACV is a substrate for IPNS, and there is evidence that, in P. chrysogenum, LLD-ACV is maintained in the reduced form by a thioredoxin reductase system.173 The pcbAB and pcbC174–178 genes, which encode for ACVS and IPNS, respectively, are always adjacent in biosynthesis gene clusters (Fig. 10), suggesting potential co-evolution. IPNS is one of few identified non-heme iron enzymes that do not require a co-factor for oxygen activation (unlike other studied 2OG oxygenases involved in BLA biosynthesis; Section 4.7).24 Reducing agents (e.g. DTT and ascorbate) stimulate the activity of IPNS which is sensitive to thiol-specific inhibitors (e.g. N-ethylmaleimide).179,180 Metal ions (e.g. CoII, ZnII and MnII) inhibit IPNS activity probably by competition with FeII.180,181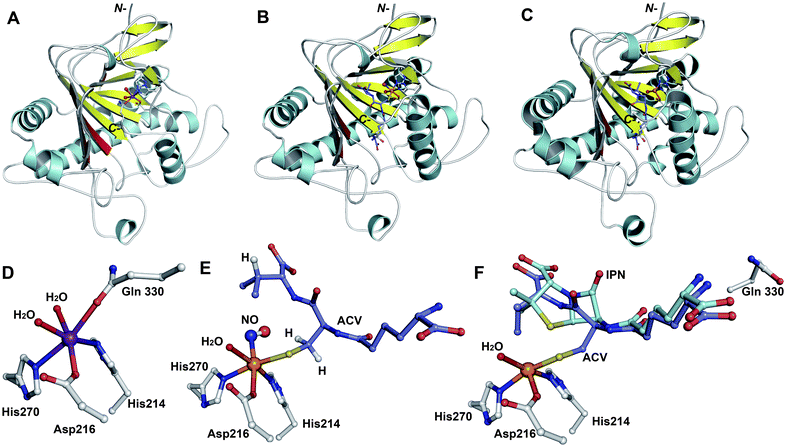 | ||
Fig. 12 Structural views of isopenicillin N synthase (IPNS). A and D: IPNS-MnII complex (PDB 1IPS). MnII, which substitutes for FeII, is in violet; B and E: IPNS-FeII-ACV-nitric oxide complex (PDB 1BLZ).189 FeII is in orange; C and F: IPNS-FeII-IPN complex (with ACV superimposed in case of F, PDB 1QJE). Note that the double stranded β-helix core fold (in yellow) – which supports the 2-histidine-1-aspartate iron binding motif – is a conserved structural feature for the non-heme iron(II) oxygenase/oxidase superfamily. The coordination site occupied by NO is highly likely to be the same as that for O2. It is proposed that the presence of O2trans to Asp216 prompts the valinyl-isopropyl group rotation about its Cα-Cβ bond (before/coupled to β-lactam formation) to direct the valine β-proton towards the FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O for reaction.182,189,196,214 The contracted bicyclic structure of IPN relative to that of ACV is proposed to reduce the efficiency of interactions including at the carboxylate termini, and so promote product release.707 O for reaction.182,189,196,214 The contracted bicyclic structure of IPN relative to that of ACV is proposed to reduce the efficiency of interactions including at the carboxylate termini, and so promote product release.707 | ||
O] species; concomitantly, the valine nitrogen can nucleophilically attack the cysteinyl β-carbon, resulting in formation of a monocyclic β-lactam (Fig. 13A).204 Generation of a high-valent FeIVO species is a common theme among non-heme iron oxidases/oxygenases and is normally generated by oxidative decarboxylation of the stereotypical co-substrate 2OG.205 Uniquely in IPNS catalysis, the FeIVO species is generated by the 2-electron oxidative cyclisation of a thiol-containing substrate, i.e. ACV. After β-lactam ring formation, the FeIVO species is proposed to react with the C–H bond of the valine β-carbon to generate a carbon-radical206 and a ferric-hydroxy [FeIII–OH] species. Notably, structural studies imply that the valine β-C–H bond is directed away from the iron centre (Fig. 12E, 13A) and rotation of the valine isopropyl group must occur (likely coupled to β-lactam ring formation) to direct the valine β-proton towards the FeIVO for productive reaction.207,208 The valine-β-carbon radical can then react with the cysteine-thiolate to form the thiazolidine ring of isopenicillin N with retention of configuration at the valinyl β-carbon (Fig. 13A).209,210 Independent deuterium kinetic isotope effects on both the cysteinyl β-carbon and the valinyl β-carbon suggest that both C–H bond cleavage steps are (at least partially) rate-limiting.211 The ring formation sequence is proposed to be determined in part by the relative strengths of the two C–H bonds with the FeIVO species being preferred for cleavage of the stronger valine β-C–H bond.195 The question of why IPNS catalyses ring formation rather than hydroxylation of C–H bonds has been addressed by density functional modelling studies, which suggest that the barrier for substrate hydroxylation is ∼4 kcal mol−1 higher than that for ring closure.195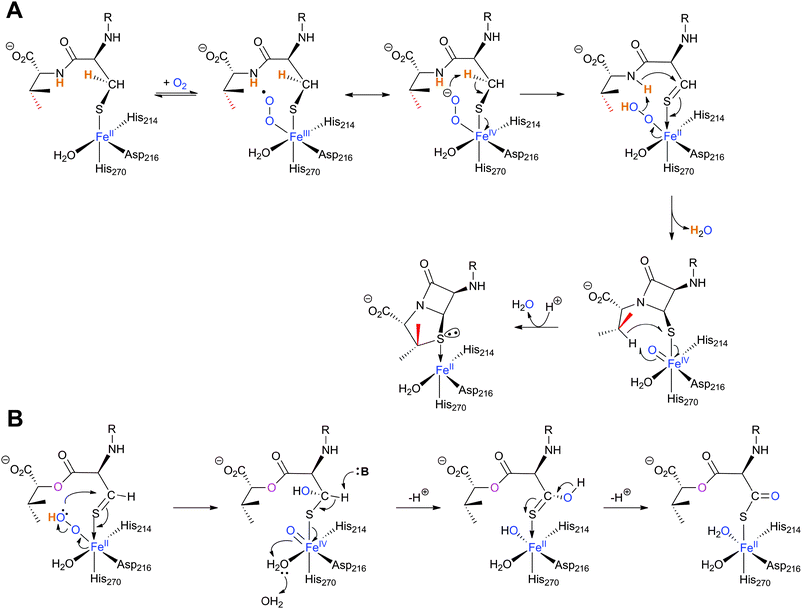 | ||
| Fig. 13 Proposed mechanism for isopenicillin N synthase catalysed conversion of the natural substrate LLD-ACV into isopenicillin N (A) and that of the substrate analogue δ-(L-α-aminoadipoyl)-L-cysteine D-α-hydroxyisovaleryl ester (ACOV) into a thiocarboxylate product (B).196,224,708 R = δ-(L-α-aminoadipoyl). ACOV is near isosteric to LLD-ACV. Note that in the absence of its natural reaction partner (the N–H proton of the L-cysteinyl-D-valine amide bond), the proposed hydroperoxide intermediate can react with the putative thioaldehyde intermediate. | ||
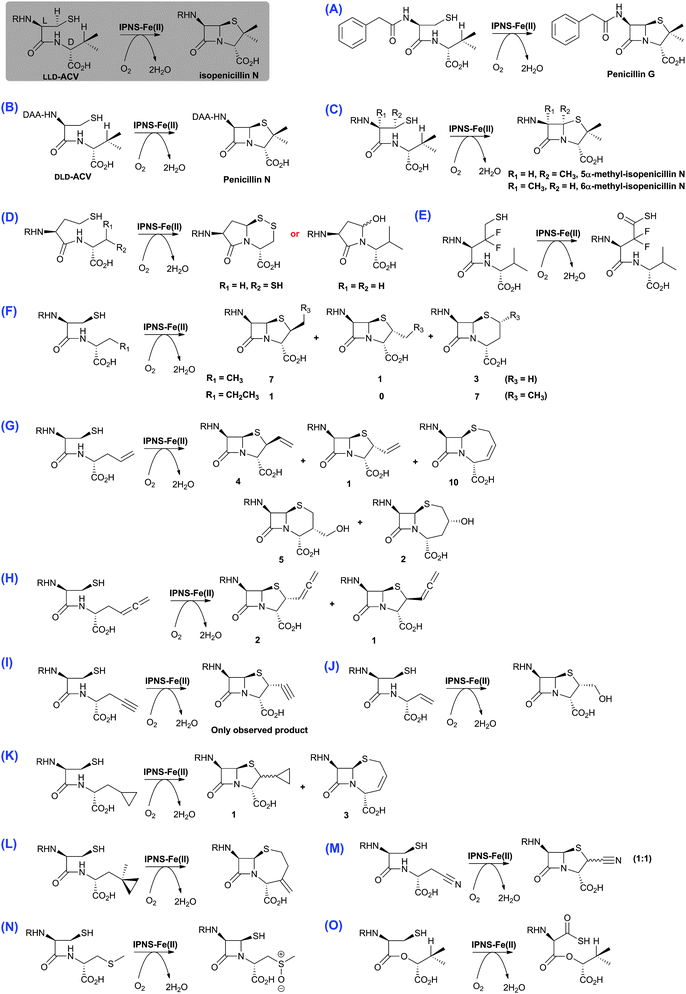 | ||
| Fig. 14 The biocatalytic versatility of isopenicillin N synthase.191,196,210,213,215–218,222–224 These reactions exemplify the oxidase and oxygenase activities of IPNS. The approximate ratios of observed products are given below/beside the structures; in some cases, these ratios were perturbed by use of deuterated substrate analogues. Note in the cases of (N) and (O), the shown products were observed in crystal as part of IPNS-product complexes.708,709 The reaction of IPNS with its natural substrate is boxed. R = δ-(L-α-aminoadipoyl); DAA = δ-(D-α-aminoadipoyl). | ||
Although studies on oxygenases related to IPNS reveal that they too can accept a variety of substrate analogues (e.g. DAOCS and CAS, Sections 4.7 and 5.3, respectively), the available evidence is that IPNS is unusually promiscuous; this may be because, unlike the 2OG oxygenases, it only uses O2 as a co-substrate, and that its prime substrate (i.e.LLD-ACV) is actually complexed to the active site iron. It should also be noted that almost all the substrate analogue studies on IPNS have been carried out on wildtype proteins, thus there is considerable chance for mutational studies to further expand the scope of IPNS reactivity, as well to optimise the formation of specific products (as has been demonstrated for DAOCS, Section 4.7).
4.5 Biosynthesis of the hydrophobic penicillins – isopenicillin amidohydrolase/acyltransferase
The final stages in the biosynthesis of penicillin G and other hydrophobic penicillins in P. chrysogenum are catalysed by an acyl-coenzyme A: isopenicillin N acyltransferase (AT, encoded by penDE) (Fig. 15A). AT catalyses the exchange of the hydrophilic α-aminoadipoyl side chain of isopenicillin N for an aromatic acyl side chain via 6-APA (Fig. 9 and 15A).225–228 In order for the acyl-transfer to occur, prior activation of the (aromatic) acid to a CoA thioester by a specific ligase is required229–231 (AT also accepts glutathione-activated aromatic acids232). Note that AT is distinct from a penicillin amidohydrolase (from P. chrysogenum) which catalyses the hydrolysis of penicillin V, and to a lesser extent penicillin G, to give 6-APA.233 AT thus has (IPN) amidohydrolase, (6-APA) acyl-transferase, as well as penicillin transacylase activities228,234 (Fig. 15). The intermediacy of 6-APA in AT catalysis is supported by the finding that 6-APA is a better substrate than isopenicillin N to produce penicillin G, in the presence of phenylacetyl-CoA.225 7-Aminocephalosporanic acid (7-ACA) is not a substrate for AT.225 AT accepts a range of acyl-CoA derivatives of both aromatic and aliphatic nature.235–237 Only aliphatic carboxylic acids substrates with 6–8 carbon chain length were accepted in vitro by AT, consistent with the observation that penicillin K, F and dihydropenicillin F (Fig. 8) occur in the fermentation broths of P. chrysogenum together with penicillin G, when no phenylacetic acid was added as a precursor.236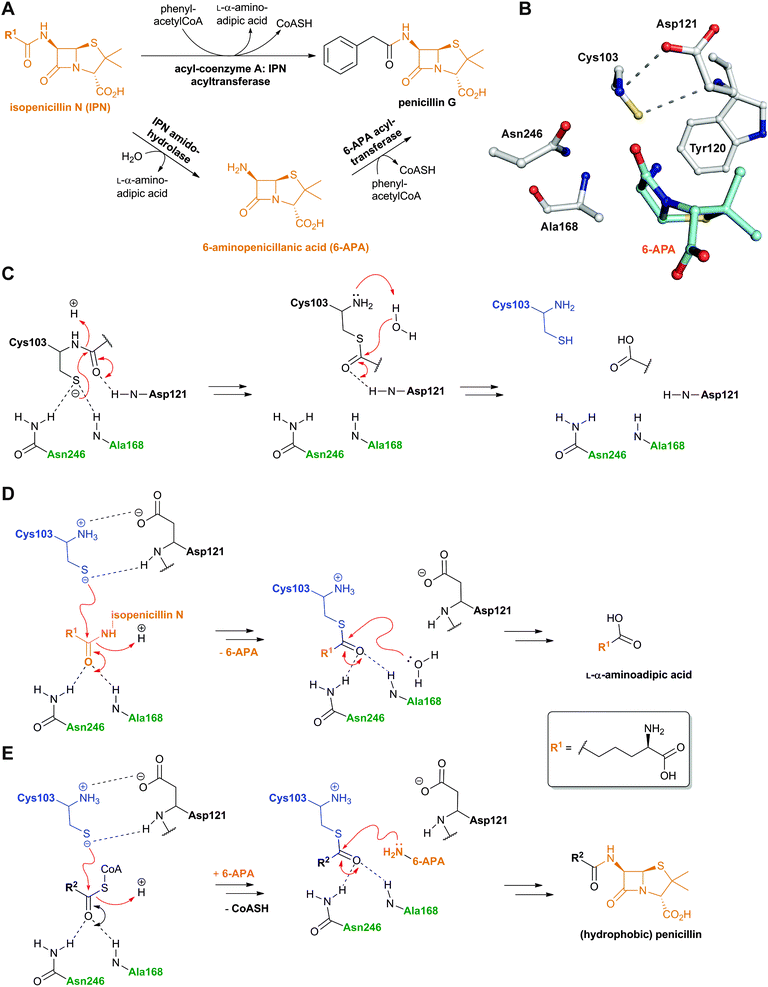 | ||
| Fig. 15 Reactions catalysed by acyl-coenzyme A: isopenicillin N acyltransferase (AT) and outline AT mechanisms. A: AT catalyses amidohydrolase and 6-APA acyl-transferase reactions;228,234B: View from mature AT:6-APA complex (PDB 2X1E);241C: Outline mechanism of AT autoproteolysis; D: The amidohydrolase activity of AT; E: The acyltransferase activity of AT. The oxyanion hole forming residues are in green and the Ntn residue is in blue. R2 can be a variety of aliphatic or aromatic side chains. See Fig. 43 and 62 for related Ntn enzyme mechanisms. | ||
Like the N-acetylornithine:glutamic acid acetyltransferase OAT2238 and the pantetheine hydrolase ThnT,239 from the clavulanic acid and thienamycin biosynthesis pathways (Sections 5.11 and 6.4.3, respectively), AT is a member of the N-terminal nucleophile (Ntn)-hydrolase superfamily.73,74 Despite the often low sequence homology among Ntn-hydrolases, the enzymes share a conserved αββα-core structure (Ntn fold, Fig. 16) and conserved catalytic residues.74 AT, like other Ntn-hydrolases, is produced as a single polypeptide chain (40 kDa inactive precursor);240 autocatalytic cleavage of the Gly102–Cys103 peptide bond produces the active α,β-heterodimeric protein (11 kDa α- and 29 kDa β-subunits) in which the N-terminal cysteine of the β-subunit acts as a nucleophile during AT-catalysis (Fig. 15B).240,241 It is notable that the penicillin acylases (which are used industrially to produce 6-APA from penicillin G) and cephalosporin acylases (which produce 7-ACA)242–245 (Section 4.10) are also members of the Ntn-hydrolase superfamily.74
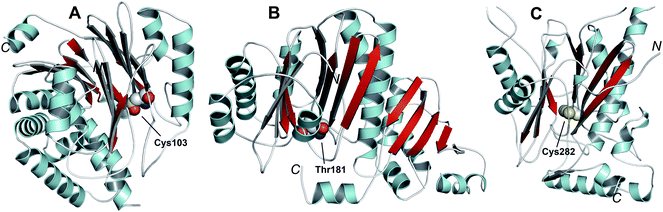 | ||
| Fig. 16 Members of the N-terminal nucleophile (Ntn) family of hydrolases involved in β-lactam antibiotic biosynthesis. A: α,β-Monomer of the mature acyl coenzyme A:isopenicillin N acyltransferase (AT, PDB 2X1E) from Penicillium chrysogenum;241B: 1 subunit/2 chains of ornithine acetyltransferase (OAT-2, PDB 2YEP, Section 5.11) from Streptomyces clavuligerus;491C: Monomer of the uncleaved ThnT T282C (PDB 3S3U, Section 6.4.3) from S. cattleya.587 Note the conserved αββα-core structure.74 The N-terminal nucleophilic residue (or T282C variant for C) is shown in space-filling mode. | ||
A recently reported crystal structure of P. chrysogenum AT241 (Fig. 16A) is of interest because it suggests how AT can accommodate a variety of substrate side chains, and, importantly, why it does not accept cephalosporins.241 Modelling studies based on the AT structure suggest that there is insufficient room to accommodate the cephalosporin carboxylate which is located at the sp2-hybridised C-2 of the cephem nucleus, but there is room for the penicillin carboxylate which is located at the sp3-hybridised C-2 of the penam nucleus (Fig. 5 in ref. 241). It is proposed that the structural work may guide protein engineering studies aiming at producing AT variants that accept cephalosporin substrates.241
4.6 Isopenicillin N/penicillin N epimerase
Prokaryotic isopenicillin N epimerase (CefD) catalyses the reversible epimerisation of the L-α-aminoadipoyl side chain in isopenicillin N to the D-α-aminoadipoyl side chain in penicillin N, in a pyridoxal phosphate dependent manner, likely via a mechanism involving imine/enamine type intermediates, as proposed for related epimerases (Fig. 17).246–248 CefD catalyses an essential step in cephalosporin biosynthesis because isopenicillin N is not a substrate for DAOCS/DACS (CefEF) or DAOCS (CefE), and thus may have acted to enable a “branch point” between the biosyntheses of cephalosporins and the hydrophobic penicillins (Fig. 9). The CefD encoding gene (cefD) is located adjacent to that encoding for DAOCS (cefE) in both S. clavuligerus (Fig. 10) and Amycolatopsis lactamdurans.249–251 The two genes are co-transcribed so as to, it is proposed, ensure that penicillin N availability is not limiting for DAOCS-catalysed ring expansion.249,251 The net result is proposed to shift the isopenicillin N/penicillin N equilibrium towards the latter. The conversion of ACV analogues to cephalosporins using partially purified S. clavuligerus extracts provides evidence that the prokaryotic epimerase may tolerate modifications in the penam nucleus.252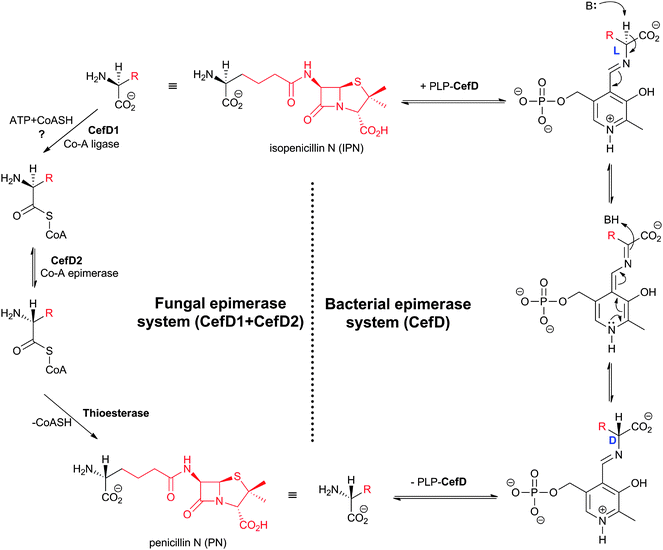 | ||
| Fig. 17 Comparison of the proposed mechanisms for conversion of the L-aminoadipoyl side chain of isopenicillin N into the D-aminoadipoyl side chain of penicillin N in bacterial and fungal cephalosporin-producers. The isopenicillin N epimerase in cephalosporin-producing bacteria (i.e. CefD) is pyridoxal phosphate (PLP)-dependent; an alternative isopenicillin N epimerisation system (comprising both CefD1 and CefD2) operates in fungi.248,253 CefD1 is homologous to long chain acyl-CoA synthetases710 and CefD2 is homologous to acyl-CoA racemases.711,712 Penicillin N release is proposed to be catalysed by an unidentified thioesterase.713 The fungal isopenicillin N epimerisation system is likely similar to those involved in phytanic acid and ibuprofen racemisation.253,714 | ||
The fungal epimerisation system (e.g. from A. chrysogenum) is proposed to be different from that involved in prokaryotic BLA biosynthesis and to involve three enzyme-catalysed steps (Fig. 17): activation of isopenicillin N to isopenicillinyl-CoA by a CoA-ligase (encoded by the cefD1gene), epimerisation to penicillinyl-CoA by an acyl CoA-epimerase (encoded by the cefD2 gene), and finally hydrolysis to penicillin N by, as yet, an unidentified thioesterase.253,254
4.7 Deacetoxycephalosporin C synthase (DAOCS) and deacetylcephalosporin C synthase (DACS)
In the eukaryote A. chrysogenum, penicillin N ring expansion (to give deacetoxycephalosporin C, DAOC) and the subsequent C-3′-hydroxylation (to give deacetylcephalosporin C, DAC) are catalysed by a single bifunctional enzyme DAOCS/DACS (encoded by cefEF) (Fig. 9).255–258 In contrast, in the prokaryote S. clavuligerus, the ring expansion and hydroxylation reactions are catalysed by two distinct but closely related enzymes (DAOCS and DACS, respectively),259–262 suggesting that a gene duplication event may have occurred during evolution which resulted in two homologous enzymes catalysing different reactions.262,263 In fact DAOCS displays a low level of DACS activity and vice versa.262 Further, the genes encoding for DAOCS (cefE) and DACS (cefF) from S. clavuligerus show extensive homology to cefEF from A. chrysogenum187 consistent with a proposed horizontal transfer of the entire pathway from bacteria to fungi.101,148,264–266O intermediate is generated by the oxidative decarboxylation of 2OG to succinate which leaves the active site before penicillin N binding and reaction (see below).269,273 For more detailed descriptions of structural and mechanistic studies on 2OG oxygenases, see ref. 24, 25.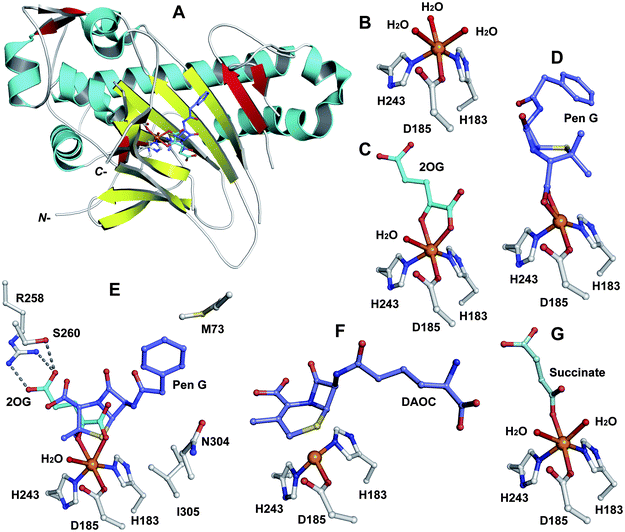 | ||
| Fig. 18 Views from crystal structures of DAOCS. A: view from the overall fold of a monomer of DAOCS:FeII:2OG:penicillin G (a substrate analogue) complex showing the conserved double stranded β-helix (in yellow) (PDB 1UOB); B to G: views from a DAOCS:FeII complex (B, PDB 1RXF), a DAOCS:FeII:2OG complex (C, PDB 1RXG), a DAOCS:FeII:penicillin G complex (D, PDB 1UOF), a DAOCS:FeII:2OG:penicillin G complex (E, PDB 1UOB), a DAOCS:FeII:DAOC complex (F, PDB 1UOG) and a DAOCS:FeII:succinate complex (G, PDB 1UO9). FeII (in orange), conserved active site residues, substrates/co-factors and products are shown. Note that in case of (E) the reported analysis of the electron density reveals that the penicillin G and 2OG binding sites overlap, leading to the proposal of an atypical 2OG oxygenase mechanism (Fig. 19).273 | ||
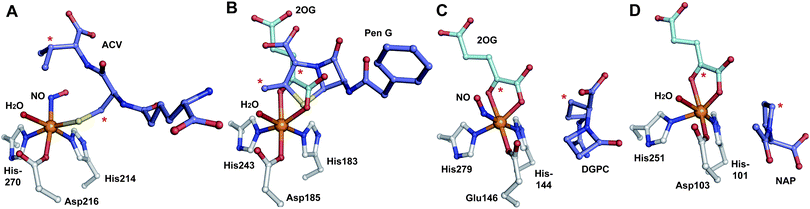 | ||
| Fig. 19 Views from crystal structures of FeII-dependent oxidases/oxygenases involved in β-lactam biosynthesis. In all cases, note the highly conserved 2-histidine-1-carboxylate motif. A: IPNS active site with ACV and NO (an O2 analogue) complexed (PDB 1BLZ);189B: DAOCS active site with penicillin G and 2OG showing the reported substrate/cosubstrate overlapping binding sites (PDB 1UNB);273C: Clavaminic acid synthase (CAS, Section 5.3) active site with NO and its substrate for hydroxylation (PDB 1GVG);420D: The carbapenem synthase (CarC, Section 6.3.3) active site with the substrate analogue, N-acetyl-proline (NAP), and 2OG bound (PDB 1NX8).542 Overall, the figure illustrates how a conserved binding chemistry can be used to mediate different types of oxidation reactions. *The carbon(s) to be oxidised. | ||
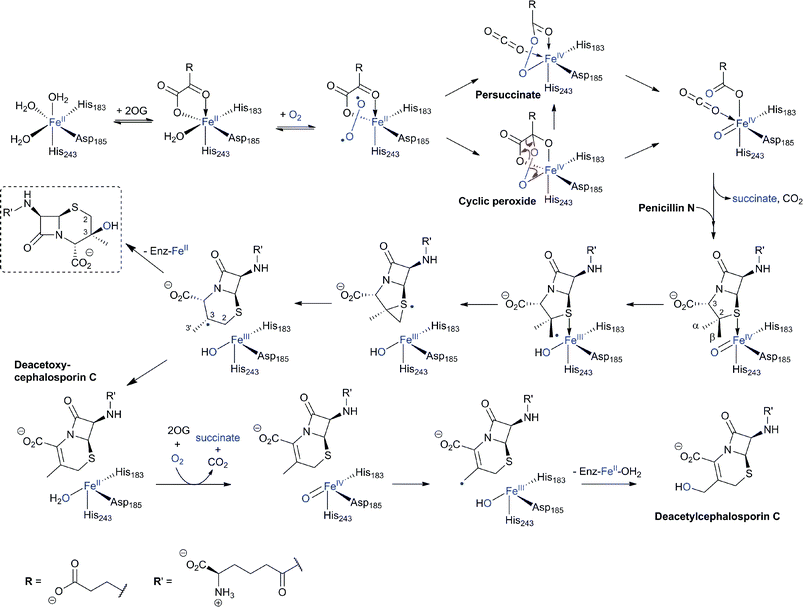 | ||
| Fig. 20 Proposed outline mechanisms of DAOCS/DACS catalysis. In the ring-expansion of penicillin N to DAOC, an FeIVO intermediate is proposed to be generated in a “typical” 2-electron oxidation process characteristic for 2OG oxygenases. In the modified proposed mechanism for ring expansion as catalysed by DAOCS, succinate is proposed to leave the active site before penicillin N binding.269,273 Further validation is required for this mechanism. The 3β-hydroxy-3α-methylcepham (in the dashed box) is formed by DAOCS/DACS catalysis as a minor “shunt” product in case of the ring expansion of penicillin N. Its yield is increased by the introduction of a deuterium atom at the C-2 position of penicillin N (due to a kinetic isotope effect) indicating a common intermediate prior to the branch point.299,300 In the DAOC hydroxylation by DACS, another 2-electron process results in the C-3′ hydroxylation of DAOC to produce DAC. | ||
Cell-based feeding studies employing (3-pro-R)-valine, with a stereogenic methyl group at the position that forms the endocyclic C-3 of cephalosporin C, reveal complete loss of stereochemical integrity during ring expansion (Fig. 21IIA).294–296 In contrast, experiments employing (3-pro-S)-valine, with a stereogenic methyl group at the position that forms the C-3′ exocyclic methylene of cephalosporin C, reveal (partial) retention of stereochemistry (Fig. 21IIB).294–296 The observed loss of stereochemistry during the ring expansion process suggested the existence of a penicillin N-derived β-methylene radical intermediate, which can undergo rotation before ring expansion. This suggestion is supported by biomimetic studies.297,298 Although it may be related to the allylic nature of the intermediate, the retention of stereochemistry in the hydroxylation reaction is of interest with regard to 2OG oxygenases catalysis because it implies that hydroxylation can occur via a process not involving a “long-lived” methylene radical (e.g. direct insertion or a very fast recombination process). Incubation of [3-2H]-penicillin N and DAOCS/DACS has also shown that, in the ring expansion process, one hydrogen from the β-methyl group is lost prior to the hydrogen at C-3 position.299,300 Notably, the incubation resulted in an increased level of 3-β-hydroxy-3-α-methylcepham,299,300 a normally minor by-product, which has been previously isolated from A. chrysogenum fermentation broths.301 A summary of mechanistic proposals for DAOCS/DACS is given in Fig. 20.
![I: The biocatalytic versatility of DAOCS/DACS catalysis. For further examples see ref. 304 and ref. 280 for wildtype and engineered DAOCS-catalysed reactions, respectively. The reaction of DAOCS/DACS with its natural substrate is boxed; II: Feeding studies employing valine labeled with a chiral methyl group at the (3-pro-R) position reveal loss of stereochemical integrity during ring expansion;294–296 experiment using valine with a chiral methyl group at the (3-pro-S) position imply retention of stereochemistry.294–296 Where reported, the approximate ratios of observed products are given below the structures; in the case of the [4-2H]-exomethylene analogue reaction (H), the ratio of products varies over time.313,317](/image/article/2013/NP/c2np20065a/c2np20065a-f21.gif) | ||
| Fig. 21 I: The biocatalytic versatility of DAOCS/DACS catalysis. For further examples see ref. 304 and ref. 280 for wildtype and engineered DAOCS-catalysed reactions, respectively. The reaction of DAOCS/DACS with its natural substrate is boxed; II: Feeding studies employing valine labeled with a chiral methyl group at the (3-pro-R) position reveal loss of stereochemical integrity during ring expansion;294–296 experiment using valine with a chiral methyl group at the (3-pro-S) position imply retention of stereochemistry.294–296 Where reported, the approximate ratios of observed products are given below the structures; in the case of the [4-2H]-exomethylene analogue reaction (H), the ratio of products varies over time.313,317 | ||
Here we highlight some of the interesting substrate analogue studies. Mutant strains of A. chrysogenum are reported to produce a cephem-3-aldehyde, probably originating from DAC oxidation.303,304 DAOCS/DACS has been shown to catalyse the oxidation of DAC to the corresponding aldehyde (Fig. 21IA).304 Incubation of 6α-methylpenicillin N with DAOCS/DACS resulted in the formation of the corresponding cephem aldehyde (Fig. 21IA); however, incubation with DACS resulted only in uncoupled turnover of 2OG.305 While IPNS accepts peptide substrates with both L- and D- N-terminal δ-(α-aminoadipoyl) side chains, DAOCS/DACS does not accept isopenicillin N (with δ-(L-α-aminoadipoyl) side chain), but does accept the adipoyl-penicillin analogues (Fig. 21IB).252,306–308 A 2β-methyl-penam derivative (equivalent to penicillin N without the 2α-methyl group) was converted by DAOCS/DACS into a C-3-demethylated DAOC (Fig. 21IC).143 As for IPNS, chemically interesting substrate analogues studies have comprised alkene (Fig. 21ID and E),309 C-3 exo-alkene (Fig. 21IF, H and I),310–314 and cyclopropane (Fig. 21IG)315 containing substrate analogues. In case of the exomethylene substrate analogue reaction, DAC was the main observed product (Fig. 21IF);312 however, in case of the [4-2H]-exomethylene analogue reaction, DAC, a 3β-spiro-epoxide cepham, and an aldehyde were also detected (Fig. 21IH), suggesting that the first irreversible event in this conversion involves FeIVO addition to the double bond (and not C-4 hydrogen abstraction).310,313,316,317 In case of the Z-3-ethylidene substrate analogue reaction, two diastereomeric alcohols were detected (Fig. 21II).318 In the case of a cyclopropane-containing analogue, a rearranged product was obtained (Fig. 21IG).315
4.8 Introduction of the 7α-methoxy and formamido-groups
Two types of 7α-functionalised cephalosporins have been identified: the cephamycins,319–321 which contain a 7α-methoxy group, and (some) cephabacins, which contain a 7α-formylamino group (Fig. 8).322 Both sub-classes show potent antibacterial activity and enhanced resistance to serine β-lactamases.323,324 Whilst the origin of the 7α-formylamino group is unknown, pioneering labeling studies by Abraham et al. demonstrated that the methyl group and the oxygen of the 7α-methoxy group are derived from methionine and O2, respectively.325 The methoxylation reaction, in cell-free extracts of S. clavuligerus, was shown to be dependent on FeII, 2OG and S-adenosylmethionine (SAM).326O-Carbamoyl DAC was found to be a better substrate for 7α-methoxylation than DAOC; DAC was apparently not methoxylated.326 These results suggest that the sequence of events in cephamycin C biosynthesis proceeds via conversion of DAOC to DAC to O-carbomoyl DAC327,328 to cephamycin C326 (Fig. 9). Hood et al. provided evidence that the methoxylation reaction is a two-step process, by isolation and characterisation of the 7α-hydroxy-O-carbamoyl DAC using S. clavuligerus extracts but excluding SAM.329 Bioinformatic analyses of the cephamycin biosynthesis gene cluster have revealed that cmcI and cmcJ likely encode for a methyl-transferase and a 7α-hydroxylase, respectively.330 However, as yet, no activity has been reported for the recombinant enzymes, and it may be that they work in concert.331,332 A crystal structure has been reported for CmcI in complex with SAM (Fig. 22).109 CmcI was initially annotated as a cephalosporin hydroxylase,333 but no evidence for FeII/2OG binding could be found.109 Despite its low sequence identity (8–23%) to SAM-dependent methyltransferases, the CmcI structure is related to that of catechol O-methyltransferase with overlapping nucleotide-binding residues.109 Attempts to obtain a CmcI structure in complex with cephalosporin C or O-carbamoyl DAC have, to date, been unsuccessful and potential substrates were therefore docked (Fig. 7 in109); the docking results show a binding mode for 7α-hydroxy-O-carbamoyl DAC which would allow methyl transfer to the C-7 hydroxyl group.109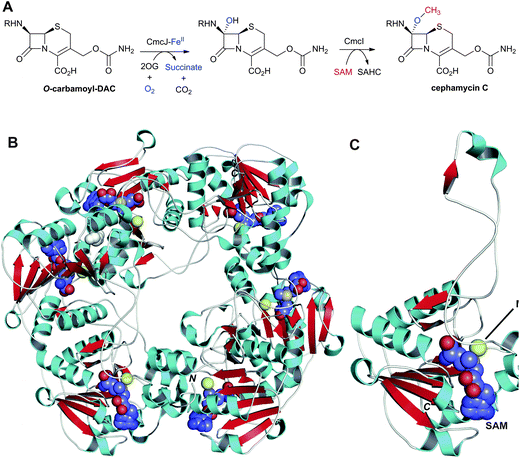 | ||
| Fig. 22 Enzymes responsible for the C-7 methoxylation of cephalosporins. A: Enzymes proposed to catalyse C-7 methoxylation of O-carbamoyl-DAC; B and C: Views from a crystal structure of CmcI109 from S. clavuligerus (PDB 2BR4). B: The hexameric quaternary structure of CmcI; C: A CmcI monomer. The S-adenosylmethionine (SAM) cofactor, shown in space-filling mode, is employed by CmcI to methylate the 7α-hydroxyl group of the O-carbamoyl-DAC to give cephamycin C and S-adenosylhomocysteine (SAHC). Magnesium(II) is in yellow. The 7α-hydroxyl group is likely introduced by a 2OG oxygenase, CmcJ.330 The geometry of the SAM/MgII binding site is similar to that found in cathechol O-methyltransferases.109 R = δ-(D-α-aminoadipoyl). | ||
4.9 Modifications at the cephem C-3′ position
Studies on the biosynthesis of cephalosporins and cephamycins substituted at the 3′-position have revealed DAC as a common precursor.334 However, in case of the cephamycins, the sequence of events leading to 3′-hydroxyl and C-7 modifications is as yet unclear. Docking studies employing a CmcI crystal structure (Section 4.8) suggest that, in the biosynthesis of cephamycin C, the 3′-hydroxyl carbamoylation may occur prior to the C-7 methoxylation.109Acetylation of the C-3-hydroxymethyl function of DAC, the final step in cephalosporin C biosynthesis, is catalysed by acetyl-CoA:DAC O-acetyltransferase (DAC-AT/CefG, encoded by cefG, Fig. 9). The presence of an acetyltransferase in the cephalosporin C biosynthesis pathway was initially predicted on the basis of accumulation of DAC in the culture broth of A. chrysogenum mutants.335,336 The CefG activity was first demonstrated in cell-free extracts of A. chrysogenum where DAOC was converted into cephalosporin C with incorporation of [14C]-label when acetyl-1-14C-CoA was added.337 Subsequent studies enabled purification of CefG338–341 and the cloning of cefG.342,343 Initially CefG was reported as a heterodimer of two subunits with molecular weights of ∼27 and 14 kDa;340 however, the enzyme has been shown later to be active only as a single polypeptide chain (i.e. is not processed into subunits).341 Crystal structures of CefG (Fig. 23) reveal that the enzyme belongs to the α/β-hydrolase class of acetyltransferases.344 Sequence334 and structural comparisons indicate that CefG is similar to homoserine-O-acetyltransferases. The CefG active site (Fig. 23B) has a conserved His362-Asp333-Ser149 catalytic triad involved in the acetyl group transfer; Ser149 is proposed to be acetylated by acetyl coenzyme A followed by transfer of the acetyl group to DAC through a tetrahedral intermediate stabilised in an oxyanion hole formed by the backbone –NHs of Leu59 and Met150 (Fig. 23C).344
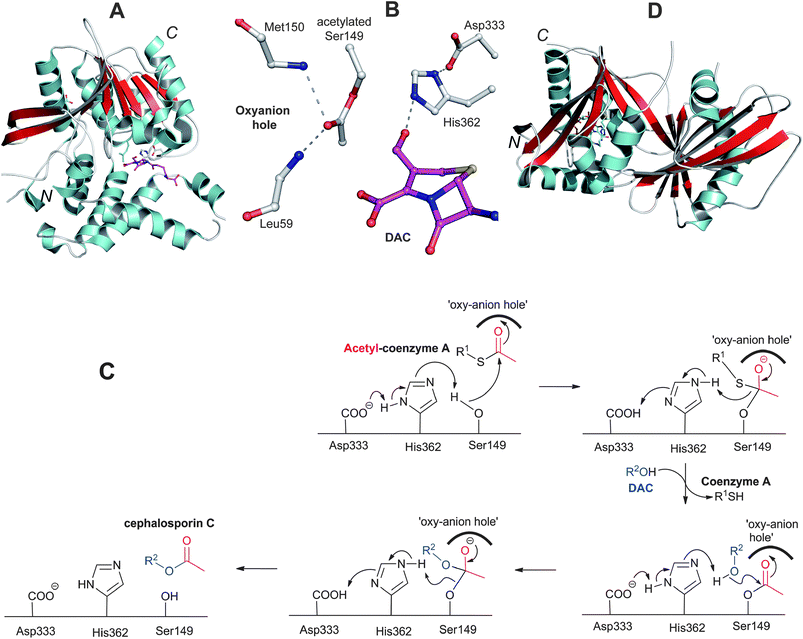 | ||
| Fig. 23 Deacetylcephalosporin C acetyltransferase (CefG). A: View of the overall structure of the CefG monomer; B: View from the active site of CefG showing the catalytic triad (Asp333-His362-Ser149) with acetylated Ser149, the oxyanion hole forming residues (Leu59 and Met150) and part of deacetylcephalosporin C (DAC);344C: Proposed mechanism for CefG catalysis.344D: View of the monomer of the GCN5-related acetyltransferase Orf14 involved in clavulanic acid biosynthesis.480 | ||
Carbamoylation of the C-3-hydroxymethyl function of DAC gives the intermediate O-carbamoyl-DAC en route to cephamycin C (Fig. 9), likely in an ATP-dependent manner, as catalysed by CmcH (encoded by cmcH).327,328 CmcH sequences analyses imply the presence of a conserved carbamoyl-phosphate-binding amino acid motif.108
Cephamycins A and B, and oganomycins (A, B, GA and GB, Fig. 8) contain a 3′-p-coumaric acid ester group. The biosynthesis of p-coumaric acids can occur either from L-phenylalanine or L-tyrosine (Fig. 24). The production of oganomycins can be stimulated by the addition of p-coumaric acid to S. oganonensis cultures.345 To our knowledge, there have been no studies on the enzymology of this modification nor on the enzymology of the formylamino moiety at C-7 of some cephabacins115 (Fig. 8). Identification of the genes encoding for the proteins responsible for both modifications should open up the way for the biosynthetic introduction of new functionalities to the cephem nucleus.
