Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature
Paul G.
Arnison
a,
Mervyn J.
Bibb
b,
Gabriele
Bierbaum
c,
Albert A.
Bowers
d,
Tim S.
Bugni
e,
Grzegorz
Bulaj
f,
Julio A.
Camarero
g,
Dominic J.
Campopiano
h,
Gregory L.
Challis
i,
Jon
Clardy
j,
Paul D.
Cotter
k,
David J.
Craik
l,
Michael
Dawson
m,
Elke
Dittmann
n,
Stefano
Donadio
o,
Pieter C.
Dorrestein
p,
Karl-Dieter
Entian
q,
Michael A.
Fischbach
r,
John S.
Garavelli
s,
Ulf
Göransson
t,
Christian W.
Gruber
u,
Daniel H.
Haft
v,
Thomas K.
Hemscheidt
w,
Christian
Hertweck
x,
Colin
Hill
y,
Alexander R.
Horswill
z,
Marcel
Jaspars
aa,
Wendy L.
Kelly
ab,
Judith P.
Klinman
ac,
Oscar P.
Kuipers
ad,
A. James
Link
ae,
Wen
Liu
af,
Mohamed A.
Marahiel
ag,
Douglas A.
Mitchell
ah,
Gert N.
Moll
ai,
Bradley S.
Moore
aj,
Rolf
Müller
ak,
Satish K.
Nair
al,
Ingolf F.
Nes
am,
Gillian E.
Norris
an,
Baldomero M.
Olivera
ao,
Hiroyasu
Onaka
ap,
Mark L.
Patchett
aq,
Joern
Piel
aras,
Martin J. T.
Reaney
at,
Sylvie
Rebuffat
au,
R. Paul
Ross
av,
Hans-Georg
Sahl
aw,
Eric W.
Schmidt
ax,
Michael E.
Selsted
ay,
Konstantin
Severinov
az,
Ben
Shen
ba,
Kaarina
Sivonen
bb,
Leif
Smith
bc,
Torsten
Stein
bd,
Roderich D.
Süssmuth
be,
John R.
Tagg
bf,
Gong-Li
Tang
bg,
Andrew W.
Truman
bh,
John C.
Vederas
bi,
Christopher T.
Walsh
bj,
Jonathan D.
Walton
bk,
Silke C.
Wenzel
bl,
Joanne M.
Willey
bm and
Wilfred A.
van der Donk
*bn
aPrairie Plant Systems Inc, Botanical Alternatives Inc, Suite 176, 8B-3110 8th Street E, Saskatoon, SK S7H 0W2, Canada
bDepartment of Molecular Microbiology, John Innes Centre, Norwich Research Park, Norwich NR4 7UH, UK
cInstitute of Medical Microbiology, Immunology and Parasitology, University of Bonn, Bonn, Germany
dDivision of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy University of North Carolina, Chapel Hill, NC 27599, USA
ePharmaceutical Sciences Division, University of Wisconsin-Madison, Wisconsin, 53705, USA
fDepartment of Medicinal Chemistry, University of Utah, Salt Lake City, UT, USA
gDepartment of Pharmacology and Pharmaceutical Sciences & Department of Chemistry, University of Southern California, 1985 Zonal Avenue, Los Angeles, CA 90033, USA
hSchool of Chemistry, EaStCHEM, University of Edinburgh, West Mains Road, Edinburgh, EH9 3JJ, UK
iDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, United Kingdom
jDepartment of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, 240 Longwood Avenue, Boston, MA 02115, USA
kTeagasc Food Research Centre, Moorepark, Fermoy, Co. and Alimentary Pharmabiotic Centre, Cork, Ireland
lInstitute for Molecular Bioscience, The University of Queensland, Brisbane, Queensland 4072, Australia
mNovacta Biosystems Ltd, UH Innovation Centre, College Lane, Hatfield, Herts AL10 9AB, United Kingdom
nUniversity of Potsdam, Institute for Biochemistry and Biology, Karl-Liebknecht-Str. 24/25, 14476 Potsdam-Golm, Germany
oNAICONS Scrl, Via G. Fantoli 16/15, 20138 Milan, Italy
pDepartment of Chemistry and Biochemistry, University of California at San Diego, La Jolla, California 92093, USA
qInstitute Molecular Biosciences, Excellence Cluster: Macromolecular Complexes, Goethe University Frankfurt, Max-von-Laue Str. 9, 60438 Frankfurt/M., Germany
rDepartment of Bioengineering and Therapeutic Sciences, California Institute for Quantitative Biosciences, University of California, San Francisco, California 94158, USA
sCenter for Bioinformatics & Computational Biology, Delaware Biotechnology Institute, University of Delaware, 15 Innovation Way, Suite 205, Newark, DE 19711-5449, USA
tDivision of Pharmacognosy, Dept Medicinal Chemistry, Uppsala University, BMC Box 574, SE 75123 Uppsala, Sweden
uMedical University of Vienna, Center for Physiology and Pharmacology, Schwarzspanierstr.17, A-1090 Vienna, Austria
vJ. Craig Venter Institute, 9704 Medical Center Drive, Rockville, Maryland 20850, USA
wDepartment of Chemistry, University of Hawai'i at Manoa, 2545 McCarthy Mall, Honolulu, HI 96822-2275, USA
xLeibniz Institute for Natural Product Research and Infection Biology (HKI), Beutenbergstr. 11a, 07745 Jena, Germany
yDepartment of Microbiology and Alimentary Pharmabiotic Centre, University College Cork, Ireland
zDepartment of Microbiology, Carver College of Medicine, University of Iowa, Iowa City, IA 52242, USA
aaDepartment of Chemistry, University of Aberdeen, Meston Walk, Aberdeen AB24 3UE, Scotland, UK
abSchool of Chemistry and Biochemistry and the Parker H. Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, Georgia 30332, USA
acDepartment of Chemistry, Department of Molecular and Cell Biology, University of California, Berkeley, CA 94720, USA
adMolecular Genetics, University of Groningen, Nijenborgh 7, 9747 AG, Groningen, The Netherlands
aeDepartment of Chemical and Biological Engineering, A207 Engineering Quadrangle, Princeton University, Princeton, New Jersey 08544, USA
afState Key Laboratory of Bioorganic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
agFachbereich Chemie/Biochemie der Philipps Universität Marburg, Hans-Meerwein-Str., D-35032 Marburg, Germany
ahDepartments of Chemistry and Microbiology and the Institute for Genomic Biology, University of Illinois at Urbana-Champaign, Urbana, Illinois 61801, USA
aiLanthiopharma, Nijenborgh 4, 9747 AG Groningen, The Netherlands
ajCenter for Marine Biotechnology and Biomedicine, Scripps Institution of Oceanography, University of California at San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA
akHelmholtz-Institute for Pharmaceutical Research Saarland (HIPS), Helmholtz Center for Infectious Research (HZI), Department Microbial Natural Products, Saarland University, Campus C2 3, 66123 Saarbrücken, Germany
alDepartment of Biochemistry, Center for Biophysics and Computational Biology, University of Illinois, 600 S. Mathews Ave, Urbana, IL 61801, USA
amLaboratory of Microbial Gene Technology Department of Chemistry, Biotechnology and Food Science, Norwegian University of Life Sciences, Biotechnology Building, N1432 Aas, Norway
anInstitute of Molecular BioSciences, Massey University, Private Bag 11222, Palmerston North, New Zealand
aoDepartment of Biology, University of Utah, Salt Lake City, Utah 84112, USA
apBiotechnology Research Center, Toyama Prefectural University, Toyama, Japan
aqInstitute of Molecular BioSciences, Massey University, Private Bag 11222, Palmerston North, New Zealand
arKekulé Institute of Organic Chemistry and Biochemistry, University of Bonn, Gerhard-Domagk-Str. 1, 53121 Bonn, Germany
asInstitute of Microbiology, Eidgenössische Technische Hochschule (ETH) Zurich, Wolfgang-Pauli-Str. 10, 8093 Zurich, Switzerland
atDepartment of Plant Sciences, University of Saskatchewan, Saskatoon, SK, Canada S7N 5A8
auLaboratory of Communication Molecules and Adaptation of Microorganisms, Muséum National d’Histoire Naturelle, Paris, France
avTeagasc Food Research Centre, Moorepark, Fermoy, Co., and Alimentary Pharmabiotic Centre, Cork, Ireland
awUniversity of Bonn, Institute for Medical Microbiology, Immunology and Parasitology, Pharmaceutical Microbiology Section, Meckenheimer Allee 168, D-53115 Bonn, Germany
axDepartment of Medicinal Chemistry and Department of Biology, University of Utah, Salt Lake City, Utah 84112, USA
ayDepartment of Pathology, Keck School of Medicine of USC, 2011 Zonal Ave., HMR 204, Los Angeles, California 90033, USA
azWaksman Institute, Department of Genetics, Rutgers University, Piscataway, New Jersey 08854, USA
baDepartments of Chemistry and Molecular Therapeutics, The Natural Products Library Initiative at the Scripps Research Institute, The Scripps Research Institute, 130 Scripps Way, #3A1, Jupiter, Florida 33458, USA
bbDepartment of Applied Chemistry and Microbiology, University of Helsinki, P.O. Box 56, Viikki Biocenter, 00014 Helsinki, Finland
bcDepartment of Biological Sciences, Texas A&M University, College Station, TX 77843, USA
bdInstitute of Molecular Biotechnology, Department of Chemistry, University of Aalen, 73430 Aalen, Germany
beTechnische Universität Berlin, Fakultät II—Institut für Chemie, Strasse des 17. Juni 124, 10623 Berlin, Germany
bfBLIS Technologies Limited Level 1, Centre for Innovation, 87 St David Street, PO Box 56, Dunedin 9054, New Zealand
bgState Key Laboratory of Bio-organic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China
bhDepartment of Chemistry, University of Cambridge, Cambridge, United Kingdom CB2 1EW
biDepartment of Chemistry, University of Alberta, Edmonton, Alberta, Canada T6G 2G2
bjDepartment of Biological Chemistry & Molecular Pharmacology, Harvard Medical School, 240 Longwood Avenue, Boston, Massachusetts 02115, USA
bkDepartment of Energy, Plant Research Laboratory, Michigan State University, East Lansing, Michigan 48824, USA
blHelmholtz-Institute for Pharmaceutical Research Saarland (HIPS), Helmholtz Center for Infectious Research (HZI), Department Microbial Natural Products, Saarland University, , Campus C2 3, 66123 Saarbrücken, Germany
bmDepartment of Biology and Science Education, Hofstra University-North Shore/LIJ, School of Medicine, Hempstead, New York 11549, USA
bnDepartment of Chemistry and Howard Hughes Medical Institute, University of Illinois at Urbana–Champaign, 600 S. Mathews Avenue, Urbana, Illinois 61801, USA
First published on 19th November 2012
Abstract
Covering: 1988 to 2012
This review presents recommended nomenclature for the biosynthesis of ribosomally synthesized and post-translationally modified peptides (RiPPs), a rapidly growing class of natural products. The current knowledge regarding the biosynthesis of the >20 distinct compound classes is also reviewed, and commonalities are discussed.
 Wilfred A. van der Donk | Wilfred van der Donk received a M.S. from Leiden University, the Netherlands, with Prof. Jan Reedijk, and a Ph.D. in organic chemistry with Kevin Burgess at Rice University. He was a Jane Coffin Childs postdoctoral fellow with JoAnne Stubbe at MIT, and joined the faculty at the University of Illinois at Urbana-Champaign in 1997, where he holds the Richard E. Heckert Chair in Chemistry. Since 2008, he is an Investigator of the Howard Hughes Medical Institute. His laboratory aims to understand the molecular mechanisms of enzyme catalysis. Of particular interests are enzymatic reactions in the biosynthesis of natural products, including several classes of RiPPs. |
1 Introduction
Natural products have played key roles over the past century in advancing our understanding of biology and in the development of medicine. Research in the 20th century identified many classes of natural products with four groups being particularly prevalent: terpenoids, alkaloids, polyketides, and non-ribosomal peptides. The genome sequencing efforts of the first decade of the 21st century have revealed that another major class is formed by ribosomally synthesized and post-translationally modified peptides. These molecules are produced in all three domains of life, their biosynthetic genes are ubiquitous in the currently sequenced genomes1–8 and transcriptomes,9,10 and their structural diversity is vast.11 The extensive post-translational/co-translational modifications endow these peptides with structures not directly accessible for natural ribosomal peptides, typically restricting conformational flexibility to allow better target recognition, to increase metabolic and chemical stability, and to augment chemical functionality.Because the common features of their biosynthetic pathways have only recently been recognized, at present the nomenclature used in different communities investigating subgroups of natural products of ribosomal origin is non-uniform, confusing, and in some cases even contradictory. In order to define a uniform nomenclature, the authors of this review engaged in a discussion over the summer and fall of 2011, and the consensus recommendations of these discussions are presented herein. In addition, this review provides an overview of the structures of and the biosynthetic processes leading to this large group of natural products. Select examples are covered from bacteria, fungi, plants, and cone snails. Post-translational in the context of this review is defined as any peptide chain modification occurring after the initiation of translation.