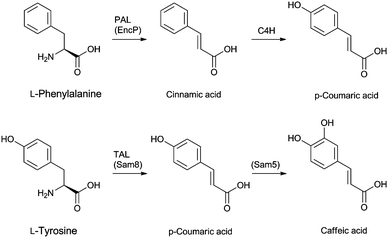 | ||
| Fig. 24 Putative biosynthetic routes to the p-coumaric- and caffeic acid moieties at the 3′-position of cephamycins (Fig. 8). In Streptomyces maritimus, L-phenylalanine is deaminated to trans-cinnamic acid in a reaction catalysed by phenylalanine ammonia lyase (PAL) EncP.715,716 In plants, trans-cinnamic acid can then be hydroxylated at the C-4 position, as catalysed by cinnamate 4-hydroxylase (C4H), but no homologous enzyme has been described in prokaryotes. Alternatively, p-coumaric acid can be derived from L-tyrosine through the activity of the tyrosine ammonia lyase Sam8.717 The presence of a caffeic acid ester in C-2081 X697 (Fig. 8) suggests that a p-coumaric acid hydroxylase, similar to Sam5,717 may be present in the producer strains S. heteromorphus and S. panayensis. | ||
4.10 Production of semisynthetic penicillins and cephalosporins
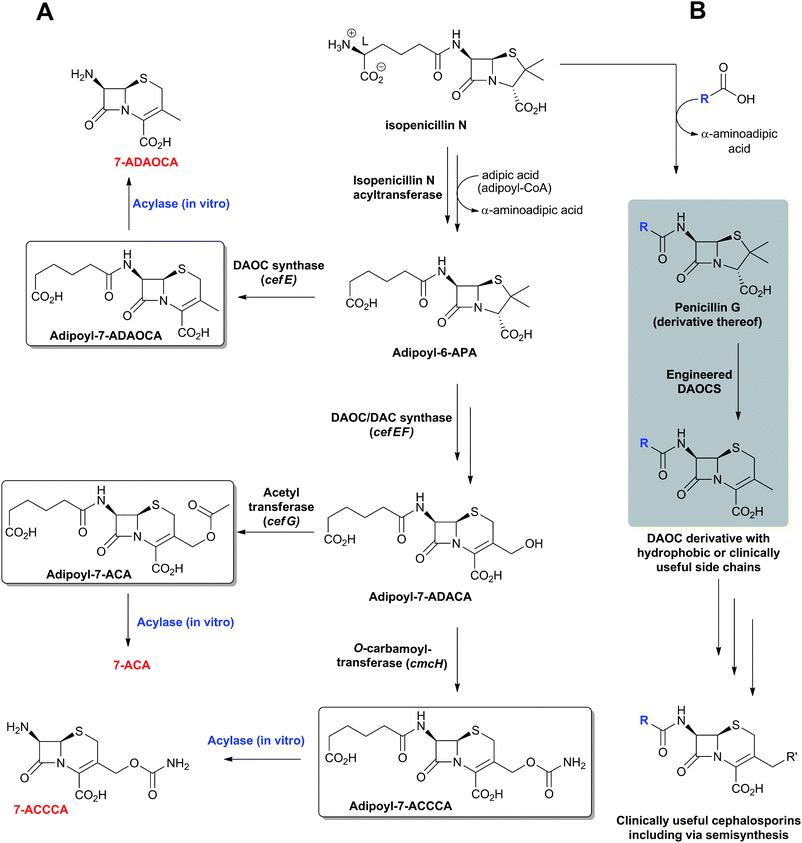
Strains used industrially for the production of penicillins and cephalosporins (i.e. P. chrysogenum and A. chrysogenum, respectively) secrete their products into the fermentation medium. The recovery process starts by filtration of the fermentation broth followed by solvent extraction steps. Whilst penicillins are mainly purified via multiple re-extractions in buffer and solvent at varying pH (employing counter-current chromatography), cephalosporins are mainly purified via solid-phase extraction due to their hydrophilic nature.26-APA, 7-ACA and, to a lesser extent, other C-7 deacylated cephalosporin intermediates are key intermediates for the production of semi-synthetic penicillins and cephalosporins, respectively, and are obtained by removal of the acyl side chain of the purified fermented products. The originally used chemical deacylation method346 has now been superseded by enzymatic processes for economic and ecological reasons.2 The enzymatic deacylation of penicillins to 6-APA is an established process, employing acylases, including from genetically manipulated E. coli or Bacillus megaterium.67 Like the biosynthetic enzyme AT, penicillin acylases are Ntn-hydrolases (Section 4.5). The D-α-aminoadipoyl side chain of the cephalosporins is less susceptible to direct in vitro enzyme-catalysed deacylation than that of the hydrophobic side chain of fermented penicillins,70 and efforts have been made to develop efficient enzyme-catalysed routes in vitro or in cells. A two-step enzymatic method has been developed wherein cephalosporins are first subjected to oxidative deamination by a D-amino acid oxidase (DAO, e.g. from Trigonopsis variabilis), then the resulting α-keto-adipoyl-7-ACA undergoes decarboxylation (induced by the peroxide released during the oxidase reaction) into glutaroyl-7-ACA. Addition of small quantities of peroxide following DAO treatment ensures full decarboxylation of α-keto-adipoyl-7-ACA into 7-ACA72 (DAO is a flavin oxido-reductase which catalyses the oxidation of D-amino acids to their corresponding keto-acids and ammonia). In the second step, the side chain of glutaroyl-7-ACA is deacylated by a glutarylamidase (e.g. from Pseudomonas diminuta,347,348 for review on industrial aspects of the enzymatic cleavage of cephalosporins, see ref. 70, 72). It is noteworthy that the glutarylamidases from P. diminuta also exhibit a low cephalosporin C acylase activity (5% of their glutarylamidase activity).349 Enhancing the activity of these amidases for “direct” (one step) splitting of the C-7 side chain of cephalosporins, employing various protein engineering strategies, may help optimise the enzymatic deacylation processes; however, a more efficient approach may be to directly ferment cephalosporins with hydrophobic side chains (Section 4.11).
4.11 Protein and metabolic engineering studies on the cephalosporin/cephamycin biosynthesis pathway
Classical strain improvements technologies (via iterative random strain mutagenesis and screening for improved productivity) have resulted in significant improvements in industrial fermentation titres of BLAs;102,350,351 however, these technologies do not (at least normally) result in the directed production of new products. One goal of “post-genomic” metabolic and protein engineering studies on BLA biosynthesis is to achieve rational optimisation of the production of BLAs of choice.352–3554.11.2.1 Cephalosporins production in engineered A. or P. chrysogenum. Overexpression of another copy of cefG in A. chrysogenum resulted in a 2–3 fold increase in cephalosporin C production, consistent with the effect of “gene dosage” on cephalosporin production.370,371 Overexpression of cefE (S. clavuligerus) simultaneously with disruption of cefEF gene in A. chrysogenum results in the production of DAOC at levels comparable to the total BLA biosynthesised by A. chrysogenum; the resulting DAOC can then be isolated and deacylated (as discussed in Section 4.10) to give 7-ADAOCA (Fig. 25A).372,373
 | ||
| Fig. 25 Metabolic and protein engineering studies aimed at modifying the penicillin-cephalosporin pathways to directly ferment cephalosporins with side chains useful for efficient purification or direct deacylation (e.g. by Pseudomonas diminuta glutarylamidase). A: production of 7-adipoyl-cephems (boxed) via adipoyl-6-aminopenicillanic acid (adipoyl-6-APA) in engineered P. chrysogenum strains fed with adipic acid; B: Proposed pathway for the production of cephems with hydrophobic or clinically useful side chains employing engineered DAOCS variants which have altered substrate specificities280,283 (see ref. 280 for review). Adipoyl-7-ACCCA: adipoyl-7-amino-3-carbamoyloxymethyl-3-cephem-4-carboxylic acid. | ||
Overexpression of cefD1, cefD2, cefEF and cefG (all from A. chrysogenum) in P. chrysogenum lacking AT activity results in the production and release of significant amounts of DAC into the culture broth as well as intracellular accumulation of cephalosporin C (for the acronyms used in this section, see Table 1).374
4.11.2.2 Adipoyl-cephalosporin production in engineered P. chrysogenum. Together with feeding adipic acid into the culture medium, overexpression of cefE (from S. clavuligerus)375,376 or cefEF with or without cefG (from A. chrysogenum)376,377 in P. chrysogenum results initially in the production of adipoyl-6-APA and subsequently, depending on the cloned activities, adipoyl-7-aminodeacetoxycephalosporanic acid (adipoyl-7-ADAOCA, in case of cefE), adipoyl-7-aminodeacetylcephalosporanic acid (adipoyl-7-ADACA in the case of cefEF), or adipoyl-7-ACA (in the case of cefEF and cefG, Fig. 25A). The removal of the adipoyl side chain, which is more susceptible to glutarylamidase cleavage than the α-aminoadipoyl side chain of natural cephalosporins,377 yields the corresponding 7-ADAOCA, 7-ADACA, and 7-ACA, respectively (Fig. 25A). Overexpression of the cefEF gene (A. chrysogenum) and the cmcH gene (S. clavuligerus) in P. chrysogenum, together with feeding of adipic acid, results in the production of adipoyl-7-amino-3-carbamoyloxymethyl-3-cephem-4-carboxylic acid (adipoyl-7-ACCCA, Fig. 25A).378 The precursor adipic acid fed to the culture medium is proposed to be introduced into the modified biosynthesis pathway via the activity of a promiscuous acyl-CoA ligase (encoded by aclA in P. chrysogenum).379 Overexpression of cefT (A. chrysogenum, which encodes for a cephalosporin C transporter belonging to the major facilitator superfamily) in the adipoyl-7-ACCCA-producing P. chrysogenum variant strain results in an ∼2-fold increase in cephalosporin production with a concomitant decrease in penicillin formation.379
Taken together, these results suggest that cephalosporin production by engineered P. chrysogenum strains is possible but may be limited by factors including the ability of the fungus to secrete “unnatural” cephalosporins.
5 Clavulanic acid biosynthesis
Clavulanic acid (CA) and its β-hydroxypropionyl derivative (isolated as its benzyl ester)380 are unusual amongst the family of naturally-occurring clavams because they have the (5R)-ring junction stereochemistry, rather than the (5S)-stereocentre observed in other naturally-occurring clavams (Fig. 26). Previous reviews have summarised pioneering labeling and enzymology studies on CA biosynthesis.32,33,143,381 Here we summarise the current state of knowledge on the pathways to CA and the (5S)-clavams, with an emphasis on the role of the enzymes involved. From the perspective of biosynthesis of the β-lactam ring itself, the work on CA biosynthesis was pioneering because it established that nature can synthesise β-lactams by routes other than the oxidative cyclisation chemistry employed by IPNS (Section 4.4).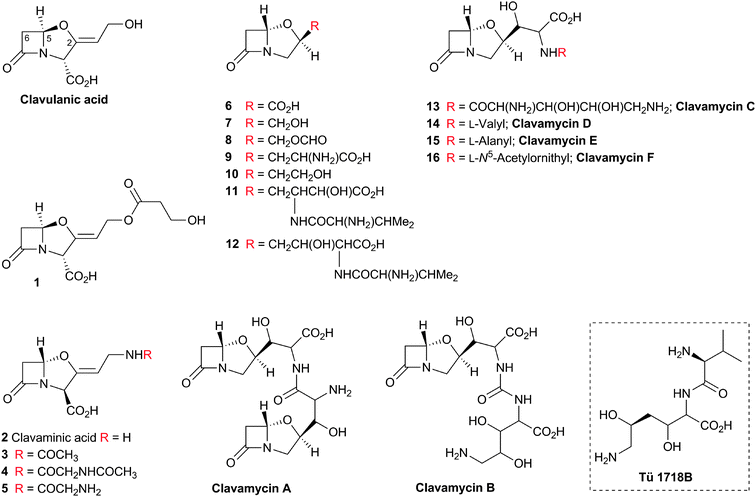 | ||
| Fig. 26 Clavams isolated from natural sources.32 In addition to clavulanic acid (CA, with (5R)-stereochemistry) and its β-hydroxypropionyl derivative 1, Streptomyces clavuligerus produces the clavams 6–9 (with the (5S)-stereochemistry).718 CA has also been isolated from S. jumonjinensis444 and S. katsurahamanus.445 The N-acyl derivatives 3–5 of clavaminic acid 2 accumulate in a S. clavuligerus mutant blocked in CA production. The hydroxyethyl clavam 10, valclavam 12, and Tü 1718B were isolated from S. antibioticus ssp. antibioticus Tü 1718 which does not produce CA. Valclavam was initially assigned the structure 11437,438 but this was reassigned to the regio-isomer 12.439 Tü 1718B (boxed) is a degradation product of 12.439,440 The structure of valclavam is related to that of clavamycins A–F which are produced by S. hygroscopicus.32 | ||
The biosynthesis route to CA can be divided into two stages (Fig. 27A) – those leading to (3S,5S)-clavaminic acid, which are reasonably well established, and those involved in the conversion of clavaminic acid to CA, which are much less well understood. A combination of labeling,382–388in vitro enzyme assays and genetic studies388,389 established the six steps leading to clavaminic acid. Fig. 28 displays the genes in the three clusters involved in the biosynthesis of CA and clavams in S. clavuligerus; Table 2 shows the proposed roles of the proteins encoded by these genes.
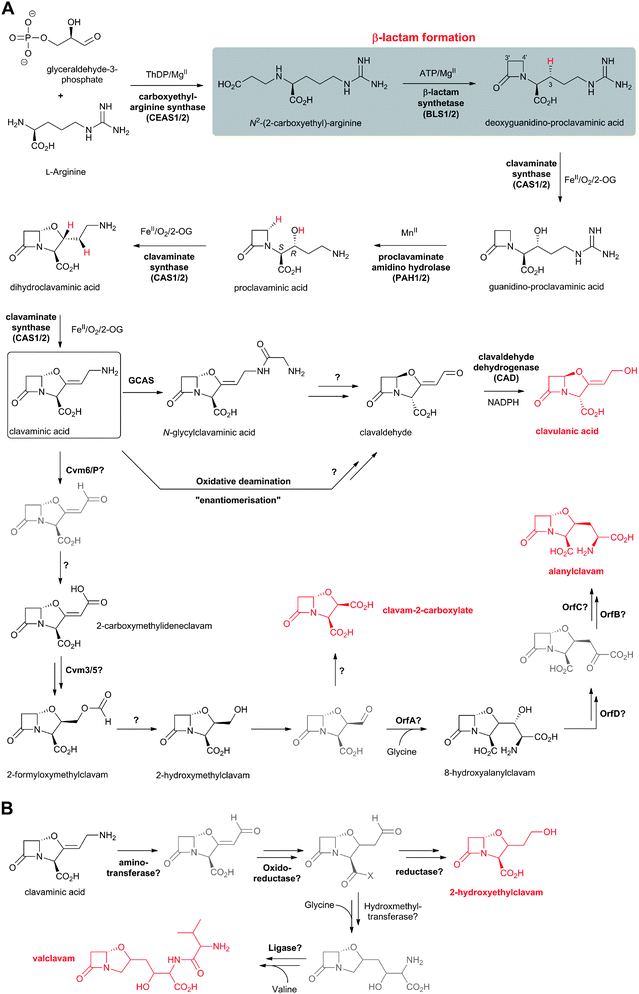 | ||
| Fig. 27 Proposed biosynthetic pathways leading to (5R)-clavulanic acid and (5S)-clavams in Streptomyces clavuligerus (A), and those leading to (5S)-clavams in S. antibioticus (B).32,381,516 Note that clavaminic acid (boxed) acts as a branch point between clavulanic acid and clavam biosynthesis. Selected cofactors and co-substrates are shown. The hydrogens to be removed during the CAS-catalysed oxidations are in red. Putative (i.e. not isolated) intermediates are in grey. | ||
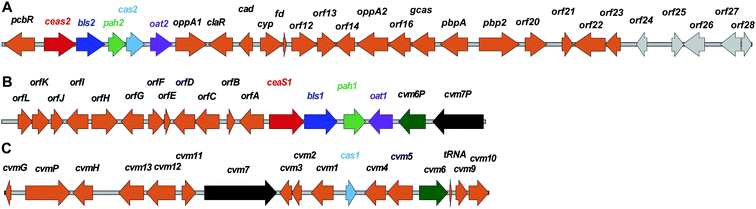 | ||
| Fig. 28 The three gene clusters encoding for proteins involved in the biosynthesis of clavulanic acid (CA) and clavams in Streptomyces clavuligerus. A: “CA” gene cluster; B: “Paralog” gene cluster; C: “Clavam” gene cluster.353,381,503 For the (predicted) role of the (putative) proteins encoded by the genes shown, see Table 2 and Fig. 27A. The potential involvement of orf24 to orf28 in CA/clavam biosynthesis is to be assessed.506 The pcbR gene (CA gene cluster) is part of the penicillin/cephalosporin gene cluster (Fig. 10). | ||
| Gene | AA | (Proposed) function of encoded protein |
|---|---|---|
| ceas1b/2a | 547/586 | Carboxyethylarginine synthase (CEAS).390–392 |
| bls1b/2a | 527/513 | β-Lactam synthetase (βLS).381,382 |
| cas1c/2a | 324/352 | Clavaminic acid synthase (CAS).420 |
| pah1/2b,a | 457/313 | Proclavaminic acid amidinohydrolase (PAH).412 |
| oat1/2b,a | 452/399 | Ornithine acetyltransferase (OAT2).238 |
| cvm6c/6Pb | 452/507 | Pyridoxal phosphate-dependant aminotransferase.503 |
| cvm7c/7Pb | 1114/915 | Transcriptional regulator.503,515 |
| oppA1a | 564 | ABC-type dipeptide transport system.475 |
| claRa | 432 | Transcriptional activator. |
| cada | 247 | Clavaldehyde dehydrogenase (CAD).451 |
| cypa | 407 | Cytochrome P-450.454 |
| fda | 71 | Ferredoxin.454 |
| orf12a | 458 | β-Lactamase-like protein.454 |
| orf13a | 340 | Amino acid (derivative) export pump.438,442 |
| orf14a | 339 | Acetyltransferase.438,442,480 |
| oppA2a | 562 | Peptide transport protein.475,476 |
| orf16a | 401 | N-Acetyltranferase.453 |
| gcasa | 529 | ATP-dependent ligase (GCAS).459 |
| pbpAa | 484 | Penicillin-binding protein.719 |
| pbp2a | 696 | Penicillin-binding protein.719 |
| orf20a | 389 | Cytochrome P-450.506 |
| orf21a | 201 | RNA polymerase σ factor.508 |
| orf22a | 571 | Two-component system histidine kinase.506 |
| orf23a | 270 | Two-component system response regulator.506,507 |
| orfLb | 229 | Transcriptional regulator.489,504 |
| orfKb | 277 | Hydrophobic ligand-binding SRPBCC domain of Micromonospora echinospora.477,494 |
| orfJb | 200 | Dihydrofolate reductase.477,494 |
| orfIb | 356 | LysR-type transcriptional regulator.477,494 |
| orfHb | 416 | Arabinose efflux permease.477,494 |
| orfGb | 364 | Kinase.477,494 |
| orfFb | 253 | Amino acid permease.477,494 |
| orfEb | 102 | Amino acid permease.477,494 |
| orfDb | 351 | Dehydratase.477,494 |
| orfCb | 393 | Aminotransferase.477,494 |
| orfBb | 126 | Amino acid biosynthesis regulator.477,494 |
| orfAb | 390 | Hydroxymethyltransferase.477,494 |
| cvmGc | 240 | Unknown.503 |
| cvmPc | 687 | Arginine deiminase.503 |
| cvmHc | 318 | Hydrolase.503 |
| cvm13c | 390 | β-Aspartyl peptidase.503 |
| cvm12c | 445 | Transcriptional regulator.503 |
| cvm11c | 216 | Efflux protein.503 |
| cvm3c | 170 | Flavin reductase.503 |
| cvm2c | 151 | Isomerase.514,515 |
| cvm1c | 344 | Aldo/keto reductase.503 |
| cvm4c | 328 | Acetyltransferase.503 |
| cvm5c | 394 | Oxido-reductase.503 |
| cvm9c | 182 | Transcriptional regulator.503 |
| cvm10c | 308 | Kinase.514,515 |
5.1 Carboxyethylarginine synthase
The first step in clavulanic acid (CA) biosynthesis is catalysed by carboxyethylarginine synthase (CEAS), a member of the thiamine diphosphate (ThDP)-dependent superfamily.390–392 CEAS catalyses the condensation of L-arginine with glyceraldehyde-3-phosphate to give N2-(2-carboxyethyl)arginine (CEA), the β-amino acid precursor for β-lactam formation (Fig. 27A, 29). Crystal structures of CEAS reveal its structural homology to pyruvate decarboxylase and related ThDP-dependent enzymes (Fig. 30).391,392 The CEAS active site is located at a dimer interface across which ThDP is bound in a V-shaped conformation, as is characteristic for many ThDP-dependent enzymes. CEAS is unusual amongst the superfamily of ThDP-utilising enzymes because it catalyses a C–N bond formation in a mechanism proposed to occur via Michael-type reaction (Fig. 29). Analogous to other ThDP-dependent enzymes (e.g. pyruvate decarboxylase), the initial step in CEAS-catalysis is proposed to involve reaction of a ThDP ylide with the aldehyde group of glyceraldehyde-3-phosphate. Following proton transfer (from the aldehyde-derived carbon at the position α- to the thiamine ring), it is proposed that elimination of the β-hydroxyl group occurs to give an enol, which undergoes further elimination of the γ-phosphate group to give a ThDP-bound acryloyl-group.390 The latter then undergoes Michael reaction with the α-amino group of L-arginine. Finally, hydrolysis of the ThDP-bound Michael addition product occurs to give CEA. This mechanism is consistent with the available labeling and crystallographic information,390–392 though kinetic analyses are needed to verify the proposed intermediates in catalysis. It should also be noted that, at least in our hands, isolated CEAS is not highly active; it cannot be ruled out that there is an alternative to glyceraldehyde-3-phosphate as the 3C-component of the reaction. One interesting proposal arising from the structural work on CEAS is that the acid–base catalysis required for the reaction is derived either from ThDP itself or from a species (e.g. hydroxide) generated from substrates – this is because of an apparent lack of suitably-positioned general acid/base side chains in the active site of CEAS.391,392 To date, the CEAS-catalysed reaction represents a novel and unique route to β-amino acids. Engineering studies on CEAS may be productive with respect to producing modified β-amino acids for conversion into β-lactams.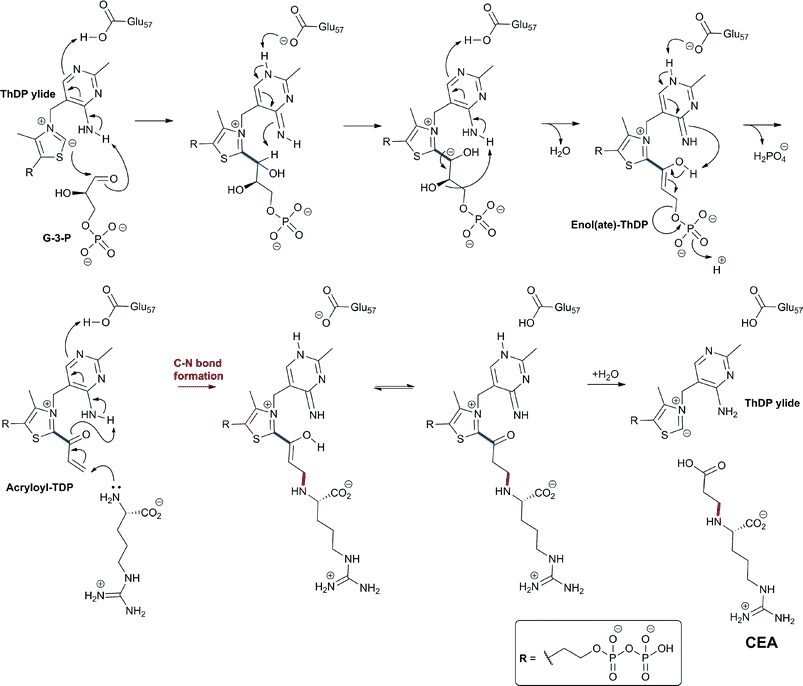 | ||
| Fig. 29 Proposed outline mechanism for carboxyethylarginine synthase (CEAS). The enol(ate)-ThDP intermediate has been provisionally assigned in crystalline CEAS following soaking with DL-glyceraldehyde-3-phosphate (G3P) (Fig. 30E).391 Details of proton transfers and eliminations are uncertain.390,391 | ||
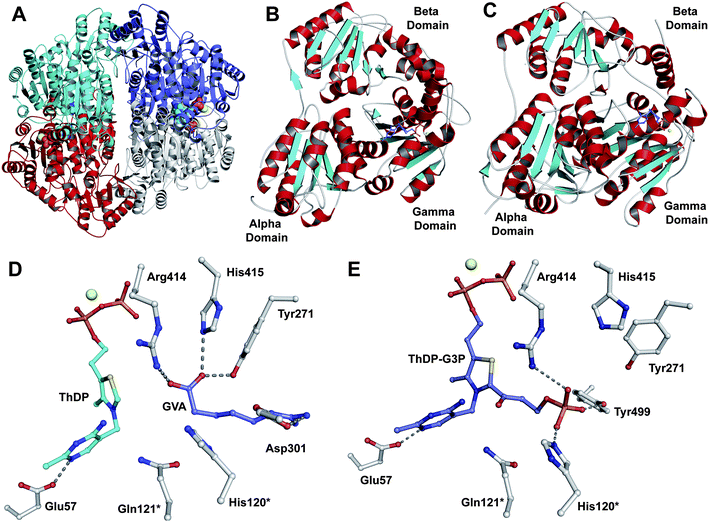 | ||
| Fig. 30 Carboxyethylarginine synthase (CEAS), a thiamine diphosphate (ThDP)-dependent enzyme. A: The tetrameric oligomerisation of CEAS.392 The CEAS active site is located at a dimer interface across which ThDP (shown in space-filling mode) is bound; B and C: Views of CEAS (B) and yeast pyruvate decarboxylase (C) monomers reveal their structural homology. Note the similar three-domain structure of each monomer;392D: View from subunit A of a crystal structure of CEAS, with ThDP bound in V-shaped conformation, following soaking with 5-guanidinovaleric acid (GVA) (PDB 2IHV).391 GVA is a nonreactive L-arginine analogue that lacks an α-amino group;391E: A view from a crystal structure of CEAS (PDB 2IHU) showing the putative trapped enol(ate)-ThDP intermediate (TDP-G3P, Fig. 29) that has been provisionally assigned in subunit C following soaking with DL-glyceraldehyde-3-phosphate (G3P).391 The structures (D and E) suggest (partially) overlapping binding sites for the substrates D-G3P and L-arginine. This is consistent with the proposed CEAS mechanism (Fig. 29). MgII is shown as a yellow sphere. *Residues from a neighbouring monomer. | ||
5.2 β-Lactam synthetases in clavam and carbapenem biosynthesis
The second step in the clavam biosynthesis pathway is catalysed by the first identified β-lactam synthetase (β-LS) which catalyses the cyclisation of N2-(2-carboxyethyl)arginine (CEA) to give the monocyclic β-lactam deoxyguanidino-proclavaminic acid (DGPC, Fig. 27A).393,394 β-LS is closely related to the carbapenam synthetases (CarA395,396 and ThnM,397Fig. 31) which are involved in carbapenem biosynthesis via catalysing the cyclisation of (2S,5S)-carboxymethylproline (t-CMP) to yield the (2S,5S)-carbapenam nucleus (Section 6.3.2). In this section, we consider structural, mechanistic, and substrate analogue studies for both β-LS and CarA/ThnM. | ||
| Fig. 31 Partial sequence alignment for known and putative β-lactam synthetases involved in the biosynthesis of clavams (β-LS), carbapenems (CarA and ThnM), and monobactams (TbIS). Catalytically important residues are highlighted. The highlighted lysine residue is proposed720 to assist in ring cyclisation via stabilisation of the proposed “tetrahedral” intermediate (in case of clavams and carbapenams, Fig. 33). The highlighted Tyr-Glu dyad (in case of clavams and carbapenems) is proposed to deprotonate the amine involved in intramolecular β-lactam formation.721 α-Helices (cyan cylinders) and β-strands (red arrows) represent the assigned secondary structure of β-LS (PDB 1MB9).399 | ||
 | ||
| Fig. 32 Views from crystal structures of β-lactam synthetases. A: Views from the structures of the β-lactam synthetases CarA and β-LS (PDB 1Q19 and 1MB9, respectively) in comparison to that of asparagine synthetase (PDB 1CT9)398 showing the N- and C-terminal domains; B: View from a crystal structure of β-LS (PDB 1MBZ) showing the acyl-adenylate N2-(2-carboxymethyl)arginine-AMP (CMA-AMP) trapped species generated by reaction of ATP with the substrate analogue N2-(2-carboxymethyl)arginine.399 The latter is one carbon shorter than the natural substrate; thus, CMA-AMP does not undergo cyclisation to give the highly strained 3-membered ring; C: View from a crystal structure of CarA (PDB 1Q19)395 complexed with the substrate (2S,5S)-5-carboxymethylproline (t-CMP) and an ATP analogue α,β-methyleneadenosine-5′-triphosphate (AMP-CPP) with t-CMP positioned in apparently “productive” conformation for adenylation and subsequent β-lactam formation. | ||
 | ||
| Fig. 33 How nature converts modified amino acids/peptides into β-lactams employing oxidative and non-oxidative mechanisms. A: Outline proposed mechanisms for CarA and β-LS (involving intramolecular nucleophilic attack) in comparison to that of Asn-B (which involves intermolecular nucleophilic attack),398 showing the common mechanism of carboxylate activation, via adenylation, followed by formation of a tetrahedral intermediate; B: Outline mechanism for the oxidase isopenicillin N synthase showing the enzyme-bound monocyclic ferryl intermediate; R = δ-(L-α-aminoadipoyl). | ||
Crystallographic analyses of Asn-B398 (Fig. 32) reveal that it comprises two domains: an N-terminal nucleophile (Ntn) glutaminase domain responsible for production of ammonia from glutamine, and a C-terminal synthetase domain which catalyses formation of β-aspartyl-AMP. It is proposed that ammonia travels from the N-terminal hydrolase domain through a tunnel that links the N- and C-terminal domains where it reacts with the β-aspartyl-AMP to form asparagine (Fig. 33A).398,401 Structural comparisons of the β-lactam synthetases and Asn-B (Fig. 32) reveal that the formers have maintained the characteristic two domain fold;400 however, as β-lactam ring formation is an “intramolecular” process, it does not require the release of ammonia from glutamine, rendering the glutaminase reaction of the N-terminal domain redundant. This analysis is consistent with the replacement of the, catalytically important, nucleophilic Cys1 of Asn-B with Phe1 in β-LS, together with the presence of nine additional N-terminal residues in β-LS which occupy the corresponding glutamine binding pocket in Asn-B.400 In CarA, the Cys1 of Asn-B is replaced by a serine which occupies a similar position; however, other important residues for glutamine binding are missing, reflecting the lack of a glutamine binding pocket.395 In the C-terminal domain of all three enzymes, the substrate-carboxyl group is activated, via ATP-mediated adenylation, at a largely conserved active site. Subsequent nucleophilic attack by NH3/secondary amine yields the amide/β-lactam product (respectively, Fig. 33A).395,398–400
Whilst both β-LS and Asn-B crystallise as dimers, CarA crystallises as a tetramer.395 For each of the three proteins: (i) the N-terminal domain consists of two antiparallel β-sheets that form a sandwich, flanked on each side by (relatively) short α-helices; (ii) the C-terminal domain comprises (11–14) α-helices surrounding a 5-stranded parallel β-sheet; and (iii) the active site is located in a cleft in the C-terminal domain formed by 4 β-strands and 5 α-helices; however, the β-LS/CarA substrate binding cleft is relatively extended to accommodate CEA (β-LS)/t-CMP (CarA) which are larger than aspartic acid (Asn-B).
The close structural and mechanistic relation between Asn-B, β-LS and CarA/ThnM coupled to their different substrate specificities suggests that it may have been relatively easy to evolve different types of β-lactam (or indeed potentially other lactams/lactones) synthetases based on the Asn-B structural platform. Whether β-LS and CarA/ThnM evolved separately from Asn-B or from one another is unknown.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of (7R)-7-methyl-t-CMPi and (7S)-7-methyl-t-CMPi, where selective conversion of the former to give (4S,6S,7R)-7-methyl-carbacepham was observed (Fig. 34C).404 Further, in the CarA-catalysed reaction of a 1:1 mixture of (4R)-4-methyl-t-CMP and (4S)-4-methyl-t-CMP, preferential (1:2) conversion of the latter to give (1S,3S,5S)-1-methyl-carbapenam was observed (Fig. 34B and C).402 Combined with studies on CarB/ThnE (Section 6.3.1), these investigations suggest that the carbapenem biosynthesis pathways are amenable to engineering.
:1 mixture of (7R)-7-methyl-t-CMPi and (7S)-7-methyl-t-CMPi, where selective conversion of the former to give (4S,6S,7R)-7-methyl-carbacepham was observed (Fig. 34C).404 Further, in the CarA-catalysed reaction of a 1:1 mixture of (4R)-4-methyl-t-CMP and (4S)-4-methyl-t-CMP, preferential (1:2) conversion of the latter to give (1S,3S,5S)-1-methyl-carbapenam was observed (Fig. 34B and C).402 Combined with studies on CarB/ThnE (Section 6.3.1), these investigations suggest that the carbapenem biosynthesis pathways are amenable to engineering.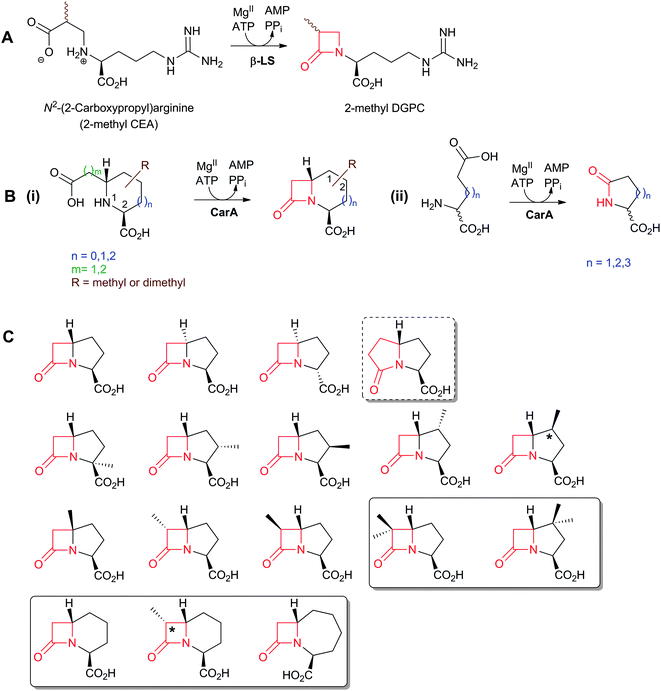 | ||
| Fig. 34 The biocatalytic versatility of the β-lactam synthetases β-LS (A)405,406 and CarA (B, C).396,402,404 In the case of substituted t-CMP substrate analogues, CarA can accept derivatives methylated at C-2, C-3, C-4, C-5 or C-6 to produce the corresponding β-lactams.402 CarA can also accept at least three of the four possible isomers of CMP.396 Compounds which are reported to be of higher stability compared to the unsubstituted carbapenams are boxed.402,404 The compounds with asterisked stereocentre represent the preferred stereoisomer at that position for CarA catalysis.402,404 The incubation of t-carboxyethylproline, a substrate analogue of t-CMP, with ATP/MgII/CarA results in the formation of a γ-lactam (shown in a dashed box).396 | ||
5.3 The trifunctional role of CAS in clavaminic acid biosynthesis
Four steps are involved in the conversion of deoxyguanidino-proclavaminic acid (DGPC) to clavaminic acid; three of these steps are catalysed by a single 2OG oxygenase, clavaminic acid synthase (CAS, Fig. 27A). It is not unprecedented that 2OG oxygenases catalyse sequential two electron oxidations, as exemplified by the bifunctional role of DAOC/DAC synthase in cephalosporin biosynthesis (Section 4.7), the TET enzymes in the hydroxylation and formylation of human DNA,408 and in the 3-stage oxidation of a methyl group catalysed by the plant gibberellin C-20 oxidase.409–411 However, in case of CAS, two of the three oxidative steps are separated from the first by the action of proclavaminic acid amidino-hydrolase (PAH, Fig. 27A), which is structurally and mechanistically unrelated to CAS.412 Another feature of CAS catalysis is that the three reactions that it catalyses involve three different types of oxidative reactions (i.e. hydroxylation, oxidative cyclisation, and desaturation) (Fig. 27A).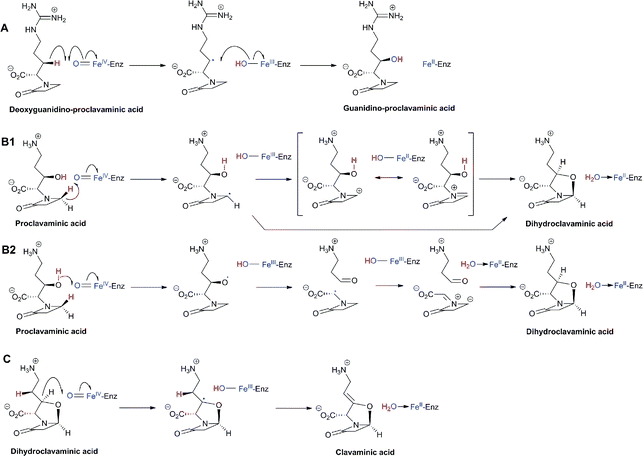 | ||
| Fig. 35 Proposed outline mechanisms for the three clavaminic acid synthase (CAS)-catalysed reactions (i.e. hydroxylation (A), oxidative ring cyclisation (B1/2), and desaturation (C), respectively). The hydroxylation reaction is separated from the other two reactions by the activity of proclavaminic acid amidino-hydrolase which catalyses the conversion of guanidino-proclavaminic acid to proclavaminic acid (Fig. 38). For the cyclisation reaction, the alternative B2 mechanism has been proposed in part on the basis of computational studies;722 it involves (1) oxidation of the hydroxyl group of proclavaminic acid to give an O-radical, (2) retro-aldol-like decomposition of the O-radical to an aldehyde and a C-centered radical, which is stabilised by the captodative effect, (3) abstraction of a hydrogen atom from the C-4 (pro-S) position of the C-centered radical by the FeIII–OH species to yield an azomethine ylide, and (4) 1,3-dipolar cycloaddition to the ylide with the aldehyde acting as a dipolarophile. There is synthetic precedent for the B2 mechanism as in the synthesis of oxapenams via 1,3-dipolar cycloaddition reactions of aldehydes and ketones.723 | ||
The second CAS-catalysed step comprises dihydroclavaminic acid formation by a desaturative ring closure reaction, which involves removal of the β-lactam 4′-pro-S hydrogen and the hydrogen of the hydroxyl group at C-3 of PCA (Fig. 35B). Evidence that the dihydroclavaminic acid oxazolidine ring is formed prior to formation of the exocyclic alkene of clavaminic acid came initially from NMR analyses of the mixture of products resulting upon incubation of PCA and partially purified CAS, which revealed the presence of a minor resonance at δ ∼ 5.4 corresponding to the C-5 proton of 2,8-dihydroclavaminic acid.415,417 Repeating the experiment with C-3-deuterated PCA enabled the isolation of the saturated bicyclic [2H2]-dihydroclavaminic acid, because of the operation of a primary isotope effect which slows down the exocyclic double bond formation. Upon incubation with CAS, the purified dihydroclavaminic acid was converted to clavaminic acid demonstrating the intermediacy of dihydroclavaminic acid in the cyclisation of PCA to clavaminic acid. Non-deuterated dihydroclavaminic acid was subsequently shown to be converted into clavaminic acid using recombinant CAS.416,418 The CAS-catalysed oxidative cyclisation reaction that forms the bicyclic ring structure of dihydroclavaminic acid is reminiscent of the IPNS-catalysed cyclisation of ACV (Section 4.4). However, the CAS-catalysed cyclisation is a two-electron oxidation in which only one ring is formed, whereas the IPNS reaction is a four-electron bicyclisation reaction.
The third CAS-catalysed step (i.e. dihydroclavaminic acid desaturation to give clavaminic acid) occurs (at least predominantly) with loss of hydrogens via a syn-elimination (Fig. 35C).419
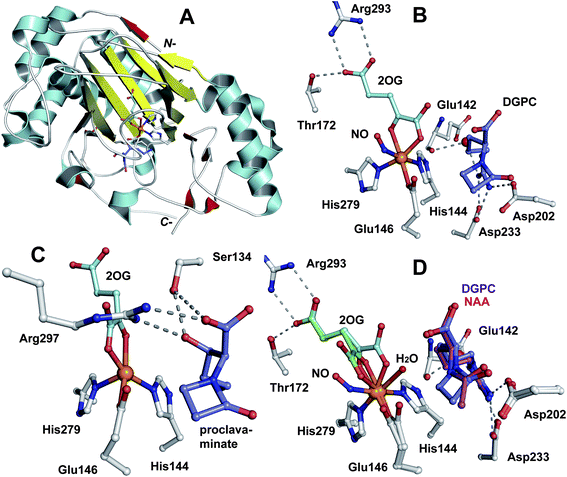 | ||
| Fig. 36 Views from crystal structures of clavaminic acid synthase (CAS).420,421A: The CAS monomer (PDB 1DRT); B: The active site of CAS (PDB 1GVG) showing the binding sites for FeII, the CAS-substrate for hydroxylation (deoxyguanidino-proclavaminic acid, DGPC), nitric oxide (NO, an O2 analogue), and 2OG; C: The active site of CAS (PDB 1DRT) with proclavaminic acid bound; D: Superimposition of the active sites of CAS:FeII:2OG:DGPC:NO complex (PDB 1GVG) and that with the substrate analogue N-α-acetylarginine (NAA, without NO but with a water molecule ligating FeII). Note: the superimposed structures (D) reveal that the C1-carboxylate oxygen of 2OG (green) undergoes a “flip” to the other possible coordination site (cyan) in the presence of NO (B).420,421 | ||
The reported structures also provide detailed insights into how CAS catalyses its three oxidative reactions.420,421 Interestingly, studies using nitric oxide (NO), as an O2 analogue, reveal the potential for the 2OG 1-carboxylate to rearrange its iron binding position (Fig. 36D). Thus, in the absence of NO as in case of the NAA complex (Fig. 36C), a water molecule occupies the coordination position trans to His279 and adjacent to the substrate C–H bond to be oxidised. Coordination of O2 to this site should generate the FeIVO in an appropriate position to effect substrate oxidation.24,420,421 However, in the presence of NO, the 2OG 1-carboxylate moves to this position enabling NO to bind trans to His144 (Fig. 36B). If O2 binds similarly to NO, the subsequently generated FeIVO would be in an inappropriate position for substrate oxidation, and it must rearrange.410,411 Alternatively, if a 5-coordinate persuccinate intermediate is formed (Fig. 20),422 simultaneously with CO2 loss, it may collapse to produce the FeIVO in the (apparently) correct position for substrate oxidation. The incorporation of oxygen from water as well as from O2 into the alcohol products in some 2OG oxygenase-catalysed hydroxylations may reflect accessibility of a five coordinate FeIVO to react with water.423 Therefore, the position of O2 binding in CAS, and in other 2OG oxygenases, is unclear.