Historically, ribosomally synthesized and post-translationally modified peptides have been subdivided based on either the producing organisms (e.g. microcins produced by Gram-negative bacteria)12 or their biological activities (e.g. bacteriocins).13 Unlike the designations of the other four major classes of natural products listed above, an overarching designation for these peptide natural products based on structural and biosynthetic commonality is currently lacking. We propose that the biosynthetic pathway to these compounds be referred to as Post-Ribosomal Peptide Synthesis (PRPS), in line with the currently used designation of Non-Ribosomal Peptide Synthetase (NRPS) enzymes that catalyze modular assembly line biosynthesis of peptide natural products.14,15 Furthermore, we propose to designate the resulting ![[r with combining low line]](https://www.rsc.org/images/entities/char_0072_0332.gif)
![[i with combining low line]](https://www.rsc.org/images/entities/char_0069_0332.gif) bosomally-synthesized and
bosomally-synthesized and ![[p with combining low line]](https://www.rsc.org/images/entities/char_0070_0332.gif) ost-translationally-modified eptides as RiPPs, irrespective of their biological functions. The name “Post-Ribosomal Peptides” was also considered as a logical extension of NRPs, but because this designation would not capture the critical post-translational modifications, the name RiPPs was ultimately preferred. Similarly, the name “Ribosomal Natural Products” was considered but the resulting acronym, RNPs, was deemed too close to NRP, the acronym for non-ribosomal peptides. Finally “Ribosomal Peptides” was discussed, but since confusion could arise with peptides that make up the ribosome, RiPPs was again preferred. A size limit of 10 kDa is imposed on RiPPs to exclude post-translationally modified proteins. We note that a small number of post-translationally modified natural metabolites are not well described by this nomenclature because the final products lack any peptide bonds, such as the cofactor pyrroloquinoline quinone (PQQ), and the thyroid hormones (see section 20).
ost-translationally-modified eptides as RiPPs, irrespective of their biological functions. The name “Post-Ribosomal Peptides” was also considered as a logical extension of NRPs, but because this designation would not capture the critical post-translational modifications, the name RiPPs was ultimately preferred. Similarly, the name “Ribosomal Natural Products” was considered but the resulting acronym, RNPs, was deemed too close to NRP, the acronym for non-ribosomal peptides. Finally “Ribosomal Peptides” was discussed, but since confusion could arise with peptides that make up the ribosome, RiPPs was again preferred. A size limit of 10 kDa is imposed on RiPPs to exclude post-translationally modified proteins. We note that a small number of post-translationally modified natural metabolites are not well described by this nomenclature because the final products lack any peptide bonds, such as the cofactor pyrroloquinoline quinone (PQQ), and the thyroid hormones (see section 20).
1.1 Common features of RiPP biosynthesis
Nearly all compounds produced by PRPS are initially synthesized as a longer precursor peptide, typically ∼20–110 residues in length, encoded by a structural gene. In the past, the various segments of this precursor peptide have been given different, sometimes conflicting names for different RiPP subclasses (Table 1). We propose here a uniform naming scheme in which the segment of the precursor peptide that will be transformed into the natural product is called the core peptide or core region (Fig. 1). A distinction can be made with respect to the unmodified core peptide (UCP) in the precursor peptide and the modified core peptide (MCP) after the post-translational modifications. In most RiPPs, a leader peptide or leader sequence is appended to the N-terminus of the core peptide that is usually important for recognition by many of the post-translational modification enzymes and for export.16 In some more rare examples such as the bottromycins (section 8),17–20 a leader peptide is not attached at the N-terminus, but rather at the C-terminus of the core peptide and has been termed a “follower” peptide. For eukaryotic peptides such as the cyclotides and conopeptides discussed in this review, a signal sequence is often found N-terminal to the leader peptide that directs the peptide to the specific cellular compartments where the post-translational modifications will take place. Finally, some peptides have C-terminal recognition sequences that are important for excision and cyclization.21–23 The unmodified precursor peptide is generally designated “A” (encoded by the xxxA gene), but for some classes different designations have been used historically (see discussion of individual RiPP families). The![[m with combining low line]](https://www.rsc.org/images/entities/char_006d_0332.gif)
![[o with combining low line]](https://www.rsc.org/images/entities/char_006f_0332.gif)
![[d with combining low line]](https://www.rsc.org/images/entities/char_0064_0332.gif)
![[f with combining low line]](https://www.rsc.org/images/entities/char_0066_0332.gif)
![[e with combining low line]](https://www.rsc.org/images/entities/char_0065_0332.gif) precursor peptide prior to proteolytic removal of the leader peptide can be abbreviated as XxxA (e.g. mLanA; Fig. 1).
precursor peptide prior to proteolytic removal of the leader peptide can be abbreviated as XxxA (e.g. mLanA; Fig. 1).
| Nomenclature proposed herein | Previous designations | ||
|---|---|---|---|
| Precursor peptide | Prepeptide (lantibiotics, microviridins) | Prepropeptide (lantibiotics, cyclic bacteriocins) | Structural peptide (translation product of the structural gene; lantibiotics, thiopeptides) |
| Leader peptide | Propeptide (microcins, conopeptides, cyclotides) | Pro-region (conopeptides) | Intervening region (conopeptides) |
| Core peptide | Propeptide (lantibiotics, mycotoxins, cytolysins, cyclic bacteriocins) | Structural peptide (lantibiotics, thiopeptides, cyanobactins) | Toxin region (conopeptides) |
| Recognition sequences | NTR, N-terminal repeats (cyclotides) | ||
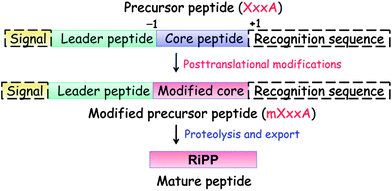 | ||
| Fig. 1 General biosynthetic pathway for RiPPs. The precursor peptide contains a core region that is transformed into the mature product. Many of the post-translational modifications are guided by leader peptide and the recognition sequences as discussed in this review for subclasses of RiPPs. Products of eukaryotic origin also often contain an N-terminal signal peptide that directs the peptide to specialized compartments for modification and secretion. C-terminal recognition sequences are sometimes also present for peptide cyclization (e.g. see sections on cyanobactins, amatoxins, cyclotides, and orbitides). In the case of the bottromycins, a leader-like peptide is appended at the C-terminus of the core peptide.17–20 | ||
It is noteworthy that the leader peptide-guided strategy for the biosynthesis of RiPPs results in highly evolvable pathways because many of the post-translational processing enzymes recognize the leader peptide and are highly permissive with respect to mutations in the core peptide. Indeed, the core regions are naturally hypervariable for subsets of the RiPP classes discussed in this review,3,22,24–26 and engineering studies have further demonstrated the plasticity of the biosynthetic enzymes.27–44 The relatively small number of enzymes involved in the maturation pathways also facilitates natural evolution, and the multiplicity of pathways towards the same types of chemical structures illustrates the convergent evolution of efficient, ribosome-based biosynthetic strategies. Collectively, these features highlight the potential evolutionary advantage of accessing high chemical diversity at low genetic cost. We note that these very same attributes also prove advantageous with respect to genome mining strategies as heterologous production of RiPPs is aided by short pathways and is not limited by supply of precursors.
The explosion in sequence information has also revealed that many biosynthetic pathways utilize common enzymes for a subset of the post-translational modifications. For instance, the Ser/Thr dehydratases involved in the biosynthesis of ![[l with combining low line]](https://www.rsc.org/images/entities/char_006c_0332.gif)
![[a with combining low line]](https://www.rsc.org/images/entities/char_0061_0332.gif)
![[n with combining low line]](https://www.rsc.org/images/entities/char_006e_0332.gif)
![[t with combining low line]](https://www.rsc.org/images/entities/char_0074_0332.gif)
![[h with combining low line]](https://www.rsc.org/images/entities/char_0068_0332.gif) onine-containing
onine-containing ![[s with combining low line]](https://www.rsc.org/images/entities/char_0073_0332.gif) (lanthipeptides) are also used in the biosynthetic pathways to the proteusins,45 thiopeptides, and some linear azole-containing peptides (LAPs),44,46–49 while some of the enzymes involved in oxazol(in)e and thiazol(in)e formation in thiopeptides46–50 are also used for the biosynthesis of LAPs50–52 and bottromycins17–20 and are also often found in cyanobactin biosynthetic gene clusters.21 Similarly, genes encoding radical-SAM dependent methyltransferases are found in the biosynthetic gene clusters of thiopeptides,46 proteusins,45 and bottromycins.17–20 Thus, acquisition of new post-translational modification enzymes appears to drive evolution to new structures. On the other hand, some post-translational modifications appear to have evolved convergently, such as the different ways to achieve head-to-tail (N-to-C) cyclization in the cyanobactins,53 amatoxins,54 circular bacteriocins,55 and cyclotides,56 and the different ways in which thioether crosslinks are formed in various classes of lanthipeptides.57 Post-translational modifications involving Cys residues occur especially frequently in RiPPs. Sulfur chemistry converts the thiols of cysteines to disulfides (cyclotides, conopeptides, lanthipeptides, cyanobactins, lasso peptides, sactipeptides, and glycocins), thioethers (lanthipeptides, sactipeptides, phalloidins, some thiopeptides), thiazol(in)es (thiopeptides, LAPs, cyanobactins, bottromycins), and sulfoxides (lanthipeptides, amatoxins). Additional common features are macrocyclization to increase metabolic stability and decrease conformational flexibility, and modifications to the N- and C-termini to limit the susceptibility to degradation by exoproteases.
(lanthipeptides) are also used in the biosynthetic pathways to the proteusins,45 thiopeptides, and some linear azole-containing peptides (LAPs),44,46–49 while some of the enzymes involved in oxazol(in)e and thiazol(in)e formation in thiopeptides46–50 are also used for the biosynthesis of LAPs50–52 and bottromycins17–20 and are also often found in cyanobactin biosynthetic gene clusters.21 Similarly, genes encoding radical-SAM dependent methyltransferases are found in the biosynthetic gene clusters of thiopeptides,46 proteusins,45 and bottromycins.17–20 Thus, acquisition of new post-translational modification enzymes appears to drive evolution to new structures. On the other hand, some post-translational modifications appear to have evolved convergently, such as the different ways to achieve head-to-tail (N-to-C) cyclization in the cyanobactins,53 amatoxins,54 circular bacteriocins,55 and cyclotides,56 and the different ways in which thioether crosslinks are formed in various classes of lanthipeptides.57 Post-translational modifications involving Cys residues occur especially frequently in RiPPs. Sulfur chemistry converts the thiols of cysteines to disulfides (cyclotides, conopeptides, lanthipeptides, cyanobactins, lasso peptides, sactipeptides, and glycocins), thioethers (lanthipeptides, sactipeptides, phalloidins, some thiopeptides), thiazol(in)es (thiopeptides, LAPs, cyanobactins, bottromycins), and sulfoxides (lanthipeptides, amatoxins). Additional common features are macrocyclization to increase metabolic stability and decrease conformational flexibility, and modifications to the N- and C-termini to limit the susceptibility to degradation by exoproteases.
Although the details of recognition of leader peptides by the biosynthetic enzymes are still mostly unknown, many leader peptides have a propensity to form α-helices,58–64 either in solution or when bound to the biosynthetic proteins.65,66 The leader peptides are thought to play multiple roles in post-translational modification, export, and immunity. Furthermore, several pieces of evidence suggest that different biosynthetic enzymes in a pathway recognize different segments of the leader peptides.59,64,67,68 Leader peptide removal can take place in one proteolytic step or in multiple proteolytic steps. In regards to the numbering of the residues of the leader peptide, it is recommended to count backwards from the final cleavage site and add a minus sign before the number, e.g. the last residue that is not incorporated in the final RiPP is numbered −1 and counting then commences −2, −3 etc towards the N-terminus (Fig. 1). Similarly, C-terminal recognition sequences/follower peptides can be numbered with a plus sign from the site of final cleavage (e.g. the N-terminal residue of the C-terminal recognition sequence would be numbered +1, Fig. 1) counting up from this site towards the C-terminus. Counting schemes in the case of multiple core peptides and recognition sequences (e.g. in the cyanobactins, cyclotides, and orbitides) are case dependent.
The sections below briefly discuss the salient features of these various compound classes including their defining structural motifs, common biosynthetic pathways, subdivisions into subclasses, and recommendations regarding nomenclature. This review covers most bacterial RiPPs that have been identified as well as select examples from fungi (amatoxins and phallotoxins), plants (cyclotides and orbitides), and metazoans (conopeptides from cone snails). Not covered, but often made by a similar biosynthetic logic if they are post-translationally modified, are defensins69–71 and the venom peptides from insects and reptiles.72–75
2 Lanthipeptides
2.1 Biosynthesis
First reported in 1928,76 nisin is one of the longest known RiPPs, but its structure was not elucidated until 1971 (Fig. 2A).77 It contains the meso-lanthionine and 3-methyllanthionine residues that define the lanthipeptide (for lanthionine-containing peptides) class of molecules. Lanthipeptides that have antimicrobial activity are called lantibiotics.78 Lanthionine (Lan) consists of two alanine residues crosslinked via a thioether linkage that connects their β-carbons; 3-methyllanthionine (MeLan) contains one additional methyl group (Fig. 2B). The ribosomal origin of lanthionine-containing peptides was first proposed in 197079 and experimentally verified in 1988 when the biosynthetic gene cluster for epidermin was sequenced.78 Shortly thereafter, the precursor genes for nisin,80,81 subtilin,82 and Pep583 were identified. The biosynthetic genes have been designated the generic locus symbol lan, with a more specific genotypic designation for each lanthipeptide member (e.g., nis for nisin,81gar for actagardine,84mrs for mersacidin,85cin for cinnamycin86). The Lan and MeLan residues are introduced in a two-step post-translational modification process. In the first step, Ser and Thr residues in the precursor peptide are dehydrated to dehydroalanine (Dha) and dehydrobutyrine (Dhb) residues, respectively, usually via a phosphorylated intermediate (Fig. 2B).87–89 The thioether crosslinks are formed subsequently via a Michael-type addition by Cys residues onto the dehydro amino acids.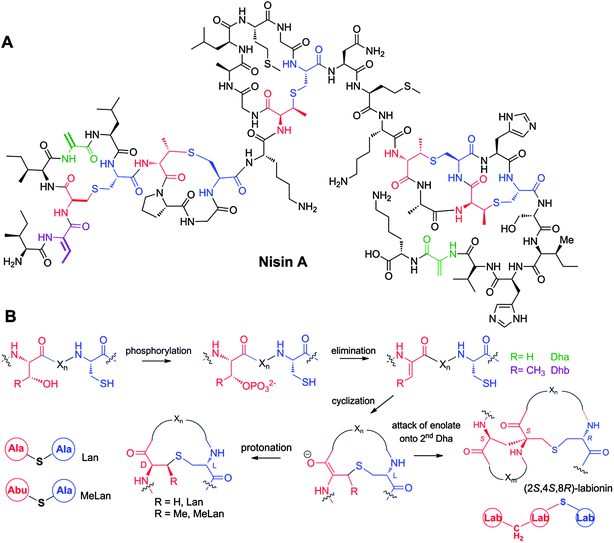 | ||
| Fig. 2 A. Structure of nisin A. Dha residues are shown in green, Dhb residues in purple. Segments of Lan and MeLan originating from Ser/Thr are depicted in red and segments of (Me)Lan originating from Cys in blue. B. Biosynthetic pathway to (Me)Lan and labionin formation. Ser and Thr residues are first phosphorylated and the phosphate is subsequently eliminated to generate Dha and Dhb residues, respectively. Phosphorylation prior to elimination has been experimentally confirmed for class II–IV lanthipeptides and is postulated for class I lanthipeptides. Subsequent intramolecular conjugate addition by the side chain of Cys generates an enolate. Protonation of the enolate produces the (Me)Lan structures whereas attack of the enolate onto another Dha generates the labionin crosslinks. | ||
At present lanthipeptides are classified into four different classes depending on the biosynthetic enzymes that install the Lan and MeLan motifs (Fig. 3).57 For class I lanthipeptides, the dehydration is carried out by a dedicated dehydratase generically called LanB,90–93 with a more specific designation for each lanthipeptide (e.g. NisB for nisin). Cyclization of class I lanthipeptides is catalyzed by a LanC cyclase.94–96 Several pieces of evidence suggest these proteins form a multienzyme complex.97 For class II,98,99 III,88 and IV89 lanthipeptides, dehydration and cyclization is carried out by bifunctional lanthionine synthetases. The class II LanM lanthionine synthetases have N-terminal dehydration domains that do not display sequence homology with other enzymes in the databases. However, the C-terminal cyclization domains have homology with the LanC cyclases of class I (Fig. 3). For both class III and IV lanthipeptides, dehydration is carried out by successive actions of a central kinase domain and an N-terminal phosphoSer/phosphoThr lyase domain,89,100 but the class III and IV synthetases differ in their C-terminal cyclization domains (Fig. 3).101 Although these domains show sequence homology to each other as well as to the LanC proteins, three metal binding residues95,102 that are fully conserved in class I, II, and IV cyclases are absent in the class III cyclization domains.101 A subset of class III enzymes generates an additional carbon–carbon crosslink, putatively by attack of the initially generated enolate onto a second dehydroalanine (Fig. 2B). The structure thus formed is called a labionin (Lab), first detected in the labyrinthopeptins.103
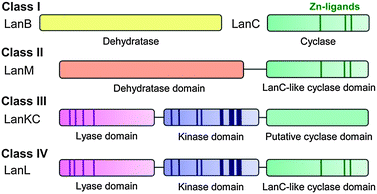 | ||
| Fig. 3 Four different classes of lanthipeptides are defined by the four different types of biosynthetic enzymes that install the characteristic thioether crosslinks. The dark areas show conserved regions that are important for catalytic activity. The cyclase domain of class III enzymes104 has homology with the other cyclase domains/enzymes but lacks the three zinc ligands. | ||
Lanthipeptide biosynthetic enzymes have demonstrated low substrate specificity, allowing substitutions of those residues in the core peptide that are not post-translationally modified with both proteinogenic27–30,33–35,105–109 and non-proteinogenic amino acids.34,110,111 The enzymes that generate the characteristic crosslinks are dependent on the presence of the leader peptide,59,63,78,93,95,99,112–116 but several of the enzymes that install other post-translational modifications do not require the leader peptide.117–119 Furthermore, non-lanthipeptide sequences attached to LanA leader peptides have been modified by the lanthipeptide biosynthetic machinery.92,120–123 For the class II lantibiotic lacticin 481116,124 and the class I lantibiotic nisin,125 it has been demonstrated that the leader peptide does not need to be attached to the core peptide. The results of these studies better fit a model in which leader peptide binding results in a shift in the equilibrium between an inactive and active form of the synthetase towards the latter, rather than models in which the leader peptide pulls the core peptide through the active site or in which leader peptide binding induces a conformational change.59,116
2.2 Structure and biological activities of lanthipeptides
Recent years have revealed that lanthionine-containing peptides can have functions beyond antimicrobial activities,101,104,126 and therefore the name lantipeptide was initially introduced for all lanthionine and methyllanthionine containing peptides,89 with lantibiotics forming a large subgroup. As described above, we revise the term lantipeptide here to lanthipeptide as a more faithful representation for lanthionine-containing peptides regardless of their biological activities.Nisin (class I) is the best studied lantibiotic and has been used in the food industry to combat food-borne pathogens for more than 40 years.127 It binds to lipid II, an essential intermediate in peptidoglycan biosynthesis, resulting in inhibition of cell wall biosynthesis and pore formation.128,129 Other notable lanthipeptides include the class II lantibiotic actagardine, a semi-synthetic derivative of which is in development against Clostridium difficile,84 the class II lantibiotic duramycin that binds phosphatidylethanolamine and is being evaluated for treatment of cystic fibrosis,130 and labyrinthopeptin, a class III compound with antiallodynic activity (Fig. 4).103 An interesting group of class II lantibiotics are the two-component peptides such as lacticin 3147 and haloduracin.131–134 These peptides synergistically kill bacteria with the α-peptide binding to lipid II.135,136 The β-peptide is then believed to bind to the complex formed between the α-peptide and lipid II and induce pore formation.137
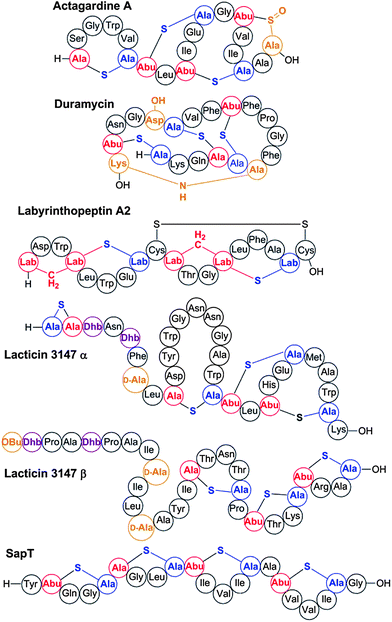 | ||
| Fig. 4 Some representative lanthipeptides. The same shorthand notation is used as in Fig. 2. Segments of crosslinks originating from Cys are in blue, segments originating from Ser/Thr are in red. Additional post-translational modifications are shown in orange.138 OBu, 2-oxobutyric acid; Asp-OH, (3R)-hydroxy-aspartate; Lys-NH-Ala, lysinoalanine. | ||
The first lanthipeptides shown to have functions other than antimicrobial agents were the morphogenetic peptides SapB and SapT from streptomycetes.101,126 These peptides are believed to function as biosurfactants during the formation of aerial hyphae.139 Lanthipeptides are defined by the presence of the characteristic (Me)Lan residues, but as many as 15 other post-translational modifications have been documented,57,128,138 with additional modifications likely to be uncovered in the future. Unlike (Me)Lan, Dha, and Dhb formation, these additional modifications appear not to rely on the presence of a leader peptide for the examples investigated thus far.117–119,140
Genome database mining has illustrated that the biosynthetic genes for lanthipeptides are distributed much more widely than the Firmicutes and Actinobacteria to which biosynthesis of these compounds was believed to be restricted. Their genes are now also found in certain bacteroidetes, chlamydiae, proteobacteria, and cyanobacteria.26,89,141,142 In all, over 90 lanthipeptides are known with hundreds more identified in genomes.2,3,26,141–143
2.3 Specific recommendations for the lanthipeptide family
The name lanthipeptides describes all lanthionine- and methyllanthionine-containing peptides made by PRPS. Lantibiotics form a large subgroup with antimicrobial activities. The gene designations for the various post-translational modification enzymes involved in lanthipeptide biosynthesis are well established and we recommend continued use of the generic name LanA for the precursor peptide, LanBC for the dehydratases and cyclases of class I lanthipeptides, and LanM and LanL for the bifunctional enzymes involved in the biosynthesis of class II and IV compounds, respectively. The class III bifunctional enzymes should be called LanKC irrespective of whether they result in formation of labionines or lanthionines because at present the final products cannot be predicted from the sequences of the synthetases.104,144 The suffix -peptin is recommended for naming new members of class III peptides.104,144Further recommendations are the continued use of LanP for Ser proteases that remove part of or the entire leader peptide, LanT for transporters that secrete the lanthipeptides (with or without an N-terminal Cys protease domain), LanEFG for the transporters that are involved in self-resistance, LanI for additional immunity proteins, and LanKR for two-component response regulators. With respect to the enzymes that install the less common PTMs, LanD should be used for the enzymes that oxidatively decarboxylate C-terminal Cys residues,145 LanO for various oxidation enzymes,84,140,146 LanX for hydroxylases,118,147 and LanJ for enzymes that convert Dha to D-Ala.148 This recommendation leaves only a small series of letters remaining (lanHNQSWYZ), some of which have already been used to name individual, non-common genes in lanthipeptide gene clusters.149 Hence, we recommend the use of a five-letter designation, such as that used for lanKC, for genes that may be uncovered in future studies and that have no sequence homology with currently known lanthipeptide biosynthetic genes.