:alkene, 3:1) (Fig. 37C); (ii) in contrast, the β-lactam deoxy-PCA gave predominantly the alkene product (alkene:alcohol, >10:1) (Fig. 37B); and (iii) incubation of NAA, like DGPC, gave only the alcohol product (Fig. 37D).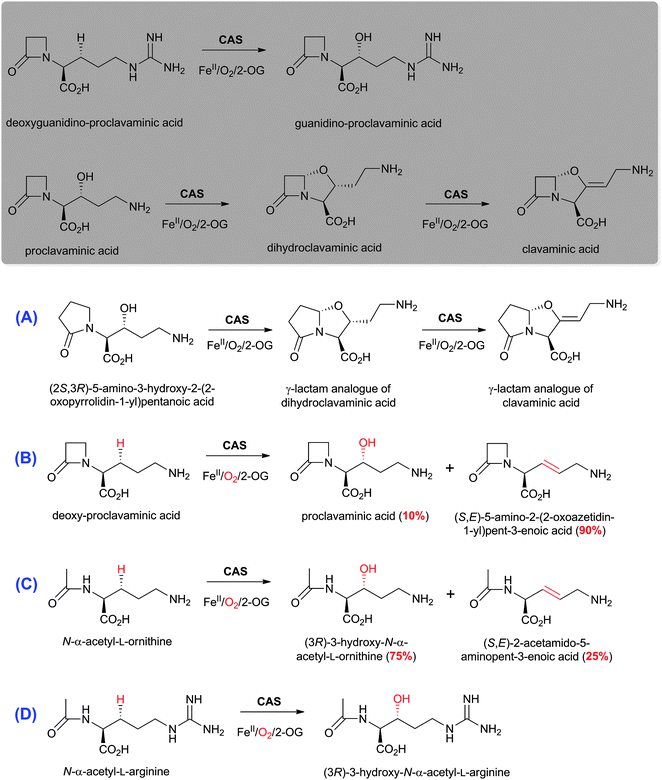 | ||
| Fig. 37 The biocatalytic versatility of clavaminic acid synthase (CAS).413,424–426 The reactions of CAS with its natural substrates are boxed. | ||
Arginine hydroxylation is a common feature in the biosynthesis of some secondary metabolites. Subsequent to the work on CAS, VioC, a 2OG oxygenase from S. vinaceus, has been found to catalyse the hydroxylation of L-arginine to give (2S,3S)-3-hydroxyarginine during viomycin biosynthesis.427 Arginine hydroxylation, likely catalysed by 2OG oxygenases, plays a role in the biosynthesis of streptothricin F428,429 and the heptapeptide antibiotic K-582 which consists of two components with threo-γ-hydroxy-L-arginine as a constituent of both.430
5.4 Proclavaminic acid amidino hydrolase (PAH)
Proclavaminic acid amidino hydrolase (PAH, Orf4)412 catalyses the hydrolysis of the guanidino side chain of guanidino-proclavaminic acid (GPC) to give proclavaminic acid and urea (Fig. 38A). Disruption of pah leads to loss of CA production, reflecting its obligatory requirement in CA biosynthesis.431 PAH is a member of the arginase family of enzymes which hydrolyse arginine to ornithine and urea; however, PAH does not accept arginine as a substrate.432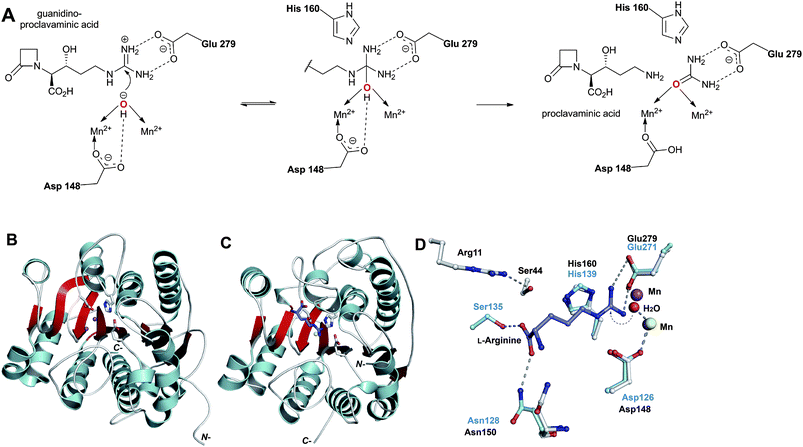 | ||
| Fig. 38 Mechanism and crystal structures of amidino-hydrolases.432,433A: Proposed outline mechanism for guanidino-proclavaminic acid (GPC) hydrolysis by proclavaminic acid amidino-hydrolase (PAH).432B: Monomer of PAH from S. cattleya432 (PDB 1GQ7); C: Monomer of the arginase from Bacillus caldovelox433 (PDB 3CEV); D: Superimposed active site views of the two amidino-hydrolases showing the residues involved in catalysis (blue for arginase), the conserved MnII-binding site (violet for PAH, and yellow for arginase), and the guanidino moiety of the substrate (L-arginine, the substrate for arginase). Note that neither Ser44 nor Arg11 of PAH, which are proposed to be responsible for binding the carboxylate and hydroxyl groups of GPC, respectively, are conserved in the arginase active site.422,423 | ||
PAH catalyses the hydrolysis of the substrate analogues deoxyguanidino-proclavaminic acid, N-acetyl-L-arginine and (3R)-hydroxy-N-acetyl-L-arginine, albeit at a lower rate than the natural substrate GPC.432 These results, together with comparison of the structures of PAH and the B. caldovelox arginase, reveal that the lack of arginine binding to PAH may be rationalised by (i) the presence of the more polar amino group rather than an amide and/or the absence of the C-3 hydroxyl group;432 (ii) Arg11 (from an adjacent PAH monomer) and Ser44 are proposed to be suitably positioned to bind the PAH-substrate carboxylate and hydroxyl groups, respectively. Neither Ser44 nor Arg11 of PAH is conserved in the arginase from B. caldovelox.433
Comparing the crystal structures of PAH and CAS also reveals an apparent convergence of the arginine and serine residues involved in binding the C-3 alcohol of their substrates. In a crystal structure of CAS complexed with proclavaminic acid (Fig. 36C), the product of PAH, the hydroxyl of proclavaminic acid is positioned to hydrogen-bond the side chains of Arg297 and Ser134 (CAS numbering). In PAH, the side chains of Arg11 and Ser44 (Fig. 38D) are similarly positioned and Ser44 is a likely candidate for substrate hydroxyl binding.432
5.5 Clavaminic acid, a branch point in (2R,5R) clavulanic acid and (3S,5S) clavam biosynthesis
A series of labeling studies434–436 has provided evidence that clavaminic acid is a branch point between the (5R)-CA and (5S)-clavams biosynthesis pathways in S. clavuligerus (Fig. 27A). For valclavam437,438 and Tü 1718B (Fig. 26)439,440 biosynthesis, labeling studies and the determination of the presence of PAH and CAS in S. antibioticus ssp antibioticus Tü 1718B436,441–443 also provided evidence for the common steps with CA biosynthesis up to the point of clavaminic acid. A biosynthetic path to the clavams has been proposed441– a modified version is shown in Fig. 27A. As genomic sequences become available for more β-lactam producers, it is likely that differences between (3R,5R)-CA and (3S,5S)-clavam producers will become clearer.5.6 From clavaminic acid to clavaldehyde: Stereoisomerisation and oxidative deamination of clavaminic acid
It seems that CA producers (S. clavuligerus, S. jumonjinesis,444 and S. katsurahamanus445), but not (5S)-clavam only producers, possess the ability to catalyse the “double-epimerisation” of (3S,5S)-clavaminic acid to (3R,5R)-clavaldehyde. The mechanism of this unprecedented process is unknown; one possibility is via C-9 oxidation, followed by oxazolidine ring opening to form an iminium ion and ring closure to give the (3R,5R)-clavulanate system (Fig. 39).446,447 Evidence has been reported for the exchange of the C-8-hydrogen during CA biosynthesis from clavaminic acid.448S. clavuligerus growth in an 18O2-rich atmosphere results in the incorporation of 18O into both the oxazolidine ring and the allylic hydroxyl group of CA.449 The incorporation of 18O into the oxazolidine ring likely results from CAS-activity (CAS-catalysed hydroxylation of deoxoguanidinoproclavaminic acid results in ∼70% incorporation of 18O from 18O2 into proclavaminic acid).425 The incorporation of 18O into the allylic group of CA rules out the possibility of a water-mediated deamination and suggests the involvement of an oxygenase (possibly a P450 enzyme, see below) in the conversion of clavaminic acid to CA. However, this is unlikely to involve an oxygenase-catalysed conversion of clavaminic acid to (3S,5S)-clavaldehyde because there is only evidence for the production of (3R,5R)-clavaldehyde.450 The (3R,5R)-stereochemistry of clavaldehyde was assigned by synthesis.450,451 Clavaldehyde, the aldehyde of which may act as an electron sink for decarboxylation, was suggested as an intermediate in (5S)-non-carboxylated clavam biosynthesis on the basis of the observation that proclavaminic acid is a common precursor of both CA and (5S)-non-carboxylated clavams.434,435,452 It is less certain that (3R,5R)-clavaldehyde is involved in the biosynthesis of the (5S)-clavams.450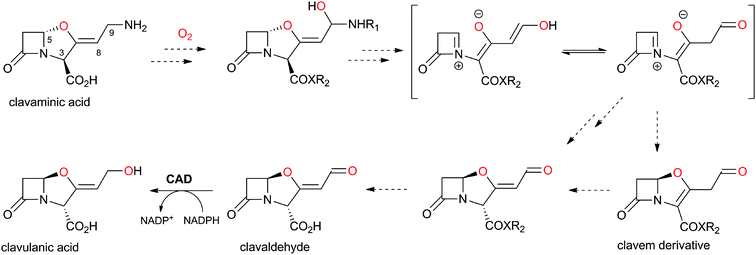 | ||
| Fig. 39 Possible outline mechanism for the double epimerisation process involved in clavulanic acid (CA) biosynthesis.446,447 Note that the oxygen at C-9 of CA originates from O2.449 R1 could be H, glycyl or N-acetyl-glycyl. | ||
Presently, it is unclear as to how many genes are essential for the conversion of clavaminic acid to CA. The products of the (orf10–orf17) genes of the CA biosynthesis gene cluster are proposed to be involved in the conversion of (3S,5S)-clavaminic acid to (3R,5R)-clavaldehyde.29,32,453 The genes orf10 (cyp) and orf11 (fd) apparently encode for a cytochrome P-450 and a ferredoxin, respectively. Inactivation of cyp resulted in complete loss of CA production, while production was only reduced in the case of an fd-disrupted mutant, suggesting that cyp is essential and fd is beneficial, but not absolutely required, for CA biosynthesis.381,453–455 It is possible that Orf10 acts on a derivative of clavaminic acid (e.g. N-glycyl-clavaminic acid or N-acetyl-glycyl-clavaminic acid) to introduce the O2-derived oxygen at C-9 (Fig. 39). Disruption of orf15 or orf16 blocks CA production and leads to accumulation of N-acetyl-glycyl-clavaminic acid and N-glycyl-clavaminic acid.453 Accumulation of clavaminic acid was observed in case of a claR disrupted mutant; claR encodes for a transcriptional regulatory protein that is proposed to regulate genes involved in the late steps of CA biosynthesis (e.g. oppA1, cyp and cad).456–458
5.7 N-Glycyl-clavaminic acid synthetase, GCAS (Orf17)
S. clavuligerus mutants with defects in orf17 are unable to produce detectable levels of CA.453 The gene product of orf17 (N-glycyl-clavaminic acid synthetase, GCAS/Orf17) catalyses the ATP-dependent ligation of the amino group at C-9 of clavaminic acid to the carboxylate moiety of glycine to give N-glycyl-clavaminic acid (Fig. 40). Under the same conditions, N-acetyl-glycine did not react suggesting that CA biosynthesis pathway may proceed via clavaminic acid to N-glycyl-clavaminic acid and then to N-acetyl-glycyl-clavaminic acid.459 GCAS shares homology with proteins from the ATP grasp fold superfamily (which includes, e.g., E. coli biotin carboxylase).460,461 The mechanism of GCAS, similarly to other members of the ATP grasp fold superfamily,460,461 likely proceeds via an enzyme-bound O-glycyl-phosphate intermediate (Fig. 40). GCAS seems to be selective for the combination of glycine and clavaminic acid;459 this is unlike some members of the ATP grasp fold superfamily (e.g. the D-alanine-D-alanine ligases, which, despite being specific for D-amino acids, do display some flexibility in their substrate specificity462). It is proposed that N-glycyl-clavaminic acid and N-acetyl-glycyl-clavaminic acid are intermediates en route to CA, but there is no firm evidence yet to support this.459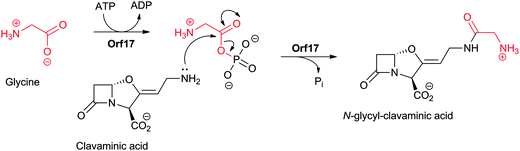 | ||
| Fig. 40 Proposed outline mechanism for the ATP-grasp family enzyme Orf17 that catalyses the condensation of the amino group of clavaminic acid to the (activated) carboxylate of glycine.459 | ||
5.8 From clavaldehyde to clavulanic acid: clavulanic acid dehydrogenase
The last step in CA biosynthesis in S. clavuligerus is catalysed by CA dehydrogenase (CAD/Orf9), an NADP(H)-dependent oxido-reductase that catalyses the (reversible) conversion of (3R,5R)-clavaldehyde into (3R,5R)-CA (Fig. 41B).451 Sequence analyses of CAD reveal homology to proteins belonging to the short chain dehydrogenases/reductases (SDR) family.463–466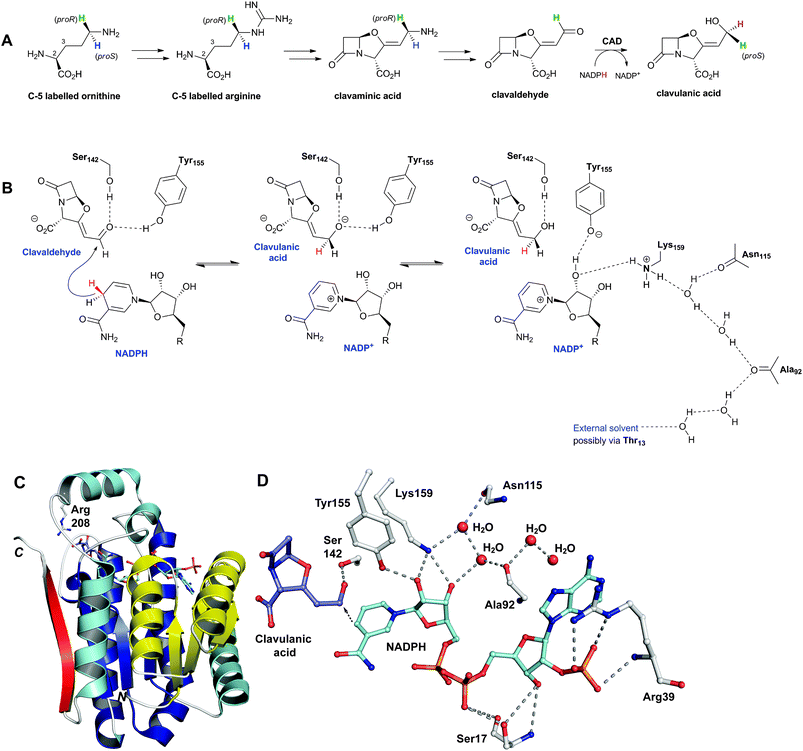 | ||
| Fig. 41 Mechanism and structural views of the NADPH-dependent reductase clavulanic acid dehydrogenase (CAD). A: Overview of the labeling studies474 aiming to reveal the origin of the C-9 hydrogens of clavulanic acid with respect to its precursor ornithine/arginine; B: Proposed reaction mechanism for CAD showing a possible proton relay involving water molecules. R = adenosine 2′-phosphate-5′-diphosphate;469C: A CAD monomer showing the Rossmann fold, which is characteristic of dinucleotide-binding enzymes (two repeats of βαβαβ motifs, one shown in yellow and the other in blue, each binds one NADPH nucleotide);469D: View from the CAD active site showing the possible proton relay involving water molecules (B). | ||
5.9 The oligopeptide binding proteins (Orf7 and Orf15) of clavulanic acid biosynthesis
Two of the CA biosynthesis cluster genes, orf7 and orf15, are closely related in sequence (∼48% identity) but apparently have little functional cross complementation.475 Orf7 and Orf15 (referred to as OppA1 and OppA2, respectively) belong to the oligopeptide-binding protein (OPP)/substrate-binding protein (SBP) family which are involved in the import of peptides in S. clavuligerus.475 Mutants of S. clavuligerus defective in orf7 or orf15 show loss of CA production, which can be restored by complementation.453 Genetic studies on CA biosynthesis453 led to the proposal that they might be involved in sensing signalling peptides that regulate CA biosynthesis.475 On the other hand, peptide binding assays suggest that OppA1/2 could be involved in binding and/or transport of small-molecules/peptides across the cell membrane of S. clavuligerus, as they have been found to bind di-/tri-peptides containing arginine and certain nona-peptides including bradykinin.476Crystal structures of apo-OppA2 and in complex with arginine or bradykinin have been reported.476 OppA2 crystallises as a pear-shaped protein (Fig. 3 in476) comprising two loops and three domains, with a large cleft (between the two loops) running the entire length of the molecule, as observed for other oligopeptide-binding proteins.476,477 The cleft is proposed to capture the substrate with subsequent domain movement, where lobes 1 and 2 close around the ligand (peptide/arginine) in a mechanism that has been likened to a “Venus flytrap”.478 Crystal structures of OppA2 in complex with arginine or bradykinin reveal that the C-terminal arginine of bradykinin binds similarly to arginine. At present, the exact role of OppA1 and OppA2 in CA biosynthesis is unclear, but they may play a role in transport of CA/intermediates or in the conversion of clavaminic acid to CA.
5.10 Orf13 and Orf14 of clavulanic acid biosynthesis
The putative proteins encoded for by orf13 and orf14453,455 show similarity to (amino acid) export pump proteins and acetyltransferases, respectively. Mutation (or deletion) resulted in substantial reduction (orf13) or ablation (orf14) of CA and (5S)-clavam production. It is proposed that Orf14 catalyses the formation of acylated derivatives (e.g. N-acetyl-glycyl clavaminic acid) and that Orf13 may be involved in their transport/excretion.455,4795.11 Role of ornithine acetyl transferase in clavulanic acid biosynthesis
The oat2 (orf6) gene of the clavulanic acid (CA) gene cluster (Fig. 28) encodes for an ornithine acetyl transferase (OAT2). Ornithine acetyltransferases (OATs) catalyse the reversible transfer of an acetyl group from N-α-acetyl-L-ornithine to L-glutamate with the corresponding production of L-ornithine and N-α-acetyl-L-glutamate.238 Although a direct role in CA biosynthesis is possible, especially given that N-α-acetyl-L-ornithine is a substrate for CAS (Section 5.3), it seems likely that OATs are involved in optimising arginine production484 as a “feedstock” for CA biosynthesis likely via increased channeling of glutamic acid towards the arginine/CA pathway (Fig. 42). The occurrence of a functional arg box (arginine-controlled regulatory sequence), which is characteristic of genes regulated by the arginine repressor ArgR, upstream of oat2 is supportive of such a role for oat2.485–488 The disruption of argJ, which encodes for another ornithine acetyl transferase (ArgJ, the fifth enzyme of the arginine biosynthesis pathway in S. clavuligerus),487 reduced OAT activity in S. clavuligerus by 24%; however, disruption of oat2 had almost no effect on OAT activity (but slightly reduced CA production), suggesting that the OAT2 activity may be of little relevance to the total OAT cellular activity. Disruption of both argJ and oat2 resulted in residual 69% of the total OAT activity in the cellular extracts, suggesting that uncharacterised enzyme(s), e.g. OAT1 of the clavaminic acid gene cluster (Fig. 28),489 might additionally contribute to the acetyltransferase reaction in S. clavuligerus.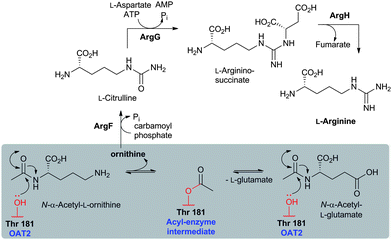 | ||
| Fig. 42 The proposed role of OAT2 in clavulanic acid (CA) biosynthesis. OAT2 is an Ntn-hydrolase proposed to be involved in optimising arginine production as a “feedstock” for CA biosynthesis. Note that N-α-acetyl-L-glutamate can be converted to N-α-acetyl-L-ornithine, via N-α-acetyl-γ-L-glutamyl phosphate and N-α-acetyl-L-glutamate semialdehyde, during L-arginine biosynthesis.724 The ornithine acetyl-transfer reaction (boxed) as catalysed by OAT2 is proposed to proceed via an acyl-enzyme intermediate involving a ping-pong bi–bi mechanism.492,724 | ||
13C NMR and IR analyses imply the presence of an acetyl-enzyme intermediate in OAT catalysis; MS and crystallographic studies have identified Thr181 as the residue being acetylated. The carbonyl oxygen of the acyl-enzyme complex is located in an oxyanion hole and positioned to hydrogen bond with the backbone amide NH of Gly112 and the alcohol of Thr111 (Fig. 43, 44II).491,492 There is evidence that the ester link of the acetyl-enzyme intermediate, similarly to those for other reported acyl-enzyme complexes,494–496 adopts the thermodynamically preferred Z-geometry.497 While the crystallographic analyses reveal only one structure, IR studies demonstrate the presence of two distinct acyl-enzyme complexes; modeling studies suggest that one structure correlates with that observed crystallographically and the other with only a single oxyanion hole hydrogen bond to the backbone amide of Gly112.492 The two structures can interconvert by movement of the Thr111 side-chain away from the oxyanion hole to hydrogen bond with the backbone carbonyl of the acetylated residue Thr181.491,492 The combined crystallographic and modeling studies reveal that whilst the side chains of L-Glu and L-Orn likely bind in different orientations, the L-Orn and L-Glu α-amino groups occupy similar positions relative to the acetylated Thr181 (Fig. 44III), consistent with a common mechanism for N-acetyl transfer.491–493 The L-Glu α-amino group is positioned close to the N-terminal α-amino group of Thr181, consistent with the proposed role of the Thr181 α-amino group in general acid–base catalysis during acetylation/deacetylation (Fig. 43).491,492 From a molecular enzymology perspective, OAT2 is proposed to serve as a model for studying the stereoelectronics of catalysis via acyl-enzyme complexes, which occur as intermediates for many enzymes including proteases, transpeptidases and esterases.
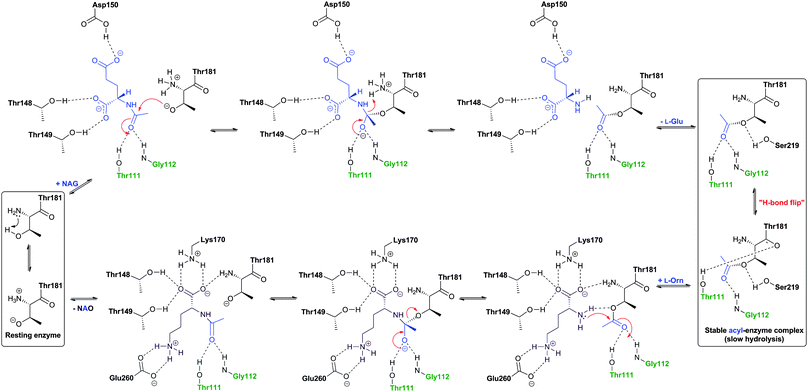 | ||
| Fig. 43 Mechanistic proposal for the Ntn-hydrolase ornithine acetyltransferase (OAT2). The oxyanion hole forming residues are in green. There is evidence for conformational changes during catalysis. NAG: N-α-acetyl-L-glutamate; NAO: N-α-acetyl-L-ornithine.491 | ||
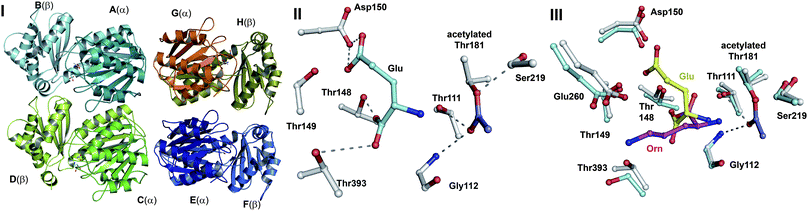 | ||
| Fig. 44 Structural views of ornithine acetyltransferase (OAT2). I: The tetrameric oligomerisation of an acetyl-OAT2-glutamate crystal structure (PDB 2YEP) showing the four subunits/eight chains of OAT2 acyl-enzyme complex.492 The AB, CD, EF and GH molecules are shown in different colours corresponding to the eight different chains; II: View from the active site of an acyl-OAT2-glutamate complex (AB molecule). The acetylated Ntn-residue Thr181 and the oxyanion hole forming residues Thr111 and Gly112 are shown; III: Superimposition of the acetyl-OAT2-glutamate complex (AB molecule) and non-acetylated OAT from Mycobacterium tuberculosis in complex with L-Orn (PDB 3IT4)493 comparing the binding modes for L-Orn and L-Glu (OAT2 numbering is employed). Note that the positions of the α-amino groups of L-Glu and L-Orn are similar, suggesting a closely related mechanism for N-acetylation/deacetylation of the two substrates, but that the binding of their side chains is different. Backbone (main chain) of some residues are omitted for clarity. | ||
Employing N-acetyl-ornithine as an acetyl donor, D-glutamate, L-aspartate, L-leucine, L-alanine, and L-α-aminoadipic acid were not substrates for OAT2. A low acetyltransferase activity was observed with L-arginine, L-glutamine and L-lysine as acceptors.238 The OAT2-catalysed acetylation of L-arginine is of interest because N-acetyl-L-arginine is a good substrate for CAS425 (Fig. 37D). These results suggest that OAT2 might be amenable to protein engineering approaches to modify its substrate specificity/acceptance range, e.g. to resolve racemic mixtures of N-acetylated α-amino acids.
5.12 Summary of genetic studies on clavulanic acid biosynthesis
Genetic studies have been instrumental in deciphering known steps in CA biosynthesis – for detailed descriptions of these, the reader is referred to other reports.381,498–500 In summary, in S. clavuligerus, three gene clusters are apparently involved in CA/clavam biosynthesis; these have been labeled the “clavulanic acid (CA)”, “clavam” and alanyl-clavam/“Paralog” gene clusters, which are located in 3 unlinked loci in the genome (Fig. 28).501 Interestingly, the CA gene cluster is located adjacent to that encoding for cephamycin C biosynthesis forming a “super-cluster”.502 Like the clavam gene cluster, the super-cluster is located on the chromosome, but the paralog gene cluster is located on a megaplasmid (pSCL4),381,503,504 which is densely packed with a large number of gene clusters for the potential production of secondary metabolites, including putative antibiotics, such as moenomycin, β-lactams, and enediynes. Interestingly, cross-regulation occurs between chromosomal and plasmid-encoded genes.503The CA gene cluster likely contains all the non-primary metabolic genes essential for CA production. Homologues of genes encoding for CEAS, β-LS, PAH, and OAT2 are found on the paralog gene cluster, and a cas homologue (CAS1) is present on the clavam gene cluster (Fig. 28). Mutants defective in only one copy of the paired genes (i.e. cas1/cas2, ceas1/ceas2, bls1/bls2, oat1/oat2, and pah1/pah2) are able to produce CA and (5S)-clavams to variable degrees. Double mutants, defective in both copies of the paired genes, are blocked in CA and (5S)-clavam biosynthesis, except for the oat1/oat2 double mutant,453 which produces reduced levels of CA and some (5S)-clavams, suggesting that OAT1/OAT2 serve only to increase arginine levels available for the biosynthesis of these metabolites (Section 5.11). Mutants defective in oppA1 (orf7, Section 5.9), claR (Section 5.6), cad (Section 5.8), or cyp (orf 10, Section 5.6) produced no CA, while production of (5S)-clavam metabolites was relatively unaffected, suggesting the absence of paralogous genes encoding functionally equivalent proteins.505 Recently, orf21, orf22, and orf23, were identified to encode a putative sigma factor, a sensor kinase, and a response regulator, respectively.506–508 Disrupted mutants of these genes displayed modest reductions of CA production, while their overexpression resulted in increased CA production.506–508 Overexpression of ceas2, bls2, pah2, and cas2 resulted in >8 fold increase in CA production;509 for other attempts aiming to improve CA production, see ref. 381, 506, 508, 510, 511.
The clavam gene cluster of S. antibioticus, which is reported to produce only two clavam metabolites, 2-hydroxyethylclavam and valclavam (Fig. 27B, Table 3), but not CA, has recently been sequenced, analysed and shown to resemble the clavam gene cluster of S. clavuligerus.516S. antibioticus cluster also contains genes encoding for apparently new/different proteins, e.g. a putative oxido-reductase and a putative ligase (Table 3). Deletion of the putative oxido-reductase encoding gene abolished clavam production.516
| Gene | AA | (Proposed) function of encoded protein |
|---|---|---|
| psr | 843 | Pathway specific transcriptional regulator (Cvm7p). |
| atr | 467 | Aminotransferase (Cvm6p). |
| akr | 340 | Aldo-keto reductase (Cvm1). |
| hmt | 416 | Hydroxymethyl transferase (OrfA). |
| ceaS3 | 571 | Carboxyethylarginine synthase (CEAS1/CEAS2). |
| bls3 | 507 | β-Lactam synthetase (βls1/βls2). |
| pah3 | 320 | Proclavaminic acid amidinohydrolase (PAH1/PAH2). |
| cas3 | 324 | Clavaminic acid synthase (CAS1/CAS2). |
| oat3 | 394 | Ornithine acetyltransferase (OAT1OAT2). |
| oxr | 214 | Enoyl-reductase. |
| trn | 405 | Clavam transporter. |
| lig | 478 | Ligase. |
| pah4 | 309 | Proclavaminic acid amidinohydrolase (PAH1/PAH2). |
| ctr | 456 | Transcriptional regulator. |
6 Carbapenem biosynthesis
6.1 Subfamilies of naturally-occurring carbapenems
Since the discovery of thienamycin, the number of naturally-occurring carbapenems has risen to over 45. Naturally-occurring carbapenems can be sub-divided into the thienamycins, olivanic acids, epi-thienamycins, carpetimycins, asparenomycins, pluracidomycins, carbapenems of the PS group, and those of the OA-6129 group (Table 4, Fig. 45).517 The majority of identified carbapenems were isolated from the fermentation broths of Streptomyces spp. with two notable exceptions: the simplest carbapen(am/em)s, which were isolated from the fermentation broths of some Enterobacteriaceae spp. e.g. the plant pathogen Pectobacterium carotovorum,518 and AB-110-D, the only known carbapenem with (Z)-geometry at its exo-alkenyl C-2 side chain (Fig. 45), which was isolated from the actinomycete Kitasatosporia papulosa.519
| |||||
|---|---|---|---|---|---|
| Group | R1 | R2 | H5/H6 | C-6 | C-8 |
| Thienamycins | OH | H | trans | S | R |
| Olivanic acids | OH | H | cis | R | S |
| Pluracidomycins | OSO3− | H | cis | R | S |
| Carpetimycins | OH | CH3 | cis | R | — |
| PS | H | H/CH3 | trans | R | — |
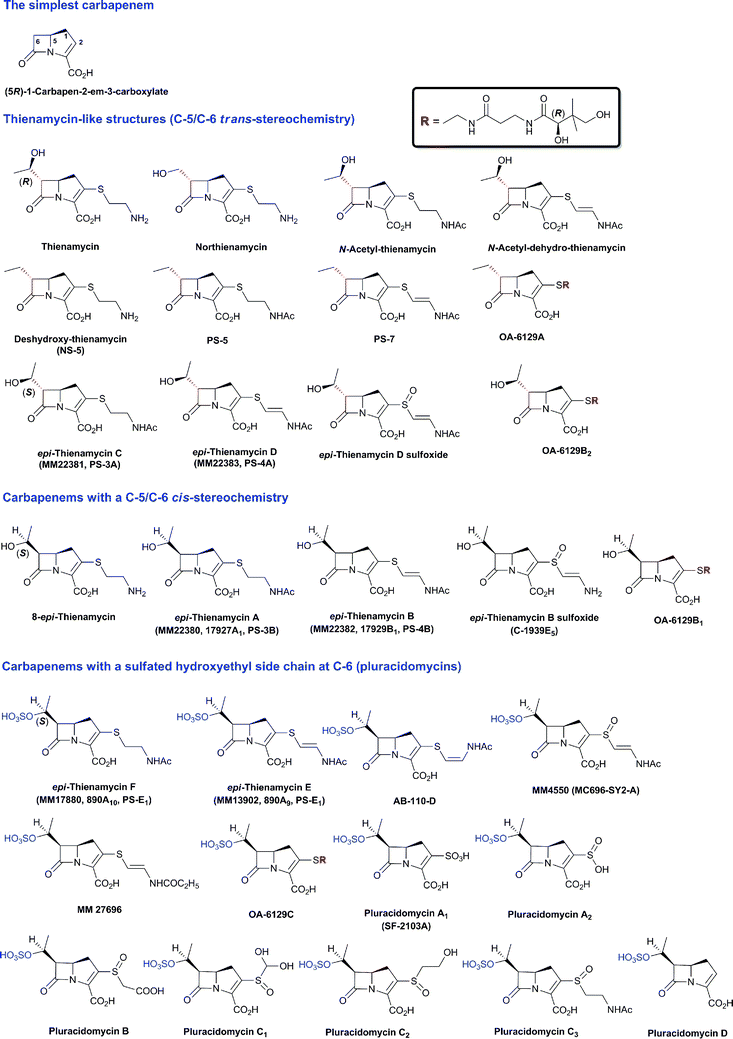 | ||
| Fig. 45a | ||
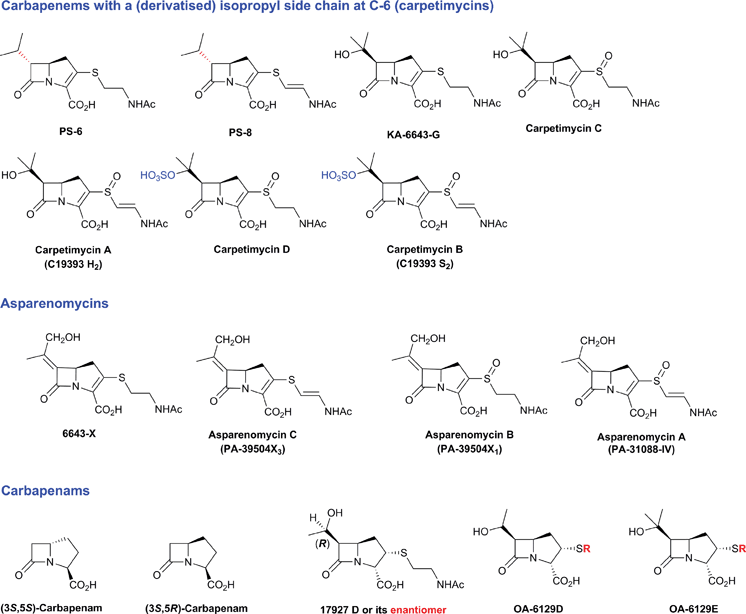 | ||
| Fig. 45b Carbapen(em/am)s isolated from natural sources.525 Some stereochemical assignments are provisional and some of the sulfoxide stereochemistries are unknown. Thienamycin, N-acetyl-thienamycin, N-acetyl-dehydro-thienamycin, 8-epi-thienamycin, northienamycin, and NS-5 were all isolated from wildtype or a mutant of S. cattleya.81,82,726–728 Thienamycin was also isolated from S. penemijaciens.729epi-Thienamycins A–F were isolated from S. flavogriseus571,730 and from S. olivaceus.572,731–735 The olivanic acids MM 4550572,733,734 and MM 27696736 were isolated from S. olivaceus. The PS subfamily of carbapenems (PS-5 to PS-8) were isolated from S. cremeus subsp. auratilis.737–739 Carpetimycins A–D and KA-6643-G were isolated from Streptomyces sp. KC-6643.740–744 Asparenomycins A–C were isolated from S. tokumonensis, S. argentealus and another Streptomyces sp.745,746 Mutation of the carpetimycin producer Streptomyces sp. KC-6643 resulted in a strain that no longer produces carpetimycins but instead produces 6643-X.747 The OA-6129 subfamily of carbapenems (A, B1, B2, C, D, E), characterised with a pantetheinyl moiety at C-2, were isolated from Streptomyces sp. OA-6129 and of S. fulvoviridis mutant.582,748–750 The seven pluracidomycins (A1, A2, B, C1, C2, C3 and D), characterised by containing 2- or 3-acidic moieties in their structures, were isolated from S. pluracidomyceticus.751–753 In addition, the culture co-produced epithienamycin A, epi-thienamycin B sulfoxide, epi-thienamycin D sulfoxide, epi-thienamycin F, MM 4550.752 The carbapenam 17927 D (or its enantiomer) was isolated from S. fulvoviridis, and exhibited no antimicrobial activity.754 | ||
Medicinal chemistry efforts have resulted in the development of the current commercially available carbapenems (see Fig. 46 for examples) which differ from thienamycin in, at least, two important aspects: (i) capping of the nucleophilicity of the C-2 amino side chain, e.g. as in imipenem,520,521 which reduces the intermolecular hydrolysis of the β-lactam; (ii) the introduction of a 1β-methyl substituent, e.g. as in ertapenem, meropenem, and doripenem (Fig. 46), which results in derivatives with superior antibacterial activity and increased resistance to inactivation by renal dehydropeptidases.91,522 The α-methyl isomers are also resistant to dehydropeptidase hydrolysis but their antibacterial activities are reduced.522
![Examples of clinically used carbapenems and the common intermediate used in their preparation, (3R,4R)-4-acetoxy-3-[(R)-1-(tert-butyldimethylsilyloxy)ethyl]azetidin-2-one (AOSA). The part of skeleton corresponding to that of thienamycin is in blue.](/image/article/2013/NP/c2np20065a/c2np20065a-f46.gif) | ||
| Fig. 46 Examples of clinically used carbapenems and the common intermediate used in their preparation, (3R,4R)-4-acetoxy-3-[(R)-1-(tert-butyldimethylsilyloxy)ethyl]azetidin-2-one (AOSA). The part of skeleton corresponding to that of thienamycin is in blue. | ||
6.2 Overview of carbapen-2-em-3-carboxylate biosynthesis
The isolation and characterisation of carbapen-2-em-3-carboxylate (C3C, the simplest carbapenem) was an important advance in understanding carbapenem biosynthesis due to the complexity of the pathways leading to the C-2 and C-6 functionalised carbapenems (e.g. thienamycin, Fig. 45). Pioneering labeling studies in the 1980s established that labeled acetate and glutamate are incorporated into the β-lactam carbons and the fused pyrroline ring, respectively, of both C3C and thienamycin.523,524 Labeled cysteine and methionine have been reported to be incorporated into the C-2 and C-6 side chains of thienamycin, respectively.523 For previous reviews on carbapenem biosynthesis, see ref. 525, 526. We begin by summarising recent studies on the biosynthesis of C3C, then return to studies on thienamycin.C3C was first isolated, in 1982, from P. carotovorum and from a Serratia sp.518 and was subsequently found to be a product of the insect pathogen Photorhabdus luminescens.527 C3C co-occurs with two saturated carbapenams: (3S,5S)- and (3S,5R)-carbapenam-3-carboxylate.528 The isolation, sequencing, and initial assignment of the P. carotovorum C3C biosynthetic gene cluster have revealed 9 genes (carA-H and carR) that are potentially involved in C3C biosynthesis and regulation (Fig. 47B, Table 5).529,530 CarA and CarC show homology to the clavam biosynthesis enzymes β-LS and CAS (Sections 5.2 and 5.3, respectively).29 After some uncertainty with respect to the stereochemistry of the carbapenam intermediates,528,531–533 it was established that only three enzymes are absolutely required for C3C biosynthesis (i.e. CarA, CarB and CarC).529,534
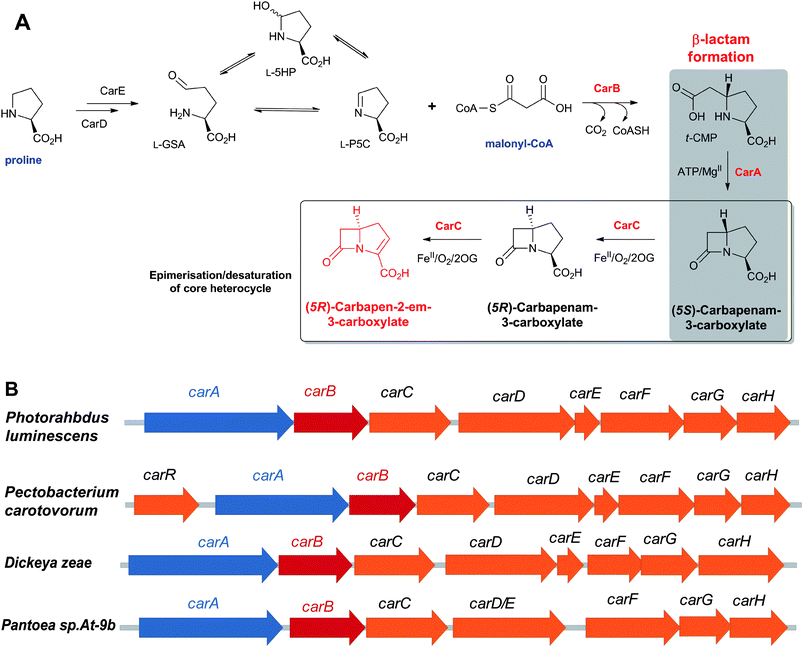 | ||
| Fig. 47 The biosynthetic pathway leading to the simplest carbapenem, (5R)-carbapen-2-em-3-carboxylate (C3C), in Pectobacterium carotovorum (A) and the gene clusters (potentially) involved in C3C biosynthesis (B). The production of C3C by D. zeae and Pantoea sp. is yet to be assessed. Note that in case of Pantoea sp., carD and carE exist as a single gene, unlike other producers of C3C where separate genes exist for carD and carE. See Table 5 for the proposed role of the encoded proteins. Selected co-substrates/co-products are shown. | ||
| Gene | AA | (Proposed) function of encoded protein |
|---|---|---|
| carR | 244 | Transcriptional regulator (CarR).535 |
| carA | 503 | CarA; β-Lactam synthetase.395 |
| carB | 250 | CarB (crotonase); t-CMP formation.537,538 |
| carC | 273 | Carbapenem synthase (CarC).542 |
| carD | 376 | Proline dehydrogenase (CarD).530 |
| carE | 92 | 2Fe-2S ferredoxin (CarE). |
| carF | 288 | Protein involved in resistance to C3C (CarF).529 |
| carG | 177 | Protein involved in resistance to C3C (CarG).529 |
| carH | 184 | Unknown. |
CarR has been identified as a DNA-binding LuxR homologue transcriptional regulator that functions in the presence of N-(3-oxohexanoyl)-L-homoserine lactone (OHHL).535 The CarR:OHHL complex binds to the carA promoter and activates the expression of the carA–H cluster. CarF and CarG are proposed to be involved in intrinsic resistance to C3C via an uncharacterised mechanism.529 CarI is involved in a quorum sensing system involving the synthesis of OHHL. The reader is referred to reviews526,536 for the details of pioneering studies on the regulation of carbapenem biosynthesis, which were of importance far beyond the BLA field. CarD (a putative proline dehydrogenase) and CarE (a putative 2Fe-2S ferredoxin) may act in concert to catalyse proline oxidation530 to give an equilibrating mixture of isomers (L-glutamate semi-aldehyde, L-GSA/L-5-hydroxyproline, L-5HP/L-pyrroline-5-carboxylate, L-P5C), collectively abbreviated as L-GHP, which is a co-substrate for the carboxymethylproline synthase CarB. The crotonase CarB catalyses the formation of (2S,5S)-carboxymethylproline (t-CMP) from malonyl-CoA and L-GHP (L-P5C).537,538 CarB (and likely ThnE in thienamycin biosynthesis) are proposed to act as a “stereochemical gateway” for carbapenem biosynthesis; only in the presence of L-GHP is t-CMP formed with concomitant CoASH production; D-GHP stimulates malonyl-CoA uncoupled turnover and CoASH production but no C–C bond formation product can be detected.539–541 The ATP-dependent carbapenam synthetase CarA (see Section 5.2) then catalyses the β-lactam closure.395 Finally, the carbapenem synthase CarC, a 2OG oxygenase, catalyses the highly unusual C-5 epimerisation of (2S,5S)-carbapenam to a (2S,5R)-derivative which then undergoes a CarC-catalysed desaturation reaction to afford the biologically active antibiotic C3C (Fig. 47A).29,542
6.3 The role of individual enzymes in C3C biosynthesis
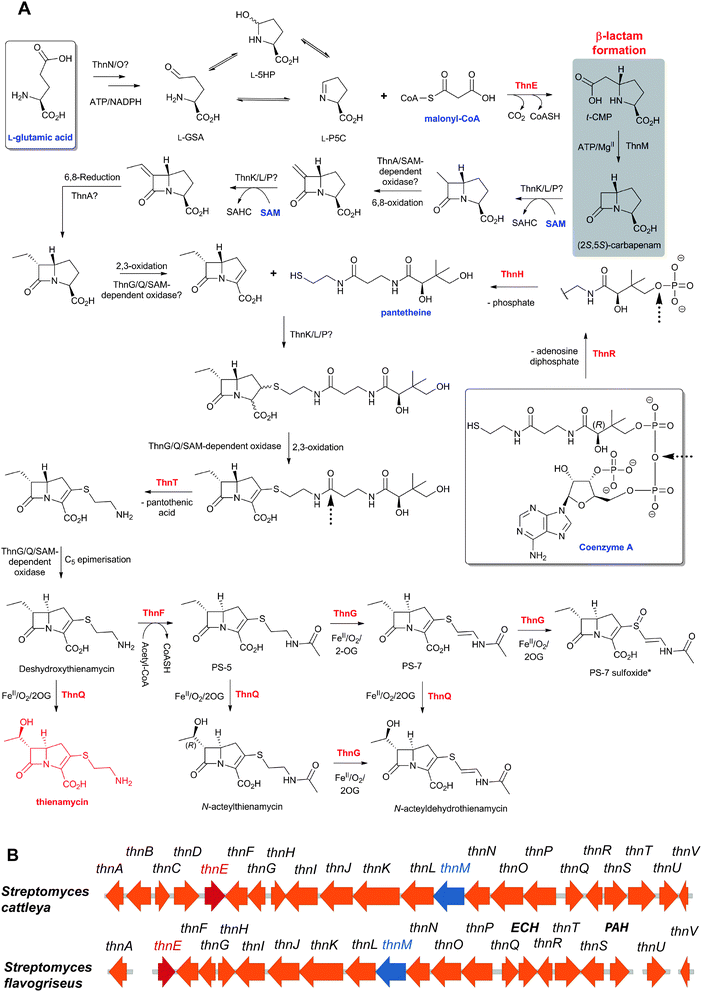 | ||
| Fig. 48 Proposed biosynthetic pathway leading to thienamycin (A) and the reported thienamycin gene cluster in Streptomyces cattleya565 (B). The thienamycin-like gene cluster identified in S. flavogriseus (B) has been reported to be unable to produce thienamycin under laboratory conditions.755 SAHC: S-adenosylhomocysteine. The enzymes catalysing the experimentally-reported steps are in red in (A). The precursors for the skeleton of thienamycin are in blue. PS-7 sulfoxide was detected only in in vitro assays with ThnG (i.e. it was not isolated from S. cattleya). See Table 6 for (proposed) roles of the proteins. Selected co-substrates/co-products are shown. | ||
 | ||
| Fig. 49 Partial sequence alignment for known and putative CMPSs catalysing the formation of t-CMP in carbapenem biosynthesis. The oxyanion hole forming residues are in pink and the catalytically important (at least in CarB)546 glutamate residue in orange (see Fig. 50). The figure was generated using Clustal W756 and Genedoc.757 α-Helices (cyan cylinders) and β-strands (red arrows) represent the assigned secondary structure of CarB.541 | ||
The CarB-catalysed formation of t-CMP is considered as the committed step in C3C biosynthesis in P. carotovorum.29 In the early stages of thienamycin biosynthesis in S. cattleya, the role of ThnE is likely to provide t-CMP, or a derivative thereof.397,539,545In vitro, in addition to its ability to catalyse t-CMP formation (from malonyl-CoA and L-P5C), ThnE catalyses the conversion of epimeric methylmalonyl-CoA to give (6R)-methyl-t-CMP as the major product, with 60% diastereomeric excess, compared to 10% in case of CarB, suggesting that the C-6 alkyl group could be introduced at an early stage in thienamycin biosynthesis (see below). However, the lack of in vitro conversion of ethylmalonyl-CoA to 6-ethyl-t-CMP by ThnE suggests subsequent further methylation and hydroxylation steps are required if ThnE were to be responsible for initial methylation at C-6; though, it seems probable that the C-6 ethyl group is introduced at a later stage (see below).539
6.3.1.1 CarB Structure. CarB crystallises as a homotrimer, a dimer of trimers and a trimer of trimers. However, in solution, both CarB and ThnE exist predominantly as trimers.539,541 While sequence similarities between individual CS proteins are often low, all members share a similar overall fold formed by repeated ββα-motifs that assemble into two approximately perpendicular β-sheets surrounded by α-helices (Fig. 50).543 The catalytic machinery of most CS enzymes operates to stabilise enolate/oxyanion intermediates via an “oxyanion hole” (OAH) comprising two backbone NH groups.543 In the case of CarB and ThnE, the active site residues forming the canonical OAH are proposed to be Met108CarB/Val153ThnE and Gly62CarB/Gly107ThnE (Fig. 49, 50B).539,541 A structure of the CarB:acetyl CoA complex541 reveals the CoA thioester derivative bound in a characteristic U-shaped conformation (Fig. 50B) and a conserved tunnel that binds the pantetheine part of coenzyme A leading to the binding pocket of L-GHP. The L-GHP (L-P5C) co-substrate is predicted to be anchored – via its carboxylate group – into the bottom of the active site pocket.541 Trp79 forms part of the hydrophobic face of the CarB active site541 (Fig. 50C) and was important in CarB engineering studies402,403 (see below). The conserved side chain of Glu131 is suitably positioned to catalyse the hydrolysis of the t-CMP-CoA thioester intermediate541,546 (Fig. 50B).
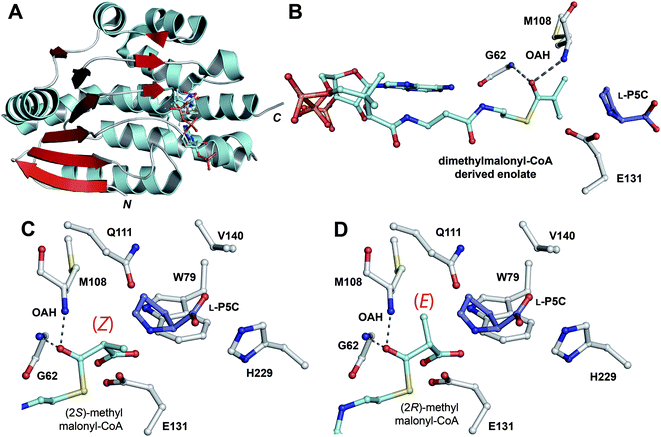 | ||
| Fig. 50 Structural views of carboxymethylproline synthase CarB.541A: A CarB monomer (PDB 2A81) displaying the repeated ββα-motif which is characteristic of the crotonase superfamily;543B: The substrate binding pocket with dimethylmalonyl-CoA derived enolate and L-pyrroline-5-carboxylate (L-P5C) modelled in. Note the characteristic U-shaped conformation of CoA, the location of the thioester carbonyl in the oxyanion hole (OAH), and that the side chain of Glu131 is suitably positioned (∼4 Å from the thioester carbonyl) to activate a water molecule for hydrolysis of a t-CMP-CoA intermediate (Path 3, Fig. 51); C and D: Models of the C-2 epimers of methylmalonyl-CoA (2S epimer: C, and 2R epimer: D) in the substrate binding pocket. Note that, in both cases, an orthogonal relationship between the methylmalonyl-CoA carboxylate and the OAH-stabilised carbonyl is maintained, as required for the stereo-electronically favoured decarboxylation. The enolate (E/Z) that would be generated following decarboxylation is shown in brackets, in each case.403 | ||
6.3.1.2 CarB and ThnE mechanism. CarB/ThnE catalysis is proposed to proceed via decarboxylation of malonyl-CoA to give an OAH-stabilised enolate (Fig. 50B). C–C bond formation can then proceed via reaction of the enolate with potentially any of the forms of L-GHP (Fig. 51): (i) with L-P5C (Path E); (ii) with L-5HP in an SN2 reaction (Path D); or (iii) with L-GSA in an aldol reaction followed by a substitution reaction (Path B), or via elimination of water across C4–C5 (Path A), or via elimination of water across C5–C6 followed by Michael addition (Path C). The ability of CarB to catalyse the formation of 6,6-dimethyl-[2H]6-t-CMP from [2H]6-L-GHP and dimethylmalonyl-CoA argues against Path A and Path C.546,547 Path B cannot be entirely eliminated but seems unlikely (at least in case of the CarB-catalysed formation of 6,6-dimethyl-[2H]6-t-CMP) because it involves a substitution reaction at the sterically hindered “neopentylic” position (C-5). Reaction of the enolate directly with L-5HP (Path D) seems unlikely because: (i) an SN2 type mechanism would have to occur on only one of the two epimeric hemiaminals of L-5HP to produce t-CMP; (ii) both of the C-5 hemiaminal epimers of L-5HP can readily eliminate water to form protonated L-P5C likely giving a lower energy reaction path (Path E).546 The observation that the hindered dimethylmalonyl-CoA derived enolate reacts (at all) is also an indirect evidence for this mechanism because the SN1 iminium ion route (Path E) is sterically less demanding than an SN2 route (Path B/D). Another supportive observation for Path E is that at pH 7.7 (the pH of the final CarB assay mixture), the imine form of L-GHP (L-P5C) is the predominant one (>95% by 1H-NMR).404,548 Analysis of the CarB structure (Fig. 50B) coupled to the observation that t-CMP is the only detectable diastereisomer imply that nucleophilic attack of the enolate anion occurs stereospecifically on the re face of L-P5C to give the intermediate t-CMP-CoA (Fig. 50 and 51).538,546
![Possible C–C bond forming reactions leading to the formation of the t-CMP-CoA intermediate as catalysed by carboxymethylproline synthases. The green box highlights the most likely path.546 For [2H]6-l-GHP, R′ = 2H.](/image/article/2013/NP/c2np20065a/c2np20065a-f51.gif) | ||
| Fig. 51 Possible C–C bond forming reactions leading to the formation of the t-CMP-CoA intermediate as catalysed by carboxymethylproline synthases. The green box highlights the most likely path.546 For [2H]6-L-GHP, R′ = 2H. | ||
For hydrolysis of the t-CMP-CoA intermediate, three mechanisms were considered (Fig. 52): a mechanism involving a ketene intermediate (Path 1), an anhydride intermediate (Path 2), or through direct attack of water (activated by Glu131) onto the thioester carbonyl of t-CMP-CoA (Path 3). The CarB-catalysed conversion of dimethylmalonyl-CoA to 6,6-dimethyl-t-CMP rules out a ketene intermediate (Path 1) at least for this substrate.546,549 Incubations in buffered H218O led to the incorporation of a single 18O into the 6,6-dimethyl-t-CMP product; trypsin digest analyses on CarB from the same incubations revealed no 18O incorporation from solvent into the side chains of Glu131. The lack of 18O incorporation from solvent into the side chains of Glu-131 eliminates Path 2B, and reveals that CarB thioester hydrolysis occurs by a different mechanism than that proposed for the crotonase 3-hydroxyisobutyryl-CoA hydrolase.550 Path 2A cannot be entirely ruled out, but the available evidence argues against it because, at least with 6,6-dimethyl-t-CMP-CoA, this path requires hydrolysis adjacent to a sterically hindered quaternary carbon atom.546 Therefore, the CarB-catalysed thioester hydrolysis is proposed to occur via direct attack of a water molecule on the carbonyl carbon of the t-CMP-CoA intermediate (Path 3).546
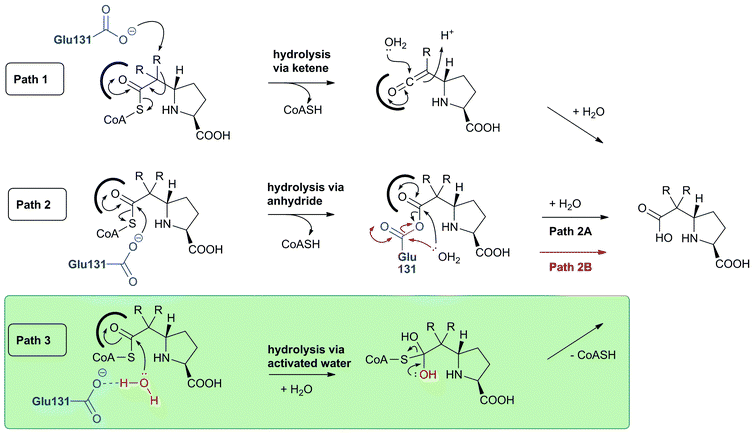 | ||
| Fig. 52 Possible thioester hydrolysis mechanisms for CarB catalysis to produce 6-alkyl-t-CMP derivatives. The green box highlights the most likely mechanism.546 R = H or CH3. | ||
6.3.1.3 CarB/ThnE engineering studies. Structure- and homology-guided CMPS engineering studies were carried out with the aim of deepening understanding of the CMPS mechanism of catalysis as well as to produce new CMPS variants that can catalyse reactions inaccessible for wildtype enzymes, e.g. to catalyse stereoselective quaternary centre formation.402
The CarB E131 A/Q/C/S variants are unable to catalyse the production of t-CMP or t-CMP-CoA from L-GHP and malonyl-CoA, but can catalyse decarboxylation of malonyl-CoA to acetyl-CoA.546 In contrast, the more conservative CarB E131D variant catalyses the formation of t-CMP, albeit in a low yield. These results reveal Glu131 as likely important for both the C–C bond formation and thioester hydrolysis steps but not important for decarboxylation.546
Analogues of L-GHP (L-P5C), substituted at C-2, C-3, C-4, and C-5 of the L-P5C skeleton, are accepted by wildtype and engineered CMPSs to produce the corresponding functionalised N-heterocycles in a stereoselective fashion (Fig. 53B). Notable results include the formation of products with quaternary centres at C-2 or C-5 of t-CMP and the stereoselective formation of (4S)-4-methyl-t-CMP which shares the same pattern of substitution and C-4 stereochemistry as many clinically used carbapenems (e.g. meropenem, Fig. 46).402
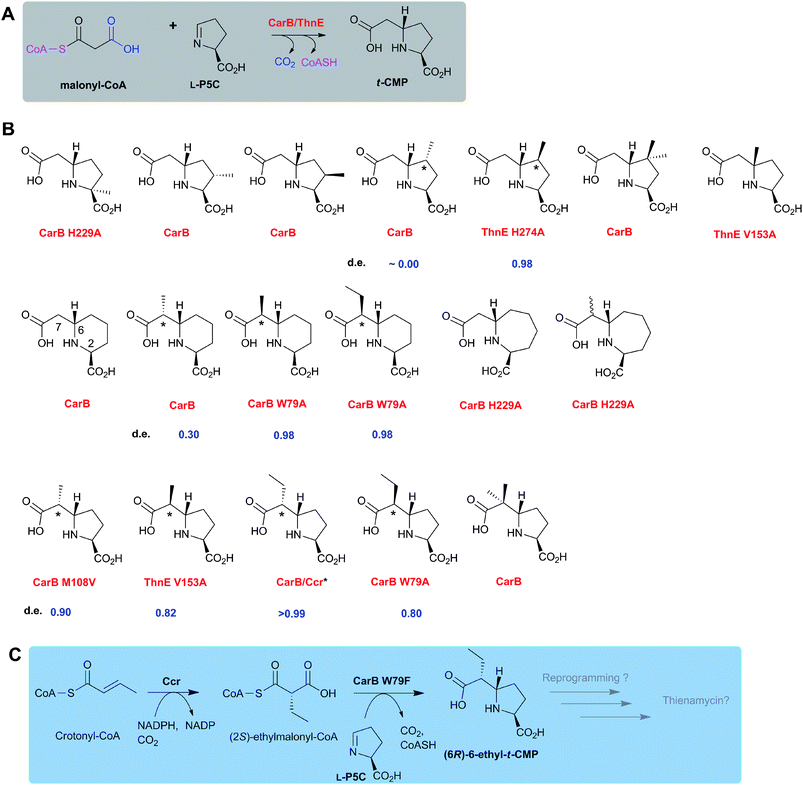 | ||
| Fig. 53 The biocatalytic versatility of wildtype and engineered carboxymethylproline synthases (CMPSs). A: The reaction of wildtype CarB and ThnE; B: A variety of functionalised N-heterocycles is produced from appropriate analogues of L-GHP (L-P5C) and analogues of malonyl-CoA as catalysed by wildtype and engineered CMPSs;402–404,538,539,546C: Coupling CarB catalysis to that of crotonyl-CoA carboxylase reductase (Ccr, which produces (2S)-ethylmalonyl-CoA551,552) results in the stereoselective formation of (6R)-6-ethyl-t-CMP.403 d.e. refers to the diastereomeric excess observed with the shown CMPSs at the asterisked positions. | ||
Analogues of L-GHP (L-P5C) with different chain lengths (i.e.L-aminoadipate semialdehyde and L-aminopimelate semialdehyde) are accepted by wildtype and/or variant CMPSs to stereoselectively produce the corresponding 6- and 7-membered N-heterocycles (Fig. 53B).404
The proposed enolate intermediacy in CMPS catalysis led to the suggestion that the catalytic machinery of these enzymes can be employed to generate “tri-substituted enolates” (from alkyl-malonyl-CoA derivatives), which can react with L-GHP/L-GHP analogues in a stereoselective manner (Fig. 54).403 While wildtype CarB and ThnE display weak to mild stereoselectivity, structure-guided engineering has resulted in CMPS variants that can exercise near complete stereocontrol at C-6/C-7 of the produced functionalised N-heterocycles (Fig. 53B).403 In this regard, two active site residues have played a crucial role: (i) Met108CarB (one of the OAH forming residues) where its substitution with β-branched residues (i.e. Val or Ile) yields variants that favour formation of products with the (6R)-stereochemistry through stereoselective formation of the (Z)-enolate (Fig. 50C, 54); and (ii) Trp79CarB (one of the residues in the hydrophobic part of the CarB active site) where its substitution with a less bulky residue (i.e. Phe or Ala) yields variants that favour formation of products with the (6S)-stereochemistry through stereoselective formation of the (E)-enolate (Fig. 50D, 54). It has been demonstrated that coupling CMPS catalysis to that of crotonyl-CoA carboxylase reductase551–553 (Ccr, from Rhodobacter sphaeroides, which produces the single epimer (2S)-ethylmalonyl-CoA) leads to stereoselective formation of (6R)-6-ethyl-t-CMP (Fig. 53C).403 Although the available evidence is that (6R)-6-ethyl-t-CMP is not an intermediate in thienamycin biosynthesis, these results suggest ways in which the pathway may be re-engineered. It is notable that Ccrs are present in some Streptomyces spp.554–557
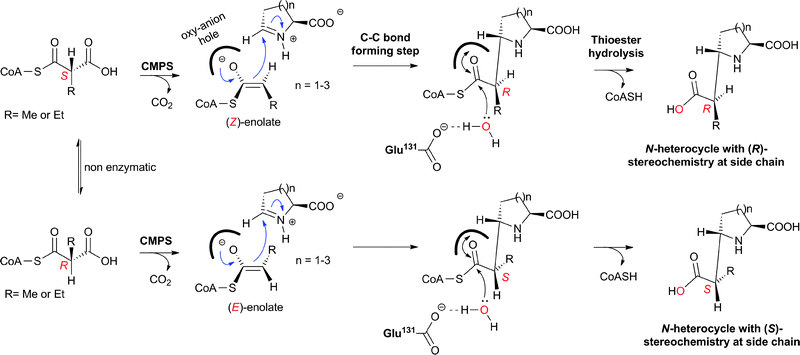 | ||
| Fig. 54 Stereoselective enolate formation and reaction by wildtype and engineered CMPSs.403,404,546 In the case of alkylmalonyl-CoA substrate analogues, the reaction is proposed to proceed via the decarboxylation of a specific epimer to give either the (E)- or (Z)-enolate, which reacts with the imine form of L-amino acid aldehyde to give a specific diastereomer of the CoA-thioester intermediate. Hydrolysis of the CoA-thioester intermediate via an “activated” water molecule (by Glu131 in case of CarB)546 results in the formation of a specific diastereomer of carboxymethyl-substituted N-heterocycles.403 For a model of the possible modes of binding of alkylmalonyl-CoA to CMPS, see Fig. 50. | ||
Overall, the results of CMPS engineering studies have pioneered the applicability of crotonases in biocatalysis and suggest that crotonases may provide a useful platform technology for catalysing synthetically challenging reactions, e.g. the stereoselective generation of enolates and their reaction with electrophiles of choice.
 | ||
| Fig. 55 Sequence alignments for known and putative 2OG and iron-dependent carbapenem synthases catalysing the formation of (5R)-carbapen-2-em-3-carboxylate. The residues involved in the conserved motif of a 2-histidine-1-carboxylate triad758 are in orange. Other residues proposed to be important in carbapenem synthase catalysis are in pink.542 The figure was generated using Clustal W756 and Genedoc.757 Note the high degree of homology between the proteins (≥78% identity). | ||
6.3.3.1 CarC structure and mechanism. The overall CarC structure is similar to those of other 2OG oxygenases and contains a double stranded β-helix core fold (Fig. 19). A crystal structure of CarC in complex with N-acetyl-L-proline (as an analogue of (3S,5S)-carbapenam) and modelling studies have provided insights into CarC substrate binding (Fig. 56). Two orientations of (3S,5S)-carbapenam in the CarC active site are proposed: orientation I positions C-3 and C-2 close to the iron centre with C-5 more exposed to the solvent (analogous to Fig. 56B); and orientation II which positions C-5 close to FeII.542
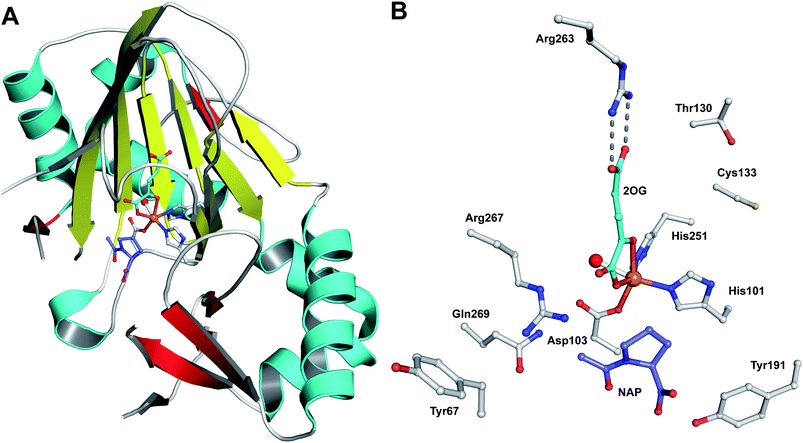 | ||
| Fig. 56 Structural views of the carbapenem synthase CarC.542A: A CarC monomer (PDB 1NX8) displaying the conserved double stranded β-helix core (yellow strands) that supports ligands binding a single FeII to which 2OG complexes in a bidentate manner; B: The active site of CarC (monomer B) showing the binding sites for iron, the prime substrate analogue (N-acetyl-L-proline) and 2OG. Note that another orientation of N-acetyl-L-proline, related by a ∼180° rotation to the one shown, is possible. | ||
Various mechanistic possibilities have been considered for the CarC-catalysed reactions; all of the proposed mechanisms involve a radical intermediate (Fig. 57). Labeling studies have shown loss of C-5 hydrogen during CarC catalysis.560 It should be noted that the epimerised (3S,5R)-carbapenam can be released from the CarC active site and that the source of the C-5 hydrogen of (3S,5R)-carbapenam is unknown. Ab initio calculations on the radicals derived by H abstraction from C-1, C-2, C-3, C-5, and C-6 of (3S,5S)-carbapenam suggest that the C-3 radical is the lowest in energy, because of the captodative effect.561 Loss of C-3-H is likely the favoured scenario in orientation I; C-5 epimerisation could then occur through β-scission of C-3˙ and ring opening to eventually give (5R)-C3C (Fig. 57i). However, in case of C-5 epimerisation mechanisms involving initial C-3, C-1, C-2 and C-6 radical formation, the large barrier associated with ring opening and the unfavourable entropy for ring closure imply that significant “enzyme catalysis” would be required. In case of C-5 hydrogen abstraction mechanisms, C-5 epimerisation is predicted to be exothermic. If a ferryl intermediate was to both abstract the C-5 hydrogen and re-hydrogenate at the same position, re-orientation of the substrate would be required (along with a mechanism for hydrogen exchange). Conformational changes upon substrate binding have been observed for other 2OG oxygenases,268,562 and modelling studies also suggest that (3S,5R)-carbapenam could be accommodated in the active site,561,563 but there is no clear driving force for such a conformational change in the case of CarC catalysis. Therefore, further studies on the CarC mechanism are required.
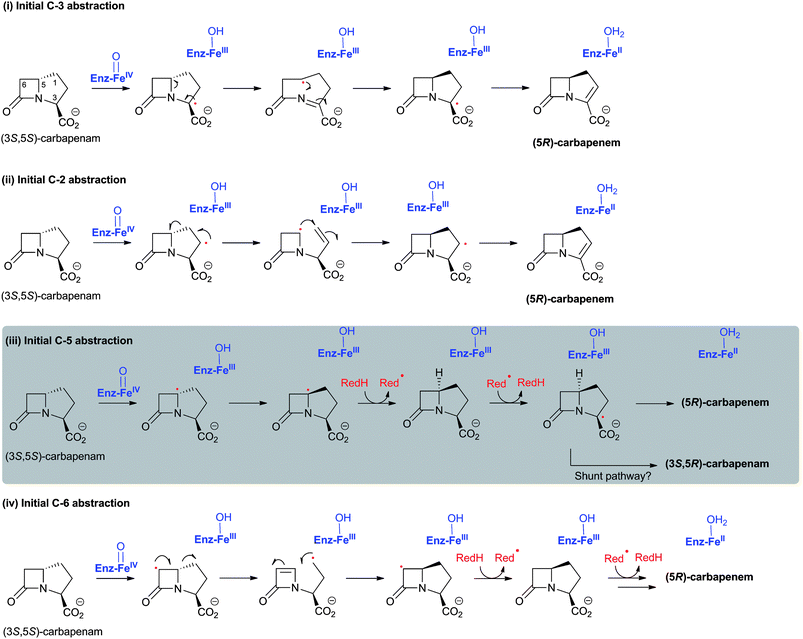 | ||
| Fig. 57 Proposed possibilities for the CarC-catalysed epimerisation/desaturation process.542 The 5-endo-trig (or 4-exo-trig) radical cyclisations in (i) and (ii) have synthetic precedent,759 including with an imine substrate.760 Note the possible intermediacy of datively stabilised radicals. Epimerisation via hydrogen abstraction at C-1 (as is initial electron loss from nitrogen) in a process analogous to (i) is also a possibility, but modelling studies suggest this is less likely. The mechanism proceeding via initial C-5 abstraction (iii) is considered most likely. | ||
6.4 Thienamycin biosynthesis
Labeling studies,523,525,564 and the structures of isolated C-2- and C-6-functionalised carbapenems (Fig. 45) led to early proposals for the pathway of thienamycin biosynthesis.523 Sequencing of the thienamycin biosynthesis gene cluster (from S. cattleya)354,565 has opened up the pathway for delineated genetic and biochemical investigations. In this section we describe the thienamycin gene cluster (Fig. 48B, Table 6) and then describe the reported labeling studies and recent biochemical investigations239,397,539,566 in the light of the genetic information545,565,567 (Fig. 48A).| Gene | AA | (Proposed) function of encoded protein |
|---|---|---|
| thnA | 259 | Oxidoreductase, similar to 3-oxoacyl-[acyl-carrier-protein] reductase. |
| thnB | 259 | Lactone-dependent transcriptional regulator.565 |
| thnC | 259 | Lactone-efflux transmembrane protein.565 |
| thnD | 259 | Alcohol dehydrogenase.565 |
| thnE | 294 | ThnE (crotonase); t-CMP formation.397,539 |
| thnF | 327 | ThnF (N-Acetyltransferase); N-acetylthienamycin formation.239 |
| thnG | 263 | ThnG (2OG oxygenase); oxidation of C-2 side chain of thienamycins.566 |
| thnH | 224 | ThnH cleaves 4-phosphopantetheine to produce pantetheine.239 |
| thnI | 476 | Transcriptional activator essential for thienamycin biosynthesis.567 |
| thnJ | 483 | Transport protein involved in thienamycin secretion.565 |
| thnK | 681 | Methyltransferase.565 |
| thnL | 474 | Methyltransferase.565 |
| thnM | 458 | ThnM; β-Lactam synthetase.397 |
| thnN | 367 | Carboxylate reductase component (similar to griC).568 |
| thnO | 472 | Carboxylate reductase component (similar to griD).568 |
| thnP | 484 | Methyltransferase.565 |
| thnQ | 259 | ThnQ (2OG oxygenase); hydroxylation of C-6 side chain of thienamycins.566 |
| thnR | 240 | ThnR cleaves CoA to produce 4-phosphopantetheine.239 |
| thnS | 329 | β-Lactamase; probably involved in a resistance mechanism.565 |
| thnT | 399 | ThnT; pantetheine hydrolase.239 |
| thnU | 268 | Transcriptional activator for cephamycin C biosynthesis genes.567 |
| thnV | 137 | Cysteine transferase.565 |
The transcriptional activator ThnI, encoded for by the thnI gene, is essential for thienamycin biosynthesis and regulates the expression of nine genes involved in thienamycin assembly and export (thnH, thnJ, thnK, thnL, thnM, thnN, thnO, thnP and thnQ); deletion of thnI ablates thienamycin biosynthesis but increases cephamycin C biosynthesis (through an up-regulation of pcbAB and cmcI transcription).567 ThnU activates cephamycin C biosynthesis genes which are not part of the thienamycin gene cluster.567 These results not only reveal some levels of cross talk between thienamycin and cephamycin C biosynthesis pathways, but also raise a question regarding the precise boundaries of the two gene clusters.567
 | ||
| Fig. 58 The possible role of the reductase GriC/GriD in grixazone biosynthesis in Streptomyces griseus.568 | ||
6.4.3.1 C-6 side chain labeling studies. The methyl group of [methyl-l4C]methionine is incorporated into northienamycin and thienamycin.523 In a double labeling experiment, it has been demonstrated that [methyl-l4C,3H]methionine is incorporated into thienamycin with 58% tritium retention, relative to 14C, corresponding to 87% of the maximum value for retention of 4 of the 6 hydrogens of the two methyl groups.523 Feeding [methyl-2H313C]-methionine to S. cattleya resulted in 13C-incorporation into thienamycin C-9 with retention of all three deuterium atoms and into C-8 with retention of one deuterium atom.564 Feeding (methyl-R)- and (methyl-S)-[methyl-2Hl,3H1]-L-methionine to S. cattleya resulted in the production of thienamycin in which the methyl group has the same configuration as that in the starting methionine (Fig. 59),564 implying that the transfer of the C-9 methyl group (from methionine/S-adenosylmethionine) occurs with net overall retention of configuration. This result is unusual because most of the studied S-adenosylmethionine transferases catalyse methyl transfer with inversion of configuration, apparently, via an SN2-type process.569 One possibility is the introduction of C-9 of thienamycin via a process involving two sequential methyl transfers, each proceeding with inversion.564 Since thienamycin fermentation has been reported to require cobalt,523 it was proposed564 that the two sequential methyl-transfers probably take place from 5-methyltetrahydrofolate, by a B12-dependent synthase, analogous to the biosynthesis of methionine from 5-methyltetrahydrofolate, which also proceeds with net retention of configuration.570–573 Vitamin B12 has been reported to be excreted from a blocked mutant that does not produce thienamycin, and also to restore the thienamycin producing capacity to other blocked mutants.574 It is also noteworthy that methylmalonyl-CoA mutase, which interconverts (2R)-methylmalonyl-CoA and succinyl-CoA, is a vitamin B12 dependent enzyme;575–577 the dependence of thienamycin biosynthesis on cobalt/vitamin B12 may imply that methylmalonyl-CoA is a potential in vivo substrate. However, there is no obvious vitamin B12-dependent protein in the thienamycin gene cluster. Thus it seems probable that the methyl transfer occurs by a different mechanism (see below).
![Summary of the labeling studies on the side chains at C-2 (A) and C-6 (B) of thienamycin in Streptomyces cattleya. *Also, feeding l-[methyl-13C,2H3]methionine to S. cattleya resulted in retention of all 3 deuterium atoms at 13C-9 and retention of one deuterium atom at 13C-8.523,525,564](/image/article/2013/NP/c2np20065a/c2np20065a-f59.gif) | ||
| Fig. 59 Summary of the labeling studies on the side chains at C-2 (A) and C-6 (B) of thienamycin in Streptomyces cattleya. *Also, feeding L-[methyl-13C,2H3]methionine to S. cattleya resulted in retention of all 3 deuterium atoms at 13C-9 and retention of one deuterium atom at 13C-8.523,525,564 | ||
6.4.3.2 Putative roles of ThnK, ThnL and ThnP. In thienamycin biosynthesis, at least two of the three related putative radical SAM superfamily enzymes (i.e. ThnK, ThnL, and ThnP) are proposed to be involved in the methyl-transfer catalysis to afford the C-6 ethyl side chain of thienamycin.545,565 SAM-dependent enzymes catalyse diverse reactions (including e.g. functionalisation of relatively unactivated C–H bonds, isomerisation, sulfur insertion, and elimination; for review on radical SAM enzymes, see ref. 578–580) employing the oxidising power of the generated 5′-deoxyadenosyl radical to abstract a hydrogen atom. Thus, ThnK, ThnL or ThnP may catalyse reactions beyond the predicted methyl-transfer in thienamycin biosynthesis (e.g. C-5 epimerisation, C2-C3 desaturation, ligation of the C-2 cysteaminyl/pantetheinyl side chain). Insertional inactivation of thnL results in a thienamycin non-producing strain.565 Insertional inactivation of thnP resulted in a thienamycin non-producing strain that accumulates a compound with a mass corresponding to that of carbapenam-3-carboxylate.545 This result, if confirmed by definitive characterisation of the accumulated metabolite, suggests the intermediacy of carbapenam-3-carboxylate in thienamycin biosynthesis, and also suggests that ThnP catalysis precedes that of ThnL.
6.4.3.3 ThnQ: hydroxylation of the C-6 (m)ethyl side chain. Recombinant ThnQ, a 2OG oxygenase, catalyses the stereoselective hydroxylation of the C-6 ethyl side chain of (i) PS-7 to produce N-acetyl-dehydro-thienamycin; and (ii) PS-5 to produce N-acetyl-thienamycin (Fig. 48A).566
6.4.4.1 C-2 side chain labeling studies. Feeding radiolabeled cystine to resting cells of S. cattleya resulted in a high level of incorporation of radioactivity into the cysteaminyl side chain of thienamycin.523 On the other hand, [35S]cystamine and [35S]pantethine are poorly incorporated compared to [35S]cystine,523 implying that they are not (direct) precursors of the C-2 cysteaminyl side chain. Feeding a mixture of [3,3′-3H2]-cystine and [35S]cystine to S. cattleya resulted in the production of thienamycin with the same 3H/35S ratio as the starting mixture (Fig. 59),523 consistent with the proposal that a dehydro-thienamycin compound is not an intermediate in thienamycin biosynthesis. Radiolabeled β-alanine is incorporated into the C-2 pantetheinyl side chain of the OA-6129 carbapenems, but labeled pantothenate was not incorporated.581 On the basis of these findings and biochemical studies on ThnR, ThnH and ThnT (see below) it would seem likely that β-alanine is taken up by the mycelia and converted sequentially into (phospho)pantothenate, then (phospho)pantetheine and finally to coenzyme A (see below).
6.4.4.2 The acylase from Streptomyces fulvoviridis. Several naturally-occurring carbapenems contain a C-2 pantetheinyl side chain (Fig. 45). It is proposed that these are likely precursors of the analogous 2-cysteaminyl-carbapenems.582 Mutation of S. fulvoviridis, which mainly produces the N-acetyl-2-cysteaminyl-carbapenems PS-5, and epi-thienamycins A, C, and F, has resulted in a strain that instead produces the 2-pantetheinyl-carbapenems OA-6129A, B1, B2 and C (Fig. 60A).582 The mutant strain also lacks the acylase activity which removes the N-pantothenyl side chain of OA-6129A to give NS-5 (Fig. 60B).582 The acylase in S. fulvoviridis (A933 acylase) which is involved in exchange of the pantothenyl moiety of OA-6129 carbapenems with acetyl-CoA (Fig. 60A) has been partially purified and characterised.583,584 The A933 acylase also catalyses (i) the depantothenylation of OA-6129A to NS5 (Fig. 60B);582 (ii) the acylation of 2-cysteaminyl-carbapenems with acyl-CoA (Fig. 60B); (iii) the acylation of 6-aminopenicillinic acid with acyl-CoAs;583 and (iv) the deacylation of N-acetyl-L-amino acids, but not that of N-acetyl-D-amino acids.584 The A933 acylase does not catalyse the deacetylation of PS-5 to NS-5.583 The acylase is inhibited by cobalt ions and p-chloromercunbenzoate, implying that it has an active thiol.584 A similar acylase activity has also been detected in the following carbapenem producers: S. cattleya (see below), S. cremeus and S. argentcolis.583 The acylase from S. cattleya has been reported, by Kubo et al., as labile and not amenable to purification, but with a similar pattern of specificity to that of S. fulvoviridis acylase.583
 | ||
| Fig. 60 Transacylase-catalysed reactions with substituted carbapenems. A and B: A933 (Streptomyces fulvoviridis) catalysed reactions;582–584 and C: ThnT (S. cattleya) catalysed reaction.239 | ||
6.4.4.3 (Putative) roles of ThnR, ThnH, ThnT, ThnF and ThnG. Efforts aiming at elucidating the enzymology of the C-2 side chain assembly have shown that recombinant ThnR, ThnH and ThnT incrementally cleave CoA to 4-phosphopantetheine, pantetheine, and cysteamine, respectively (Fig. 48A).239 ThnR, a CoA pyrophosphatase, shows similarity to the Nudix hydrolase family.585 ThnH, which hydrolyses the phosphate of 4-phosphopantetheine, shows weak similarity to the haloacid dehalogenase superfamily of hydrolases.586 ThnT, an Ntn hydrolase587 (Section 4.5), catalyses the depantothenylation of both cis- and trans- 2-pantetheinyl-carbapenams into the 2-cysteaminyl analogues239 (Fig. 60C), suggesting that ThnT might be a homologue of the A933 acylase (see above). However, Blanco et al. reported that insertional inactivation of thnR and thnT does not affect thienamycin production in the mutant strains, raising the question whether they are involved in thienamycin biosynthesis;545 one possibility is that the mutant strains have another set of CoA-processing enzymes. The mechanism and timing of ligation of CoA/CoA-derivative into the bicyclic carbapen(am/em) remain to be determined.
ThnF, a putative acetyltransferase, which is weakly related to the GNAT superfamily (Section 5.10), has been predicted to catalyse the conversion of thienamycin to N-acetyl-thienamycin. ThnF has been shown to catalyse N-acetylation of a model compound containing cysteamine in the presence of acetyl-CoA as a co-substrate (Fig. 61).239
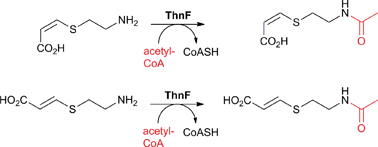 | ||
| Fig. 61 ThnF-catalysed reaction with a substrate analogue.239 Note that the putative substrate for ThnF is thienamycin (Fig. 44).239 | ||
ThnG, a 2OG oxygenase, catalyses the oxidation of the N-acetyl-2-cysteaminyl side chain of (i) N-acetyl-thienamycin to produce N-acetyl-dehydro-thienamycin (desaturation); and (ii) PS-5 to produce PS-7 and PS-7 sulfoxide (desaturation followed by sulfoxidation, Fig. 48A).566 Insertional inactivation of thnG resulted in a mutant strain that showed (i) 2.5-fold increase in thienamycin production; and (ii) accumulation of a metabolite with a mass corresponding to that of 2,3-dihydro-thienamycin in the mutant strain.545 Overall, these results imply that ThnG is not essential for thienamycin biosynthesis, but is required for C-2 oxidation of thienamycin derivatives.