3 Linaridins
Linaridins are a recently discovered family of RiPPs that share the presence of thioether crosslinks with the lanthipeptide family but that are generated by a different biosynthetic pathway. Cypemycin is the founding member of the family and it was considered a lanthipeptide because of the presence of a C-terminal aminovinyl cysteine (Fig. 5).150 These same structures are found in several lantibiotics such as epidermin and mersacidin and are believed to be generated by Michael-type addition of an oxidatively decarboxylated C-terminal Cys onto a Dha,151 with the latter formed by dehydration of Ser. However, when the biosynthetic gene cluster of cypemycin was identified, it became clear that none of the four types of lanthipeptide dehydratases were present in the cluster.152 Furthermore, the sequence of the core peptide illustrates that the AviCys structure is formed from two Cys residues. Based on these differences from the biosynthetic route to AviCys in lanthipeptides, cypemycin and related peptides found in the genome databases6,152,153 have been classified as a separate group, the linaridins. If future research identifies linaridins containing Lan and/or MeLan, it may be that linaridins are better classified as another subclass of the lanthipeptides, but as long as that is not the case, they are grouped separately.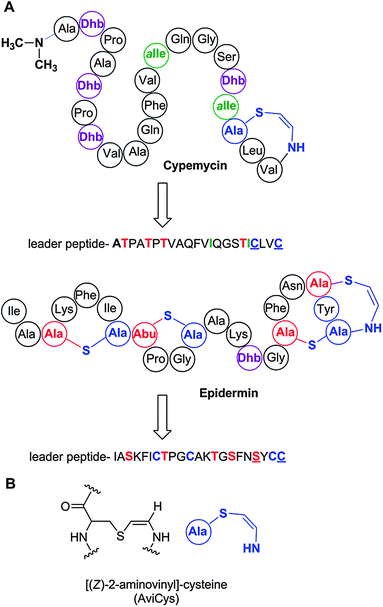 | ||
| Fig. 5 A. Structure of cypemycin, a representative member of the linaridins. The linear sequence of the precursor peptide is shown below the retrosynthetic arrow to illustrate the biosynthetic origin of each PTM: allo-Ile from Ile, Dhb from Thr, and AviCys from two Cys residues (underlined). For comparison, the structure of the lanthipeptide epidermin, which also contains an AviCys structure, is shown but its AviCys is formed from a Ser and Cys residue (underlined). B. Chemical structure of AviCys as well as the shorthand notation used in panel A; the absolute configuration of the AviCys and allo-Ile in cypemycin is not known. | ||
4 Proteusins
Polytheonamides (Fig. 6) were reported in 1994 as extremely cytotoxic constituents of a Japanese Theonella swinhoei sponge,154 which harbors a large diversity of symbiotic bacteria.155 These initial studies and a structural revision in 2005 by the same group156 established the compounds as highly complex 48-mer peptides containing, in addition to an unprecedented N-acyl moiety, an unusually large number of nonproteinogenic residues, including many tert-leucine and other C-methylated amino acids. One of the most interesting features is the presence of multiple D-configured units that are localized in near perfect alternation with L-amino acids, reminiscent of the nonribosomally produced linear gramicidins.157 Like these compounds, polytheonamides form membrane channels, which is at least in part the basis of their cytotoxicity.158 Channel formation occurs by adoption of a β-helical secondary structure and directional insertion into the membrane aided by the lipophilic N-acyl unit.158,159 However, unlike the bimolecular gramicidin pores, the much larger polytheonamides are able to span the entire membrane and thus form minimalistic ion channels as single molecules. Because of the highly modified polytheonamide structures it was generally assumed that they are nonribosomal peptides and, as such, the largest known members of this family. Recently, however, metagenomic studies were conducted on the sponge involving screening of a ∼1 million clone DNA library, which revealed that polytheonamides are biosynthesized via a ribosomal pathway.45 Such an origin is notable, since it implies the existence of 48 postribosomal modification steps excluding removal of the leader region, making polytheonamides the most extensively modified RiPPs known to date. Particularly noteworthy is the regiospecific epimerization of 18 structurally diverse L-amino acids, which suggests that there is no significant biochemical limitation to the stereo- and regiospecific biosynthesis of configurationally mixed ribosomal products. These structural peculiarities as well as the unusually large leader region of the precursor (see below) classified polytheonamides as members of a new RiPP family, termed proteusins. The metagenomic study also showed that an as-yet uncultivated bacterial symbiont of the sponge is the actual producer, highlighting the potential of environmental bacteria as sources of structurally novel RiPPs.45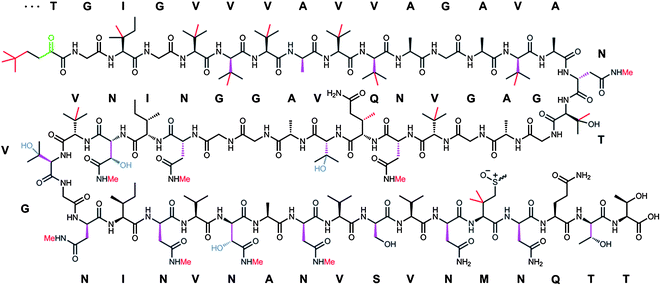 | ||
| Fig. 6 Structure of polytheonamide A and B and sequence of the precursor core region. The two congeners differ by the configuration of the sulfoxide moiety. Sulfur oxidation is an artifact occurring during isolation. Color code for posttranslations modifications: purple, epimerization; green, dehydration; red, methylation; blue, hydroxylation. | ||
4.1 Biosynthesis and structural classifications of proteusins
Biosynthesis of polytheonamides involves a precursor protein (PoyA) with a large leader region that exhibits similarity to nitrile hydratase-like enzymes. By coexpressing the precursor gene with a variety of genes for posttranslational modification, the function of several modifying enzymes was established.45 Remarkably, in a first step a single epimerase, PoyD, generates most, and possibly all, D-residues. The enzyme is homologous to members of the radical S-adenosyl methionine superfamily160 and likely acts by abstracting an α-hydrogen to form a stabilized amino acyl radical. Hydrogen donation from the backside would then result in epimerization. The source of the second hydrogen is currently unknown. Epimerization occurs unidirectional, which is in contrast to the equilibrium-forming, as-yet uncharacterized epimerases associated with, e.g., several modified peptides from animals.161 Only few homologues of PoyD are currently listed in GenBank. All contain a characteristic N-terminal domain-like region of unknown function and are associated with nitrile hydratase-like precursors. The second step in polytheonamide biosynthesis is dehydration of a Thr residue that seems to be the biosynthetic source of the unusual N-acyl residue (Fig. 7A).45 This reaction is performed by PoyF, an enzyme resembling the N-terminal dehydratase domain of LanM-type class II bifunctional lanthionine synthetases (see section 2). Further conversion to the N-acyl unit likely proceeds via net t-butylation catalyzed by one or more class B162 radical-SAM methyltransferases (PoyB and/or C are candidates), which would add a total of four methyl groups to the methyl group originating from Thr (Fig. 7A). Hydrolytic removal of the leader region would then generate an enamine moiety that would spontaneously form the α-keto unit found in polytheonamides, similar to the ketone generation at the N-terminus of Pep5-like lanthipeptides.83,140,146,163 Regiospecific generation of the 8 N-methylated Asn residues was shown to be catalyzed by a single N-methyltransferase, PoyE.45 A general hallmark of the pathway is the extremely streamlined gene cluster that encodes several highly iterative enzymes (Fig. 7B). For all 48 modifications, only 6 enzyme candidates encoded in a 14 kb cluster were identified. However, it has to be tested whether the remaining as-yet uncharacterized modifications (4 hydroxylations and 17 C-methylations) are indeed catalyzed by enzymes belonging to the cluster or whether additional genes might be located elsewhere on the symbiont genome. If the cluster is sufficient for biosynthesis, the putative Fe(II)/α-ketoglutarate-dependent oxygenase PoyI and the two radical-SAM methyltransferase-like enzymes PoyC and PoyD would be candidates for these 23 transformations.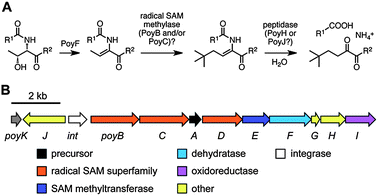 | ||
| Fig. 7 A. Proposed biosynthesis of the N-acyl unit of polytheonamides. The dehydration step was experimentally verified. B. Polytheonamide biosynthetic gene cluster. | ||
Prior to the discovery of the polytheonamide gene cluster, the existence of a new RiPP family with unusually long nitrile hydratase-like leader peptides (NHLP) sequences was postulated based on bioinformatic analyses.3 These sequences are found in genome sequences of diverse bacteria, although the identity of the peptides was unknown. This prediction demonstrates the power of in silico genome mining methods to discover new RiPP classes. In addition to NHLP homologues, a second precursor type that contains a leader region resembling Nif11 nitrogen-fixing proteins (N11P) was identified in the bioinformatic study. For both groups of leader peptide, one or multiple radical-SAM genes (predicted to encode amongst others, epimerases, and C-methyltransferases) are frequently present in the biosynthetic gene clusters.5 Thus, although polytheonamides are currently the only known characterized proteusins, a unifying feature of this family seems to be NHLP or N11P sequences and modifications involving radical-SAM enzymes. Interestingly, the N11P-like leader peptides are also used to produce lanthipeptides in marine cyanobacteria.3,26 Radical-SAM proteins are also used for the biosynthesis of two other RiPP families, bottromycins and sactipeptides, and bioinformatic analyses suggest that these radical-SAM mediated transformations may be as common as the ubiquitous (cyclo)dehydration reactions discussed in sections 5–8.5
4.2 Specific recommendations for the proteusin family
One common feature at the genetic level is the conspicuous and characteristic nitrile hydratase- or Nif11-like leader peptide of the precursor gene. Similar to other RiPPs it is recommended to use the descriptor “A“ for the precursor peptide. Regarding posttranslational modification, it is currently premature to propose unifying recommendations, since polytheonamides are the only members known to be associated with this family, and there is generally little overlap with the biosynthetic enzymes of other RiPP types.5 Linear azol(in)e-containing peptides (LAPs)
5.1 Historical perspective
The earliest work on the linear azol(in)e-containing peptides (LAPs) dates back to at least 1901 with the observation that certain pathogenic isolates of streptococci secrete a factor responsible for what later became known as the β-hemolytic phenotype.164,165 Despite a flurry of work carried out on the hemolytic streptococci in the opening decade of the 20th century, studies in the 1930s provided the first significant insight into the β-hemolytic factor, streptolysin S (SLS).166,167 Because of the problematic physicochemical properties of SLS, it was not until 2000 that researchers realized the defining chemical attributes of this RiPP subfamily.168 This realization was enabled by a rapid advancement in the understanding of the biosynthesis of another LAP member, microcin B17, the first RiPP to have its biosynthesis reconstituted in vitro (vide infra).50,51 To this day, the exact chemical structure of SLS remains elusive. However, the classification of SLS as a LAP and an integral component of the pathogenic mechanism of Streptococcus pyogenes was confirmed by a series of recent genetic and biochemical studies.7,37,169 Bioinformatic and biochemical investigations have shown that a number of pathogens from the Firmicutes phylum contain SLS-like gene clusters, including lineage I strains of Listeria monocytogenes and Clostridium botulinum.170–1725.2 LAP biosynthesis
LAPs are decorated with various combinations of thiazole and (methyl)oxazole heterocycles, which can sometimes also be found in their corresponding 2-electron reduced azoline state (Fig. 8).52 As demonstrated for microcin B17 in 1996, the azol(in)e heterocycles derive from cysteine, serine, and threonine residues of a ribosomally synthesized precursor peptide.51 The critical components in LAP biosynthesis are the inactive precursor peptide (referred to as “A”) and the heterotrimeric synthetase complex comprised of a dehydrogenase (B) and cyclodehydratase (C/D). In many LAP biosynthetic gene clusters, the C–D proteins are fused in a single polypeptide, highlighting the importance of their enzymatic collaboration.52,173 The first step in LAP biosynthesis is substrate recognition, driven through an interaction of the N-terminal leader peptide of the precursor (Fig. 8 and 9), which remains unmodified.37,58 After formation of the enzyme-substrate complex, ATP-dependent cyclodehydration occurs with the expulsion of water from the preceding amide carbonyl giving rise to an azoline heterocycle.173 ATP is used in this step to phosphorylate the amide oxygen to facilitate the elimination of a water equivalent from the substrate.173 In a second step, a flavin mononucleotide (FMN)-dependent dehydrogenase oxidizes a subset (sometimes all) of the azolines to the aromatic azole heterocycles.51,174 A typical LAP will then undergo proteolytic processing to remove the leader peptide with subsequent transport out of the producing cell by a dedicated ATP binding cassette (ABC) type transporter.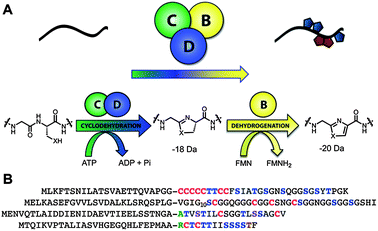 | ||
| Fig. 8 LAP biosynthetic scheme and precursor peptide sequences. A, Upon recognition of the precursor peptide (black line) by the heterocycle-forming BCD enzymatic complex, select Cys and Ser/Thr undergo a backbone cyclodehydration reaction with a preceding amino acid. Catalyzed by a cyclodehydratase (C/D-proteins, green and blue), net loss of water from the amide carbonyl during this reaction yields an azoline heterocycle. Further processing by a FMN-dependent dehydrogenase (B-protein, yellow) affords the aromatic azole heterocycle. B, Amino acid sequence of several LAP precursors. Like most RiPPs, these consist of a leader peptide (to facilitate recognition by the modifying enzymes) and core region that encodes for the resultant natural product. From top to bottom are the LAP precursors for SLS, microcin B17, goadsporin, and plantazolicin. Post-translational modifications are color-coded (Cys resulting in thiazoles, red; Ser/Thr resulting in oxazoles and methyloxazoles, blue; Thr leading to methyloxazoline, brown; Ser converted to dehydroalanine, light blue; N-terminal acetylated/methylated residues, green). The leader peptide cleavage site is indicated with a dash (predicted for SLS). | ||
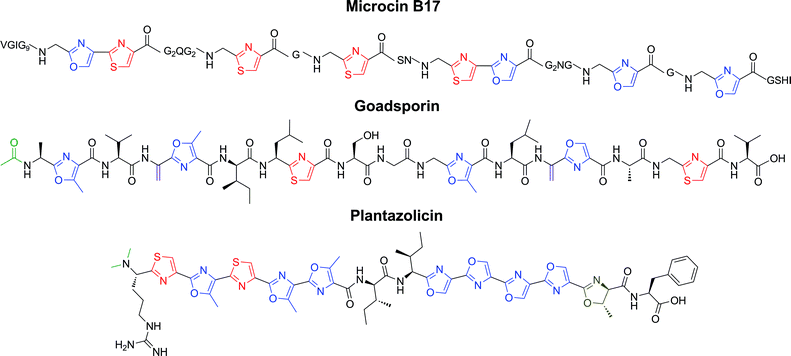 | ||
| Fig. 9 Representative structures of LAPs. Microcin B17 is a DNA gyrase inhibitor (antibiotic), goadsporin induces secondary metabolism and morphogenesis in actinomycetes, and plantazolicin is a selective antibiotic with an unknown biological target. Color coding as in Fig. 8. | ||
While the defining feature of a LAP is the genetically and biochemically conserved installation of azol(in)e rings on non-macrocyclized natural products (as opposed to the cyanobactins and thiopeptides discussed in the following sections), other post-translational modifications are often found. These ancillary tailoring modifications include acetylation, methylation, and dehydration.7,44,52
5.3 Structures and bioactivities of LAPs
Aside from microcin B17 and SLS, a number of additional LAPs have been identified by mining publicly available genome sequences. Only those LAPs with a fully elucidated chemical structure are discussed here, but it is probable that trifolitoxin and some nitrile hydratase/nif11-derived natural products are also LAPs.3,175,176 Goadsporin is produced by Streptomyces sp. TP-A0584 and contains two thiazoles, four (methyl)oxazoles, two dehydroalanines, and an acetylated N-terminus (Fig. 9).177,178 The genes responsible for the chemical maturation of this LAP were reported in 2005.44 Goadsporin provides a link between the LAP family and the lanthipeptides as the dehydroalanines are generated by proteins with homology to the class I lanthipeptide dehydratases (LanBs). The precise biological target(s) of goadsporin has not yet been determined, but it is known to induce secondary metabolism and morphogenesis in a wide variety of streptomycetes. Goadsporin also exhibits antibiotic activity against Streptomyces, including potent activity against Streptomyces scabies, the causative agent of potato scab.Plantazolicin (PZN) was predicted by bioinformatics to be an excreted metabolite from the soil-dwelling microbe Bacillus amyloliquefaciens FZB42.7 Genetic evaluation of the PZN biosynthetic pathway, molecular weight determination, and confirmation of antibiotic activity were reported in early 2011.179 Later that year, two groups independently reported the structure of PZN.180,181 Cyclodehydration was shown to precede dehydrogenation in vivo,181 as hypothesized from earlier work on microcin B17 and azol(in)e-containing cyanobactins.51,182–184 PZN has striking antimicrobial selectivity for Bacillus anthracis, and its biosynthetic genes are found in several other Gram-positive bacterial species.181
5.4 Relationship to other RiPP subfamilies
The genes responsible for installing the azol(in)e heterocycles are the defining characteristic of the LAP subfamily of RiPPs. It is important to note that the BCD heterocycle-forming genes are genetically similar with respect to biochemical function to those that install heterocycles on macrocyclized RiPPs (e.g. the thiopeptides, cyanobactins, and bottromycins, vide infra).50,52,1855.5 Specific recommendations for the LAP family
To avoid future confusion with the letter assignment of biosynthetic genes for LAPs, we recommend that “A” always be reserved for the precursor peptide, regardless of its location within a biosynthetic gene cluster. Likewise, we encourage the use of “C/D” for the cyclodehydratase and “B” for clusters that encode a dehydrogenase.6 Cyanobactins
6.1 Structures and biosynthesis
In the 1970s, growing interest in bioactive components of marine animals prompted Ireland and Scheuer to investigate the tunicate Lissoclinum patella, collected on tropical coral reefs in the Western Pacific. In 1980, they reported the structure elucidation of two small, cytotoxic peptides, ulicyclamide and ulithiacyclamide, revealing a then-unprecedented combination of chemical features, including N-to-C macrocyclization and heterocyclization to form thiazol(in)e and oxazoline motifs (Fig. 10).186L. patella harbors an abundant, uncultivated symbiotic cyanobacterium, Prochloron didemni, leading the authors to propose that symbiotic cyanobacteria make the compounds. Indeed, 12 years after this initial report, the homologous compound westiellamide was isolated from cultivated cyanobacteria and from ascidians.187,188 Overall, approximately 100 related compounds were described from marine animals and cultivated cyanobacteria as of the early 2000s.185 Compounds in the group were N-to-C macrocyclic, and many (but not all) were further modified by heterocyclization. Several family members were prenylated on Ser, Thr, or Tyr, while others were N-methylated on His.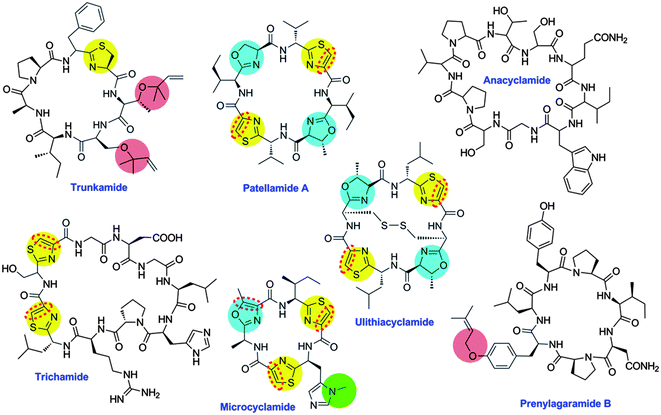 | ||
| Fig. 10 Representative cyanobactins. Anacyclamide is modified only by N–C cyclization, while others are modified by heterocyclization of Cys (yellow) or Ser/Thr (blue), oxidation to azole (red dashed line), prenylation (pink) or N-methylation (green). | ||
The family relationship of these structurally and functionally diverse metabolites was not universally appreciated until their gene clusters were sequenced. Genes were reported for the patellamide group in 200521 and for ulithiacyclamide itself in 2006,25 confirming that the “animal” compounds were in fact produced by P. didemni.21,189 Biosynthetic genes from many different free-living and symbiotic cyanobacteria have since been reported, leading to the proposal for a family name: cyanobactins (Fig. 10).32,64,190–195 Cyanobactins are defined as N–C macrocyclic peptides encoded on a precursor peptide (designated as “E”, as in PatE in patellamide biosynthesis), with proteolytic cleavage and macrocyclization catalyzed by homologous serine proteases “A” and “G”, as in PatA and PatG.32,185,196 Optional, but widely-occurring, cyanobactin biosynthetic proteins include “D”, as in PatD, the cyclodehydratase from the patellamide pathway; “F”, as in LynF, the tyrosine prenyltransferase from the lyn pathway; and conserved proteins of unknown function (“B” and “C”). Additional elements include thiazoline/oxazoline dehydrogenases that aromatize the heterocycles to thiazoles and oxazoles, methyltransferases, and several other uncharacterized proteins. Of note, the D cyclodehydratase consists of the C–D domains found in LAP biosynthesis (vide supra), and the azoline dehydrogenase is often, although not always, part of the G protein and is homologous to the dehydrogenase referred to as “B” in the LAP family.
About 200 cyanobactins have been identified, mostly via traditional chemical methods but at least one-third by genome mining.64,185,196 All known cyanobactins derive from cyanobacteria (or marine animals, where a cyanobacterial origin is likely) and are between 6–20 amino acids in length. Phylogenetic analysis has grouped the gene clusters into several families that are predictive of the resulting chemical structures.64 Other types of N-to-C macrocyclic peptides (circular peptides) abound in nature and include the cyclotides and orbitides from plants,197,198 the amatoxins and phallotoxins from fungi,22,199 and circular bacteriocins200 among others (see their respective sections in this review). However, these compounds are so far not known to be produced using PatA, PatG, and PatE-like proteins, and they contain different secondary modifications. Cyanobactin gene clusters have been identified only in cyanobacteria to date, and evidence suggests that they might be present in ∼30% of cyanobacterial strains.191,192 However, it is likely that cyanobactins exist in other organisms as well. For example, structurally related peptides such as telomestatin are well known from Streptomyces spp. These compounds are head-to-tail cyclized and contain heterocycles, but, in contrast to known cyanobactins, multiple heterocycles are found in tandem, some of which derive from non-proteinogenic amino acids such as β-hydroxytyrosine. Recently, a gene cluster for the telomestatin family compound YM-216391 was reported.201 Interestingly, while YM-216391 is made by LAP-like cyclodehydratases, the route to macrocyclization is convergent and utilizes different machinery.