6.4.4.4 ThnT mechanism of autoproteolysis and structure. The autoproteolysis of the Ntn hydrolase ThnT has been studied in detail, and is proposed to be initiated by attack of the activated alcohol of Thr282 onto the carbonyl carbon of the scissile bond, which is located in an oxyanion hole (OAH, Fig. 62C).587 Two water molecules in ThnT active site are proposed to facilitate autoproteolysis via deprotonation of Thr282-OH, protonation of the amine leaving group in the oxazolidine intermediate, and hydrolysis of the ester intermediate to release the Ntn nucleophile Thr282 (Fig. 62A).587 A crystal structure of ThnT Thr282Cys variant led to the observation of two conformations of the backbone sequence in the region of the scissile bond (Fig. 62B); one of the two conformations has the carbonyl oxygen of the scissile bond in the OAH (cleavage-competent form, Fig. 62C).588
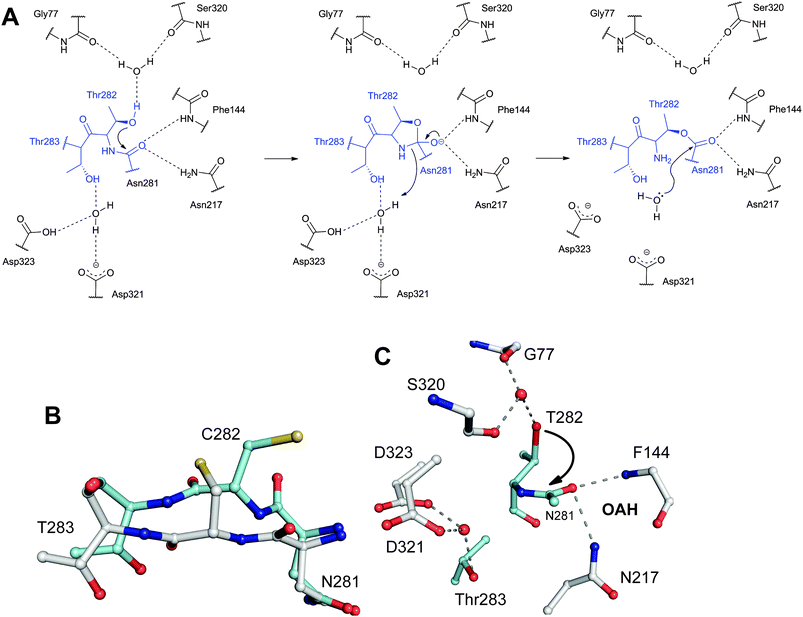 | ||
| Fig. 62 Proposed mechanism and structural views of the pantetheine hydrolase ThnT.587A: Proposed autoproteolysis mechanism of ThnT; B: Dual occupancy of the ThnT Thr282Cys variant active site by shown residues (residues in cyan represent the proposed cleavage-competent conformer, while those in white represent the inactive conformer); C: Model of the cleavage-competent form of ThnT. Note the position of the carbonyl-oxygen of the scissile amide bond in the oxyanion hole (OAH). | ||
7 Monocyclic β-lactam biosynthesis
The two most populated subfamilies of naturally-occurring monocyclic β-lactams are the monobactams (3-aminomonobactamic acid derivatives, Fig. 63A) and the nocardicins (3-aminonocardicinic acid derivatives, Fig. 63B). In addition, there are the tabtoxins (with unsubstituted nitrogen of the β-lactam core, Fig. 63C), and the “conjugate” β-lactams (where the β-lactam nucleus is N-linked to a terpenoid, Fig. 63D). The discovery of conjugate β-lactams is notable as it marks the apparent ability of higher plants to produce β-lactams.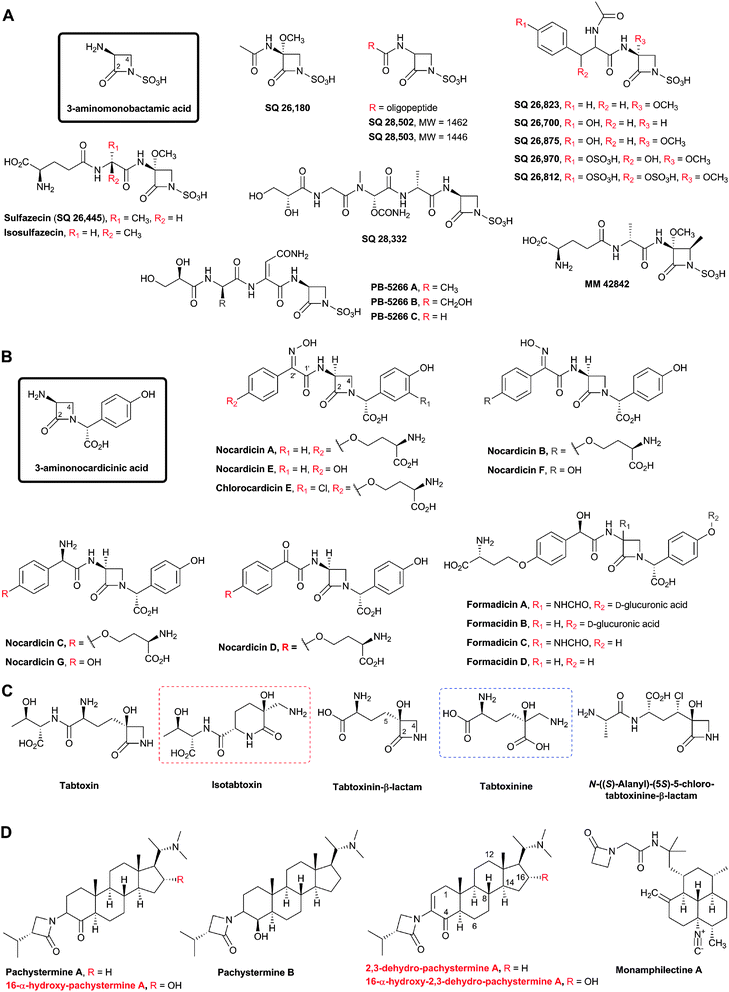 | ||
| Fig. 63 Monocyclic β-lactams isolated from natural sources: The monobactams (A), the nocardicins (B), the tabtoxins (C), and the conjugate β-lactams (D). Some stereochemical assignments are provisional. The nuclei for monobactam and nocardicin subfamilies are boxed. Sulfazecin and its epimer isosulfazecin are produced by Pseudomonas acidophila and P. mesoacidophila, respectively.761,762 Sulfazecin has also been isolated from Gluconobacter and Acetobacter.763,764 SQ 26,180 has been isolated from Chromobacterium violaceum ATCC 31532.763–765 SQ 26,700, SQ 26,812, SQ 26,823, SQ 26,875 and SQ 26,970 (all characterised by a C-3-substituent with an aromatic side chain) were isolated from Agrobacterium radiobacter ATCC 31700.763,766,767 The monobactams SQ 28,332,768 SQ 28,502 and SQ 28,503, with the latter two having longer C-3 oligo-peptide side chains, were isolated from Flexibacter sp.769 PB-5266 A, B and C were isolated from Cytophaga johnsonae.770,771 MM 42842, the only known naturally-occurring monobactam with a 4β-methyl-substituent, was isolated from P. cocovenenans.772,773 Nocardicins A–G were isolated from Nocardia uniformis.78,609,610,774,775 Nocardicins are also produced by Actinosynnema mirum,776Nocardiopsis atra777 and Microtetraspora caesia.778 Chlorocardicin was isolated from a Streptomyces sp.605,606 Formadicins A–D were isolated from Flexibacter alginoliquefaciens.607,608 Nocardicin A and B are stereoisomers differing only in the oxime stereochemistry.611,775 Isotabtoxin (red dashed box) is a stable product of tabtoxin rearrangement; tabtoxinine (blue dashed box) is the hydrolysis product of tabtoxinine-β-lactam. | ||
The monobactams and nocardicins are inhibitors of bacterial cell wall biosynthesis.589 Only three members of the tabtoxin subfamily have been isolated: tabtoxinine β-lactam, its precursor tabtoxin, and N-((S)-alanyl)-(5S)-5-chloro-tabtoxinine-β-lactam (Fig. 63C). Tabtoxin and its hydrolysis product, tabtoxinine β-lactam, are involved in the induction of the wildfire disease of tobacco plant likely through inhibition of glutamine synthetase (see below). N-((S)-Alanyl)-(5S)-5-chloro-tabtoxinine-β-lactam has been isolated from a previously unidentified Streptomyces species 372A590 and found to inhibit the growth of strains of gram-positive and gram-negative bacteria. This inhibition can be relieved by addition of L-glutamine,590 suggesting that, like tabtoxin, it is likely a glutamine synthetase inhibitor. There are few examples of the conjugate β-lactams (Fig. 63D); however, nothing has been reported yet about the biosynthesis of their β-lactam part.591 The antiulcer592,593 and anticancer594 steroidal-β-lactams pachystermine A and pachystermine B have been isolated from Pachysandra terminalis.595,596 16α-Hydroxy-pachystermine A has been isolated from Pachysandra procumbens.597 The antiestrogen-binding site inhibitor dehydro-pachystermine A and its 16α-hydroxy-derivative have also been isolated from Pachysandra procumbens.598 The antimalarial diterpene-β-lactam monamphilectine A has been isolated from the marine sponge Hymeniacidon sp.599
In the following sections, we outline biosynthetic studies on monobactams, nocardicins, and tabtoxins.
7.1 Monobactam biosynthesis
All of the identified monobactams possess an N-sulfonated-β-lactam, but differ in their C-3 acylamido side chain and in the presence/absence of a C-3-α-methoxy group (Fig. 63A). The N-sulfonic acid group is proposed to activate the β-lactam carbonyl for reaction with nucleophiles and enable binding to the PBP active site in an analogous way to the penicillin C-2 carboxylate.600,601 The 4-methyl group in MM 42842 (Fig. 63A) is proposed to enhance the stability of the β-lactam ring towards β-lactamase attack and to improve antibacterial activity (as occurs in the case of aztreonam).600Pioneering studies on the biosynthesis of the β-lactam of sulfazecin, SQ 26,180 and SQ 26,812 using double-labeled amino acids imply that the carbon atoms of the β-lactam of these compounds are derived from serine.602 Feeding experiments using 3-[3H]2-serine indicate that ring closure occurs without change of oxidation state of the serine β-carbon as little [3H]-loss was observed.602 These observations led to the proposed mechanism for β-lactam formation (Fig. 64), analogous to that of nocardicin biosynthesis (see below), including an SN2-type displacement of an activated serine hydroxyl group.
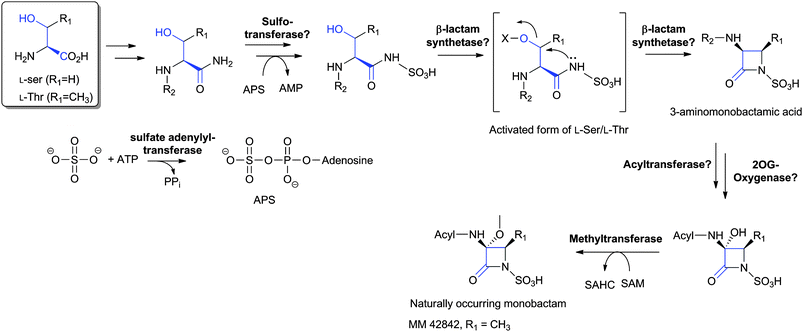 | ||
| Fig. 64 Proposed biosynthetic pathway leading to naturally-occurring monobactams (on the basis of labeling studies as reviewed in ref. 589). The order of steps is uncertain. APS: adenosine-5′-phosphosulfate. SAM: S-adenosylmethionine; SAHC: S-adenosylhomocysteine. | ||
Experiments to investigate the origin of the sulfamate sulfur of some naturally-occurring monobactams have revealed that inorganic sulfur is the only source utilised in the studied organisms.603 It is proposed that the sulfamate group of monobactams is produced via an activated sulfate ion; sulfate activation might be achieved via the formation of adenosine-5′-phosphosulfate (APS, which is produced from ATP and sulfate as catalysed by sulfate adenylyl-transferase) or 3′-phosphoadenosine-5′-phosphosulfate (PAPS, as catalysed by adenylyl-sulfate kinase). Cell free extracts of Agrobacterium radiobacter produce APS upon incubation of sulfate and ATP with the appropriate cofactors, in support of sulfate activation preceding sulfamate formation.603 The question of whether sulfur introduction precedes β-lactam formation is unanswered. The observation of a sulfated peptide in a sulfazecin producer suggests sulfation might occur prior to ring closure; the peptide contains N-terminal glutamate, alanine, serine (incorporated with retention of its C-3 hydrogens), and sulfur.589 The MS and electrophoretic data of the peptide suggest a molecule with properties that fit a substrate for a β-lactam synthetase (Fig. 64). However, incubation with cell free extracts of Acetobacter did not lead to β-lactam formation.589
The absence of the 3α-methoxy group in some naturally-occurring monobactams suggests that the methoxy group is introduced after β-lactam formation, as occurs in cephamycin biosynthesis (Section 4.8). The methyl of the 3α-methoxy group has been shown to originate from methionine,602 likely via methylation of the 3α-hydroxyl group with S-adenosylmethionine (SAM). Again, by analogy to cephamycin biosynthesis, it is reasonable to propose that hydroxylation of the β-lactam is catalysed by a 2OG oxygenase.
The isolation of isosulfazecin, which differs from sulfazecin in the presence of an L- rather than D-alanyl residue in its C-3 side chain, may suggest that isosulfazecin is an intermediate in the biosynthesis of sulfazecin, and that epimerisation occurs, at a late stage, following both β-lactam formation and C-3α-methoxylation. If so, this represents a further example of production of structural diversity by epimerisation in β-lactam biosynthesis, as demonstrated in the penicillin, cephalosporin, clavam, carbapenem, and nocardicin (see below) biosynthesis pathways (Fig. 9, 27, 47 and 66, respectively).
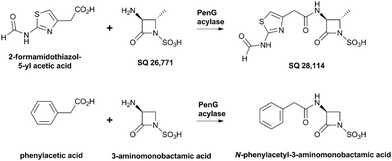 | ||
| Fig. 65 A penicillin G acylase from E. coli catalyses the acylation of 3-amino-monobactamic acid and its 4-methyl derivative.604 | ||
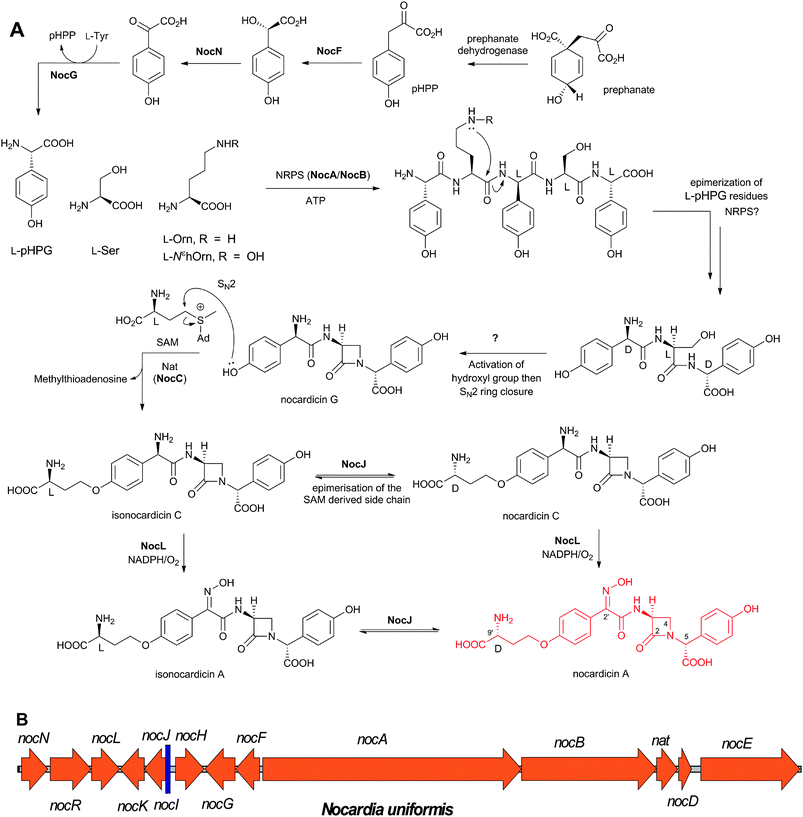 | ||
| Fig. 66 Nocardicin A biosynthesis. A: Proposed biosynthetic pathways leading to naturally-occurring nocardicins; B: The cluster of genes624 encoding for the proteins proposed to be involved in nocardicin A biosynthesis in Nocardia uniformis. See Table 7 for the proposed role of the encoded proteins. pHPP: p-hydroxyphenylpyruvic acid; pHPG: p-hydroxyphenylglycine. | ||
The variety of C-3 acyl-amido side chains observed in naturally-occurring monobactams has led to the hypothesis that they are derived from a common biosynthetic precursor. As yet, however, a monobactam acyl-transferase, similar to the one in penicillin biosynthesis, has not been identified.589 O'Sullivan and Aklonis604 have reported that an immobilised penicillin acylase from E. coli can catalyse the acylation of both 3-aminomonobactamic acid and 4α-methyl-3-aminomonobactamic acid (SQ 26771), when incubated with a variety of acylating agents (e.g. phenylacetic acid and 2-formamidothiazol-5-yl acetic acid, Fig. 65) at pH 4.5. However, in a deacylase reaction, none of the naturally-occurring monobactams has been found to be a substrate for the acylase,604 consistent with biosynthetic reports that addition of possible side chain precursors failed to enhance the levels of monobactam produced.602
7.2 Nocardicin biosynthesis
The nocardicin subfamily of monocyclic β-lactams bear two characteristic aromatic side chains, one attached through the β-lactam nitrogen, and the other is part of a p-hydroxy-phenylacetamido system attached to the β-lactam at C-3. The p-hydroxy group can be functionalised by an ether linked L- or D-homoserinyl moiety (Fig. 63B). The structural diversity of nocardicins originates partly from the nature of the substituents at C-2′ which can be an amine, a ketone, or syn/anti-oxime function (Fig. 63B). The p-hydroxy-phenylacetic acid moiety, attached at its α-carbon to the β-lactam nitrogen, is another source of introducing diversity into the nocardicins (Fig. 63B): (i) Chlorocardicin605,606 is identical to nocardicin A except for the presence of a m-chlorine; (ii) formadicins A and B are p-O-glycosylated nocardicins with formadicin A bearing a 3α-formamido group; formadicins C and D are non-glycosylated analogues with formadicin C bearing a 3α-formamido group.607,608The antibacterial activities of nocardicins B, C, D and E, and formadicins B and D are weaker than those of nocardicin A, chlorocardicin, and formadicins A and C, while nocardicins F and G are devoid of antibiotic activity.609–611 Therefore, the presence of an oxime, and its syn-relationship to the acylamino group, as well as the presence of a D-homoserinyl ether side chain are important for antimicrobial activity.609 Note that the formadicins and some cephabacins (Fig. 8) have an α-formamido substituent at analogous positions on the β-lactam ring; the 6α-/7α-formamido substituent has been shown to improve the β-lactamase stability and activity of some penicillins and cephalosporins.612,613
Experiments employing (2S,4R)- and (2S,4S)-[4-2H]methionine revealed that the ether-linked D-homoserinyl moiety of nocardicin A is efficiently precursored by L-methionine via direct nucleophilic displacement of S-adenosylmethionine (SAM, Fig. 66A).615,619,620
Feeding of racemic [2-13C,15N]-pHPG demonstrated that pHPG provides the nitrogen atoms of both the β-lactam and of the oxime in nocardicin A.621 Employing L-[2-3H, 1-14C]-pHPG, it was found that tritium is not incorporated into the 5-position of nocardicin A.619 Feeding a labeled sample of (DLD)-nocardicin G618 and (LLD)-epinocardicin G revealed intact incorporation of nocardicin G into nocardicin A,622 suggesting the intermediacy of the tripeptide D-pHPG-L-Ser-D-pHPG and consistent with epimerisation of the pHPG residues during/after peptide formation (Fig. 66A), possibly in an analogous fashion to that of the valinyl residue by ACV synthetase during the tripeptide LLD-ACV formation in penicillin biosynthesis (Fig. 11). Furthermore, it was reported that nocardicin E is converted into isonocardicin A (with L-configuration in its homoserinyl side chain) and, consequently, via epimerisation, into nocardicin A in cell-free extracts supplemented with SAM.623 Taken together, these observations imply that the simplest nocardicin, nocardicin G, is the first formed nocardicin β-lactam, and that other nocardicins are derived from nocardicin G following the introduction of the homoserinyl moiety (Fig. 66A).
| Gene | AA | (Proposed) function of encoded protein |
|---|---|---|
| nocN | 376 | p-Hydroxymandelate oxidase. |
| nocR | 582 | Transcriptional regulator.628 |
| nocL | 398 | NocL (Cytochrome P-450 oxidase).623,636 |
| nocK | 344 | Hydrolase/esterase. |
| nocJ | 327 | NocJ (Epimerase).626 |
| nocI | 73 | Unknown. |
| nocH | 408 | Membrane transport protein.628 |
| nocG | 431 | p-Hydroxyphenylglycine transaminase. |
| nocF | 345 | p-Hydroxymandelate synthase. |
| nocA | 3692 | Non-ribosomal peptide synthetase.624,779 |
| nocB | 1925 | Non-ribosomal peptide synthetase.624,779 |
| nat | 301 | 3-Amino-3-carboxypropyl transferase.625 |
| nocD | 185 | Acetyltransferase. |
| nocE | 1414 | Unknown. |
7.2.2.1 NocF, NocN and NocG (pHPG biosynthesis genes). NocF, NocG, and NocN are p-hydroxymandelate synthase, putative p-hydroxyphenylglycine transaminase, and p-hydroxymandelate oxidase, respectively.624 By analogy to other clusters which encode for proteins involved in pHPG biosynthesis,629–631 the reported nocardicin biosynthesis cluster is lacking a gene encoding for a prephenate dehydrogenase.632 NocF stereospecifically catalyses conversion of p-hydroxyphenylpyruvate to (S)-p-hydroxymandelate; NocN oxidises the latter into p-hydroxybenzoylformate (Fig. 66A).624 The predicted transaminase activity of NocG632 awaits experimental testing.
7.2.2.2 NocA and NocB (NRPSs enzymes). On the basis of initial labeling studies (see above) and by analogy to ACVS catalysis (Fig. 11), NocA and B were predicted to catalyse the formation of the tripeptide L-pHPG-L-Ser-L-pHPG and epimerisation into its DLD-isomer, which is a proposed precursor for an SN2 ring closure reaction to give nocardicin G (Fig. 66A).
Analyses of the predicted NocA and NocB sequences (3692 and 1925 amino acid residues, respectively) for NRPS motifs150 imply that together they possess a 5-module structure (3 in NocA and 2 in NocB) including 5 adenylation domains (A1–A5), 4 condensation domains, and a single epimerisation domain, with the 4th module being split between NocA and NocB: the condensation domain being located in NocA while the adenylation and thiolation domains are located in NocB.624 This organisation is unusual as it implies that the NRPS machinery of NocA and NocB does not follow the “colinearity paradigm”.633 On the basis of sequence analyses and homologies, the substrate specificities of the NocA and NocB adenylation domains have been predicted as follows: A1 L-pHPG, A2 L-ornithine (L-Orn) or L-Nε-hydroxyornithine (L-NεhOrn), A3 L-pHPG, A4 L-serine and A5 L-pHPG.634,635 Consequently, a pentapeptide product with the sequence pHPG-X-pHPG-Ser-pHPG has been predicted (X refers to Orn or NεhOrn). However, the proposed precursor for the predicted SN2-type ring closure is the tripeptide D-pHPG-L-Ser-D-pHPG. Further analysis of the NocA sequence has revealed two short unusual amino acid repeats near A1 and T2, suggesting that the first two modules of NocA might be inactive due to a change in their tertiary structure, and that the remaining three modules are involved in the formation of the proposed precursor.624 Another possibility is that the pentapeptide is processed to yield a tripeptide precursor, either through self-editing via intramolecular attack of the (hydroxy)ornithine side chain onto the X-pHPG peptide bond, via a 6-exo trig cyclisation, (Fig. 66A) or as a result of peptidase activity. A similar cyclisation reaction had originally been proposed for coelichelin biosynthesis636 prior to elucidation of the correct structure of coelichelin.637
Only one epimerisation domain has been identified in module 3, and this may catalyse epimerisation of the L-pHPG substrate of A3 upon peptide formation. The necessary epimerisation of the L-pHPG substrate of A5 has been proposed to occur624via acid–base catalysis, exploiting the base-labile nature of the C-5 benzylic proton, or following completion of the peptide product, by a separate peptide epimerase, possibly as in case of serine epimerisation in a 48-amino acid peptide in funnel web spider venom, Agelenopsis aperta.638
To date, attempts to produce active NocA and NocB by recombinant expression have been unsuccessful.624 Two endogenous proteins of 200 kDa (identified as NocB, calculated mass of NocB is 206 kDa) and 150 kDa (identified as a fragment of NocB) have been purified from N. uniformis.624 NocB catalyses ATP/PPi exchange substantially faster in the presence of L-pHPG than in the case of D-pHPG, supporting the proposal that L-pHPG is the substrate for the A5 adenylation domain and suggesting that epimerisation takes place during or after, but not prior to, peptide formation.624 Exchange in the presence of L-Ser was less efficient. NocB has been shown to contain a reactive sulfhydryl group and 4′-phosphopantetheine prosthetic group; however the nocardicin gene cluster is missing a gene encoding for a phosphopantetheinyl transferase required in the conversion of apo-NocA/B to holo-NocA/B.624
Thus, important aspects of NocA/B catalysis remain unclear including the stereochemical course of tripeptide formation and, of particular interest, whether NocA/B are directly involved in β-lactam formation.
7.2.2.3 Nat (NocC). Nat, S-adenosylmethionine (SAM):nocardicin 3-amino-3-carboxypropyltransferase, catalyses the transfer of the 3-amino-3-carboxypropyl moiety of SAM to the phenolic –OH of nocardicin G to form isonocardicin C (Fig. 66A).625 This SN2-type of transfer of the sterically hindered methionine-derived moiety of SAM, rather than the methyl group, is unusual; the structural basis for this preference is proposed to reside on the orientation of the substrate binding to fulfil the geometric requirements for an SN2 reaction, possibly via shielding the methyl group while exposing the 3-amino-3-carboxypropyl moiety for nucleophilic attack.625 Nat displays weak sequence similarity to bacterial SAM-utilising enzymes.625 The C-9′ epimerisation of isonocardicin C to nocardicin C is unrelated to Nat activity (see below). Kinetic analyses revealed a sequential mechanism for Nat and that nocardicin G is a better substrate than nocardicins E/F, supporting the proposed sequence of events in nocardicin A biosynthesis (Fig. 66A).
7.2.2.4 NocJ. The pyridoxal 5′-phosphate (PLP)-dependent enzyme NocJ catalyses the reversible C-9′ epimerisation at the homoserinyl side-chain of nocardicins (Fig. 66A).626 In order to avoid the limitation that isonocardicin A and nocardicin A are indistinguishable by some techniques, e.g. reverse phase HPLC, and to confirm the epimerisation at C-9′ of nocardicin A, the NocJ-reaction has been carried out in deuterated buffer and resulted in a loss of the H-9′ signal in the 1H NMR spectrum.626 Insertional inactivation of nocJ abolishes nocardicin A production but does not affect that of isonocardicin A, consistent with the proposed function of NocJ.626 Despite their functional and co-factor dependence similarity, NocJ shows no significant sequence similarity to prokaryotic isopenicillin N epimerases (Section 4.6),626 but shows similarity to PLP-dependent deaminases.639
7.2.2.5 NocL. NocL is a cytochrome P450 oxidase which catalyses oxidation of the nocardicin C C-2′ amine to produce the syn-oxime of nocardicin A (Fig. 66A) in the presence of spinach ferredoxin, spinach ferredoxin-NADP+ reductase and NADPH.627,640 The reported nocardicin A cluster does not possess genes encoding for NAD(P)H-dependent reductase or a ferredoxin likely required for NocL catalysis. The NocL sequence possesses both a putative heme binding motif (FGHGxHxCLG),640 and an oxygen binding motif (LLxAGHET).641,642 Nocardicin G is not a substrate for NocL in vitro, suggesting the importance of the homoserinyl side chain for NocL activity, and raising the question of how nocardicins E and F are biosynthesised;640 one possibility is through the removal of the homoserinyl side chains of nocardicins A and B, respectively. The order of the C-9′ epimerisation/oxime formation is unclear. Disruption of nocL results in a strain unable to produce nocardicin A but still able to produce nocardicin C.627 The mechanism of NocL catalysis is proposed to proceed via two successive 2′-N-hydroxylations, followed by elimination of water, facilitated by the delocalisation of the neighbouring amide bond, and tautomerisation of the resulting nitroso species to yield an oxime (Fig. 67).640 The greater abundance of the syn-oxime (nocardicin A) relative to the anti-oxime (nocardicin B) isolated from N. uniformis fermentation broth78,610 is proposed to result from intramolecular hydrogen bonding, during the course of the NocL-catalysed oxidation, which can facilitate the dehydration reaction and stabilise the conformation of the nitroso group leading to preferential formation of the syn oxime (Fig. 67).627
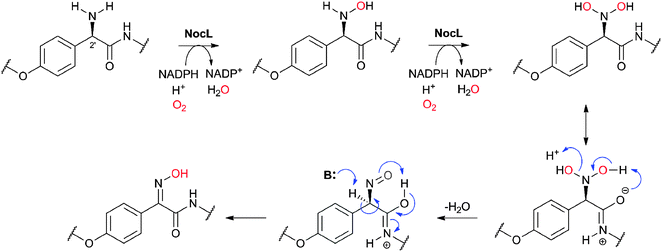 | ||
| Fig. 67 Proposed outline mechanism for the cytochrome P450 NocL-catalysed oxime formation.627,640 | ||
7.3 Tabtoxin biosynthesis
Tabtoxin is a structurally unique monocyclic β-lactam dipeptide because it is not functionalised at its β-lactam nitrogen (Fig. 63C). Elucidation of the tabtoxin structure643 and stereochemistry590,644–646 took a relatively long time, largely due to its instability; tabtoxin undergoes a facile intramolecular transacylation to give the stable, inactive δ-lactam, isotabtoxin (Fig. 63C). Tabtoxin is an exotoxin precursor produced by the phytopathogenic bacterium Pseudomonas syringae pv. tabaci and is involved in the induction of the wildfire (infectious leafspot) disease of tobacco (Nicotiana tabacum, which occasionally obliterated whole fields of tobacco plants).643,646,647 In the infected host, tabtoxin is proposed to be hydrolysed by a peptidase648 to release the tabtoxinine-β-lactam (TBL, the actual wildfire toxin,649Fig. 63C) which inhibits the host glutamine synthetase, consequently resulting in the accumulation of toxic concentrations of ammonia and the characteristic chlorosis.649–653 It should be noted that TBL production is linked to the presence of zinc in the growth media654 and zinc is required for the aminopeptidase activity leading to the cleavage of tabtoxin to yield TBL.648 Self-protection of the producing bacterium has been associated with the adenylation of its glutamine synthetase, which renders the modified enzyme less susceptible to inactivation by TBL.655 A second potential self-resistance mechanism involves the production of β-lactamases which hydrolyse the β-lactam of TBL to liberate the nontoxic metabolite, tabtoxinine (Fig. 63C).656,657 Recently, a tabtoxin-resistance gene (ttr) has been characterised658 and used to enable the development of transgenic tobacco cultivars resistant to P. syringae.659 TTR is an acetyltransferase and contains the four motifs conserved in the GNAT superfamily (Section 5.10).483The mechanism of β-lactam ring closure in tabtoxin biosynthesis is of interest; analyses of the biosynthetic gene cluster of tabtoxin660 (see below) suggests that β-lactam formation is likely mediated by a β-lactam synthetase, TblS, which is less closely related to the identified β-lactam synthetases than they are to each other (Fig. 31). The β-lactam C-3 hydroxylation is likely catalysed by a 2OG oxygenase. It has been proposed that an N-formylated α-amino carbonyl compound might be an intermediate during the formation of the 3-hydroxy-β-lactam nucleus of tabtoxin;661 such compounds have been shown to undergo ring closure to yield 3-hydroxy-β-lactams in a potential biomimetic fashion.662
 | ||
| Fig. 68 Tabtoxin biosynthesis. A: Proposed biosynthetic pathway leading to tabtoxin (on the basis of reported labeling studies661,663,664); B: The gene cluster encoding for the proteins proposed660 to be involved in tabtoxin biosynthesis in Pseudomonas syringae. The enzymes involved in lysine biosynthesis are in red. See Table 8 and text for full description of the cluster. Steps in lysine biosynthesis are shown to illustrate that 2-amino-6-oxopimelate (AOP) is proposed as a “branch point” between lysine and tabtoxin biosynthesis. | ||
| Gene | AA | Proposed function of encoded protein |
|---|---|---|
| tabP | 388 | Zinc metallo-peptidase. |
| tabD | 397 | PLP-dependent aminotransferase. |
| tabB | 276 | Acetyltransferase. |
| tabA | 420 | PLP-dependent decarboxylase. |
| tblA | 252 | SAM-dependent methyltransferase. |
| tabC | 412 | Zinc-binding protein. |
| tablS | 621 | β-Lactam synthetase. |
| tblC | 287 | 2OG oxygenase. |
| tblD | 694 | Fused oxido-reductase and GNAT-acetyltransferase. |
| tblE | 129 | Membrane protein. |
| tblF | 213 | D-Ala-D-Ala ligase. |
| tblR | 873 | Major facilitator superfamily transporter. |
The orf6–8 genes are important for tabtoxin biosynthesis. The tabA (orf7) gene665 is not required for lysine formation despite the significant sequence similarity of its product to prokaryotic diaminopimelate decarboxylase, suggesting that the TabA substrate may be a DAP analogue (Fig. 68A).665 It has also been demonstrated that the dapB gene product is required for both DAP and tabtoxin formation, providing evidence for an overlap between the tabtoxin and lysine biosynthesis pathways, and suggesting that the branch point of the pathways is likely after THDPA, but prior to DAP, biosynthesis (Fig. 68A).666 The tabB (orf6) gene product667 shows sequence similarity to N-succinyl-transferase which catalyses N-succinyl-2-amino-6-oxopimelate formation in lysine biosynthesis (Fig. 68A), suggesting that TabB might be an acetyltransferase;667 acetylated intermediates as part of tabtoxin biosynthesis have already been proposed.668 The tblA (orf8) gene is required for tabtoxin formation, though its function is unclear.669 Regulation of tblA has been shown to be controlled by the lemA gene in P. syringae, probably at the transcriptional level.670–672
A preliminary proposal for tabtoxin biosynthesis, consistent with the reported labeling studies, is shown in Fig. 68A. Key steps are β-lactam synthetase (TblS)-mediated β-lactam formation and 2OG oxygenase (TblC)-catalysed hydroxylation at C-3 of the β-lactam.
8 Summary, conclusions and future prospects
In this article, we have reviewed studies on the enzymology of BLA biosynthesis which have been conducted over the past half century or so. Given the compact nature of the β-lactam nucleus and the plethora of synthetic methods for β-lactam preparation, it is perhaps surprising that only two β-lactam biosynthesis strategies have been identified to date – the oxidative peptide cyclisation reaction of IPNS (in penicillin/cephalosporin biosynthesis) and the ATP-dependent β-amino acid cyclisation reaction of the β-lactam synthetases (in clavam/carbapenem biosynthesis). It seems most probable that nocardicin biosynthesis presents a third strategy involving a synthetase catalysed N-1/C-4 bond formation. Thus, although it seems that (some of) the pathways may have arisen from (a) common evolutionary origin(s), the available evidence suggests that certain types of enzymes may be particularly suited to β-lactam formation. In contrast to the limited ways for β-lactam formation, there are multiple ways by which structural diversity of BLAs is achieved.8.1 Mechanisms of structural diversity introduction into β-lactam biosynthesis pathways
As highlighted previously,29 once the β-lactam has been biosynthesised, diversity is achieved both by branching from common intermediates and by multiple sequential reactions (Table 9). 6-APA acts as a branch point for the biosynthesis of various penicillins with different C-6 acetamido side chains, as does deacetylcephalosporin C (DAC, or an analogue thereof) in the formation of C-3′ cephalosporins. A multiplicity of carbapenems is produced by modification of the C-2 and C-6 substituents, though the details are only emerging. Common themes in the modification of the β-lactam include methylation, acetylation, hydrolysis, addition of peptides by NRPS catalysis, and redox reactions (Table 9). The latter are often catalysed by 2OG oxygenases, though dehydrogenases and P450 enzymes also play roles. The use of epimerisation (isomerisation is the most efficient way of creating diversity from an atom economy perspective) is also notable as it occurs at least in the penicillin/cephalosporin, carbapenem, clavulanic acid and nocardicin pathways, and possibly others. For example, the inversion of the α-stereochemistry of L-valine during LLD-ACV formation and isopenicillin N/penicillin N side chain epimerisation are central to the formation of the penicillins and cephalosporins.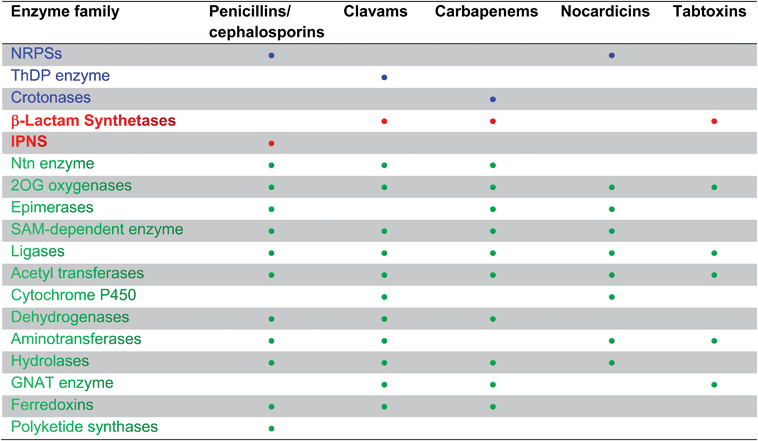 |
8.2 The future of β-lactam biosynthesis studies
Pioneering studies on BLA fermentation were vital by enabling commercially available production procedures. Subsequent studies on the molecular basis of BLA biosynthesis, which came after the “golden age” of BLAs, have been of (bio)chemical interest and have had impact far beyond the BLA biosynthesis, in fields including quorum sensing, oxygen sensing, DNA repair, and epigenetics. Studies on β-lactam biosynthesis have also been a testing ground for new techniques, particularly in molecular biology. However, the studies on BLA biosynthesis enzymes have not yet had a major impact on BLA production or on the development of new BLAs. This is probably a question of timing.There remain chemically interesting questions in β-lactam biosynthesis; these include the mechanisms of the epimerisation in CA biosynthesis, the detailed mechanisms of CarC and DAOCS catalysis, and as to how the reactions of the NRPS machinery of ACVS are coordinated. The mechanisms by which the β-lactam of the monocyclic BLAs is formed are still unclear. No doubt genome sequencing of (new) β-lactam producers will also reveal other interesting enzymes/reactions yet to be considered. It would be rather interesting if new ways for Nature to make β-lactams were to be discovered.
Although there are many secondary metabolites of chemical interest, we (and a few others) like to think of BLAs as special and believe that there is still considerable scope for the development of the BLAs. Most, if not all, clinically used BLAs were developed prior to structural and functional insights into their PBP targets. Thus, there is interest in applying “modern” medicinal chemistry approaches to PBP inhibition. Whether this should be centred around β-lactams, or other chemical templates, including alternative acylating agents, is unclear. However, the track record of BLAs is exceptional, despite resistance issues, as for other antibiotics. If there were renewed interest in a new generation of BLAs, biosynthetic studies could be useful. The scarce discovery of new BLAs over the past 30 years coupled to the need to secure green and sustainable routes for the production of clinically useful ones implies that new strategies need to be pursued.673 One possibility is to make the fermentation of carbapenems, or carbapenem intermediates, viable with a view to avoiding reliance on the AOSA intermediate (Fig. 46). Another would be the development of methods for biocatalytic production of “hybrid” antibiotics, e.g. penams with carbapenem side chains and carbapenems with acetamido side chains, possibly employing engineered enzymes and “mixing and matching” of components from different pathways. The ACVS/IPNS coupled system is one unexplored opportunity, as is building on recent studies on the engineering of carbapenem biosynthesis. The ability of β-lactam biosynthesis enzymes to accept alternative substrates, to date best exemplified by the range of heterocycles produced by IPNS and CMPSs (Fig. 14 and 53, respectively), also presents them as candidates for combinatorial biosynthesis approaches. Other opportunities include, for example, exploiting metagenomic cloning,674,675 biasing pathways via epigenetic control676,677 and the activation of apparently silent gene clusters.678
9 Acknowledgements
We gratefully acknowledge the efforts of all the scientists and their supporters who have worked on the β-lactam biosynthesis story. We apologise for lack of comprehensive citations and any errors or omissions. We also thank the agencies who have funded our work, especially the Biotechnology and Biological Sciences Research Council and the Wellcome Trust. CD thanks the Deutsche Akademie für Naturforscher Leopoldina, Germany, for a postdoctoral fellowship.10 References
- R. P. Elander, Appl. Microbiol. Biotechnol., 2003, 61, 385–392 CAS.
- F.-R. Schmidt, in Industrial Applications, ed. M. Hofrichter, SpringerBerlin Heidelberg, 2010, vol. 10, pp. 101–121 Search PubMed.
- D. M. Livermore, J. Antimicrob. Chemother., 2011, 66, 1941–1944 Search PubMed.
- C. T. Walsh and G. Wright, Chem. Rev., 2005, 105, 391–394 CrossRef CAS.
- I. Chopra, C. Schofield, M. Everett, A. O'Neill, K. Miller, M. Wilcox, J.-M. Frère, M. Dawson, L. Czaplewski, U. Urleb and P. Courvalin, Lancet Infect. Dis., 2008, 8, 133–139 Search PubMed.
- L. Freire-Moran, B. Aronsson, C. Manz, I. C. Gyssens, A. D. So, D. L. Monnet and O. Cars, Drug Resist. Updates, 2011, 14, 118–124 Search PubMed.
- A. R. M. Coates and Y. Hu, Br. J. Pharmacol., 2007, 152, 1147–1154 Search PubMed.
- D. J. Waxman and J. L. Strominger, Annu. Rev. Biochem., 1983, 52, 825–869 CrossRef CAS.
- E. Sauvage, F. Kerff, M. Terrak, J. A. Ayala and P. Charlier, FEMS Microbiol. Rev., 2008, 32, 234–258 CrossRef CAS.
- W. Vollmer, D. Blanot and M. de Pedro, FEMS Microbiol. Rev., 2008, 32, 149–167 CrossRef CAS.
- W. Vollmer and J.-V. Holtje, J. Bacteriol., 2004, 186, 5978–5987 CrossRef CAS.
- J. B. Doherty, B. M. Ashe, L. W. Argenbright, P. L. Barker, R. J. Bonney, G. O. Chandler, M. E. Dahlgren, C. P. Dorn, P. E. Finke, R. A. Firestone, D. Fletcher, W. K. Hagmann, R. Mumford, L. O'Grady, A. L. Maycock, J. M. Pisano, S. K. Shah, K. R. Thompson and M. Zimmerman, Nature, 1986, 322, 192–194 CAS.
- J. Hamilton-Miller, J. Antimicrob. Chemother., 1999, 44, 729–734 CrossRef CAS.
- T. Sperka, J. Pitlik, P. Bagossi and J. Tözsér, Bioorg. Med. Chem. Lett., 2005, 15, 3086–3090 CrossRef CAS.
- C. Yoakim, W. W. Ogilvie, D. R. Cameron, C. Chabot, C. Grand-Maitre, I. Guse, B. Hache, S. Kawai, J. Naud, J. A. O'Meara, R. Plante and R. Deziel, Antiviral Chem. Chemother., 1998, 9, 379–387 Search PubMed.
- O. A. Pierrat, K. Strisovsky, Y. Christova, J. Large, K. Ansell, N. Bouloc, E. Smiljanic and M. Freeman, ACS Chem. Biol., 2010, 6, 325–335 Search PubMed.
- W. D. Vaccaro, R. Sher and H. R. Davis, Bioorg. Med. Chem. Lett., 1998, 8, 319–322 Search PubMed.
- J. Earl and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2003, 2, 97–98 CrossRef CAS.
- M. Garcia-Calvo, J. M. Lisnock, H. G. Bull, B. E. Hawes, D. A. Burnett, M. P. Braun, J. H. Crona, H. R. Davis, D. C. Dean, P. A. Detmers, M. P. Graziano, M. Hughes, D. E. MacIntyre, A. Ogawa, K. A. O'Neill, S. P. N. Iyer, D. E. Shevell, M. M. Smith, Y. S. Tang, A. M. Makarewicz, F. Ujjainwalla, S. W. Altmann, K. T. Chapman and N. A. Thornberry, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8132–8137 Search PubMed.
- B. K. Banik, F. F. Becker and I. Banik, Bioorg. Med. Chem., 2004, 12, 2523–2528 CrossRef CAS.
- B. K. Banik, I. Banik and F. F. Becker, Eur. J. Med. Chem., 2010, 45, 846–848 CrossRef CAS.
- P. Perez-Faginas, M. T. Aranda, M. T. Garcia-Lopez, A. Francesch, C. Cuevas and R. Gonzalez-Muniz, Eur. J. Med. Chem., 2011, 46, 5108–5119 Search PubMed.
- J. Rothstein, M. Dykes-Hoberg, C. Pardo, L. Bristol, L. Jin, R. Kuncl, Y. Kanai, M. Hediger, Y. Wang and J. Schielke, Neuron, 1996, 16, 675–686 CrossRef CAS.
- I. J. Clifton, M. A. McDonough, D. Ehrismann, N. J. Kershaw, N. Granatino and C. J. Schofield, J. Inorg. Biochem., 2006, 100, 644–669 CrossRef CAS.
- M. A. McDonough, C. Loenarz, R. Chowdhury, I. J. Clifton and C. J. Schofield, Curr. Opin. Struct. Biol., 2010, 20, 659–672 CrossRef CAS.
- C. Loenarz and C. J. Schofield, Trends Biochem. Sci., 2011, 36, 7–18 Search PubMed.
- R. Chowdhury, A. Hardy and C. J. Schofield, Chem. Soc. Rev., 2008, 37, 1308–1319 RSC.
- K. S. Hewitson, N. Granatino, R. W. D. Welford, M. A. McDonough and C. J. Schofield, Philos. Trans. R. Soc. London, Ser. A, 2005, 363, 807–828 CrossRef CAS.
- N. J. Kershaw, M. E. C. Caines, M. C. Sleeman and C. J. Schofield, Chem. Commun., 2005, 4251–4263 RSC.
- P. Liras and A. L. Demain, in Methods Enzymol., ed. A. H. David, Academic Press, 2009, vol. 458, pp. 401–429 Search PubMed.
- C. J. Schofield, J. E. Baldwin, M. F. Byford, I. Clifton, J. Hajdu, C. Hensgens and P. Roach, Curr. Opin. Struct. Biol., 1997, 7, 857–864 CrossRef CAS.
- K. H. Baggaley, A. G. Brown and C. J. Schofield, Nat. Prod. Rep., 1997, 14, 309–333 RSC.
- C. A. Townsend, Curr. Opin. Chem. Biol., 2002, 6, 583–589 CrossRef CAS.
- C. A. Townsend, Chem. Biol., 1997, 4, 721–730 CrossRef CAS.
- S. Harada, S. Tsubotani, T. Hida, K. Koyana, M. Kondo and H. Ono, Tetrahedron, 1988, 44, 6589–6606 CrossRef CAS.
- S. Harada, S. Tsubotani, T. Hida, H. Ono and H. Okazaki, Tetrahedron Lett., 1986, 27, 6229–6232 CrossRef CAS.
- Y. Nozaki, N. Katayama, H. Ono, S. Tsubotani, S. Harada, H. Okazaki and Y. Nakao, Nature, 1987, 325, 179–180 CrossRef CAS.
- D. J. Tipper and J. L. Strominger, Proc. Natl. Acad. Sci. U. S. A., 1965, 54, 1133–1141 CAS.
- N. J. Westwood, T. D. W. Claridge, P. N. Edwards and C. J. Schofield, Bioorg. Med. Chem. Lett., 1997, 7, 2973–2978 CrossRef CAS.
- R. C. Wilmouth, N. J. Westwood, K. Anderson, W. Brownlee, T. D. W. Claridge, I. J. Clifton, G. J. Pritchard, R. T. Aplin and C. J. Schofield, Biochemistry, 1998, 37, 17506–17513 CrossRef CAS.
- P. Macheboeuf, D. S. Fischer, T. Brown, A. Zervosen, A. Luxen, B. Joris, A. Dessen and C. J. Schofield, Nat. Chem. Biol., 2007, 3, 565–569 CrossRef CAS.
- T. Brown, P. Charlier, R. l. Herman, C. J. Schofield and E. Sauvage, J. Med. Chem., 2010, 53, 5890–5894 CrossRef.
- W. M. M. Kirby, Science, 1944, 99, 452–453 CrossRef CAS.
- K. Poole, Cell. Mol. Life Sci., 2004, 61, 2200–2223 Search PubMed.
- B. G. Spratt, Science, 1994, 264, 388–393 CAS.
- K. Poole, Ann. Med., 2007, 39, 162–176 Search PubMed.
- A. H. Delcour, Biochim. Biophys. Acta, Proteins Proteomics, 2009, 1794, 808–816 CrossRef CAS.
- A. Matagne, A. Dubus, M. Galleni and J. M. Frere, Nat. Prod. Rep., 1999, 16, 1–19 RSC.
- S. Bratu, S. Brooks, S. Burney, S. Kochar, J. Gupta, D. Landman and J. Quale, Clin. Infect. Dis., 2007, 44, 972–975 Search PubMed.
- J.-M. Frere, Beta-Lactamases, Nova Science Pub Inc, New York, US, 2012 Search PubMed.
- I. Massova and S. Mobashery, Curr. Pharm. Des., 1999, 5, 929–937 Search PubMed.
- Antimicrobial Drug Resistance: Mechanisms of Drug Resistance, ed. D. L. Mayers, Human Press, 2009 Search PubMed.
- S. O. Meroueh, G. Minasov, W. Lee, B. K. Shoichet and S. Mobashery, J. Am. Chem. Soc., 2003, 125, 9612–9618 CrossRef CAS.
- T. R. Walsh, M. A. Toleman, L. Poirel and P. Nordmann, Clin. Microbiol. Rev., 2005, 18, 306–325 CrossRef CAS.
- D. L. Paterson and R. A. Bonomo, Clin. Microbiol. Rev., 2005, 18, 657–686 Search PubMed.
- S. M. Drawz, M. Babic, C. R. Bethel, M. Taracila, A. M. Distler, C. Ori, E. Caselli, F. Prati and R. A. Bonomo, Biochemistry, 2010, 49, 329–340 CrossRef CAS.
- M. G. P. Page, Drug Resist. Updates, 2000, 3, 109–125 Search PubMed.
- S. M. Drawz and R. A. Bonomo, Clin. Microbiol. Rev., 2010, 23, 160–201 CrossRef CAS.
- C. E. Jamieson, P. A. Lambert and I. N. Simpson, Antimicrob. Agents Chemother., 2003, 47, 1652–1657 Search PubMed.
- A. Endimiani, K. M. Hujer, A. M. Hujer, M. E. Pulse, W. J. Weiss and R. A. Bonomo, Antimicrob. Agents Chemother., 2011, 55, 82–85 Search PubMed.
- T. Stachyra, M.-C. Pechereau, J.-M. Bruneau, M. Claudon, J.-M. Frere, C. Miossec, K. Coleman and M. T. Black, Antimicrob. Agents Chemother., 2010, 54, 5132–5138 CrossRef CAS.
- T. Stachyra, P. Levasseur, M.-C. Péchereau, A.-M. Girard, M. Claudon, C. Miossec and M. T. Black, J. Antimicrob. Chemother., 2009, 64, 326–329 Search PubMed.
- O. K. Behrens, J. Corse, J. P. Edwards, L. Garrison, R. G. Jones, Q. F. Soper, F. R. Van Abeele and C. W. Whitehead, J. Biol. Chem., 1948, 175, 793–809 CAS.
- J. Nayler, Proc. R. Soc. London, Ser. B, 1971, 179, 357–367 Search PubMed.
- O. K. Behrens, J. Corse, J. P. Edwards, L. Garrison, R. G. Jones, Q. F. Soper, F. R. Van Abeele and C. W. Whitehead, J. Biol. Chem., 1948, 175, 771–792 Search PubMed.
- F. R. Batchelor, F. P. Doyle, J. H. C. Nayler and G. N. Rolinson, Nature, 1959, 183, 257–258 CrossRef CAS.
- A. K. Chandel, L. V. Rao, M. L. Narasu and O. V. Singh, Enzyme Microb. Technol., 2008, 42, 199–207 CrossRef.
- G. G. F. Newton and E. P. Abraham, Nature, 1955, 175, 548–548 Search PubMed.
- R. B. Morin, B. G. Jackson, R. A. Mueller, E. R. Lavagnino, W. B. Scanlon and S. L. Andrews, J. Am. Chem. Soc., 1969, 91, 1401–1407 CrossRef CAS.
- V. Sonawane, Crit. Rev. Biotechnol., 2006, 26, 95–120 Search PubMed.
- J. Berdy, in Bioactive Metabolites from Microorganisms, ed. M. E. Bushell and U. Grafe, Elsevier, Amsterdam, 1989, vol. 27, pp. 3–25 Search PubMed.
- M. S. Barber, U. Giesecke, A. Reichert and W. Minas, Adv. Biochem. Eng. Biotechnol., 2004, 88, 179–215 CAS.
- Ö. D. Ekici, M. Paetzel and R. E. Dalbey, Protein Sci., 2008, 17, 2023–2037 CrossRef CAS.
- C. Oinonen and J. Rouvinen, Protein Sci., 2000, 9, 2329–2337 CAS.
- R. B. Sykes and J. S. Wells, J. Antibiot., 1985, 38, 119–121 Search PubMed.
- K. Kitano, K. Kintaka, S. Suzuki, K. Katamoto, K. Nara and Y. Nakao, J. Ferment. Technol., 1975, 29, 668–669 Search PubMed.
- A. Brown, D. Butterworth, M. Cole, G. Hanscomb, J. Hood, C. Reading and G. Rolinson, J. Antibiot., 1976, 29, 668–669 CAS.
- H. Aoki, H. Sakai, M. Kohsaka, T. Konomi and J. Hosoda, J. Antibiot., 1976, 29, 492–500 Search PubMed.
- R. B. Sykes and D. P. Bonner, Am. J. Med., 1985, 78, 2–10 Search PubMed.
- S. Coulton and E. Hunt, in Prog. Med. Chem., ed. G. P. Ellis and D. K. Luscombe, Elsevier, 1996, vol. 33, pp. 99–145 Search PubMed.
- J. S. Kahan, F. M. Kahan, R. Goegelman, S. A. Currie, M. Jackson, E. O. Stapley, T. W. Miller, A. K. Miller, D. Hendlin, S. Mochales, S. Hernandez, H. B. Woodruff and J. Birnbaum, J. Antibiot., 1979, 32, 1–12 CAS.
- G. Albers-Schönberg, B. H. Arison, O. D. Hensens, J. Hirshfield, K. Hoogsteen, E. A. Kaczka, R. E. Rhodes, J. S. Kahan, F. M. Kahan, R. W. Ratcliffe, E. Walton, L. J. Ruswinkle, R. B. Morin and B. G. Christensen, J. Am. Chem. Soc., 1978, 100, 6491–6499 CrossRef.
- A. L. Demain and R. P. Elander, Antonie van Leeuwenhoek, 1999, 75, 5–19 CrossRef CAS.
- M. Sunagawa and A. Sasaki, Heterocycles, 2001, 54, 497–528 Search PubMed.
- G. Brooks, G. Bruton, M. F. Finn, J. B. Harbridge, M. A. Harris, T. T. Howarth, E. Hunt, I. Stirling and I. I. Zomaya, in Recent Advances in the Chemistry of β-Lactam Antibiotics, ed. A. G. Brown and S. M. Roberts, Royal Society of Chemistry, London, 1985, p. 221 Search PubMed.
- J. S. Davies, Tetrahedron Lett., 1982, 23, 5089–5092 CrossRef CAS.
- C. E. Newall, in Recent Advances in the Chemistry of β-Lactam Antibiotics, ed. G. I. Gregory, Royal Society of Chemistry, London, 1980, p. 151 Search PubMed.
- P. C. Cherry and E. Newall, in Chemistry and Biology of β-Lactam Antibiotics, ed. R. B. Morin and M. Gorman, Academic Press, New York, 1982, p. 361 Search PubMed.
- C. Reading and M. Cole, J. Enzyme Inhib. Med. Chem., 1986, 1, 83–104 Search PubMed.
- A. Sasaki and M. Sunagawa, Chem. Heterocycl. Compd., 1998, 34, 1249–1265 Search PubMed.
- D. H. Shih, F. Baker, L. Cama and B. G. Christensen, Heterocycles, 1984, 21, 29–40 CAS.
- A. H. Berks, Tetrahedron, 1996, 52, 331–375 CrossRef CAS.
- J. Shoji, T. Kato, R. Sakazaki, W. Nagata, Y. Terui, Y. Nakagawa, M. Shiro, K. Matsumoto and T. Hattori, J. Antibiot., 1984, 37, 1486–1490 CAS.
- J. Shoji, R. Sakazaki, T. Kato, Y. Terui, K. Matsumoto, T. Tanimoto, T. Hattori, K. Hirooka and E. Kondo, J. Antibiot., 1985, 38, 538–540 Search PubMed.
- P. D. Singh, M. G. Young, J. H. Johnson, C. M. Cimarusti and R. B. Sykes, J. Antibiot., 1984, 37, 773–780 CAS.
- S. Tsubotani, T. Hida, H. Ono and S. Harada, J. Antibiot., 1985, 38, 1152–1165 Search PubMed.
- Y. Nozaki, N. Katayama, S. Tsubotani, H. Ono and H. Okazaki, J. Antibiot., 1985, 38, 1141–1151 Search PubMed.
- S. Tsubotani, T. Hida, F. Kasahara, Y. Wada and S. Harada, J. Antibiot., 1984, 37, 1546–1554 CAS.
- S. Harada, S. Tsubotani, H. Ono and H. Okazaki, J. Antibiot., 1984, 37, 1536–1545 CAS.
- T. R. M. Barends, H. Yoshida and B. W. Dijkstra, Curr. Opin. Biotechnol., 2004, 15, 356–363 Search PubMed.
- A. A. Brakhage, Q. Al-Abdallah, A. Tüncher and P. Spröte, Phytochemistry, 2005, 66, 1200–1210 CrossRef CAS.
- S. S. Weber, R. A. L. Bovenberg and A. J. M. Driessen, Biotechnol. J., 2012, 7, 225–236 Search PubMed.
- R. D. G. Cooper, Bioorg. Med. Chem., 1993, 1, 1–17 CrossRef CAS.
- T. R. van der Lende, M. van de Kamp, M. v. d. Berg, K. Sjollema, R. A. L. Bovenberg, M. Veenhuis, W. N. Konings and A. J. M. Driessen, Fungal Genet. Biol., 2002, 37, 49–55 CrossRef CAS.
- M. F. Byford, J. E. Baldwin, C. Y. Shiau and C. J. Schofield, Chem. Rev., 1997, 97, 2631–2650 CrossRef.
- P. L. Roach, I. J. Clifton, V. Fulop, K. Harlos, G. J. Barton, J. Hajdu, I. Andersson, C. J. Schofield and J. E. Baldwin, Nature, 1995, 375, 700–704 CrossRef CAS.
- P. Liras, Antonie van Leeuwenhoek, 1999, 75, 109–124 CrossRef CAS.
- J. J. R. Coque, F. J. Perez-Llarena, F. J. Enguita, J. L. Fuente, J. F. Martin and P. Liras, Gene, 1995, 162, 21–27 CrossRef CAS.
- L. M. Öster, D. R. Lester, A. Terwisscha van Scheltinga, M. Svenda, M. van Lun, C. Généreux and I. Andersson, J. Mol. Biol., 2006, 358, 546–558 Search PubMed.
- S. Inouye, M. Kojima, T. Shomura, K. Iwamatsu, T. Niwa, Y. Kondo, T. Niida, Y. Ogawa and K. Kusama, J. Antibiot., 1983, 36, 115–124 Search PubMed.
- T. Kanzaki, T. Fukita, H. Shirafuji, Y. Fujisawa and K. Kitano, J. Antibiot., 1974, 27, 361–362 Search PubMed.
- T. Kanzaki, T. Fukita, K. Kitano, K. Katamoto, K. Nara and Y. Nakao, J. Ferment. Technol., 1976, 54, 720–725 Search PubMed.
- T. Osono, S. Watanabe, T. Saito, H. Gushima, K. Murakami, I. Takahashi, H. Yamaguchi, T. Sasaki, K. Susaki, S. Takamura, T. Miyoshi and Y. Oka, J. Antibiot., 1980, 33, 1074–1078 Search PubMed.
- A. V. Demirev, C. H. Lee, B. P. Jaishy, D. H. Nam and D. D. Y. Ryu, FEMS Microbiol. Lett., 2006, 255, 121–128 CrossRef CAS.
- Y. S. Sohn, D. H. Nam and D. D. Y. Ryu, Metab. Eng., 2001, 3, 380–392 CrossRef CAS.
- S. Goulden and F. Chattaway, Biochem. J., 1968, 110, 55P–56P Search PubMed.
- T. M. Zabriskie and M. D. Jackson, Nat. Prod. Rep., 2000, 17, 85–97 RSC.
- H. Xu, B. Andi, J. Qian, A. West and P. Cook, Cell Biochem. Biophys., 2006, 46, 43–64 CrossRef CAS.
- E. Valmaseda, S. Campoy, L. Naranjo, J. Casqueiro and J. Martín, Mol. Genet. Genomics, 2005, 274, 272–282 Search PubMed.
- A. L. Demain, Arch. Biochem. Biophys., 1957, 67, 244–246 Search PubMed.
- J. J. R. Coque, P. Liras, L. Laiz and J. F. Martin, J. Bacteriol., 1991, 173, 6258–6264 Search PubMed.
- K. Madduri, C. Stuttard and L. C. Vining, J. Bacteriol., 1991, 173, 985–988 CAS.
- G. Scapin and J. S. Blanchard, Adv. Enzymol. Relat. Areas Mol. Biol., 1998, 72, 279–324 Search PubMed.
- B. A. Kern, D. Hendlin and E. Inamine, Antimicrob. Agents Chemother., 1980, 17, 679–685 Search PubMed.
- F. J. Perez-Llarena, A. Rodriguez-Garcia, F. J. Enguita, J. F. Martin and P. Liras, J. Bacteriol., 1998, 180, 4753–4756 CAS.
- A. L. Leitão, F. J. Enguita, J. F. Martín and J. F. Santos Oliveira, Appl. Microbiol. Biotechnol., 2001, 56, 670–675 Search PubMed.
- V. K. Chary, J. L. De La Fuente, A. L. Leitão, P. Liras and J. F. Martín, Appl. Microbiol. Biotechnol., 2000, 53, 282–288 Search PubMed.
- M.-L. Wu, J.-H. Chen, C.-T. Ho and T.-C. Huang, J. Agric. Food Chem., 2007, 55, 1767–1772 CrossRef CAS.
- J. L. De la Fuente, A. Rumbero, J. F. Martin and P. Liras, Biochem. J., 1997, 327, 59–64 CAS.
- H. Yu, E. Serpe, J. Romero, J.-J. Coque, K. Maeda, M. Oelgeschläger, G. Hintermann, P. Liras, J.-F. Martín, A. L. Demain and J. Piret, Microbiology, 1994, 140, 3367–3377 Search PubMed.
- D. C. Alexander, C. L. Anders, L. Lee and S. E. Jensen, J. Bacteriol., 2007, 189, 5867–5874 Search PubMed.
- D. C. Alexander, M. J. Brumlik, L. Lee and S. E. Jensen, J. Bacteriol., 2000, 182, 348–356 Search PubMed.
- T. Schwecke, Y. Aharonowitz, H. Palissa, H. von Dohren, H. Kleinkauf and H. van Liempt, Eur. J. Biochem., 1992, 205, 687–694 Search PubMed.
- H. B. A. Theilgaard, K. N. Kristiansen, C. M. Henriksen and J. Nielsen, Biochem. J., 1997, 327, 185–191 CAS.
- Y. Aharonowitz, J. Bergmeyer, J. M. Cantoral, G. Cohen, A. L. Demain, U. Fink, J. Kinghorn, H. Kleinkauf, A. MacCabe, H. Palissa, E. Pfeifer, T. Schwecke, H. Van Liempt, H. von Dohren, S. Wolfe and J. Zhang, Bio/Technology, 1993, 11, 807–810 Search PubMed.
- J. Zhang and A. L. Demain, Crit. Rev. Biotechnol., 1992, 12, 245–260 Search PubMed.
- H. von Döhren, in Molecular Biotechnolgy of Fungal beta-Lactam Antibiotics and Related Peptide Synthetases, ed. A. A. Brakhage, Springer Berlin/Heidelberg, 2004, vol. 88, pp. 217–264 Search PubMed.
- U. Linne and M. A. Marahiel, Methods Enzymol., 2004, 388, 293–315 CAS.
- J. Grünewald and M. A. Marahiel, Microbiol. Mol. Biol. Rev., 2006, 70, 121–146 CrossRef.
- M. A. Marahiel and L. O. Essen, Methods Enzymol., 2009, 458, 337–351 CAS.
- T. J. Montavon and S. D. Bruner, in Comprehensive Natural Products II, ed. M. Lew and L. Hung-Wen, Elsevier, Oxford, 2010, pp. 619–655 Search PubMed.
- S. A. Sieber and M. A. Marahiel, Chem. Rev., 2005, 105, 715–738 CrossRef CAS.
- J. E. Baldwin and C. J. Schofield, in The Chemistry of β-lactams, ed. M. I. Page, Blackie Academic & Professional, Glasgow, 1992, pp. 1–78 Search PubMed.
- J. Kennedy and G. Turner, Mol. Gen. Genet., 1996, 253, 189–197 CrossRef CAS.
- G. Banko, S. Wolfe and A. L. Demain, Biochem. Biophys. Res. Commun., 1986, 137, 528–535 Search PubMed.
- J. F. Martín, T. D. Ingolia and S. W. Queener, Molecular Industrial Mycology, 1990, pp. 149–195 Search PubMed.
- J. Martin, T. Ingolia and S. Queener, in Molecular Industrial Mycology: Systems and Applications for Filamentous Fungi, ed. S. Leong and R. Berka, Marcel Dekker, New York, 1991, pp. 149–196 Search PubMed.
- Y. Aharonowitz, G. Cohen and J. F. Martin, Annu. Rev. Microbiol., 1992, 46, 461–495 CrossRef CAS.
- B. Díez, J. Barredo, E. Alvarez, J. Cantoral, P. Solingen, M. Groenen, A. Veenstra and J. Martin, MGG, Mol. Gen. Genet., 1989, 218, 572–576 Search PubMed.
- M. A. Marahiel, T. Stachelhaus and H. D. Mootz, Chem. Rev., 1997, 97, 2651–2674 CrossRef CAS.
- J. F. Martin, J. Antibiot., 2000, 53, 1008–1021.
- H. Kleinkauf and H. von Döhren, Eur. J. Biochem., 1996, 236, 335–351 CrossRef CAS.
- W. Kallow, J. Kennedy, B. Arezi, G. Turner and H. von Döhren, J. Mol. Biol., 2000, 297, 395–408 CrossRef CAS.
- J. Zhang, S. Wolfe and A. L. Demain, Biochem. J., 1992, 283, 691–698 Search PubMed.
- D. Keszenman-Pereyra, S. Lawrence, M.-E. Twfieg, J. Price and G. Turner, Curr. Genet., 2003, 43, 186–190 Search PubMed.
- J. E. Baldwin, J. W. Bird, R. A. Field, N. M. O'Callaghan, C. J. Schofield and A. C. Willis, J. Antibiot., 1991, 44, 241–248 CAS.
- J. E. Baldwin, J. W. Bird, R. A. Field, N. M. O'Callaghan and C. J. Schofield, J. Antibiot., 1990, 43, 1055–1057 CAS.
- J. E. Baldwin, C. Y. Shiau, M. F. Byford and C. J. Schofield, Biochem. J., 1994, 301, 367–372 CAS.
- C.-Y. Shiau, J. E. Baldwin, M. F. Byford, W. J. Sobey and C. J. Schofield, FEBS Lett., 1995, 358, 97–100 Search PubMed.
- C. Y. Shiau, M. F. Byford, R. T. Aplin, J. E. Baldwin and C. J. Schofield, Biochemistry, 1997, 36, 8798–8806 CrossRef CAS.
- L. Luo and C. T. Walsh, Biochemistry, 2001, 40, 5329–5337 Search PubMed.
- A. Stindl and U. Keller, Biochemistry, 1994, 33, 9358–9364 CrossRef CAS.
- X. Wu, C. García-Estrada, I. Vaca and J.-F. Martín, Biochimie, 2012, 94, 354–418 Search PubMed.
- G. Banko, A. L. Demain and S. Wolfe, J. Am. Chem. Soc., 1987, 109, 2858–2860 CrossRef CAS.
- S. E. Jensen, D. W. S. Westlake and S. Wolfe, FEMS Microbiol. Lett., 1988, 49, 213–218 CrossRef CAS.
- J. J. R. Coque, J. L. De La Fuente, P. Liras and J. F. Martín, Eur. J. Biochem., 1996, 242, 264–270 Search PubMed.
- A. Tanovic, S. A. Samel, L. O. Essen and M. A. Marahiel, Science, 2008, 321, 659–663 CrossRef CAS.
- M. Strieker, A. Tanovic and M. A. Marahiel, Curr. Opin. Struct. Biol., 2010, 20, 234–240 CrossRef CAS.
- A. Koglin and C. T. Walsh, Nat. Prod. Rep., 2009, 26, 987–1000 RSC.
- S. A. Samel, G. Schoenafinger, T. A. Knappe, M. A. Marahiel and L. O. Essen, Structure, 2007, 15, 781–792 CrossRef CAS.
- D. P. Frueh, H. Arthanari, A. Koglin, D. A. Vosburg, A. E. Bennett, C. T. Walsh and G. Wagner, Nature, 2008, 454, 903–906 CrossRef CAS.
- C. Pang, B. Chakravarti, R. Adlington, H. Ting, R. White, G. Jayatilake, J. Baldwin and E. Abraham, Biochem. J., 1984, 222, 789–795 CAS.
- G. Cohen, A. Argaman, R. Schreiber, M. Mislovati and Y. Aharonowitz, J. Bacteriol., 1994, 176, 973–984 CAS.
- B. K. Leskiw, Y. Aharonowitz, M. Mevarech, S. Wolfe, L. C. Vining, D. W. S. Westlake and S. E. Jensen, Gene, 1988, 62, 187–196 CrossRef CAS.
- O. Landman, D. Shiffman, Y. Av-Gay, Y. Aharoniwitz and G. Cohen, FEMS Microbiol. Lett., 1991, 84, 239–244 CrossRef CAS.
- J. J. R. Coque, J. F. Martin, J. G. Calzada and P. Liras, Mol. Microbiol., 1991, 5, 1125–1133 CAS.
- M. Garcia-Dominguez, P. Liras and J. F. Martin, Antimicrob. Agents Chemother., 1991, 35, 44–52 Search PubMed.
- P. Loke, C. P. Ng and T. S. Sim, Can. J. Microbiol., 2000, 46, 166–170 CrossRef CAS.
- S. E. Jensen, B. K. Leskiw, L. C. Vining, Y. Aharonowitz, D. W. Westlake and S. Wolfe, Can. J. Microbiol., 1986, 32, 953–958 Search PubMed.
- D. Perry, E. P. Abraham and J. E. Baldwin, Biochem. J., 1988, 255, 345–351 CAS.
- J. M. Castro, P. Liras, L. Laiz, J. Cortes and J. F. Martin, J. Gen. Microbiol., 1988, 134, 133–141 CAS.
- P. L. Roach, I. J. Clifton, C. M. Hensgens, N. Shibata, A. J. Long, R. W. Strange, S. S. Hasnain, C. J. Schofield, J. E. Baldwin and J. Hajdu, Eur. J. Biochem., 1996, 242, 736–740 CrossRef CAS.
- C. J. Schofield and Z. H. Zhang, Curr. Opin. Struct. Biol., 1999, 9, 722–731 CrossRef CAS.
- I. Borovok, O. Landman, R. Kreisberg-Zakarin, Y. Aharonowitz and G. Cohen, Biochemistry, 1996, 35, 1981–1987 CrossRef CAS.
- R. Kreisberg-Zakarin, I. Borovok, M. Yanko, F. Frolow, Y. Aharonowitz and G. Cohen, Biophys. Chem., 2000, 86, 109–118 Search PubMed.
- T. S. Sim and P. Loke, Appl. Microbiol. Biotechnol., 2000, 54, 1–8 Search PubMed.
- A. G. Prescott, J. Exp. Bot., 1993, 44, 849–861 Search PubMed.
- M. Sami, T. J. N. Brown, P. L. Roach, C. J. Schofield and J. E. Baldwin, FEBS Lett., 1997, 405, 191–194 CrossRef CAS.
- P. L. Roach, I. J. Clifton, C. M. Hensgens, N. Shibata, C. J. Schofield, J. Hajdu and J. E. Baldwin, Nature, 1997, 387, 827–830 CrossRef CAS.
- M. Mantri, Z. Zhang, M. A. McDonough and C. J. Schofield, FEBS J., 2012, 279, 1563–1575 Search PubMed.
- J. E. Baldwin and M. Bradley, Chem. Rev., 1990, 90, 1079–1088 CrossRef CAS.
- V. J. Chen, A. M. Orville, M. R. Harpel, C. A. Frolik, K. K. Surerus, E. Munck and J. D. Lipscomb, J. Biol. Chem., 1989, 264, 21677–21681 CAS.
- R. A. Scott, S. Wang, M. K. Eidsness, A. Kriauciunas, C. A. Frolik and V. J. Chen, Biochemistry, 1992, 31, 4596–4601 CrossRef CAS.
- C. R. Randall, Y. Zang, A. E. True, L. Que, Jr., J. M. Charnock, C. D. Garner, Y. Fujishima, C. J. Schofield and J. E. Baldwin, Biochemistry, 1993, 32, 6664–6673 CrossRef CAS.
- M. Lundberg, P. E. M. Siegbahn and K. Morokuma, Biochemistry, 2008, 47, 1031–1042 CrossRef CAS.
- N. I. Burzlaff, P. J. Rutledge, I. J. Clifton, C. M. Hensgens, M. Pickford, R. M. Adlington, P. L. Roach and J. E. Baldwin, Nature, 1999, 401, 721–724 CrossRef CAS.
- J. M. Elkins, P. J. Rutledge, N. I. Burzlaff, I. J. Clifton, R. M. Adlington, P. L. Roach and J. E. Baldwin, Org. Biomol. Chem., 2003, 1, 1455–1460 RSC.
- A. J. Long, I. J. Clifton, P. L. Roach, J. E. Baldwin, C. J. Schofield and P. J. Rutledge, Biochem. J., 2003, 372, 687–693 CrossRef CAS.
- A. J. Long, I. J. Clifton, P. L. Roach, J. E. Baldwin, P. J. Rutledge and C. J. Schofield, Biochemistry, 2005, 44, 6619–6628 CrossRef CAS.
- A. Daruzzaman, I. J. Clifton, R. M. Adlington, J. E. Baldwin and P. J. Rutledge, ChemBioChem, 2006, 7, 351–358 CrossRef CAS.
- E. I. Solomon, T. C. Brunold, M. I. Davis, J. N. Kemsley, S. K. Lee, N. Lehnert, F. Neese, A. J. Skulan, Y. S. Yang and J. Zhou, Chem. Rev., 2000, 100, 235–349 CrossRef CAS.
- C. D. Brown, M. L. Neidig, M. B. Neibergall, J. D. Lipscomb and E. I. Solomon, J. Am. Chem. Soc., 2007, 129, 7427–7438 CrossRef CAS.
- J. E. Baldwin, R. M. Adlington and S. E. Moroney, J. Chem. Soc., Chem. Commun., 1984,(15), 984–986 RSC.
- J. E. Baldwin, M. Bradley, R. M. Adlington, W. J. Norris and N. J. Turner, Tetrahedron, 1991, 47, 457–480 CrossRef CAS.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que Jr, Chem. Rev., 2004, 104, 939–986 CrossRef CAS.
- J. E. Baldwin, E. P. Abraham, R. M. Adlington, J. A. Murphy, N. B. Green, H. H. Ting and J. J. Usher, J. Chem. Soc., Chem. Commun., 1983, 1319–1320 RSC.
- L. Que Jr and R. Y. N. Ho, Chem. Rev., 1996, 96, 2607–2624 CrossRef.
- J. E. Baldwin, R. M. Adlington, D. G. Marquess, A. R. Pitt, M. J. Porter and A. T. Russell, Tetrahedron, 1996, 52, 2515–2536 CrossRef CAS.
- J. E. Baldwin, R. M. Adlington, B. P. Domaynehayman, H. H. Ting and N. J. Turner, J. Chem. Soc., Chem. Commun., 1986, 110–113 RSC.
- J. M. Blackburn, J. D. Sutherland and J. E. Baldwin, Biochemistry, 1995, 34, 7548–7562 CrossRef CAS.
- J. E. Baldwin and E. Abraham, Nat. Prod. Rep., 1988, 5, 129–145 RSC.
- J. E. Baldwin, M. Bradley, S. D. Abbott and R. M. Adlington, Tetrahedron, 1991, 47, 5309–5328 CrossRef CAS.
- A. R. Howard-Jones, J. M. Elkins, I. J. Clifton, P. L. Roach, R. M. Adlington, J. E. Baldwin and P. J. Rutledge, Biochemistry, 2007, 46, 4755–4762 CrossRef CAS.
- P. J. Rutledge, N. I. Burzlaff, J. M. Elkins, M. Pickford, J. E. Baldwin and P. L. Roach, Anal. Biochem., 2002, 308, 265–268 CrossRef CAS.
- A. L. Demain, Y. Q. Shen and S. E. Jensen, J. Antibiot., 1986, 39, 1007–1010 Search PubMed.
- J. M. Castro, P. Liras, J. Cortes and J. F. Martin, FEMS Microbiol. Lett., 1986, 34, 349–353 Search PubMed.
- J. M. Luengo, M. T. Alemany and F. Salto, Bio/Technology, 1986, 4, 44–47 Search PubMed.
- J. E. Baldwin, R. M. Adlington, N. Moss and N. G. Robinson, J. Chem. Soc., Chem. Commun., 1987, 1664–1667 RSC.
- J. E. Baldwin, J. M. Blackburn, M. Sako and C. J. Schofield, J. Chem. Soc., Chem. Commun., 1989, 970–972 RSC.
- J. E. Baldwin, G. P. Lynch and C. J. Schofield, Tetrahedron, 1992, 48, 9085–9100 CrossRef CAS.
- J. E. Baldwin, R. M. Adlington, B. P. Domayne-Hayman, G. Knight and H.-H. Ting, J. Chem. Soc., Chem. Commun., 1987, 1661–1663 RSC.
- J. E. Baldwin, W. J. Norris, R. T. Freeman, M. Bradley, R. M. Adlington, S. Long-Fox and C. J. Schofield, J. Chem. Soc., Chem. Commun., 1988, 1128–1130 RSC.
- J. E. Baldwin, R. M. Adlington, A. E. Derome, H. H. Ting and N. J. Turner, J. Chem. Soc., Chem. Commun., 1984, 1211–1214 RSC.
- J. M. Ogle, I. J. Clifton, P. J. Rutledge, J. M. Elkins, N. I. Burzlaff, R. M. Adlington, P. L. Roach and J. E. Baldwin, Chem. Biol., 2001, 8, 1231–1237 CrossRef CAS.
- E. Alvarez, J. M. Cantoral, J. L. Barredo, B. Diez and J. F. Martin, Antimicrob. Agents Chemother., 1987, 31, 1675–1682 Search PubMed.
- J. L. Barredo, P. van Solingen, B. Diez, E. Alvarez, J. M. Cantoral, A. Kattevilder, E. B. Smaal, M. A. Groenen, A. E. Veenstra and J. F. Martin, Gene, 1989, 83, 291–300 Search PubMed.
- M. B. Tobin, M. D. Fleming, P. L. Skatrud and J. R. Miller, J. Bacteriol., 1990, 172, 5908–5914 Search PubMed.
- P. A. Whiteman, E. P. Abraham, J. E. Baldwin, M. D. Fleming, C. J. Schofield, J. D. Sutherland and A. C. Willis, FEBS Lett., 1990, 262, 342–344 Search PubMed.
- F.-Q. Wang, J. Liu, M. Dai, Z.-H. Ren, C.-Y. Su and J.-G. He, Biochem. Biophys. Res. Commun., 2007, 360, 453–458 Search PubMed.
- Z.-L. Yu, J. Liu, F.-Q. Wang, M. Dai, B.-H. Zhao, J.-G. He and H. Zhang, Folia Microbiol., 2011, 56, 246–252 Search PubMed.
- M. Lamas-Maceiras, I. Vaca, E. Rodriguez, J. Casqueiro and J. F. Martin, Biochem. J., 2006, 395, 147–155 CrossRef CAS.
- M. A. Ferrero, A. Reglero, J. Martin-Villacorta, J. M. Fernandez-Canon and J. M. Luengo, J. Antibiot., 1990, 43, 684–691 CAS.
- P. A. Whiteman and E. P. Abraham, FEBS Lett., 1996, 394, 31–33 Search PubMed.
- E. Alvarez, B. Meesschaert, E. Montenegro, S. Gutierez, B. Diez, J. L. Barredo and J. F. Martin, Eur. J. Biochem., 1993, 215, 323–332 CAS.
- J. M. Luengo, J. Antibiot., 1995, 48, 1195–1212 CAS.
- J. M. Luengo, J. L. Iriso and M. J. Lopez-Nieto, J. Antibiot., 1986, 39, 1754–1759 Search PubMed.
- H. Martinez-Blanco, A. Reglero and J. M. Luengo, J. Antibiot., 1991, 44, 1252–1258 Search PubMed.
- N. J. Kershaw, H. J. McNaughton, K. S. Hewitson, H. Hernández, J. Griffin, C. Hughes, P. Greaves, B. Barton, C. V. Robinson and C. J. Schofield, Eur. J. Biochem., 2002, 269, 2052–2059 CrossRef CAS.
- M. F. Freeman, K. A. Moshos, M. J. Bodner, R. Li and C. A. Townsend, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11128–11133 CrossRef CAS.
- R. T. Aplin, J. E. Baldwin, S. C. J. Cole, J. D. Sutherland and M. B. Tobin, FEBS Lett., 1993, 319, 166–170 Search PubMed.
- M. Bokhove, H. Yoshida, C. M. H. Hensgens, J. Metske van der Laan, J. D. Sutherland and B. W. Dijkstra, Structure, 2010, 18, 301–308 Search PubMed.
- Y. Li, J. Chen, W. Jiang, X. Mao, G. Zhao and E. Wang, Eur. J. Biochem., 1999, 262, 713–719 Search PubMed.
- M. A. Arroyo, I. d. l. M. de la Mata, C. A. Acebal and M. P. C. Pilar Castillón, Appl. Microbiol. Biotechnol., 2003, 60, 507–514 CAS.
- Y. Kim, K.-H. Yoon, Y. Khang, S. Turley and W. G. J. Hol, Structure, 2000, 8, 1059–1068 Search PubMed.
- V. K. Sudhakaran, B. S. Deshpande, S. S. Ambedkar and J. G. Shewale, Process Biochem., 1992, 27, 131–143 Search PubMed.
- N. Esaki and C. T. Walsh, Biochemistry, 1986, 25, 3261–3267 CrossRef CAS.
- A. Watababe, Y. Kurokawa, T. Yoshimura, T. Kurihara, K. Soda and N. Esaki, J. Biol. Chem., 1999, 274, 4189–4194 CrossRef CAS.
- S. E. Jensen, D. W. S. Westlake and S. Wolfe, Can. J. Microbiol., 1983, 29, 1526–1531 CrossRef CAS.
- F. J. Enguita, J. J. R. Coque, P. Liras and J. F. Martin, J. Bacteriol., 1998, 180, 5489–5494 Search PubMed.
- J. J. Coque, J. F. Martin and P. Liras, MGG, Mol. Gen. Genet., 1993, 236, 453–458 Search PubMed.
- S. Kovacevic, M. B. Tobin and J. R. Miller, J. Bacteriol., 1990, 172, 3952–3958.
- R. J. Bowers, S. E. Jensen, L. Lyubechansky, D. W. S. Westlake and S. Wolfe, Biochem. Biophys. Res. Commun., 1984, 120, 607–613 Search PubMed.
- R. Ullan, J. Casqueiro, O. Banuelos, F. Fernandez, S. Gutierrez and J. Martin, J. Biol. Chem., 2002, 277, 46216–46225 CrossRef CAS.
- J. F. Martín, R. V. Ullán and J. Casqueiro, in Molecular Biotechnolgy of Fungal beta-Lactam Antibiotics and Related Peptide Synthetases, ed. A. A. Brakhage, Springer, Berlin/Heidelberg, 2004, vol. 88, pp. 91–109 Search PubMed.
- A. Scheidegger, M. T. Kuenzi and J. Nuesch, J. Antibiot., 1984, 37, 522–531 Search PubMed.
- J. E. Baldwin, R. M. Adlington, J. B. Coates, M. J. Crabbe, N. P. Crouch, J. W. Keeping, G. C. Knight, C. J. Schofield, H. H. Ting, C. A. Vallejo, M. Thorniley and E. P. Abraham, Biochem. J., 1987, 245, 831–841 CAS.
- J. E. Dotzlaf and W. K. Yeh, J. Bacteriol., 1987, 169, 1611–1618 Search PubMed.
- S. M. Samson, J. E. Dotzlaf, M. L. Slisz, G. W. Becker, R. M. Van Frank, L. E. Veal, W. K. Yeh, J. R. Miller, S. W. Queener and T. D. Ingolia, Bio/Technology, 1987, 5, 1207–1208 Search PubMed.
- S. E. Jensen, D. W. S. Westlake and S. Wolfe, Antimicrob. Agents Chemother., 1983, 24, 307–312 Search PubMed.
- S. E. Jensen, D. W. S. Westlake and S. Wolfe, J. Antibiot., 1985, 38, 262–265 Search PubMed.
- S. Kovacevic, B. J. Weigel, M. B. Tobin, T. D. Ingolia and J. R. Miller, J. Bacteriol., 1989, 171, 754–760 CAS.
- B. J. Baker, J. E. Dotzlaf and W. K. Yeh, J. Biol. Chem., 1991, 266, 5087–5093 CAS.
- S. Kovacevic and J. R. Miller, J. Bacteriol., 1991, 173, 398–400 Search PubMed.
- P. Liras, A. Rodriguez-Garcïa and J. F. Martin, Int. Microbiol., 1998, 1, 271–278 Search PubMed.
- C. García-Estrada, F. Fierro and J. F. Martín, in Current Research, Technology and Education Topics in Applied Microbiology and Microbial Biotechnology, ed. A. Mendez-Vilas, Formatex Research Center, Badajoz, Spain, 2010, vol. 1, pp. 577–588 Search PubMed.
- A. A. Brakhage, M. Thön, P. Spröte, D. H. Scharf, Q. Al-Abdallah, S. M. Wolke and P. Hortschansky, Phytochemistry, 70, pp. 1801–1811 Search PubMed.
- K. Valegård, A. C. Van Terwisscha Scheltinga, M. D. Lloyd, T. Hara, S. Ramaswamy, A. Perrakis, A. Thompson, H. J. Lee, J. E. Baldwin, C. J. Schofield, J. Hajdu and I. Andersson, Nature, 1998, 394, 805–809 CrossRef CAS.
- M. D. Lloyd, H. J. Lee, K. Harlos, Z. H. Zhang, J. E. Baldwin, C. J. Schofield, J. M. Charnock, C. D. Garner, T. Hara, A. C. T. van Scheltinga, K. Valegård, J. A. C. Viklund, J. Hajdu, I. Andersson, A. Danielsson and R. Bhikhabhai, J. Mol. Biol., 1999, 287, 943–960 CrossRef CAS.
- L. M. Öster, A. C. T. van Scheltinga, K. Valegård, A. M. Hose, A. Dubus, J. Hajdu and I. Andersson, J. Mol. Biol., 2004, 343, 157–171 Search PubMed.
- H. J. Lee, M. D. Lloyd, K. Harlos, I. J. Clifton, J. E. Baldwin and C. J. Schofield, J. Mol. Biol., 2001, 308, 937–948 CrossRef CAS.
- H. J. Lee, M. D. Lloyd, I. J. Clifton, K. Harlos, A. Dubus, J. E. Baldwin, J. M. Frere and C. J. Schofield, J. Biol. Chem., 2001, 276, 18290–18295 Search PubMed.
- G. Cicero, C. Carbonera, K. Valegård, J. Hajdu, I. Andersson and G. Ranghino, Int. J. Quantum Chem., 2007, 107, 1514–1522 Search PubMed.
- K. Valegård, A. C. Terwisscha Van Scheltinga, A. Dubus, G. Ranghino, L. M. Öster, J. Hajdu and I. Andersson, Nat. Struct. Mol. Biol., 2004, 11, 95–101 CrossRef CAS.
- P. Loke and T. S. Sim, FEMS Microbiol. Lett., 1999, 179, 423–429 Search PubMed.
- J. Zhou, M. Gunsior, B. O. Bachmann, C. A. Townsend and E. I. Solomon, J. Am. Chem. Soc., 1998, 120, 13539–13540 CrossRef CAS.
- R. B. Morin, B. G. Jackson, R. A. Mueller, E. R. Lavagnino, W. B. Scanlon and S. L. Andrews, J. Am. Chem. Soc., 1963, 85, 1896–1897 CrossRef CAS.
- J. E. Baldwin, R. M. Adlington, N. P. Crouch, N. J. Turner and C. J. Schofield, J. Chem. Soc., Chem. Commun., 1989, 1141–1143 RSC.
- H. Kluender, C. H. Bradley, C. J. Sih, P. Fawcett and E. P. Abraham, J. Am. Chem. Soc., 1973, 95, 6149–6150 CrossRef CAS.
- N. Neuss, C. H. Nash, J. E. Baldwin, P. A. Lemke and J. B. Grutzner, J. Am. Chem. Soc., 1973, 95, 3797–3798 Search PubMed.
- K.-S. Goo, C.-S. Chua and T.-S. Sim, J. Ind. Microbiol. Biotechnol., 2009, 36, 619–633 Search PubMed.
- S. G. Kian, S. C. Chun and T. S. Sim, Proteins: Struct., Funct., Genet., 2008, 70, 739–747 Search PubMed.
- K. S. Goo, C. S. Chua and T.-S. Sim, Appl. Environ. Microbiol., 2008, 74, 1167–1175 Search PubMed.
- M. D. Lloyd, S. J. Lipscomb, K. S. Hewitson, C. M. H. Hensgens, J. E. Baldwin and C. J. Schofield, J. Biol. Chem., 2004, 279, 15420–15426 CrossRef CAS.
- H. S. Chin, K. S. Goo and T. Sim, Appl. Environ. Microbiol., 2004, 70, 607–609 Search PubMed.
- H. J. Lee, Y. F. Dai, C. Y. Shiau, C. J. Schofield and M. D. Lloyd, Eur. J. Biochem., 2003, 270, 1301–1307 Search PubMed.
- S. J. Lipscomb, H. J. Lee, M. Mukherji, J. E. Baldwin, C. J. Schofield and M. D. Lloyd, Eur. J. Biochem., 2002, 269, 2735–2739 Search PubMed.