Biosynthesis of cyanobactins begins with the E-peptide, which contains at minimum a leader peptide, an N-terminal protease recognition sequence, a core peptide, and a C-terminal recognition sequence (Fig. 11).21 Both recognition sequences are generally 4–5 amino acids in length. Often, more than one core peptide (up to 3 thus far) is encoded on a single E-gene; in these cases, each core peptide is flanked by conserved N- and C-terminal recognition sequences.64 If a D-protein (cyclodehydratase) is present, this protein acts first by regioselective modification of Cys, Ser, and/or Thr residues to form oxazoline and thiazoline.183,184 The E-peptide substrate is helical,60 presumably facilitating its interaction with D,58 and a short sequence element in the E leader peptide is diagnostic of heterocycle formation.64 Subsequently, other modifications take place. The A protease clips off the N-terminal recognition sequences, leaving a free amine for macrocyclization.53 The G protease then cleaves at the C-terminal recognition sequence and catalyzes C–N macrocyclization.53,202 An X-ray structure of the macrocyclase domain of PatG demonstrated that it has a subtilisin-like fold but contains insertions not found in these proteases.203 Furthermore, the structure of an acyl enzyme intermediate in which the peptide to be cyclized is bound to an active site Ser residue provides insights into the macrocyclization process.203 At present, it appears that prenylation occurs after macrocyclization, and only if a certain type of F gene is present.204 The F-protein is usually specific for dimethylallylpyrophosphate,204 but evidence suggests that longer isoprenoids can be incorporated in some cyanobactins.192 When multiple core peptides are present on a single E-precursor, they are all modified into cyanobactin products,21 and multiple E genes are often found in single organisms or pathways.64 Cyanobactins often contain D-amino acids, which arise spontaneously because of their adjacency to azol(in)e residues.32,205 Like the biosynthetic enzymes discussed for many other compound classes in this review, the cyanobactin biosynthetic enzymes are promiscuous and accept diverse proteinogenic and non-proteinogenic substrates in vivo and in vitro.25,202,204,206
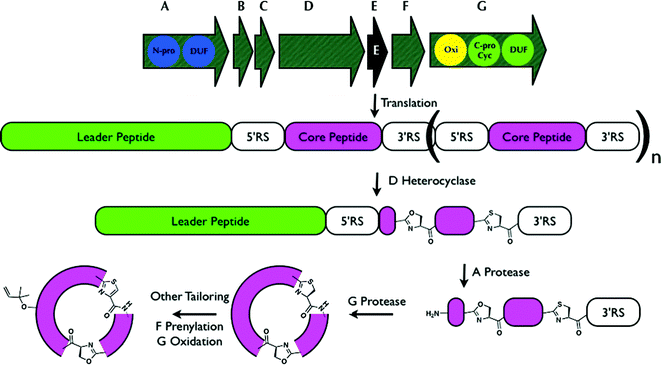 | ||
| Fig. 11 Generalized biosynthesis of cyanobactins that contain azoline heterocycles. Translation of the core peptide is followed by heterocyclization, then proteolytic tailoring to yield macrocycles. RS = recognition sequence, essential for proteolytic tailoring. Multiple copies of core peptide + recognition sequences are possible; here an example is shown in which a single core peptide exists and is modified (n = 0). Other tailoring, such as prenylation, probably takes place on the mature cycle, while the timing of oxidation / dehydrogenation to azoles is not known. Additional modifications are also sometimes present.185 Oxi = thiazoline oxidase, N-pro and C-pro are the protease domains that cleave the N-terminus and C-terminus of the core peptides, respectively, and DUF are domains of unknown function. C-pro also performs macrocyclization (Cyc). | ||
Cyanobactins represent an enormous array of sequence and functional group diversity, and many different biological activities have been attributed to the molecules, including anticancer, antiviral, anti-protease, and antibiotic activity.185,207 Notably, several compounds exhibit selective toxicity to certain human cancer cell lines,207,208 some potently block the human multidrug efflux pump, P-glycoprotein,209,210 and many cyanobactins are known for their ability to bind transition metals.207,211,212
6.2 Specific recommendations for the cyanobactin family
It is recommended that the nomenclature for proteins in this group be based upon the precedent of the patellamide pathway. Therefore, the defining peptides are precursor peptide (E), N-terminal protease (A), and C-terminal protease (G). Other commonly occurring proteins in this group include cyclodehydratase (D), prenyltransferase (F), and hypothetical proteins (B and C).7 Thiopeptides
Thiopeptides were first reported in the late 1940s with the isolation of a micrococcin,213 and have since burgeoned into a family of roughly 100 metabolites, a few of which are represented in Fig. 12A. To date, the majority of thiopeptides are produced by Actinobacteria, but several have been obtained from Firmicutes, including those from the Bacillus and Staphylococcus genera.214–216 Also referred to as thiazolyl peptides or pyridinyl polythiazolyl peptides, the thiopeptides typically feature a highly modified peptide macrocycle bearing several thiazole rings and often possess multiple dehydrated amino acid residues (for a review, see214). A defining feature of the thiopeptide macrocycle is a six-membered nitrogenous ring that can be present in one of three oxidation states: a piperidine, dehydropiperidine, or pyridine. Further architectural complexity is achieved in some thiopeptides by the addition of a second macrocycle to incorporate a tryptophan-derived quinaldic acid or an indolic acid residue, such as those found in thiostrepton A and nosiheptide (Fig. 12A).217,218 For over sixty years following the discovery of micrococcin, it was uncertain whether thiopeptides derived from the extensive post-translational modification of ribosomally-synthesized precursor peptides, or, instead, if their polypeptide backbones were the products of nonribosomal peptide synthetases. A series of publications in early 2009 revealed the biosynthetic gene clusters of five thiopeptides, leading to the formal recognition that these metabolites are indeed RiPPs.46–49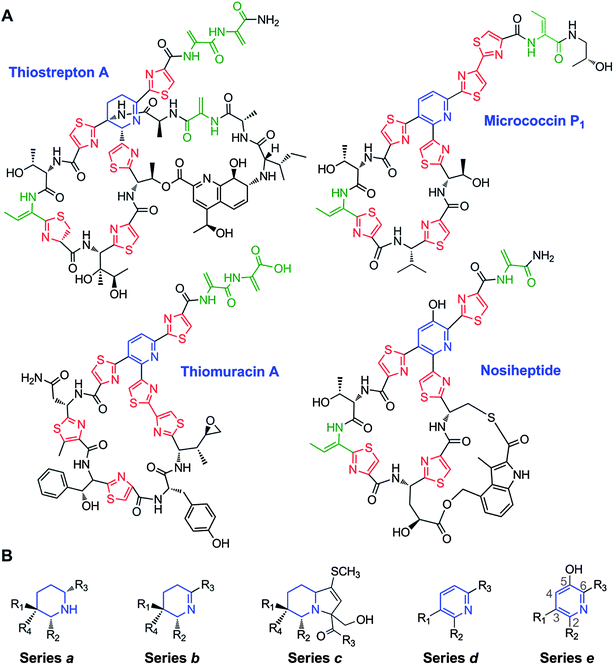 | ||
| Fig. 12 A. Representative thiopeptide structures. The central dehydropiperidine or pyridine rings are in blue, the thiazole and thiazoline rings are in red, and the Dha and Dhb residues are in green. B. The structural series of thiopeptides, showing the typical substitution pattern of the central nitrogenous heterocycle. | ||
7.1 Structural classifications of thiopeptides
Structurally, the thiopeptides can be classified into five series, based on the oxidation state and substitution pattern of the central nitrogenous heterocycle (Fig. 12B). This classification scheme originates from an initial proposal by Hensens in 1978, and was later elaborated by Bagley to accommodate a more diverse set of metabolites – nearly 80 members by 2005.214,219 Series a thiopeptides, exemplified by certain thiopeptins, present the core heterocycle as a fully saturated piperidine.219,220 Series b is classically represented by thiostrepton A and its dehydropiperidine ring.221 Series c, thus far, is comprised of a single entry, Sch 40832, in which the core nitrogen heterocycle is contained within an imidazopiperidine.222 The trisubstituted pyridine ring, in micrococcin P1 and other series d metabolites, is the most frequently encountered scaffold among the thiopeptides.214,223,224 The final sub-family, series e, also contains a pyridine, but possesses a hydroxyl group at the fifth position of the ring, as can be observed in nosiheptide (Fig. 12A).225,226 All members of the series a–c thiopeptides contain a second macrocycle in which a quinaldic acid, 4-(1-hydroxyethyl)quinoline-2-carboxylic acid, links one side chain from the core macrocycle to the core peptide N-terminus (e.g. thiostrepton A, Fig. 12A). In an analogous fashion, series e metabolites also harbor a second macrocycle, with the distinction that two of the core macrocycle's side chains are connected to an indolic acid residue.7.2 Biological activities of thiopeptides
Varied biological activities are attributed to thiopeptides, yet these RiPPs are perhaps best known for their antibacterial properties. Although thiopeptides are potent inhibitors of Gram-positive bacterial growth, little effect is generally exerted upon Gram-negative bacteria. This spectrum of antibacterial activity is attributed to an inability of thiopeptides to penetrate the outer membrane of Gram-negative species, since in vitro studies of thiopeptide function often employ the biochemical targets from Escherichia coli.227–230 Two major modes of antibacterial action are commonly reported for the thiopeptides, both of which impede protein synthesis. The first, observed of thiostrepton A, nosiheptide, micrococcin P1, and several others, results from a direct interaction with the prokaryotic 50S ribosomal subunit.231–233 These thiopeptides bind near the GTPase-associated center that engages a number of translation factors during initiation and elongation.234–236 Structural studies revealed that thiopeptides bind to a cleft between the ribosomal protein L11 and the 23S rRNA, and the precise intermolecular interactions differ according to the exact structural features of the thiopeptide.231,237 Complexation of the ribosome and a thiopeptide disrupts the conformational changes ordinarily communicated from the translation factors to the ribosome, ultimately halting translocation along the mRNA template.229,233,235,236 A subset of thiopeptides, including the thiomuracins and GE2270A, exert a separate mode of antibacterial action by the inhibition of elongation factor Tu (EF-Tu). EF-Tu ordinarily delivers aminoacylated tRNAs to the ribosome following translocation.49,228 When a thiopeptide binds to EF-Tu, a portion of the recognition site for the aminoacyl tRNA substrate is occluded and formation of an EF-Tu·aminoacyl-tRNA complex is prevented.228,238,239In addition to their well-known antibacterial properties, certain thiopeptides have demonstrated antimalarial and anticancer activities. For both of these latter cases, it appears that the mode of action may be two-fold. Thiostrepton A inhibits two opposing aspects of protein homeostasis in the malaria parasite Plasmodium falciparum: the proteasome and protein synthesis within the parasite's specialized apicoplast organelle.240–243 In cancer cell lines, thiostrepton A appears to induce apoptosis through the interplay of proteasome inhibition and direct interference with the forkhead box M1 (FOXM1) transcription factor.244–246 Other activities ascribed to thiopeptides span a range that encompasses RNA polymerase inhibition, renin inhibition, and some of these metabolites are proposed to serve as streptomycete signal molecules.247–250 The challenges now remain to decipher which aspects of the thiopeptide structure are key for each biological target, and then to apply that knowledge toward the development of novel anti-infective and anti-cancer chemotherapies.
7.3 Biosynthesis of thiopeptides
At the time of this review's preparation, ten thiopeptide biosynthetic gene clusters have been identified.46–49,251–255 In addition to the precursor peptide (e.g. TpdA of the thiomuracin cluster), at least six proteins, corresponding to TpdB–G, are found within each cluster. These six proteins presumably provide the minimal set of post-translational modifications required to construct the defining thiopeptide scaffold.49 The thiopeptides harbor structural features similar to both lanthipeptides (Dha and Dhb residues) and the linear azol(in)e-containing peptides (LAPs) and cyanobactins (thiazole structures).50,52 Not surprisingly, these backbone modifications are installed by similar enzymes. Dehydration of Ser and Thr side chains to introduce Dha and Dhb residues, respectively, is likely effected by one or two proteins, TpdB and TpdC, each bearing weak similarity to the class I lanthipeptide LanB dehydratases.90,91 Proteins similar to the cyclodehydratases (TpdG) and dehydrogenases (TpdE) that construct the polyazoline rings of the cyanobactins and LAPs are predicted to perform in a similar context for thiopeptide maturation.21,44,179 Cyclization of a linear peptide is expected to provide the central pyridine/dehydropiperidine/piperidine of the thiopeptides, and Bycroft and Gowland suggested this could ensue through a [4 + 2] cycloaddition of two dehydroalanines (Fig. 13).223 Inactivation of tclM, a tpdD homolog, in the thiocillin-producing Bacillus cereus strain led to the accumulation of a highly modified and linear peptide that contains the two anticipated dehydroalanine resides.256 The identification of this metabolite lends support to the Bycroft–Gowland proposal and implicates a key role for TclM/TpdD and their homologs in this biochemically intriguing transformation. At present, it remains to be determined whether this cycloaddition proceeds in a concerted or stepwise process. The sixth protein found in all thiopeptide biosynthetic systems, TpdF, does not display any significant similarity to proteins of known function, and its biochemical role awaits elucidation. Like other RiPPs discussed in this review, the biosynthetic machinery of thiopeptides is tolerant of many mutations in their linear substrate peptides resulting in new variants.36,38,42,43 However, thiopeptides from the d and e series may be unique among RiPPs in the manner by which the leader peptide is likely removed. Rather than relying on proteolytic cleavage, the leader peptide is thought to be eliminated from the central nitrogen-containing heterocycle (Fig. 13).50 Beyond the conserved six proteins, one or more additional enzymes are embedded within each cluster to introduce the specialized features of each metabolite, including hydroxylations, methylations, and incorporation of an indolic or quinaldic acid macrocycle.46,49,255,257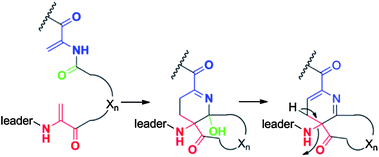 | ||
| Fig. 13 Bycroft proposal for generation of the pyridine ring in thiopeptides from two Dha residues. The aromatization step would also remove the leader peptide.50 | ||
7.4 Specific recommendations for the thiopeptide family
Given the overlap in nomenclature used between the LAP, cyanobactin, and lanthipeptide modification enzymes and the current lack of a common naming scheme, a thiopeptide-specific nomenclature for the core set of biosynthetic proteins seems appropriate. To be consistent with the bulk of RiPP gene nomenclature, it is recommended that the precursor peptide is annotated as “A”. It is also suggested that future annotations of thiopeptide gene clusters follow the nomenclature outlined for the thiomuracins, wherein the precursor peptide and the core set of six genes were collinearly organized.49 By adopting this nomenclature, proteins corresponding to the dehydratases will be designated as “B” and “C”; the azoline-synthesizing dehydrogenase and cyclodehydratase are to be designated as “E” and “G”, respectively; “D” is to be used for the protein implicated in the putative [4 + 2] cycloaddition; and “F” should be assigned for the remaining conserved protein. Although this annotation may lead to discontinuous name assignment for a cluster's components, since thiopeptide clusters do not all follow this gene organization, it will ultimately permit a uniform notation for the core set of thiopeptide modification enzymes.8 Bottromycins
The first member of a series of related compounds called bottromycins was originally isolated from the fermentation broth of Streptomyces bottropensis DSM 40262,258 but their structures were not definitively known until a total synthesis of bottromycin A2 in 2009 that settled a series of structural revisions (Fig. 14).259 These compounds contain a number of unique structural features including a macrocyclic amidine and a decarboxylated C-terminal thiazole. In addition, like the proteusins (section 4), they contain a series of C-methylated amino acids (Fig. 14). The bottromycins display potent antimicrobial activity against methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci (VRE),260 and function by blocking aminoacyl-tRNA binding to the A-site of the bacterial 50S ribosome.261–263 Recently, using various genome mining and genome sequencing methods, four studies reported that the bottromycins are RiPPs arising from an impressive series of modifications.17–20 The biosynthetic gene clusters were obtained by shot-gun sequencing of the genomes for Streptomyces sp. BC1601917 and S. bottropensis18,19 that both were known to produce bottromycin A2, B2, and C2 (Fig. 14).264–267 In an alternative strategy, a search of the non-redundant databases for a sequence encoding a putative linear core peptide GPVVVFDC provided a hit in the sequenced genome of the plant pathogen Streptomyces scabies 87.22, which was subsequently shown to produce bottromycins A2, B2, and C2.18,19 And in a final approach, a new analog, bottromycin D (Fig. 14) was detected in a metabolomics study of the marine ascidian-derived Streptomyces sp. WMMB272; subsequent genome sequencing revealed its biosynthetic gene cluster.20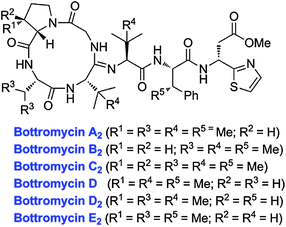 | ||
| Fig. 14 Structures of various bottromycins. | ||
8.1 Biosynthesis of the bottromycins
All four studies report the intriguing observation that the precursor peptide to the bottromycins does not contain an N-terminal leader peptide, but instead a C-terminal extension of 35–37 amino acids (Fig. 15). The exact role of this extension is at present unclear, but it likely guides the biosynthetic enzymes. It is significantly larger than the C-terminal recognition sequences used for N-to-C cyclization in cyanobactins (section 6), amatoxins (section 14), cyclotides (section 15), and orbitides (section 16), and because it likely functions in a role more similar to the leader peptides, the term follower peptide was introduced.19 Bioinformatics analysis has been used to predict the function of some of the genes that are found in the gene clusters of all four strains, which are highly similar in organization and sequence.17–20 Three putative cobalamin-dependent class B162 radical-SAM methyltransferases were predicted to catalyze the methylation at the non-nucleophilic β-carbons. The Cys constellation in these enzymes (CxxxxxxxCxxC)18 is different from the canonical CxxxCxxC motif involved in Fe-S clusters in most radical-SAM proteins, but is also found in the polytheonamide C-methyltransferases PoyB and PoyC (section 4). The predicted function of these radical-SAM methyltransferases has been experimentally validated by gene deletion experiments in a heterologous expression system using Streptomyces coelicolor A3(2),17 and by gene deletions in S. scabies.18 One of the enzymes is responsible for C-methylation of the Pro residue as its deletion resulted in the exclusive production of bottromycin B2.17,18 Similarly, a second radical-SAM methyltransferase is specific for the C-methylation of the Phe residue as its disruption resulted in the production of a new metabolite, bottromycin D2.17 Deletion of the third methyltransferase resulted in the production of a compound that lacked two methyl groups,17,18 which was identified by tandem MS as bottromycin E2 (Fig. 14).17 Hence, this third methyltransferase adds methyl groups to two of the three original Val residues of the precursor peptide (Fig. 15). In addition to the radical-SAM methyltransferases, the gene clusters also contain an O-methyltransferase that was shown to be responsible for the methylation of the Asp residue (Fig. 15).17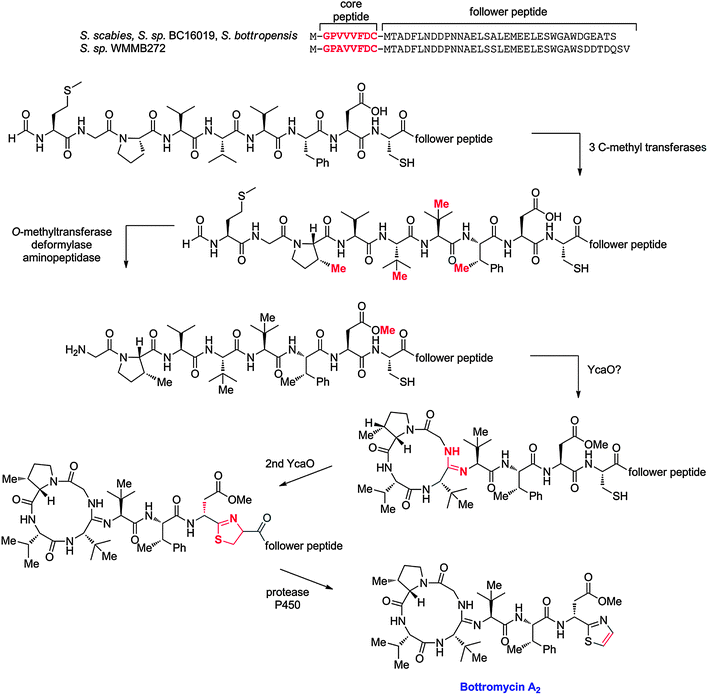 | ||
| Fig. 15 Potential biosynthetic pathway to bottromycin A2. The order of the posttranslational modifications is currently not known and hence the order depicted is arbitrary. The precursor peptide sequences for the currently known bottromycins are shown at the top. Variations in the posttranslational modifications morph these precursor sequences into the structures shown in Fig. 14. | ||
The gene clusters also contain two genes with a YcaO-like domain similar to the D-protein involved in LAP biosynthesis (section 5). This domain was shown for LAPs to use ATP to phosphorylate the amide backbone during the cyclodehydration reaction,173 and hence at least one of the two YcaO-like genes is predicted to be involved in thiazoline formation.17–20 Interestingly, the bottromycin gene cluster does not contain the C-protein that together with the D-protein promotes efficient cyclodehydration in LAPs (section 5) or cyanobactins (in which the C and D proteins are fused in one protein, see section 6). Why two YcaO-domain containing proteins are encoded in the bottromycin gene cluster is at present unclear, but one possibility is that one is involved in cyclodehydration to form a thiazoline and the second in the macrocyclodehydration to form the amidine structure (Fig. 15).17–19
Two genes encoding putative proteases/hydrolases and one gene encoding a putative aminopeptidase may be involved in removal of the follower peptide and the N-terminal Met, and possibly to aid formation of the unique macrocyclic amidine. Finally, a gene encoding a cytochrome P450 has been suggested to facilitate the oxidative decarboxylation of the C-terminal residue by hydroxylating the β-carbon of the Cys.19 The observed epimerization at the Asp α-carbon may possibly occur non-enzymatically as a consequence of its location next to a thiazole/thiazoline as has also been suggested for cyanobactins (section 6).32,205 The order of this impressive array of posttranslational modifications is currently not known and one possibility out of many is shown in Fig. 15. A putative bottromycin-transporter was also identified in the biosynthetic gene cluster,17–20 and proposed to be involved in the export of and self-resistance to bottromycins. This hypothesis was verified by an overexpression experiment with the Streptomyces coelicolor A3(2) heterologous expression system, which led to a 20 fold increase of bottromycin production yields compared to a parallel experiment using the unmodified pathway.17
8.2 Specific recommendations for the bottromycin family
It is recommended to use the term follower sequence for the unique C-terminal extension found in the bottromycin precursor peptides. Since four independent studies all used different gene annotations, at present a common gene nomenclature is not in use. In line with the recommendations for most other RiPPs, the use of the designation A is recommended for the gene encoding the precursor peptide in biosynthetic gene clusters that are uncovered in future studies.9 Microcins
Historically, the name microcins has been used for ribosomally synthesized antimicrobial peptides from Enterobacteriaceae, mainly Escherichia coli. In addition to their lower molecular masses (<10 kDa), microcins differentiate from colicins, the larger antibacterial proteins (30–80 kDa) produced by Gram-negative bacteria, by a resistance to proteases, extreme pH and temperature, and in many cases the presence of post-translational modifications. Fourteen representatives have been identified thus far (for reviews see12,268,269) and eight of them have been isolated and structurally characterized (e.g.Fig. 16).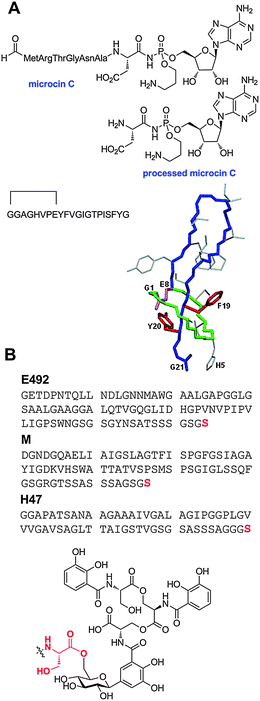 | ||
| Fig. 16 Structures of the post-translationally modified microcins. A. Structures of several class I microcins. Both the linear sequence and three dimensional structure of microcin J25 is shown (PDB 1Q71). Microcin B17, another class I microcin, is shown in Fig. 9. B. Class II microcins. The post-translational modifications of class IIb microcins attach a linear trimer of 2,3-dihydroxybenzoyl L-serine linked via a C-glycosidic linkage to a β-D-glucose. The glucose is anchored to the carboxylate of the C-terminal serine residues of the peptides (red font) through an ester linkage to O6. | ||
Most microcins are plasmid-encoded peptides. Their biosynthetic clusters display a conserved general organization, with (i) a structural gene encoding the precursor peptide, (ii) a self-immunity gene that protects the producing strain against its own toxic peptide, (iii) genes encoding the microcin export system, and (iv) often several genes encoding auxiliary proteins or modification enzymes. The classification of microcins adopted presently distinguishes two classes.12,270 Class I consists of three plasmid-encoded peptides with a molecular mass below 5 kDa that have extensive backbone post-translational modifications: microcins B17, C (also called C7, C51 or C7/C51), and J25. Two of these compounds, microcins B17 and J25 were initially believed to be unique to Enterobacteria, but the genome sequencing efforts have demonstrated that they are part of larger families of structurally related peptides that are produced by a variety of bacteria. As described in the section on the LAP family, microcin B17 is a 43 amino acid peptide that carries four thiazole and four oxazole heterocycles (Fig. 9), which result from the modification of six glycines, four serines and four cysteines in the core peptide of the 69 amino acids long microcin precursor peptide (Fig. 8).271 Microcin C is an N-formylated heptapeptide covalently linked to adenosine monophosphate via its C-terminal aspartate through an N-acyl phosphoramidate linkage;272 the phosphate group is further esterified with 1,3-propanolamine (Fig. 16A). Microcin J25 is a 21-amino acid lasso peptide.273 It consists of a macrolactam, which is formed by the N-terminal amino group and the side-chain carboxylate of an aspartate at position 8, through which the peptide tail is threaded and firmly trapped by steric interactions, thus forming a lasso. Therefore, microcin J25 belongs to both families of microcins from Enterobacteria and lasso peptides, which are mainly synthesized by Actinobacteria (section 10).273–275
Class II microcins consist of higher molecular mass peptides in the 5–10 kDa range. This class is further subdivided into two subclasses, IIa and IIb. Class IIa comprises three plasmid-encoded peptides, microcins L, V and N (also named microcin 24) that are not post-translationally modified and that will not be considered here. Class IIb consists of chromosome-encoded linear microcins that carry a C-terminal siderophore (Fig. 16B).276,277
9.1 Biological activities of microcins
Microcins display potent antibacterial activity against Gram-negative bacteria, and specifically Enterobacteria. They exhibit diverse mechanisms of antibacterial activity and appear attractive models for the design of novel bioactive peptides with anti-infective properties.12,269,278 Microcins have receptor-mediated mechanisms of antibacterial activity that improve their uptake in sensitive bacteria. Therefore, they have a narrow spectrum of activity and minimal inhibitory concentrations in the nanomolar range. Microcins hijack receptors involved in the uptake of essential nutrients, and many parasitize iron siderophore receptors or porins. Those using siderophore receptors are also dependent on the TonB-ExbB-ExbD inner-membrane protein complex, which ensures energy transduction to the outer membrane and its receptors, using the proton-motive force from the cytoplasmic membrane. Microcin J25 uses the receptor to ferrichrome (FhuA), microcins B17 and C use the porin OmpF, and microcins E492 and M use the receptors to catechol siderophores (FepA, Cir and Fiu). After crossing the outer membrane, microcins often require an inner membrane protein to exert their toxic activity, such as SbmA for microcins B17 and J25 or components of the mannose permease for microcin E492. Others require an ABC-transporter, such as YejABEF for microcin C. Many microcins inhibit vital bacterial enzymes, such as DNA gyrase (microcin B17) or RNA polymerase (microcin J25). The latter blocks transcription by binding in the secondary channel that directs NTP substrates toward the RNA polymerase active site, resulting in its obstruction in a cork-in-the-bottle fashion.A two-step processing procedure inside target bacteria is required to activate microcin C. First peptide deformylase removes the N-terminal formyl group, and then several broad-specificity aminopeptidases target the ultimate peptide bond Ala6-Asp7 and cleave off the N-terminal peptide (Fig. 16A). The resulting molecule is a mimic of aspartyl adenylate, which inhibits aspartyl tRNA synthetase and therefore blocks protein synthesis at the translation step. To exert its bactericidal activity, the class IIb microcin E492 forms channels in the inner membrane.