- H. J. Lee, C. J. Schofield and M. D. Lloyd, Biochem. Biophys. Res. Commun., 2002, 292, 66–70 Search PubMed.
- H. S. Chin and T. S. Sim, Biochem. Biophys. Res. Commun., 2002, 295, 55–61 Search PubMed.
- J. Sim and T. S. Sim, Enzyme Microb. Technol., 2001, 29, 240–245 Search PubMed.
- H. S. Chin, J. Sim and T. S. Sim, Biochem. Biophys. Res. Commun., 2001, 287, 507–513 Search PubMed.
- J. Sim and T. S. Sim, Biosci., Biotechnol., Biochem., 2000, 64, 828–832 CAS.
- H. J. Lee, M. D. Lloyd, K. Harlos and C. J. Schofield, Biochem. Biophys. Res. Commun., 2000, 267, 445–448 Search PubMed.
- A. Dubus, M. D. Lloyd, H. J. Lee, C. J. Schofield, J. E. Baldwin and J. M. Frère, Cell. Mol. Life Sci., 2001, 58, 835–843 Search PubMed.
- C. A. Townsend, A. B. Theis, A. S. Neese, E. B. Barrabee and D. Poland, J. Am. Chem. Soc., 1985, 107, 4760–4767 CrossRef CAS.
- C. A. Townsend, J. Nat. Prod., 1985, 48, 708–724 CrossRef CAS.
- C. P. Pang, R. L. White, E. P. Abraham, D. H. Crout, M. Lutstorf, P. J. Morgan and A. E. Derome, Biochem. J., 1984, 222, 777–788 CAS.
- J. E. Baldwin, R. M. Adlington, T. W. Kang, E. Lee and C. J. Schofield, J. Chem. Soc., Chem. Commun., 1987, 104–106 RSC.
- J. E. Baldwin, R. M. Adlington, T. W. Kang, E. Lee and C. J. Schofield, Tetrahedron, 1988, 44, 5953–5957 CrossRef CAS.
- J. E. Baldwin, R. M. Adlington, R. T. Aplin, N. P. Crouch, G. Knight and C. J. Schofield, J. Chem. Soc., Chem. Commun., 1987, 1651–1654 RSC.
- J. E. Baldwin, R. M. Adlington, N. P. Crouch, C. J. Schofield, N. J. Turner and R. T. Aplin, Tetrahedron, 1991, 47, 9881–9900 CrossRef CAS.
- R. D. Miller, L. L. Huckstep, J. P. McDermott, S. W. Queener, S. Kukolja, D. O. Spry, T. K. Elzey, S. M. Lawrence and N. Neuss, J. Antibiot., 1981, 34, 984–993 Search PubMed.
- J. Myllyharju and K. I. Kivirikko, EMBO J., 1997, 16, 1173–1180 CrossRef CAS.
- Y. Fujisawa and T. Kanzaki, J. Antibiot., 1975, 28, 372–378 Search PubMed.
- J. E. Baldwin, K. C. Goh and C. J. Schofield, J. Antibiot., 1992, 45, 1378–1381 CAS.
- C. S. Hamilton, A. Yasuhara, J. E. Baldwin, M. D. Lloyd and P. J. Rutledge, Bioorg. Med. Chem. Lett., 2001, 11, 2511–2514 Search PubMed.
- J. E. Baldwin, R. M. Adlington, J. B. Coates, M. J. C. Crabbe, J. W. Keeping, G. C. Knight, T. Nomoto, C. J. Schofield and H. H. Ting, J. Chem. Soc., Chem. Commun., 1987, 374–375 RSC.
- J. E. Baldwin, R. M. Adlington, M. J. Crabbe, G. Knight, T. Nomoto, C. J. Schofield and H. H. Ting, Tetrahedron, 1987, 43, 3009–3014 CrossRef CAS.
- N. Shibata, M. D. Lloyd, J. E. Baldwin and C. J. Schofield, Bioorg. Med. Chem. Lett., 1996, 6, 1579–1584 CrossRef CAS.
- J. E. Baldwin, R. M. Adlington, N. P. Crouch, J. B. Coates, J. W. Keeping, C. J. Schofield, W. A. Shuttleworth and J. D. Sutherland, J. Antibiot., 1988, 41, 1694–1695 CAS.
- N. Morgan, I. A. C. Pereira, I. A. Andersson, R. M. Adlington, J. E. Baldwin, S. C. J. Cole, N. P. Crouch and J. D. Sutherland, Bioorg. Med. Chem. Lett., 1994, 4, 1595–1600 CrossRef CAS.
- J. E. Baldwin, R. M. Adlington, N. P. Crouch and C. J. Schofield, Tetrahedron, 1988, 44, 643–650 CrossRef CAS.
- J. E. Baldwin, R. M. Adlington, C. J. Schofield, N. P. Crouch and H. H. Ting, J. Chem. Soc., Chem. Commun., 1987, 1556–1558 RSC.
- J. E. Baldwin, R. M. Adlington, N. P. Crouch and I. A. C. Pereira, Tetrahedron, 1993, 49, 4907–4922 CrossRef CAS.
- J. E. Baldwin, R. M. Adlington, N. P. Crouch and I. A. C. Pereira, Tetrahedron, 1993, 49, 7499–7518 CrossRef CAS.
- J. E. Baldwin, R. M. Adlington, N. P. Crouch, J. W. Keeping, S. W. Leppard, J. Pitlik, C. J. Schofield, W. J. Sobey and M. E. Wood, J. Chem. Soc., Chem. Commun., 1991, 768–770 RSC.
- J. E. Baldwin, R. M. Adlington, N. P. Crouch, R. J. Heath, I. A. C. Pereira and J. D. Sutherland, Bioorg. Med. Chem. Lett., 1992, 2, 669–672 Search PubMed.
- J. E. Baldwin, R. M. Adlington, N. P. Crouch and I. A. C. Pereira, J. Chem. Soc., Chem. Commun., 1992, 1448–1451 RSC.
- J. E. Baldwin, R. M. Adlington, N. P. Crouch, R. L. Hill, M. E. Wood and M. D. Lloyd, Bioorg. Med. Chem. Lett., 1997, 7, 593–596 Search PubMed.
- R. Nagarajan, L. D. Boeck, M. Gorman, R. L. Hamill, C. E. Higgens, M. M. Hoehn, W. M. Stark and J. G. Whitney, J. Am. Chem. Soc., 1971, 93, 2308–2310 CrossRef CAS.
- E. O. Stapley, M. Jackson, S. Hernandez, S. B. Zimmerman, S. A. Currie, S. Mochales, J. M. Mata, H. B. Woodruff and D. Hendlin, Antimicrob. Agents Chemother., 1972, 2, 122–131 CAS.
- G. Gauze, V. Chugasova, L. Terekhova, N. Lomakina and G. Fedorova, Antibiotiki, 1976, 21, 1059–1062 Search PubMed.
- Y. Nozaki, K. Okonogi, N. Katayama, H. Ono, S. Harada, M. Kondo and H. Okazaki, J. Antibiot., 1984, 37, 1555–1565 CAS.
- E. O. Stapley, J. Birnbaum, A. K. Miller, H. Wallick, D. Hendlin and H. B. Woodruff, Rev. Infect. Dis., 1979, 1, 73–89 Search PubMed.
- K. Okonogi, A. Sugiura, M. Kuno, H. Ono, S. Harada and E. Higashide, J. Antibiot., 1985, 38, 1555–1563 Search PubMed.
- J. O'Sullivan, R. Aplin, C. Stevens and E. Abraham, Biochem. J., 1979, 179, 47–52 Search PubMed.
- J. O'Sullivan and E. P. Abraham, Biochem. J., 1980, 186, 613–616 Search PubMed.
- S. J. Brewer, P. M. Taylor and M. K. Turner, Biochem. J., 1980, 185, 555–564 Search PubMed.
- S. J. Brewer, T. T. Boyle and M. K. Turner, Biochem. Soc. Trans., 1977, 5, 1026–1029 Search PubMed.
- J. D. Hood, A. Elson, M. L. Gilpin and A. G. Brown, J. Chem. Soc., Chem. Commun., 1983, 1187–1188 RSC.
- L. M. Iyer, S. Abhiman, R. F. de Souza and L. Aravind, Nucleic Acids Res., 2010, 38, 5261–5279 CrossRef CAS.
- J. J. R. Coque, F. J. Enguita, J. F. Martin and P. Liras, J. Bacteriol., 1995, 177, 2230–2235 Search PubMed.
- F. J. Enguita, P. Liras, A. L. Leitão and J. F. Martín, J. Biol. Chem., 1996, 271, 33225–33230 Search PubMed.
- X. Xiao, G. Hintermann, A. Häusler, P. J. Barker, F. Foor, A. L. Demain and J. Piret, Antimicrob. Agents Chemother., 1993, 37, 84–88 Search PubMed.
- S. Gutierrez, J. Velasco, F. J. Fernandez and J. F. Martin, J. Bacteriol., 1992, 174, 3056–3064 Search PubMed.
- S. W. Queener, J. J. Capone, A. B. Radue and R. Nagarajan, Antimicrob. Agents Chemother., 1974, 6, 334–337 Search PubMed.
- Y. Fujisawa, K. Kitano and T. Kanzaki, Agric. Biol. Chem., 1975, 39, 2049–2055 Search PubMed.
- Y. Fujisawa and T. Kanzaki, Agric. Biol. Chem., 1975, 39, 2043–2048 Search PubMed.
- H. R. Felix, J. Nüesch and W. Wehrli, FEMS Microbiol. Lett., 1980, 8, 55–58 Search PubMed.
- A. Scheidegger, A. Gutzwiller, M. T. Küenzi, A. Fiechter and J. Niiesch, J. Biotechnol., 1985, 3, 109–117 Search PubMed.
- K. Matsuyama, H. Matsumoto, A. Matsuda, H. Sugiura, K. Komatsu and S. Ichikawa, Biosci., Biotechnol., Biochem., 1992, 56, 1410–1412 Search PubMed.
- J. Velasco, S. Gutierrez, S. Campoy and J. F. Martin, Biochem. J., 1999, 337, 379–385 Search PubMed.
- A. Matsuda, H. Sugiura, K. Matsuyama, H. Matsumoto, S. Ichikawa and K. I. Komatsu, Biochem. Biophys. Res. Commun., 1992, 186, 40–46 Search PubMed.
- A. Matsuda, H. Sugiura, K. Matsuyama, H. Matsumoto, S. Ichikawa and K.-i. Komatsu, Biochem. Biophys. Res. Commun., 1992, 182, 995–1001 Search PubMed.
- S. Lejon, J. Ellis and K. Valegård, J. Mol. Biol., 2008, 377, 935–944 Search PubMed.
- H. Gushima, S. Watanabe, T. Saito, T. Sasaki, H. Eiki, Y. Oka and T. Osono, J. Antibiot., 1981, 34, 1507–1512 Search PubMed.
- R. B. Morin, B. G. Jackson, E. H. Flynn, R. W. Roeske and S. L. Andrews, J. Am. Chem. Soc., 1969, 91, 1396–1400 Search PubMed.
- R. G. Binder, K. Numata, D. A. Lowe, T. Murakami and J. L. Brown, Appl. Environ. Microbiol., 1993, 59, 3321–3326 Search PubMed.
- C. F. Sio and W. J. Quax, Curr. Opin. Biotechnol., 2004, 15, 349–355 Search PubMed.
- T. Isogai, M. Fukagawa, I. Aramori, M. Iwami, H. Kojo, T. Ono, Y. Ueda, M. Kohsaka and H. Imanaka, Bio/Technology, 1991, 9, 188–191 Search PubMed.
- J. Lein, in Overproduction of Microbial Metabolites: Strain Improvement and Process Control Strategies, ed. Z. Vanek and Z. Hostalek, Butterworth Publishers, Boston, 1986, pp. 105–140 Search PubMed.
- G. J. M. Hersbach, C. Van der Beek and P. VanDijck, in Biotechnology of Industrial Antibiotics. Drugs and the Pharmaceutical Sciences, ed. E. J. Vandamme, Marcel Dekker, New York, 1984, vol. 22, pp. 45–140 Search PubMed.
- M. A. van den Berg, R. Albang, K. Albermann, J. H. Badger, J. M. Daran, A. J. M. Driessen, C. Garcia-Estrada, N. D. Fedorova, D. M. Harris, W. H. M. Heijne, V. Joardar, J. A. K. W. Kiel, A. Kovalchuk, J. F. Martin, W. C. Nierman, J. G. Nijland, J. T. Pronk, J. A. Roubos, I. J. van der Klei, N. N. M. E. van Peij, M. Veenhuis, H. von Dohren, C. Wagner, J. Wortman and R. A. L. Bovenberg, Nat. Biotechnol., 2008, 26, 1161–1168 CrossRef CAS.
- J. Y. Song, H. Jeong, D. S. Yu, M. A. Fischbach, H. S. Park, J. J. Kim, J. S. Seo, S. E. Jensen, T. K. Oh, K. J. Lee and J. F. Kim, J. Bacteriol., 2010, 192, 6317–6318 Search PubMed.
- V. Barbe, M. Bouzon, S. Mangenot, B. Badet, J. Poulain, B. Segurens, D. Vallenet, P. Marlière and J. Weissenbach, J. Bacteriol., 2011, 193, 5055–5061 Search PubMed.
- M. Land, A. Lapidus, S. Mayilraj, F. Chen, A. Copeland, T. G. Del Rio, M. Nolan, S. Lucas, H. Tice, J. F. Cheng, O. Chertkov, D. Bruce, L. Goodwin, S. Pitluck, M. Rohde, M. Goker, A. Pati, N. Ivanova, K. Mavromatis, A. Chen, K. Palaniappan, L. Hauser, Y. J. Chang, C. C. Jeffries, T. Brettin, J. C. Detter, C. Han, P. Chain, B. J. Tindall, J. Bristow, J. A. Eisen, V. Markowitz, P. Hugenholtz, N. C. Kyrpides and H. P. Klenk, Stand. Genomic Sci., 2009, 1, 46–53 Search PubMed.
- T. W. Johannes and H. Zhao, Curr. Opin. Microbiol., 2006, 9, 261–267 CrossRef CAS.
- R. A. Chica, N. Doucet and J. N. Pelletier, Curr. Opin. Biotechnol., 2005, 16, 378–384 Search PubMed.
- G. J. Williams, A. S. Nelson and A. Berry, Cell. Mol. Life Sci., 2004, 61, 3034–3046 CrossRef CAS.
- R. Chen, Trends Biotechnol., 2001, 19, 13–14 CrossRef CAS.
- U. T. Bornscheuer and M. Pohl, Curr. Opin. Chem. Biol., 2001, 5, 137–143 CrossRef CAS.
- X. B. Wu, X. Y. Tian, J. J. Ji, W. B. Wu, K. Q. Fan and K. Q. Yang, Biotechnol. Lett., 2011, 33, 805–812 Search PubMed.
- J. Thykaer and J. Nielsen, Metab. Eng., 2003, 5, 56–69 CrossRef CAS.
- M. A. Van den Berg, R. A. L. Bovenberg, W. T. A. M. De Laat and A. G. Van Velzen, Antonie van Leeuwenhoek, 1999, 75, 155–161 Search PubMed.
- M. Rodríguez-Sáiz, J. L. D. L. Fuente, J. L. Barredo and M. C. Flickinger, in Encyclopedia of Industrial Biotechnology, John Wiley & Sons, Inc., 2009 Search PubMed.
- A. A. Brakhage, Microbiol. Mol. Biol. Rev., 1998, 62, 547–585 CAS.
- J. L. Barredo, B. Diez, E. Alvarez and J. F. Martin, Curr. Genet., 1989, 16, 453–459 Search PubMed.
- F. Fierro, C. García-Estrada, N. I. Castillo, R. Rodríguez, T. Velasco-Conde and J.-F. Martín, Fungal Genet. Biol., 2006, 43, 618–629 Search PubMed.
- F. Fierro, J. L. Barredo, B. Díez, S. Gutierrez, F. J. Fernández and J. F. Martín, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 6200–6204 CrossRef CAS.
- R. W. Newbert, B. Barton, P. Greaves, J. Harper and G. Turner, J. Ind. Microbiol. Biotechnol., 1997, 19, 18–27 CrossRef CAS.
- L. Mathison, C. Soliday, T. Stepan, T. Aldrich and J. Rambosek, Curr. Genet., 1993, 23, 33–41 Search PubMed.
- S. Gutiérrez, J. Velasco, A. T. Marcos, F. J. Fernández, F. Fierro, J. L. Barredo, B. Díez and J. F. Martín, Appl. Microbiol. Biotechnol., 1997, 48, 606–614 Search PubMed.
- J. Velasco, J. L. Adrio, M. A. Moreno, B. Diez, G. Soler and J. L. Barredo, Nat. Biotechnol., 2000, 18, 857–861 Search PubMed.
- M. Rodriguez-Sáiz, J.-L. d. l. Fuente and J.-L. Barredo, Methods Biotechnol., 2005, 18, 41–64 Search PubMed.
- R. V. Ullán, S. Campoy, J. Casqueiro, F. J. Fernández and J. F. Martín, Chem. Biol., 2007, 14, 329–339 Search PubMed.
- C. A. Cantwell, R. J. Beckmann, J. E. Dotzlaf, D. L. Fisher, P. L. Skatrud, W. K. Yeh and S. W. Queener, Curr. Genet., 1990, 17, 213–221 Search PubMed.
- L. Crawford, A. M. Stepan, P. C. McAda, J. A. Rambosek, M. J. Conder, V. A. Vinci and C. D. Reeves, Bio/Technology, 1995, 13, 58–62 Search PubMed.
- M. Conder, N. Almeida, S. Behrens, L. Crawford, S. Delawder, T. Hoerner, P. McAda, J. Rambosek, C. Reeves, T. Schimmell, T. Stepan, R. Stieber, I. T. Tong and V. Vinci, presented in part at the Proceedings of the Conference on Applied Biocatalysis, Brighton, UK, 1994 Search PubMed.
- D. M. Harris, I. Westerlaken, D. Schipper, Z. A. van der Krogt, A. K. Gombert, J. Sutherland, L. M. Raamsdonk, M. A. van den Berg, R. A. L. Bovenberg, J. T. Pronk and J.-M. Daran, Metab. Eng., 2009, 11, 125–137 Search PubMed.
- M. J. Koetsier, A. K. Gombert, S. Fekken, R. A. L. Bovenberg, M. A. van den Berg, J. A. K. W. Kiel, P. A. Jekel, D. B. Janssen, J. T. Pronk, I. J. van der Klei and J.-M. Daran, Fungal Genet. Biol., 2010, 47, 33–42 Search PubMed.
- C. Reading, GB Pat., 1547222, 1979.
- J. Y. Song, S. E. Jensen and K. J. Lee, Appl. Microbiol. Biotechnol., 2010, 88, 659–669 Search PubMed.
- J. Romero, P. Liras and J. F. Martin, Appl. Environ. Microbiol., 1986, 52, 892–897 CAS.
- C. A. Townsend and M. F. Ho, J. Am. Chem. Soc., 1985, 107, 1065–1066 CrossRef CAS.
- C. A. Townsend and M. F. Ho, J. Am. Chem. Soc., 1985, 107, 1066–1068 CrossRef CAS.
- J. Pitlik and C. A. Townsend, Chem. Commun., 1997, 225–226 RSC.
- J. E. Thirkettle, J. E. Baldwin, J. Edwards, J. P. Griffin and C. J. Schofield, Chem. Commun., 1997, 1025–1026 RSC.
- B. P. Valentine, C. R. Bailey, A. Doherty, J. Morris, S. W. Elson, K. H. Baggaley and N. H. Nicholson, J. Chem. Soc., Chem. Commun., 1993, 1210–1211 RSC.
- N. Khaleeli, R. Li and C. A. Townsend, J. Am. Chem. Soc., 1999, 121, 9223–9224 CrossRef CAS.
- R. Perez-Redondo, A. Rodriguez-Garcia, J. F. Martin and P. Liras, J. Bacteriol., 1999, 181, 6922–6928 CAS.
- M. Merski and C. A. Townsend, J. Am. Chem. Soc., 2007, 129, 15750–15751 Search PubMed.
- M. E. C. Caines, J. L. Sorensen and C. J. Schofield, Biochem. Biophys. Res. Commun., 2009, 385, 512–517 Search PubMed.
- M. E. C. Caines, J. M. Elkins, K. S. Hewitson and C. J. Schofield, J. Biol. Chem., 2004, 279, 5685–5692 CAS.
- H. J. McNaughton, J. E. Thirkettle, Z. Zhang, C. J. Schofield, S. E. Jensen, B. Barton and P. Greaves, Chem. Commun., 1998, 2325–2326 RSC.
- B. O. Bachmann, R. Li and C. A. Townsend, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 9082–9086 CrossRef CAS.
- M. T. Miller, B. Gerratana, A. Stapon, C. A. Townsend and A. C. Rosenzweig, J. Biol. Chem., 2003, 278, 40996–41002 CrossRef CAS.
- B. Gerratana, A. Stapon and C. A. Townsend, Biochemistry, 2003, 42, 7836–7847 CrossRef CAS.
- M. J. Bodner, R. Li, R. M. Phelan, M. F. Freeman, K. A. Moshos, E. P. Lloyd and C. A. Townsend, ChemBioChem, 2011, 12, 2159–2165 Search PubMed.
- T. M. Larsen, S. K. Boehlein, S. M. Schuster, N. G. J. Richards, J. B. Thoden, H. M. Holden and I. Rayment, Biochemistry, 1999, 38, 16146–16157 CrossRef CAS.
- M. T. Miller, B. O. Bachmann, C. A. Townsend and A. C. Rosenzweig, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14752–14757 CrossRef CAS.
- M. T. Miller, B. O. Bachmann, C. A. Townsend and A. C. Rosenzweig, Nat. Struct. Biol., 2001, 8, 684–689 CrossRef CAS.
- S. K. Boehlein, J. D. Stewart, E. S. Walworth, R. Thirumoorthy, N. G. J. Richards and S. M. Schuster, Biochemistry, 1998, 37, 13230–13238 CrossRef CAS.
- R. B. Hamed, L. Henry, J. R. Gomez-Castellanos, J. Mecinović, C. Ducho, J. L. Sorensen, T. D. W. Claridge and C. J. Schofield, J. Am. Chem. Soc., 2012, 134, 471–479 Search PubMed.
- R. B. Hamed, J. R. Gomez-Castellanos, A. Thalhammer, D. Harding, C. Ducho, T. D. W. Claridge and C. J. Schofield, Nat. Chem., 2011, 3, 365–371 Search PubMed.
- R. B. Hamed, J. Mecinovic, C. Ducho, T. D. W. Claridge and C. J. Schofield, Chem. Commun., 2010, 46, 1413–1415 RSC.
- M. C. Sleeman, C. H. MacKinnon, K. S. Hewitson and C. J. Schofield, Bioorg. Med. Chem. Lett., 2002, 12, 597–599 CrossRef CAS.
- J. W. Labonte, F. Kudo, M. F. Freeman, M. L. Raber and C. A. Townsend, Med. Chem. Commun., 2012, 3, 960 RSC.
- M. C. Sleeman, P. Smith, B. Kellam, S. R. Chhabra, B. W. Bycroft and C. J. Schofield, ChemBioChem, 2004, 5, 879–882 CrossRef CAS.
- Y.-F. He, B.-Z. Li, Z. Li, P. Liu, Y. Wang, Q. Tang, J. Ding, Y. Jia, Z. Chen, L. Li, Y. Sun, X. Li, Q. Dai, C.-X. Song, K. Zhang, C. He and G.-L. Xu, Science, 2011, 333, 1303–1307 CrossRef CAS.
- T. Lange, Planta, 1994, 195, 108–115 CAS.
- P. Hedden, Physiol. Plant., 1997, 101, 709–719 CrossRef CAS.
- P. Hedden, J. Exp. Bot., 1999, 50, 553–563 Search PubMed.
- S. W. Elson, K. H. Baggaley, M. Fulston, N. H. Nicholson, J. W. Tyler, J. Edwards, H. Holms, I. Hamilton and D. M. Mousdale, J. Chem. Soc., Chem. Commun., 1993, 1211–1212 RSC.
- J. E. Baldwin, M. D. Lloyd, B. Wha-Son, C. J. Schofield, S. W. Elson, K. H. Baggaley and N. H. Nicholson, J. Chem. Soc., Chem. Commun., 1993, 500–502 RSC.
- S. W. Elson, K. H. Baggaley, J. Gillett, S. Holland, N. H. Nicholson, J. T. Sime and S. R. Woroniecki, J. Chem. Soc., Chem. Commun., 1987, 1736–1738 RSC.
- J. E. Baldwin, R. M. Adlington, J. S. Bryans, A. O. Bringhen, J. B. Coates, N. P. Crouch, M. D. Lloyd, C. J. Schofield, S. W. Elson, K. H. Baggaley, R. Cassels and N. Nicholson, Tetrahedron, 1991, 47, 4089–4100 CrossRef CAS.
- S. P. Salowe, E. N. Marsh and C. A. Townsend, Biochemistry, 1990, 29, 6499–6508 CrossRef CAS.
- J. E. Baldwin, R. M. Adlington, J. S. Bryans, A. O. Bringhen, J. B. Coates, N. P. Crouch, M. D. Lloyd, C. J. Schofield, S. W. Elson and K. H. Baggaley, J. Chem. Soc., Chem. Commun., 1990, 617–1236 RSC.
- S. Salowe, W. Krol, D. Iwata-Reuyl and C. Townsend, Biochemistry, 1991, 30, 2281–2373 CrossRef CAS.
- J. E. Baldwin, R. M. Adlington, N. P. Crouch, D. J. Drake, Y. Fujishima, S. W. Elson and K. H. Baggaley, J. Chem. Soc., Chem. Commun., 1994, 1133–1134 RSC.
- Z. Zhang, J. Ren, D. K. Stammers, J. E. Baldwin, K. Marios and C. J. Schofield, Nat. Struct. Biol., 2000, 7, 127–133 CrossRef CAS.
- Z. Zhang, J.-S. Ren, K. Harlos, C. H. McKinnon, I. J. Clifton and C. J. Schofield, FEBS Lett., 2002, 517, 7–12 CrossRef CAS.
- T. Borowski, A. Bassan and P. E. M. Siegbahn, Chem.–Eur. J., 2004, 10, 1031–1041 CrossRef CAS.
- R. W. D. Welford, J. M. Kirkpatrick, L. A. McNeill, M. Puri, N. J. Oldham and C. J. Schofield, FEBS Lett., 2005, 579, 5170–5174 Search PubMed.
- J. E. Baldwin, R. M. Adlington, J. S. Bryans, M. D. Lloyd, T. J. Sewell, C. J. Schofield, K. H. Baggaley and R. Cassels, Tetrahedron, 1997, 53, 7011–14031 CrossRef CAS.
- M. D. Lloyd, K. D. Merritt, V. Lee, T. J. Sewell, B. Wha-Son, J. E. Baldwin, C. J. Schofield, S. W. Elson, K. H. Baggaley and N. H. Nicholson, Tetrahedron, 1999, 55, 10201–10220 CrossRef CAS.
- J. E. Baldwin, V. Lee, M. D. Lloyd, C. J. Schofield, S. W. Elson and K. H. Baggaley, J. Chem. Soc., Chem. Commun., 1993, 1694–1696 RSC.
- V. Helmetag, S. A. Samel, M. G. Thomas, M. A. Marahiel and L.-O. Essen, FEBS J., 2009, 276, 3669–3682 Search PubMed.
- K. J. Martinkus, C.-H. Tann and S. J. Gould, Tetrahedron, 1983, 39, 3493–3505 CrossRef CAS.
- M. Jackson, S. Gould and T. Zabriskie, J. Org. Chem., 2002, 67, 2934–2975 CrossRef CAS.
- H. Kawauchi, M. Tohno, Y. Tsuchiya, M. Hayashida, Y. Adachi, T. Mukai, I. Hayashi, S. Kimura and S. Kondo, Int. J. Pept. Protein Res., 1983, 21, 546–554 CAS.
- K. Aidoo, A. Wong, D. Alexander, R. Rittammer and S. Jensen, Gene, 1994, 147, 41–47 CrossRef CAS.
- J. M. Elkins, I. J. Clifton, H. Hernández, L. X. Doan, C. V. Robinson, C. J. Schofield and K. S. Hewitson, Biochem. J., 2002, 366, 423–434 CrossRef CAS.
- M. C. Bewley, P. D. Jeffrey, M. L. Patchett, Z. F. Kanyo and E. N. Baker, Structure, 1999, 7, 435–448 CrossRef CAS.
- D. Iwata-Reuyl and C. A. Townsend, J. Am. Chem. Soc., 1992, 114, 2762–2763 CrossRef CAS.
- J. W. Janc, L. A. Egan and C. A. Townsend, Bioorg. Med. Chem. Lett., 1993, 3, 2313–2316 CrossRef.
- L. A. Egan, R. W. Busby, D. Iwata-Reuyl and C. A. Townsend, J. Am. Chem. Soc., 1997, 119, 2348–2355 CrossRef CAS.
- H. Peter, J. Rabenhorst, F. Röhl and H. Zähner, in Recent advances in chemotherapy, ed. J. Ishigami, University of Tokyo Press, Tokyo, 1985, pp. 237–238 Search PubMed.
- F. Röhl, J. Rabenhorst and H. Zähner, Arch. Microbiol., 1987, 147, 315–320 Search PubMed.
- J. E. Baldwin, T. D. W. Claridge, K.-C. Goh, J. W. Keeping and C. J. Schofield, Tetrahedron Lett., 1993, 34, 5645–5648 CrossRef CAS.
- H. T. Postels and W. A. König, Tetrahedron Lett., 1994, 35, 4535–4538 CrossRef CAS.
- J. E. Baldwin, Y. Fujishima, K.-C. Goh and C. J. Schofield, Tetrahedron Lett., 1994, 35, 2783–2786 CrossRef CAS.
- J. E. Baldwin, K. C. Goh and C. J. Schofield, Tetrahedron Lett., 1994, 35, 2779–2782 CrossRef CAS.
- J. W. Janc, L. A. Egan and C. A. Townsend, J. Biol. Chem., 1995, 270, 5399–5404 CrossRef CAS.
- J. S. Box, DE 2646001, 1977.
- K. Kitano, K. Kazuaki, K. Kintaka, K. Katamoto, JP 53104796, 1978.
- S. W. Elson, in Recent Advances in the Chemistry of β-Lactam Antibiotics, ed. P. H. Bentley and R. Southgate, RSC, Cambridge, UK, 1988, p. 303 Search PubMed.
- C. E. Newal, in Recent Advances in the Chemistry of the β-Lactam Antibiotics, ed. G. I. Gregory, RSC, London, UK, 1980, p. 142 Search PubMed.
- B. W. Bycroft, A. Penrose, J. Gillett and S. W. Elson, J. Chem. Soc., Chem. Commun., 1988, 980–981 RSC.
- C. A. Townsend and W. J. Krol, J. Chem. Soc., Chem. Commun., 1988, 1234–1236 RSC.
- N. H. Nicholson, K. H. Baggaley, R. Cassels, M. Davison, S. W. Elson, M. Fulston, J. W. Tyler and S. R. Woroniecki, J. Chem. Soc., Chem. Commun., 1994, 1281–1282 RSC.
- M. Fulston, M. Davison, S. W. Elson, N. H. Nicholson, J. W. Tyler and S. R. Woroniecki, J. Chem. Soc., Perkin Trans. 1, 2001, 1122–1130 RSC.
- D. Iwata-Reuyl, A. Basak, L. S. Silverman, C. A. Engle and C. A. Townsend, J. Nat. Prod., 1993, 56, 1373–1396 CrossRef CAS.
- S. Jensen, A. Paradkar, R. Mosher, C. Anders, P. Beatty, M. Brumlik, A. Griffin and B. Barton, Antimicrob. Agents Chemother., 2004, 48, 192–202 Search PubMed.
- R. F. Li, N. Khaleeli and C. A. Townsend, J. Bacteriol., 2000, 182, 4087–4095 CrossRef CAS.
- E. Mellado, L. M. Lorenzana, M. Rodriguez-Saiz, B. Diez, P. Liras and J. L. Barredo, Microbiology, 2002, 148, 1427–1438 CAS.
- A. S. Paradkar, K. A. Aidoo and S. E. Jensen, Mol. Microbiol., 1998, 27, 831–843 CrossRef CAS.
- R. Perez-Redondo, A. Rodriguez-Garcia, J. F. Martin and P. Liras, Gene, 1998, 211, 311–321 CrossRef CAS.
- P. Liras, J. P. Gomez-Escribano and I. Santamarta, J. Ind. Microbiol. Biotechnol., 2008, 35, 667–676 Search PubMed.
- H. Arulanantham, N. J. Kershaw, K. S. Hewitson, C. E. Hughes, J. E. Thirkettle and C. J. Schofield, J. Biol. Chem., 2006, 281, 279–287 Search PubMed.
- M. Fawaz, M. Topper and S. Firestine, Bioorg. Chem., 2011, 39, 185–276 Search PubMed.
- M. Galperin and E. Koonin, Protein Sci., 1997, 6, 2639–2682 CrossRef CAS.
- F. C. Neuhaus, C. V. Carpenter, J. L. Miller, N. M. Lee, M. Gragg and R. A. Stickgold, Biochemistry, 1969, 8, 5119–5124 CrossRef CAS.
- Y. Kallberg, U. Oppermann, H. Jörnvall and B. Persson, Eur. J. Biochem., 2002, 269, 4409–4417 CrossRef CAS.
- N. Tanaka, T. Nonaka, K. T. Nakamura and A. Hara, Curr. Org. Chem., 2001, 5, 89–111 Search PubMed.
- H. Joernvall, B. Persson, M. Krook, S. Atrian, R. Gonzalez-Duarte, J. Jeffery and D. Ghosh, Biochemistry, 1995, 34, 6003–6013 CrossRef CAS.
- U. Oppermann, C. Filling, M. Hult, N. Shafqat, X. Wu, M. Lindh, J. Shafqat, E. Nordling, Y. Kallberg, B. Persson and H. Jornvall, Chem.-Biol. Interact., 2003, 143–144, 247–253 CrossRef CAS.
- M. G. Rossmann, D. Moras and K. W. Olsen, Nature, 1974, 250, 194–199 CAS.
- C. Filling, K. D. Berndt, J. Benach, S. Knapp, T. Prozorovski, E. Nordling, R. Ladenstein, H. Jornvall and U. Oppermann, J. Biol. Chem., 2002, 277, 25677–25684 CrossRef CAS.
- A. K. MacKenzie, N. J. Kershaw, H. Hernandez, C. V. Robinson, C. J. Schofield and I. Andersson, Biochemistry, 2007, 46, 1523–1533 Search PubMed.
- R. K. Wierenga, P. Terpstra and W. G. J. Hol, J. Mol. Biol., 1986, 187, 101–107 Search PubMed.
- Z. Chen, J. C. Jiang, Z. G. Lin, W. R. Lee, M. E. Baker and S. H. Chang, Biochemistry, 1993, 32, 3342–3346 Search PubMed.
- A. G. Brown, D. F. Corbett, J. Goodacre, J. B. Harbridge, T. T. Howarth, R. J. Ponsford, I. Stirling and T. J. King, J. Chem. Soc., Perkin Trans. 1, 1984, 635–650 RSC.
- T. T. Howarth, A. G. Brown and T. J. King, J. Chem. Soc., Chem. Commun., 1976, 266b–267 RSC.
- C. A. Townsend, M.-F. Ho and S.-S. Mao, J. Chem. Soc., Chem. Commun., 1986, 638–639 RSC.
- L. M. Lorenzana, R. Pérez-Redondo, I. Santamarta, J. F. Martín and P. Liras, J. Bacteriol., 2004, 186, 3431–3438 Search PubMed.
- A. K. Mackenzie, K. Valegård, A. Iqbal, M. E. C. Caines, N. J. Kershaw, S. E. Jensen, C. J. Schofield and I. Andersson, J. Mol. Biol., 2010, 396, 332–344 Search PubMed.
- J. R. H. Tame, G. N. Murshudov, E. J. Dodson, T. K. Neil, G. G. Dodson, C. F. Higgins and A. J. Wilkinson, Science, 1994, 264, 1578–1581 CrossRef CAS.
- F. A. Quiocho and P. S. Ledvina, Mol. Microbiol., 1996, 20, 17–25 CrossRef CAS.
- S. W. Elson, J. Gillett, N. H. Nicholson and J. W. Tyler, J. Chem. Soc., Chem. Commun., 1988, 979–980 RSC.
- A. Iqbal, H. Arunlanantham, T. Brown, R. Chowdhury, I. J. Clifton, N. J. Kershaw, K. S. Hewitson, M. A. McDonough and C. J. Schofield, Proteins: Struct. Funct. Bioinform., 2010, 78, 1398–1407 Search PubMed.
- M. Vetting, L. S. de Carvalho, M. Yu, S. Hegde, S. Magnet, S. Roderick and J. Blanchard, Arch. Biochem. Biophys., 2005, 433, 212–238 CrossRef CAS.
- F. Dyda, D. Klein and A. Hickman, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 81–184 CrossRef CAS.
- H. He, Y. Ding, M. Bartlam, F. Sun, Y. Le, X. Qin, H. Tang, R. Zhang, A. Joachimiak, J. Liu, N. Zhao and Z. Rao, J. Mol. Biol., 2003, 325, 1019–1049 Search PubMed.
- W. Dou, M. Xu, D. Cai, X. Zhang, Z. Rao and Z. Xu, Appl. Biochem. Biotechnol., 2011, 165, 845–900 Search PubMed.
- A. Rodríguez-García, M. Ludovice, J. F. Martín and P. Liras, Mol. Microbiol., 1997, 25, 219–228 Search PubMed.
- J. E. Hodgson, A. P. Fosberry, N. S. Rawlinson, H. N. M. Ross, R. J. Neal, J. C. Arnell, A. J. Earl and E. J. Lawlor, Gene, 1995, 166, 49–55 CrossRef CAS.
- A. Rodríguez-García, Á. de la Fuente, R. Pérez-Redondo, J. F. Martín and P. Liras, J. Mol. Microbiol. Biotechnol., 2000, 2, 543–550 Search PubMed.
- A. de la Fuente, J. F. Martin, A. Rodriguez-Garcia and P. Liras, J. Bacteriol., 2004, 186, 6501–6507 Search PubMed.
- K. Tahlan, H. U. Park, A. Wong, P. H. Beatty and S. E. Jensen, Antimicrob. Agents Chemother., 2004, 48, 930–939 CrossRef CAS.
- J. M. Elkins, N. J. Kershaw and C. J. Schofield, Biochem. J., 2005, 385, 565–573 CrossRef CAS.
- A. Iqbal, I. J. Clifton, R. Chowdhury, D. Ivison, C. Domene and C. J. Schofield, Org. Biomol. Chem., 2011, 9, 6219–6225 RSC.
- A. Iqbal, I. J. Clifton, M. Bagonis, N. J. Kershaw, C. Domene, T. D. W. Claridge, C. W. Wharton and C. J. Schofield, J. Am. Chem. Soc., 2009, 131, 749–757 CrossRef CAS.
- R. Sankaranarayanan, M. M. Cherney, C. Garen, G. Garen, C. Niu, M. Yuan and M. N. James, J. Mol. Biol., 2010, 397, 979–990 CrossRef CAS.
- R. C. Wilmouth, K. Edman, R. Neutze, P. A. Wright, I. J. Clifton, T. R. Schneider, C. J. Schofield and J. Hajdu, Nat. Struct. Biol., 2001, 8, 689–694 CrossRef CAS.
- G. Katona, R. C. Wilmouth, P. A. Wright, G. I. Berglund, J. Hajdu, R. Neutze and C. J. Schofield, J. Biol. Chem., 2002, 277, 21962–21970 CrossRef CAS.
- E. S. Radisky, J. M. Lee, C. J. Lu and D. E. Koshland, Jr, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6835–6840 CrossRef CAS.
- P. Deslongchamps, Stereoelectronic effects in organic chemistry, Pergamon Press, New York, 1983 Search PubMed.
- P. Liras, I. Santamarta and R. Perez-Redondo, in Streptomyces: Molecular Biology and Biotechnology, ed. P. Dypson, Caister Academic Press, Wymondham, UK, 2011, pp. 167–178 Search PubMed.
- P. Liras and A. Rodriguez-Garcia, Appl. Microbiol. Biotechnol., 2000, 54, 467–475 Search PubMed.
- S. E. Jensen and A. S. Paradkar, Antonie van Leeuwenhoek, 1999, 75, 125–133 CrossRef CAS.
- K. Tahlan, H. U. Park and S. E. Jensen, Can. J. Microbiol., 2004, 50, 803–810 Search PubMed.
- J. M. Ward and J. E. Hodgson, FEMS Microbiol. Lett., 1993, 110, 239–242 CrossRef CAS.
- M. H. Medema, A. Trefzer, A. Kovalchuk, M. van den Berg, U. Müller, W. Heijne, L. Wu, M. T. Alam, C. M. Ronning, W. C. Nierman, R. A. L. Bovenberg, R. Breitling and E. Takano, Genome Biol. Evol., 2010, 2, 212–224 Search PubMed.
- N. J. Zelyas, H. Cai, T. Kwong and S. E. Jensen, J. Bacteriol., 2008, 190, 7957–7965 Search PubMed.
- S. E. Jensen, K. J. Elder, K. A. Aidoo and A. S. Paradkar, Antimicrob. Agents Chemother., 2000, 44, 720–726 CrossRef CAS.
- H. Jnawali, T.-J. Oh, K. Liou, B. Park and J. Sohng, J. Antibiot., 2008, 61, 651–660 Search PubMed.
- J. Song, E. Kim, D. Kim, S. Jensen and K. Lee, J. Ind. Microbiol. Biotechnol., 2009, 36, 301–312 Search PubMed.
- H. Jnawali, K. Liou and J. Sohng, Microbiol. Res., 2011, 166, 369–448 Search PubMed.
- H. Jnawali, J. Yoo and J. Sohng, Biotechnol. Lett., 2011, 33, 1221–1227 Search PubMed.
- M. Medema, M. Alam, W. Heijne, M. van den Berg, U. Müller, A. Trefzer, R. Bovenberg, R. Breitling and E. Takano, Microb. Biotechnol., 2011, 4, 300–305 Search PubMed.
- R. F. Li and C. A. Townsend, Metab. Eng., 2006, 8, 240–252 CrossRef CAS.
- K. Tahlan, C. Anders and S. E. Jensen, J. Bacteriol., 2004, 186, 6286–6297 Search PubMed.
- S. E. Jensen, A. Wong, A. Griffin and B. Barton, Antimicrob. Agents Chemother., 2004, 48, 514–520 CrossRef CAS.
- K. Tahlan, C. Anders, A. Wong, R. H. Mosher, P. H. Beatty, M. J. Brumlik, A. Griffin, C. Hughes, J. Griffin, B. Barton and S. E. Jensen, Chem. Biol., 2007, 14, 131–142 Search PubMed.
- R. H. Mosher, A. S. Paradkar, C. Anders, B. Barton and S. E. Jensen, Antimicrob. Agents Chemother., 1999, 43, 1215–1224 CAS.
- S. Goomeshi Nobary and S. E. Jensen, Can. J. Microbiol., 2012, 58, 413–425 Search PubMed.
- W. Dürckheimer, J. Blumbach, R. Lattrell and K. H. Scheunemann, Angew. Chem., Int. Ed. Engl., 1985, 24, 180–202 CrossRef.
- W. L. Parker, M. L. Rathnum, J. S. Wells, Jr., W. H. Trejo, P. A. Principe and R. B. Sykes, J. Antibiot., 1982, 35, 653–660 CAS.
- Y. Nakamura, K. Ishii, E. Ono, M. Ishikara, T. Kohda, Y. Yokogawa and H. Shibai, J. Antibiot., 1988, 41, 707–711 Search PubMed.
- F. M. Kahan, H. Kropp, J. G. Sundelof and J. Birnbaum, J. Antimicrob. Chemother., 1983, 12, 1–35 Search PubMed.
- W. J. Leanza, K. J. Windonger, T. W. Miller and B. G. Christensen, J. Med. Chem., 1979, 22, 1435–1436 CrossRef CAS.
- D. H. Shih, L. Cama and B. G. Christensen, Tetrahedron Lett., 1985, 26, 587–590 Search PubMed.
- J. M. Williamson, E. Inamine, K. E. Wilson, A. W. Douglas, J. M. Liesch and G. Albers-Schönberg, J. Biol. Chem., 1985, 260, 4637–4647.
- B. W. Bycroft, C. Maslen, S. J. Box, A. Brown and J. W. Tyler, J. Antibiot., 1988, 41, 1231–1242 CAS.
- J. M. Williamson, Crit. Rev. Biotechnol., 1986, 4, 111–173 CrossRef CAS.
- S. J. Coulthurst, A. M. L. Barnard and G. P. C. Salmond, Nat. Rev. Microbiol., 2005, 3, 295–306 Search PubMed.
- S. Derzelle, E. Duchaud, F. Kunst, A. Danchin and P. Bertin, Appl. Environ. Microbiol., 2002, 68, 3780–3789 CrossRef CAS.
- B. W. Bycroft, C. Maslen, S. J. Box, A. G. Brown and J. W. Tyler, J. Chem. Soc., Chem. Commun., 1987, 1623–1625 RSC.
- S. J. McGowan, M. Sebaihia, S. O'Leary, K. R. Hardie, P. Williams, G. S. A. B. Stewart, B. W. Bycroft and G. P. C. Salmond, Mol. Microbiol., 1997, 26, 545–556 CrossRef CAS.
- S. J. McGowan, M. Sebaihia, L. E. Porter, G. S. A. B. Stewart, P. Williams, B. W. Bycroft and G. P. C. Salmond, Mol. Microbiol., 1996, 22, 415–426 CrossRef CAS.
- A. Stapon, R. Li and C. A. Townsend, J. Am. Chem. Soc., 2003, 125, 8486–8493 CrossRef.
- B. W. Bycroft and S. R. Chhabra, J. Chem. Soc., Chem. Commun., 1989, 423–425 RSC.
- H. Tanaka, H. Sakagami and K. Ogasawara, Tetrahedron Lett., 2002, 43, 93–96 CrossRef CAS.
- R. F. Li, A. Stapon, J. T. Blanchfield and C. A. Townsend, J. Am. Chem. Soc., 2000, 122, 9296–9297 CrossRef CAS.
- S. J. McGowan, M. Sebaihia, S. Jones, B. Yu, N. Bainton, P. F. Chan, B. W. Bycroft, G. S. A. B. Stewart, P. Williams and G. P. C. Salmond, Microbiology, 1995, 141, 541–550 Search PubMed.
- S. J. McGowan, M. T. G. Holden, B. W. Bycroft and G. P. C. Salmond, Antonie van Leeuwenhoek, 1999, 75, 135–141 CrossRef CAS.
- M. C. Sleeman and C. J. Schofield, J. Biol. Chem., 2004, 279, 6730–6736 CrossRef CAS.
- B. Gerratana, S. O. Arnett, A. Stapon and C. A. Townsend, Biochemistry, 2004, 43, 15936–15945 CrossRef CAS.
- R. B. Hamed, E. T. Batchelar, J. Mecinović, T. D. W. Claridge and C. J. Schofield, ChemBioChem, 2009, 10, 246–250 CrossRef CAS.
- J. L. Sorensen, M. C. Sleeman and C. J. Schofield, Chem. Commun., 2005, 1155–1157 RSC.
- M. C. Sleeman, J. L. Sorensen, E. T. Batchelar, M. A. McDonough and C. J. Schofield, J. Biol. Chem., 2005, 280, 34956–34965 CrossRef CAS.
- I. J. Clifton, L. X. Doan, M. C. Sleeman, M. Topf, H. Suzuki, R. C. Wilmouth and C. J. Schofield, J. Biol. Chem., 2003, 278, 20843–20850 CrossRef.
- R. B. Hamed, E. T. Batchelar, I. J. Clifton and C. J. Schofield, Cell. Mol. Life Sci., 2008, 65, 2507–2527 CrossRef CAS.
- H. M. Holden, M. M. Benning, T. Haller and J. A. Gerlt, Acc. Chem. Res., 2001, 34, 145–157 CrossRef CAS.
- M. Rodriguez, L. E. Nunez, A. F. Brana, C. Mendez, J. A. Salas and G. Blanco, Antimicrob. Agents Chemother., 2011, 55, 1638–1649 Search PubMed.
- E. T. Batchelar, R. B. Hamed, C. Ducho, T. D. W. Claridge, M. J. Edelmann, B. Kessler and C. J. Schofield, Angew. Chem., Int. Ed., 2008, 47, 9322–9325 CrossRef CAS.
- C. Ducho, R. B. Hamed, E. T. Batchelar, J. L. Sorensen, B. Odell and C. J. Schofield, Org. Biomol. Chem., 2009, 7, 2770–2779 RSC.
- S. L. Bearne and R. Wolfendon, Biochemistry, 1995, 34, 11515–11520 CrossRef CAS.
- K. T. Douglas, Acc. Chem. Res., 1986, 19, 186–192 CrossRef CAS.
- B. J. Wong and J. A. Gerlt, J. Am. Chem. Soc., 2003, 125, 12076–12077 Search PubMed.
- T. J. Erb, V. Brecht, G. Fuchs, M. Muller and B. E. Alber, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8871–8876 CrossRef CAS.
- T. J. Erb, I. A. Berg, V. Brecht, M. Muller, G. Fuchs and B. E. Alber, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10631–10636 CrossRef CAS.
- A. S. Eustáquio, R. P. McGlinchey, Y. Liu, C. Hazzard, L. L. Beer, G. Florova, M. M. Alhamadsheh, A. Lechner, A. J. Kale, Y. Kobayashi, K. A. Reynolds and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 12295–12300 CrossRef.
- M. Wilson and B. Moore, Nat. Prod. Rep., 2012, 29, 72–158 RSC.
- B. Alber, Appl. Microbiol. Biotechnol., 2011, 89, 17–42 CrossRef CAS.
- H.-G. Yoo, S.-Y. Kwon, S. Kim, S. Karki, Z.-Y. Park and H.-J. Kwon, Biosci., Biotechnol., Biochem., 2011, 75, 1191–1194 CrossRef CAS.
- N. Quade, L. Huo, S. Rachid, D. Heinz and R. Müller, Nat. Chem. Biol., 2011, 8, 117–141 Search PubMed.
- J. J. Turnbull, J. I. Nakajima, R. W. D. Welford, M. Yamazaki, K. Saito and C. J. Schofield, J. Biol. Chem., 2004, 279, 1206–1216 CAS.
- N. H. Horowitz, Proc. Natl. Acad. Sci. U. S. A., 1945, 31, 153–157 CrossRef CAS.
- A. Stapon, R. Li and C. A. Townsend, J. Am. Chem. Soc., 2003, 125, 15746–15747 CrossRef CAS.
- M. Topf, G. M. Sandala, D. M. Smith, C. J. Schofield, C. J. Easton and L. Radom, J. Am. Chem. Soc., 2004, 126, 9932–9933 CrossRef CAS.
- I. K. Leung, T. J. Krojer, G. T. Kochan, L. Henry, F. von Delft, T. D. Claridge, U. Oppermann, M. A. McDonough and C. J. Schofield, Chem. Biol., 2010, 17, 1316–1324 CrossRef CAS.
- T. Borowski, E. Broclawik, C. J. Schofield and P. E. M. Siegbahn, J. Comput. Chem., 2006, 27, 740–748 CrossRef CAS.
- D. R. Houck, K. Kobayashi, J. M. Williamson and H. G. Floss, J. Am. Chem. Soc., 1986, 108, 5365–5366 CrossRef CAS.
- L. E. Nunez, C. Mendez, A. F. Brana, G. Blanco and J. A. Salas, Chem. Biol., 2003, 10, 301–311 CrossRef CAS.
- M. J. Bodner, R. M. Phelan, M. F. Freeman, R. Li and C. A. Townsend, J. Am. Chem. Soc., 2010, 132, 12–13 CrossRef CAS.
- M. Rodriguez, L. Nunez, A. Brana, C. Mendez, J. Salas and G. Blanco, Mol. Microbiol., 2008, 69, 633–645 CrossRef CAS.
- H. Suzuki, Y. Ohnishi and S. Horinouchi, J. Antibiot., 2007, 60, 380–387 Search PubMed.
- D. O'Hagan and J. W. Schmidberger, Nat. Prod. Rep., 2010, 27, 900–918 RSC.
- T. M. Zydowsky, L. F. Courtney, V. Frasca, K. Kobayashi, H. Shimizu, L. D. Yuen, R. G. Matthews, S. J. Benkovic and H. G. Floss, J. Am. Chem. Soc., 1986, 108, 3152–3153 CrossRef CAS.
- E. O. Stapley, P. J. Cassidy, J. Tunac, R. L. Monaghan, M. Jackson, S. Hernandez, S. B. Zimmerman, J. M. Mata, S. A. Currie, D. Daoust and D. Hendlin, J. Antibiot., 1981, 34, 628–636 CAS.
- D. Butterworth, M. Cole, G. Hanscomb and G. N. Rolinson, J. Antibiot., 1979, 32, 287–294 CAS.
- A. Imada, Y. Nozaki, K. Kintaka, K. Okonogi, K. Kitano and S. Harada, J. Antibiot., 1980, 33, 1417–1424 Search PubMed.
- T. S. Chen, B. H. Arison, C. L. Ruby, A. W. Dombrowski and E. S. Inamine, J. Ind. Microbiol. Biotechnol., 1993, 12, 66–67 Search PubMed.
- A. J. Brooks, M. Vlasie, R. Banerjee and T. C. Brunold, J. Am. Chem. Soc., 2005, 127, 16522–16528 CrossRef CAS.
- A. J. Brooks, M. Vlasie, R. Banerjee and T. C. Brunold, J. Am. Chem. Soc., 2004, 126, 8167–8180 CrossRef CAS.
- B. Zagalak and J. Retey, Eur. J. Biochem., 1974, 44, 529–535 Search PubMed.
- E. Marsh, D. Patterson and L. Li, ChemBioChem, 2010, 11, 604–625 CrossRef CAS.
- P. Frey, A. Hegeman and F. Ruzicka, Crit. Rev. Biochem. Mol. Biol., 2008, 43, 63–88 CrossRef CAS.
- M. Challand, R. Driesener and P. Roach, Nat. Prod. Rep., 2011, 28, 1696–2417 RSC.
- K. Kubo, T. Ishikura and Y. Fukagawa, J. Antibiot., 1985, 38, 904–911 CAS.
- Y. Fukagawa, M. Okabe, S. Azuma, I. Kojima, T. Ishikura and K. Kubo, J. Antibiot., 1984, 37, 1388–1393 Search PubMed.
- K. Kubo, T. Ishikura and Y. Fukagawa, J. Antibiot., 1984, 37, 1394–1402 Search PubMed.
- K. Kubo, T. Ishikura and Y. Fukagawa, J. Antibiot., 1985, 38, 622–630 Search PubMed.
- L. W. Kang, S. B. Gabelli, M. A. Bianchet, W. L. Xu, M. J. Bessman and L. M. Amzel, J. Bacteriol., 2003, 185, 4110–4118 Search PubMed.
- E. V. Koonin and R. L. Tatusov, J. Mol. Biol., 1994, 244, 125–132 CrossRef CAS.
- A. R. Buller, M. F. Freeman, N. T. Wright, J. F. Schildbach and C. A. Townsend, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2308–2313 Search PubMed.
- H. B. Burgi, J. D. Dunitz and E. Shefter, J. Am. Chem. Soc., 1973, 95, 5065–5067 CrossRef CAS.
- W. L. Parker, J. O'Sullivan and R. B. Sykes, in Adv. Appl. Microbiol., ed. I. L. Allen, Academic Press, 1986, vol. 31, pp. 181–205 Search PubMed.
- J. P. Scannell, D. Pruess, J. F. Blount, H. A. Ax, M. Kellett, F. Weiss, T. C. Demny, T. H. Williams and A. Stempel, J. Antibiot., 1975, 28, 1–6 Search PubMed.
- K. P. Devkota, B. N. Lenta, P. A. Fokou and N. Sewald, Nat. Prod. Rep., 2008, 25, 612–630 RSC.
- H. Watanabe, K. Watanabe, M. Shimadzu, T. Kikuchi and Z. Liu, Planta Med., 1986, 56–58 Search PubMed.
- H. de Sousa Falcao, J. A. Leite, J. M. Barbosa-Filho, P. F. de Athayde-Filho, M. C. de Oliveira Chaves, M. D. Moura, A. L. Ferreira, A. B. de Almeida, A. R. Souza-Brito, M. de Fatima Formiga Melo Diniz and L. M. Batista, Molecules, 2008, 13, 3198–3223 Search PubMed.
- S. Funayama, T. Noshita, K. Shinoda, N. Haga, S. Nozoe, M. Hayashi and K. Komiyama, Biol. Pharm. Bull., 2000, 23, 262–264 CAS.
- T. Kikuchi and S. Uyeo, Chem. Pharm. Bull., 1967, 15, 549–570 CAS.
- T. Kikuchi and S. Uyeo, Tetrahedron Lett., 1965, 39, 3473–3485 Search PubMed.
- L. C. Chang, K. P. L. Bhat, H. H. S. Fong, J. M. Pezzuto and A. D. Kinghorn, Tetrahedron, 2000, 56, 3133–3138 CrossRef CAS.
- L. C. Chang, K. P. L. Bhat, E. Pisha, E. J. Kennelly, H. H. S. Fong, J. M. Pezzuto and A. D. Kinghorn, J. Nat. Prod., 1998, 61, 1257–1262 CrossRef CAS.
- E. Avilés and A. D. Rodríguez, Org. Lett., 2010, 12, 5290–5293 CrossRef CAS.
- R. B. Sykes and D. P. Bonner, Rev. Infect. Dis., 1985, 7, S579–593 Search PubMed.
- S. Han, N. Caspers, R. P. Zaniewski, B. M. Lacey, A. P. Tomaras, X. Feng, K. F. Geoghegan and V. Shanmugasundaram, J. Am. Chem. Soc., 2011, 133, 20536–20545 Search PubMed.
- J. O'Sullivan, A. M. Gillum, C. A. Aklonis, M. L. Souser and R. B. Sykes, Antimicrob. Agents Chemother., 1982, 21, 558–564 CAS.
- J. O'Sullivan, M. L. Souser, C. C. Kao and C. A. Aklonis, Antimicrob. Agents Chemother., 1983, 23, 598–602 Search PubMed.
- J. O'Sullivan and C. A. Aklonis, J. Antibiot., 1984, 37, 804–806 Search PubMed.
- L. J. Nisbet, R. J. Mehta, Y. Oh, C. H. Pan, C. G. Phelen, M. J. Polansky, M. C. Shearer, A. J. Giovenella and S. F. Grappel, J. Antibiot., 1985, 38, 133–138 Search PubMed.
- J. A. Chan, E. A. Shultis, J. J. Dingerdissen, C. W. DeBrosse, G. D. Roberts and K. M. Snader, J. Antibiot., 1985, 38, 139–144 Search PubMed.
- N. Katayama, Y. Nozaki, K. Okonogi, H. Ono, S. Harada and H. Okazaki, J. Antibiot., 1985, 38, 1117–1127 CAS.
- T. Hida, S. Tsubotani, N. Katayama, H. Okazaki and S. Harada, J. Antibiot., 1985, 38, 1128–1140 CAS.
- J. Hosoda, T. Konomi and N. Tani, Agric. Biol. Chem., 1977, 41, 2013–2020 Search PubMed.
- M. Kurita, K. Jomon, T. Komori, N. Miyairi and H. Aoki, J. Antibiot., 1976, 29, 1243–1245 Search PubMed.
- W. L. Parker, in Journal of Chromatography Library, ed. H. W. Gerald and C. Raymond, Elsevier, 1989, vol. 43, ch. 7, pp. 259–292 Search PubMed.
- P. H. Milner, A. W. Guest, F. P. Harrington, R. J. Ponsford, T. C. Smale and A. V. Stachulski, J. Chem. Soc., Chem. Commun., 1984, 1335–1336 RSC.
- M. J. Basker, R. A. Edmondson, S. J. Knott, R. J. Ponsford, B. Slocombe and S. J. White, Antimicrob. Agents Chemother., 1984, 26, 734–740 Search PubMed.
- J. Hosoda, N. Tani and T. Konomi, Agric. Biol. Chem., 1977, 41, 2007–2012 Search PubMed.
- C. A. Townsend and A. M. Brown, J. Am. Chem. Soc., 1981, 103, 2873–2874 Search PubMed.
- C. A. Townsend and L. T. Nguyen, J. Am. Chem. Soc., 1981, 103, 4582–4583 Search PubMed.
- C. A. Townsend, A. M. Brown and L. T. Nguyen, J. Am. Chem. Soc., 1983, 105, 919–927 CrossRef CAS.
- C. A. Townsend, G. M. Salituro, L. T. Nguyen and M. J. DiNovi, Tetrahedron Lett., 1986, 27, 3819–3822 Search PubMed.
- C. A. Townsend and A. M. Brown, J. Am. Chem. Soc., 1983, 105, 913–918 Search PubMed.
- C. A. Townsend, A. M. Reeve and G. M. Salituro, J. Chem. Soc., Chem. Commun., 1988, 1579–1581 RSC.
- C. A. Townsend and G. M. Salituro, J. Chem. Soc., Chem. Commun., 1984, 1631–1632 RSC.
- C. A. Townsend and B. A. Wilson, J. Am. Chem. Soc., 1988, 110, 3320–3321 CrossRef CAS.
- B. A. Wilson, S. Bantia, G. M. Salituro, A. M. Reeve and C. A. Townsend, J. Am. Chem. Soc., 1988, 110, 8238–8239 Search PubMed.
- M. Gunsior, S. D. Breazeale, A. J. Lind, J. Ravel, J. W. Janc and C. A. Townsend, Chem. Biol., 2004, 11, 927–938 CrossRef CAS.
- A. M. Reeve, S. D. Breazeale and C. A. Townsend, J. Biol. Chem., 1998, 273, 30695–30703 CrossRef CAS.
- W. L. Kelly and C. A. Townsend, J. Biol. Chem., 2004, 279, 38220–38227 CrossRef CAS.
- W. L. Kelly and C. A. Townsend, J. Bacteriol., 2005, 187, 739–746 Search PubMed.
- J. M. Davidsen and C. A. Townsend, J. Bacteriol., 2009, 191, 1066–1077 Search PubMed.
- A. M. van Wageningen, P. N. Kirkpatrick, D. H. Williams, B. R. Harris, J. K. Kershaw, N. J. Lennard, M. Jones, S. J. Jones and P. J. Solenberg, Chem. Biol., 1998, 5, 155–162 CrossRef.
- P. P. Chong, S. M. Podmore, H. M. Kieser, M. Redenbach, K. Turgay, M. Marahiel, D. A. Hopwood and C. P. Smith, Microbiology, 1998, 144, 193–199 CrossRef CAS.
- H. T. Chiu, B. K. Hubbard, A. N. Shah, J. Eide, R. A. Fredenburg, C. T. Walsh and C. Khosla, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8548–8553 CrossRef CAS.
- B. K. Hubbard, M. G. Thomas and C. T. Walsh, Chem. Biol., 2000, 7, 931–942 CrossRef CAS.
- S. C. Wenzel and R. Müller, Curr. Opin. Chem. Biol., 2005, 9, 447–458 CrossRef CAS.
- G. L. Challis, J. Ravel and C. A. Townsend, Chem. Biol., 2000, 7, 211–224 CrossRef CAS.
- T. Stachelhaus, H. D. Mootz and M. A. Marahiel, Chem. Biol., 1999, 6, 493–505 CrossRef CAS.
- G. L. Challis and J. Ravel, FEMS Microbiol. Lett., 2000, 187, 111–114 CrossRef CAS.
- S. Lautru, R. J. Deeth, L. M. Bailey and G. L. Challis, Nat. Chem. Biol., 2005, 1, 265–269 CrossRef CAS.
- Y. Shikata, T. Watanabe, T. Teramoto, A. Inoue, Y. Kawakami, Y. Nishizawa, K. Katayama and M. Kuwada, J. Biol. Chem., 1995, 270, 16719–16723 Search PubMed.
- P. K. Mehta and P. Christen, Adv. Enzymol. Relat. Areas Mol. Biol., 2000, 74, 129–184 Search PubMed.
- W. L. Kelly and C. A. Townsend, J. Am. Chem. Soc., 2002, 124, 8186–8187 CrossRef CAS.
- I. Molnar, J. F. Aparicio, S. F. Haydock, L. E. Khaw, T. Schwecke, A. Konig, J. Staunton and P. F. Leadlay, Gene, 1996, 169, 1–7 CrossRef CAS.
- T. L. Poulos, B. C. Finzel and A. J. Howard, J. Mol. Biol., 1987, 195, 687–700 CAS.
- W. W. Stewart, Nature, 1971, 229, 174–178 CrossRef CAS.
- D. L. Lee and H. Rapoport, J. Org. Chem., 1975, 40, 3491–3495 CrossRef CAS.
- P. A. Taylor, H. K. Schnoes and R. D. Durbin, Biochim. Biophys. Acta, Gen. Subj., 1972, 286, 107–117 Search PubMed.
- D. Woolley, R. Pringle and A. Braun, J. Biol. Chem., 1952, 197, 409–426 Search PubMed.
- F. Wolf and A. Foster, Science, 1917, 46, 361–363 Search PubMed.
- C. Levi and R. D. Durbin, Physiol. Mol. Plant Pathol., 1986, 28, 345–352 Search PubMed.
- M. D. Thomas, P. J. Langston-Unkefer, T. F. Uchytil and R. D. Durbin, Plant Physiol., 1983, 71, 912–915 Search PubMed.
- J. G. Turner, Physiol. Plant Pathol., 1981, 19, 57–67 Search PubMed.
- J. Turner and J. Debbage, Physiol. Plant Pathol., 1982, 20, 223–456 Search PubMed.
- P. L. Langston-Unkefer, P. A. Macy and R. D. Durbin, Plant Physiol., 1984, 76, 71–74 Search PubMed.
- P. J. Langston-Unkefer, A. C. Robinson, T. J. Knight and R. D. Durbin, J. Biol. Chem., 1987, 262, 1608–1613 Search PubMed.
- R. D. Durbin and T. F. Uchytil, Experientia, 1985, 41, 136–137 CrossRef CAS.
- T. J. Knight, R. D. Durbin and P. J. Langston-Unkefer, J. Bacteriol., 1986, 166, 224–229 Search PubMed.
- T. J. Knight, R. D. Durbin and P. J. Langston-Unkefer, J. Bacteriol., 1987, 169, 1954–1959 Search PubMed.
- R. H. Coleman, J. Shaffer and H. True, Curr. Microbiol., 1996, 32, 147–150 Search PubMed.
- J. Liu, Y. Le, B. Ye, Y. Zhen, C. Zhu, J. Shen and R. Zhang, Protein Expression Purif., 2002, 24, 439–444 Search PubMed.
- R. Batchvarova, V. Nikolaeva, S. Slavov, S. Bossolova, V. Valkov, S. Atanassova, S. Guelemerov, A. Atanassov and H. Anzai, Theor. Appl. Genet., 1998, 97, 986–989 Search PubMed.
- T. G. Kinscherf and D. K. Willis, J. Antibiot., 2005, 58, 817–821 CrossRef CAS.
- B. Müller, A. Hädener and C. Tamm, Helv. Chim. Acta, 1987, 70, 412–422 Search PubMed.
- H. Wehrli, Helv. Chim. Acta, 1980, 63, 1915–1919 Search PubMed.
- C. J. Unkefer, R. E. London, R. D. Durbin, T. F. Uchytil and P. J. Langston-Unkefer, J. Biol. Chem., 1987, 262, 4994–4999 CAS.
- P. Roth, A. Hädener and C. Tamm, Helv. Chim. Acta, 1990, 73, 476–482 CAS.
- K. Engst and P. D. Shaw, Mol. Plant-Microbe Interact., 1992, 5, 322–329 CAS.
- L. Liu and P. D. Shaw, J. Bacteriol., 1997, 179, 507–513 CAS.
- L. Liu and P. D. Shaw, J. Bacteriol., 1997, 179, 5922–5927 CAS.
- G. J. Feistner, T. F. Uchytil, K. K. Knoche and R. D. Durbin, J. Org. Chem., 1991, 56, 2922–2925 Search PubMed.
- T. M. Barta, T. G. Kinscherf, T. F. Uchytil and D. K. Willis, Appl. Environ. Microbiol., 1993, 59, 458–466 CAS.
- E. M. Hrabak and D. K. Willis, J. Bacteriol., 1992, 174, 3011–3020 Search PubMed.
- T. M. Barta, T. G. Kinscherf and D. K. Willis, J. Bacteriol., 1992, 174, 3021–3029 Search PubMed.
- J. J. Rich, S. S. Hirano and D. K. Willis, Appl. Environ. Microbiol., 1992, 58, 1440–1446 Search PubMed.
- J. Spizek, J. Novotna, T. Rezanka and A. L. Demain, J. Ind. Microbiol. Biotechnol., 2010, 37, 1241–1248 CrossRef CAS.
- V. Miao and J. Davies, in Uncultivated Microorganisms, ed. S. S. Epstein, SpringerBerlin/Heidelberg, 2009, vol. 10, pp. 161–180 Search PubMed.
- C. Schmeisser, H. Steele and W. R. Streit, Appl. Microbiol. Biotechnol., 2007, 75, 955–962 CrossRef CAS.
- R. H. Cichewicz, Nat. Prod. Rep., 2010, 27, 11–22 RSC.
- R. B. Williams, J. C. Henrikson, A. R. Hoover, A. E. Lee and R. H. Cichewicz, Org. Biomol. Chem., 2008, 6, 1895–1897 RSC.
- A. A. Brakhage and V. Schroeckh, Fungal Genet. Biol., 2011, 48, 15–22 Search PubMed.
- P. S. Padayatti, M. S. Helfand, M. A. Totir, M. P. Carey, P. R. Carey, R. A. Bonomo and F. van den Akker, J. Biol. Chem., 2005, 280, 34900–34907 CrossRef CAS.
- M. A. Totir, M. S. Helfand, M. P. Carey, A. Sheri, J. D. Buynak, R. A. Bonomo and P. R. Carey, Biochemistry, 2007, 46, 8980–8987 Search PubMed.
- R. Li, J.-M. Liao, C.-R. Gu, Y.-T. Wang and C.-L. Chen, J. Phys. Chem. B, 2011, 115, 10298–10310 Search PubMed.
- L. W. Tremblay, J.-E. Hugonnet and J. S. Blanchard, Biochemistry, 2008, 47, 5312–5316 Search PubMed.
- S. Han, R. P. Zaniewski, E. S. Marr, B. M. Lacey, A. P. Tomaras, A. Evdokimov, J. R. Miller and V. Shanmugasundaram, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 22002–22007 Search PubMed.
- R. P. A. Brown, R. T. Aplin and C. J. Schofield, Biochemistry, 1996, 35, 12421–12432 CrossRef CAS.
- J.-E. Hugonnet, L. W. Tremblay, H. I. Boshoff, C. E. Barry, 3rd and J. S. Blanchard, Science, 2009, 323, 1215–1218 Search PubMed.
- N. O. Concha, B. A. Rasmussen, K. Bush and O. Herzberg, Structure, 1996, 4, 823–836 CrossRef CAS.
- M. I. Page and A. Badarau, Bioinorg. Chem. Appl., 2008, 576297 Search PubMed.
- J. D. Williams, Clin. Infect. Dis., 1997, 24, 494–497 Search PubMed.
- C. M. Perry and A. Markham, Drugs, 1999, 57, 805–843 Search PubMed.
- H. Troonen, P. Roelants and B. Boon, J. Antibiot., 1976, 29, 1258–1267 CAS.
- F. Robinson, Analyst, 1947, 72, 274–276 RSC.
- K. Kitano, K. Kintaka, S. Suzuki, K. Katamoto, K. Nara and Y. Nakao, J. Ferment. Technol., 1976, 54, 705–711 Search PubMed.
- K. Kitano, Y. Fujisawa, K. Katamoto, K. Nara and Y. Nakao, J. Ferment. Technol., 1976, 54, 712–719 Search PubMed.
- J. Shoji, R. Sakazaki, K. Matsumoto, T. Tanimoto, Y. Terui, S. Kozuki and E. Kondo, J. Antibiot., 1983, 36, 167–169 Search PubMed.
- K. Tanaka, Y. Kawamura, M. Hattori, E. Kondo, K. Matsumoto, A. Terui, J. Shoji, Japan Pat., JP55143996, 1980.
- H. Aoki and M. Okuhara, Annu. Rev. Microbiol., 1980, 34, 159–181 Search PubMed.
- H. Fukase, T. Hasegawa, K. Hatano, H. Iwasaki and M. Yoneda, J. Antibiot., 1976, 29, 113–120 Search PubMed.
- A. A. Brakhage, P. Spröte, Q. Al-Abdallah, A. Gehrke, H. Plattner and A. Tüncher, in Molecular Biotechnolgy of Fungal beta-Lactam Antibiotics and Related Peptide Synthetases, ed. A. A. Brakhage, SpringerBerlin / Heidelberg, 2004, vol. 88, pp. 45–90 Search PubMed.
- P. Liras and J. F. Martin, Int. Microbiol., 2006, 9, 9–19 Search PubMed.
- D. H. Nam, S. K. Lim, M. H. Chung, E. S. Lee, Y. S. Sohn and D. D. Y. Ryu, J. Microbiol. Biotechnol., 2001, 11, 153–159 Search PubMed.
- R. Ullán, G. Liu, J. Casqueiro, S. Gutiérrez, O. Bañuelos and J. Martín, Mol. Genet. Genomics, 2002, 267, 673–683 Search PubMed.
- A. Paradkar, K. Aidoo, A. Wong and S. Jensen, J. Bacteriol., 1996, 178, 6266–6274 Search PubMed.
- M. B. Tobin, S. Kovacevic, K. Madduri, J. A. Hoskins, P. L. Skatrud, L. C. Vining, C. Stuttard and J. R. Miller, J. Bacteriol., 1991, 173, 6223–6229 Search PubMed.
- D. C. Alexander and S. E. Jensen, J. Bacteriol., 1998, 180, 4068–4079 CAS.
- J. F. Martin, J. Casqueiro and P. Liras, Curr. Opin. Microbiol., 2005, 8, 282–293 CrossRef CAS.
- M. J. Park, J. O. Yon, S. K. Lim, D. D. Y. Ryu and D. H. Nam, J. Microbiol. Biotechnol., 2004, 14, 635–638 Search PubMed.
- Y. Zhang and E. Oldfield, J. Am. Chem. Soc., 2004, 126, 9494–9495 Search PubMed.
- W. Ge, I. J. Clifton, A. R. Howard-Jones, J. E. Stok, R. M. Adlington, J. E. Baldwin and P. J. Rutledge, ChemBioChem, 2009, 10, 2025–2031 CrossRef CAS.
- W. Ge, I. J. Clifton, J. E. Stok, R. M. Adlington, J. E. Baldwin and P. J. Rutledge, J. Am. Chem. Soc., 2008, 130, 10096–10102 CrossRef CAS.
- K. Turgay, M. Krause and M. A. Marahiel, Mol. Microbiol., 1992, 6, 529–546 CAS.
- W. Schmitz, R. Fingerhut and E. Conzelmann, Eur. J. Biochem., 1994, 222, 313–323 Search PubMed.
- W. Schmitz, C. Albers, R. Fingerhut and E. Conzelmann, Eur. J. Biochem., 1995, 231, 815–822 CAS.
- R. D. Knihinicki, R. O. Day and K. M. Williams, Biochem. Pharmacol., 1991, 42, 1905–1911 CrossRef CAS.
- M. D. Lloyd, D. J. Darley, A. S. Wierzbicki and M. D. Threadgill, FEBS J., 2008, 275, 1089–1102 CrossRef CAS.
- J. Piel, C. Hertweck, P. R. Shipley, D. M. Hunt, M. S. Newman and B. S. Moore, Chem. Biol., 2000, 7, 943–955 CrossRef CAS.
- L. Xiang and B. S. Moore, J. Bacteriol., 2005, 187, 4286–4289 CrossRef CAS.
- M. Berner, D. Krug, C. Bihlmaier, A. Vente, R. Muller and A. Bechthold, J. Bacteriol., 2006, 188, 2666–2673 Search PubMed.
- D. Brown, J. R. Evans and R. A. Fletton, J. Chem. Soc., Chem. Commun., 1979, 282–283 RSC.
- K. Ishida, T. V. Hung, K. Liou, H. C. Lee, C. H. Shin and J. K. Sohng, Biotechnol. Lett., 2006, 28, 409–417 Search PubMed.
- M. L. Raber, A. Castillo, A. Greer and C. A. Townsend, ChemBioChem, 2009, 10, 2904–2912 CrossRef CAS.
- M. L. Raber, S. O. Arnett and C. A. Townsend, Biochemistry, 2009, 48, 4959–4971 CrossRef CAS.
- T. Borowski, S. de Marothy, E. Broclawik, C. J. Schofield and P. E. M. Siegbahn, Biochemistry, 2007, 46, 3682–3691 CrossRef CAS.
- M. D. Andrews, G. A. Brown, J. P. H. Charmant, T. M. Peakman, A. Rebello, K. E. Walsh, T. Gallagher and N. J. Hales, Chem. Commun., 1999, 249–250 RSC.
- F. Marc, P. Weigel, C. Legrain, Y. Almeras, M. Santrot, N. Glansdorff and V. Sakanyan, Eur. J. Biochem., 2000, 267, 5217–5226 CAS.
- K. E. Wilson, in Journal of Chromatography Library, ed. H. W. Gerald and C. Raymond, Elsevier, 1989, vol. 43, pp. 293–346 Search PubMed.
- D. Rosi, M. L. Drozd, M. F. Kuhrt, L. Terminiello, P. E. Came and S. Daum, J. Antibiot., 1981, 34, 341–343 Search PubMed.
- K. E. Wilson, A. J. Kempf, J. M. Liesch and B. H. Arison, J. Antibiot., 1983, 36, 1109–1117 Search PubMed.
- J. S. Kahan, U. S. Pat., 4,162,323, 1979.
- K. Tanaka, N. Tsuiji, E. Kondo and Y. Kawamura, Eur. Pat. Appl., 1981, 38, 534 Search PubMed.
- P. J. Cassidy, G. Albers-Schonberg, R. T. Goegelman, T. Miller, B. Arison, E. O. Stapley and J. Birnbaum, J. Antibiot., 1981, 34, 637–648 CAS.
- S. J. Box, J. D. Hood and S. R. Spear, J. Antibiot., 1979, 32, 1239–1247 CAS.
- A. G. Brown, D. F. Corbett, A. J. Eglington and T. T. Howarth, J. Antibiot., 1979, 32, 961–963 CAS.
- J. D. Hood, S. J. Box and M. S. Verrall, J. Antibiot., 1979, 32, 295–304 CAS.
- A. G. Brown, D. F. Corbett, A. J. Eglington and T. T. Howarth, J. Chem. Soc., Chem. Commun., 1977, 523–525 RSC.
- D. F. Corbett, A. J. Eglington and T. T. Howarth, J. Chem. Soc., Chem. Commun., 1977, 953–954 RSC.
- S. J. Box, D. F. Corbett, K. G. Robins, S. R. Spear and M. S. Verrall, J. Antibiot., 1982, 35, 1394–1396 Search PubMed.
- K. Okamura, S. Hirata and A. Koki, J. Antibiot., 1979, 32, 262–271.
- N. Shibamoto, A. Koki, M. Nishino, K. Nakamura, K. Kiyoshima, K. Okamura, M. Okabe, R. Okamoto, Y. Fukagawa and Y. Shimauchi, J. Antibiot., 1980, 33, 1128–1137 Search PubMed.
- N. Shibamoto, M. Nishino, K. Okamura, Y. Fukagawa and T. Ishikura, J. Antibiot., 1982, 35, 763–765 Search PubMed.
- M. Nakayama, A. Iwasaki, S. Kimura, T. Mizoguchi, S. Tanabe, A. Murakami, I. Watanabe, M. Okuchi, H. Itoh, Y. Saino, F. Kobayashi and T. Mori, J. Antibiot., 1980, 33, 1388–1390 CAS.
- M. Nakayama, S. Kimura, S. Tanabe, T. Mizoguchi, I. Watanabe, T. Mori, K. Miyahara and T. Kawasaki, J. Antibiot., 1981, 34, 818–823.
- M. Nakayama, S. Kimura, T. Mizoguchi, S. Tanabe, A. Iwasaki, A. Murakami, M. Okuchi, H. Itoh and T. Mori, J. Antibiot., 1983, 36, 943–949 Search PubMed.
- M. Okuchi, M. Nakayama, A. Iwasaki, S. Kimura, T. Mizoguchi, S. Tanabe, A. Murakami, H. Ito, T. Mori, U. S. Pat., 4468350, 1984.
- S. Harada, S. Shinagawa, Y. Nozaki, M. Asai and T. Kishi, J. Antibiot., 1980, 33, 1425–1430 Search PubMed.
- Y. Kawamura, Y. Yasuda, M. Mayama and K. Tanaka, J. Antibiot., 1982, 35, 10–14 Search PubMed.
- J. Shoji, H. Hinoo, R. Sakazaki, N. Tsuji, K. Nagashima, K. Matsumoto, Y. Takahashi, S. Kozuki, T. Hattori, E. Kondo and K. Tanaka, J. Antibiot., 1982, 35, 15–23 Search PubMed.
- S. Tanabe, M. Okuchi, M. Nakayama, S. Kimura, A. Iwasaki, T. Mizoguchi, A. Murakami, H. Itoh and T. Mori, J. Antibiot., 1982, 35, 1237–1239 Search PubMed.
- M. Okabe, S. Azuma, I. Kojima, K. Kouno, R. Okamoto, Y. Fukagawa and T. Ishikura, J. Antibiot., 1982, 35, 1255–1263.
- T. Yoshioka, I. Kojima, K. Isshiki, A. Watanabe, Y. Shimauchi, M. Okabe, Y. Fukagawa and T. Ishikura, J. Antibiot., 1983, 36, 1473–1482 Search PubMed.
- T. Yoshioka, A. Watanabe, I. Kojima, Y. Shimauchi, M. Okabe, Y. Fukagawa and T. Ishikura, J. Antibiot., 1984, 37, 211–217 CAS.
- N. Tsuji, K. Nagashima, M. Kobayashi, Y. Terui, K. Matsumoto and E. Kondo, J. Antibiot., 1982, 35, 536–540 CAS.
- N. Tsuji, M. Kobayashi, Y. Terui, K. Matsumoto, Y. Takahashi and E. Kondo, J. Antibiot., 1985, 38, 270–274 CAS.
- T. Ito, N. Ezaki, K. Ohba, S. Amano, Y. Kondo, H. S. Miyado, T. Shomura, M. Sezaki, T. Niwa, M. Kojima, S. Inouye, Y. Yamada and T. Nida, J. Antibiot., 1982, 35, 533–535 Search PubMed.
- T. Haneishi, M. Nakajima, N. Serizawa, M. Inukai, Y. Takiguchi, M. Arai, S. Satoh, H. Kuwano and C. Tamura, J. Antibiot., 1983, 36, 1581–1584 CAS.
- G. Blanco, Arch. Microbiol., 2012, 194, 549–555 Search PubMed.
- J. D. Thompson, D. G. Higgins and T. J. Gibson, Nucleic Acids Res., 1994, 22, 4673–4680 CrossRef CAS.
- K. B. Nicholas, H. B. Nicholas and D. W. Deerfield, EMBNEW. NEWS, 1997, 4, 1–4 Search PubMed.
- L. Que, Nat. Struct. Biol., 2000, 7, 182–184 CrossRef CAS.
- H. Ishibashi, T. Sato and M. Ikeda, Synthesis, 2002, 695–713 CrossRef CAS.
- D. D. Tanner and P. M. Rahimi, J. Org. Chem., 1979, 44, 1674–1677 CrossRef CAS.
- M. Asai, K. Haibara, M. Muroi, K. Kintaka and T. Kishi, J. Antibiot., 1981, 34, 621–627 CAS.
- A. Imada, K. Kitano, K. Kintaka, M. Muroi and M. Asai, Nature, 1981, 289, 590–591 CrossRef CAS.
- R. B. Sykes, C. M. Cimarusti, D. P. Bonner, K. Bush, D. M. Floyd, N. H. Georgopapadakou, W. M. Koster, W. C. Liu, W. L. Parker, P. A. Principe, M. L. Rathnum, W. A. Slusarchyk, W. H. Trejo and J. S. Wells, Nature, 1981, 291, 489–491 CrossRef CAS.
- W. L. Parker, W. H. Koster, C. M. Cimarusti, D. M. Floyd, W. C. Liu and M. L. Rathnum, J. Antibiot., 1982, 35, 189–195 Search PubMed.
- J. S. Wells, W. H. Trejo, P. A. Principe, K. Bush, N. Georgopapadakou, D. P. Bonner and R. B. Sykes, J. Antibiot., 1982, 35, 184–188 Search PubMed.
- W. L. Parker and M. L. Rathnum, J. Antibiot., 1982, 35, 300–305 Search PubMed.
- J. S. Wells, W. H. Trejo, P. A. Principe, K. Bush, N. Georgopapadakou, D. P. Bonner and R. B. Sykes, J. Antibiot., 1982, 35, 295–299 Search PubMed.
- P. D. Singh, J. H. Johnson, P. C. Ward, J. S. Wells, W. H. Trejo and R. B. Sykes, J. Antibiot., 1983, 36, 1245–1251 CAS.
- R. Cooper, K. Bush, P. A. Principe, W. H. Trejo, J. S. Wells and R. B. Sykes, J. Antibiot., 1983, 36, 1252–1257 CAS.
- T. Kato, H. Hinoo, Y. Terui, J. Nishikawa, Y. Nakagawa, Y. Ikenishi and J. Shoji, J. Antibiot., 1987, 40, 139–144 CAS.
- T. Kato, H. Hinoo, J. Shoji, K. Matsumoto, T. Tanimoto, T. Hattori, K. Hirooka and E. Kondo, J. Antibiot., 1987, 40, 135–138 CAS.
- S. J. Box, A. G. Brown, M. L. Gilpin, M. N. Gwynn and S. R. Spear, J. Antibiot., 1988, 41, 7–12 CAS.
- M. N. Gwynn, S. J. Box, A. G. Brown and M. L. Gilpin, J. Antibiot., 1988, 41, 1–6 CAS.
- M. Hashimoto, T. Komori and T. Kamiya, J. Antibiot., 1976, 29, 890–901.
- M. Hashimoto, T. A. Komori and T. Kamiya, J. Am. Chem. Soc., 1976, 98, 3023–3025 CrossRef CAS.
- K. Watanabe, T. Okuda, K. Yokose, T. Furumai and H. B. Maruyama, J. Antibiot., 1983, 36, 321–324 Search PubMed.
- W. D. Celmer, L. H. Huang, M. T. Jefferson, H. Maeda, K. Inoue and R. Shibakawa, US Pat., 4212944, 1980.
- K. Tomita, H. Tsukiura and H. Kawaguchi, US Pat., 4320199, 1982.
- J. M. Davidsen and C. A. Townsend, Chem. Biol., 2012, 19, 297–306 Search PubMed.
Footnote |
| † Present address: Department of Chemistry, University of Paderborn, Warburger Str. 100, 33 098 Paderborn, Germany. |
| This journal is © The Royal Society of Chemistry 2013 |