With the exception of the lasso structure of microcin J25 (see section 10), three-dimensional structures of microcins are not available.273,279–281 However, in the absence of precise three-dimensional structures, structure–activity relationships of class I microcins have been undertaken. A chemical strategy was developed to prepare a series of microcin C analogs, which retained the Trojan horse mechanism of antibacterial activity of the natural microcin, while targeting different aminoacyl-tRNA synthetases specified by the last amino acid that is linked to the adenosine.282 In the case of microcin B17, point mutations indicate that the number and position of the thiazole and oxazole rings in the microcin structure are essential, and that the C-terminal bis-heterocycle is critical.51
9.2 Microcin biosynthesis
All microcins except microcin C, which does not possess a leader peptide and is secreted without cleavage of a precursor, are translated from their respective mRNA as precursor peptides consisting of an N-terminal leader peptide and a C-terminal core peptide, which undergoes post-translational modifications prior to becoming the mature active microcin.12 Class II microcins result from maturation of large precursors carrying small conserved leader peptides (15–19 amino acids). Class I microcins have longer leaders of 19–37 amino acids that (at present) do not display common features, neither in length nor in amino acid sequences. Removal of the microcin leader peptides most often occurs at a specific cleavage site, the typical double-glycine motif or the glycine-alanine motif found in proteins exported through ABC (![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) TP-
TP-![[b with combining low line]](https://www.rsc.org/images/entities/char_0062_0332.gif) inding
inding ![[c with combining low line]](https://www.rsc.org/images/entities/char_0063_0332.gif) assette) transporters.283 The roles of the leader peptides have not been fully clarified to date, but they could contribute to maturation, secretion, protection of the precursor against degradation, or to keeping the precursor inactive inside the host cells during biosynthesis.16
assette) transporters.283 The roles of the leader peptides have not been fully clarified to date, but they could contribute to maturation, secretion, protection of the precursor against degradation, or to keeping the precursor inactive inside the host cells during biosynthesis.16
The biosyntheses of microcin B17 and J25 have been reconstituted in vitro.51,284,285 Mature microcin B17 was obtained from the precursor McbA in the presence of the modification enzymes McbB, McbC, and McbD that form the microcin B17 synthetase complex.51 We note that the original gene designations for the microcin B17 gene cluster were alphabetical,286 and were different from that recommended in section 5 for LAPs. In this early designation McbB corresponds to the “C” protein of LAPs and McbC was the flavin dependent dehydrogenase (designated B in other LAPs).51,52 The oxazole and thiazole heterocycles result from a series of cyclization, dehydration, and dehydrogenation reactions as described in section 5. During the modification process, the McbBCD complex recognizes the leader peptide that has a helical structure.58 Upon removal of the N-terminal 26-amino acid leader peptide, presumably by TldD/TldE,287 mature microcin B17 is exported from the cell by the dedicated ABC transporter McbEF.
The maturation process of microcin C requires five steps including three modifications of the precursor MccA and two cleavage steps that are accomplished in the target cells:278,288 (i) conversion of Asn7 to a derivatized isoAsn7, (ii) formation of an N-P bond between the amide group of isoAsn7 and the α-phosphate of ATP catalyzed by MccB, affording the peptidyl-AMP linkage, (iii) aminopropylation of the phosphoramidate using two enzymes encoded by mccD and mccE, (iv) deformylation, and (v) proteolysis at the Ala6–Asp7 peptide bond (see section 9.1).
The modification anchored at the C-terminus of microcin E492 is a C-glucosylated linear trimer of 2,3-dihydroxybenzoyl-L-serine (DHBS), which itself results from linearization of the cyclic catechol-type siderophore enterobactin.289–291 Enterobactin and salmochelin, its glycosylated counterpart, are first made by nonribosomal peptide synthetases. Modification of the 84-amino acid ribosomally produced peptide at its C-terminal serine with the sidereophore requires four genes mceCDIJ. MceC and MceD ensure enterobactin glucosylation and linearization, while MceI and MceJ are responsible for the formation of the ester linkage between the glucosylated enterobactin and the C-terminal serine of the microcin (Fig. 16B). The biosynthesis of the lasso peptide microcin J25 is discussed in section 10.2.
9.3 Specific recommendations for the microcin family
At present, the precursor peptide and common biosynthetic genes encoding proteins involved in export, immunity and modification enzymes are not always given the same gene name (e.g. mcxA for the precursor peptides to the microcins). Given the relatively small number of microcins, we recommend that a common designation of biosynthetic genes will be systematically implemented, including using the nomenclature systems implemented for other RiPPs, such as LAPs and lasso peptides.10 Lasso peptides
Lasso peptides form a growing class of bioactive bacterial peptides that are characterized by a specific knotted structure, the lasso fold.292 The producing bacteria are most often Actinobacteria (Streptomyces, Rhodococcus), but some are Proteobacteria (Escherichia, Burkholderia).273–275 They consist of 16–21 residues, with an N-terminal macrolactam, resulting from the condensation of the N-terminal amino group with a side-chain carboxylate of a glutamate or aspartate at position 8 or 9. Lactam formation irreversibly traps the C-terminal tail within the macrocycle. The net result is a highly compact and stable structure that to date has proven inaccessible by peptide synthesis. Lasso peptides are remarkably resistant to proteases and denaturing agents, and this high stability and their biological activities have attracted much interest. Indeed, the known lasso peptides inhibit enzymes or antagonize receptors, which confer antimicrobial properties on some of them.273 Furthermore, the lasso structure has recently been shown to be an efficient scaffold for epitope grafting,40 demonstrating the capacity to give novel biological functions to the lasso fold.Lasso peptides are classified into three classes (Fig. 17A). Members of the first class contain two disulfide bonds and the first residue is a cysteine. This class contains four representatives: siamycin I (also called MS-271),293,294 siamycin II and RP-71955,295,296 and SSV-2083.6 Class II lasso peptides do not contain disulfides and the first residue is a glycine. This class contains eight representatives: RES-701,297–300 propeptin,301,302 anantin,303,304 lariatins A and B,305 microcin J25,306,307 capistruin,308,309 and SRO15-2005.6 The third class, which was recently discovered and encompasses a single peptide, BI-32169,310 is characterised by the presence of a single disulfide bond and a glycine at the N-terminus. In all of the lasso peptides identified to date, the residue providing the side-chain carboxylate implicated in the macrolactam is a Glu at position 8 or an Asp at position 9 (or position 8 in one example), pointing to a stringent role of the size of the macrolactam in maintaining the lasso fold. A recent genome mining study has identified 76 potential lasso peptide producers spanning nine bacterial phyla and an archaeal phylum.311 One gene cluster from the freshwater bacterium Asticcacaulis excentricus was heterologously expressed in E. coli leading to the production of the largest lasso peptide to date, astexin-1.311 It consists of 23 amino acids and unlike most other lasso peptides is highly polar.
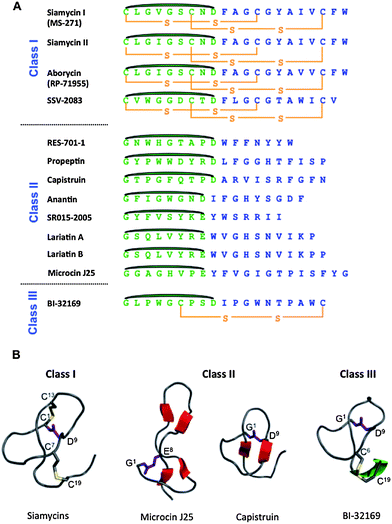 | ||
| Fig. 17 Structural features of lasso peptides. A) Sequences of lasso peptides of the three classes showing the residues involved in the macrolactam in green and those forming the tail in blue. The isopeptide linkage between the N-terminal amino group and the side chain carboxylates of Asp and Glu are shown in dark green. B) Three-dimensional structures of lasso peptides of the three classes; the structures of the class II microcin J25 and capistruin exhibit the respective sizes of the loop and tail above and below the ring. | ||
Microcin J25 produced by E. coli AY25 is the archetype of lasso peptides and the most extensively studied. Its lasso structure of type II results from linkage of the amino group of Gly1 to the side-chain carboxylate of Glu8 to give the N-terminal macrolactam; the threaded 13-residue C-terminal tail (Fig. 17A) is maintained irreversibly in the ring by the bulky side-chains of two aromatic residues, Phe19 and Tyr20, that straddle the ring and prevent the tail from escaping. Its three-dimensional structure contains two small anti-parallel β-sheets.279–281 One involves residues 6–7 located in the ring and residues 18–19 of the threading C-terminal tail. The other is located in the loop region of the lasso structure and comprises residues 10–11 and 15–16 that form a hairpin-like structure including a β-turn that involves residues 11–14 (Fig. 17B).
Capistruin is a 19-residue peptide that is produced by Burkholderia thailandensis E264 and contains a 9-residue ring resulting from a Gly1-Asp9 isopeptide bond.308 Six amino acids out of the 10-residue C-terminal segment pass through the ring, forming quite a long tail, which is trapped in the ring by the side-chains of Arg11 and Arg15. Similar to microcin J25, its three-dimensional structure contains a small antiparallel β-sheet involving residues 7–8 of the ring and residues 13–14 of the threading C-terminal tail that are connected by a β-turn involving residues 9–12. Having a shorter and tighter loop, capistruin lacks the second β-sheet present in the loop region of microcin J25.
10.1 Biological activities of lasso peptides
Lasso peptides are usually enzyme inhibitors or receptor antagonists. These properties confer some of them with antibacterial activity, which is directed against either Gram-negative bacteria or Gram-positive bacteria. The spectrum of activity is generally limited to bacteria phylogenetically related to the producing bacteria, helping them to colonize a given biotope. In class I, siamycin I (MS-271) is an inhibitor of the myosin light chain kinase and an anti-HIV agent,293,294 and siamycin II and RP-71955 are anti-HIV agents.293,294 In the class II lasso peptides, RES-701 peptides are antagonists of the type B receptor of endothelins,297–300 propeptin is a prolyl endopeptidase inhibitor,301,302 anantin is an antagonist of the atrial natriuretic factor,303,304 lariatins A and B are anti-mycobacterial agents,305 and microcins J25306,307 and capistruin308,309 are RNA polymerase inhibitors and antibacterial agents. The single class III lasso peptide BI-32169 is an antagonist of the glucagon receptor.31010.2 Lasso peptide biosynthesis
Microcin J25 and capistruin are the two lasso peptides that have been the most extensively studied with regards to their biosynthetic pathways. Both microcin J25 and capistruin arise from four-gene clusters (mcjABCD and capABCD) in E. coli and B. thailandensis, respectively. Microcin J25 has been the model for the study of the biosynthesis of lasso peptides. Its production requires McjB and McjC to control the acquisition of the lasso fold from the 58-amino acid linear precursor.284 On the basis of sequence comparisons,269,284 McjB is believed to be an ATP-dependent cysteine protease that cleaves off the leader peptide. McjC has homology with Asn synthetase B,284,285 and is hypothesized to form the isopeptide bond between the glycine at position 1 generated upon proteolytic cleavage of the leader and the Glu8 side-chain carboxylate, which is activated through an acyl-AMP intermediate. McjB and McjC are two interdependent enzymes that mutually need the physical presence of their partner to carry out their enzymatic reactions. The distinct functions of McjB and McjC were recently demonstrated by in vivo mutagenesis studies of putative active site residues,312 and in vitro studies using recombinant precursor and maturation enzymes.313 McjC was confirmed to be a lactam synthetase and McjB was shown to be a novel cysteine protease that requires ATP hydrolysis for proteolytic activity.313 In view of the prefolding step required for obtaining the lasso fold, the ATP-dependent character of McjB could be related to a chaperone function. Interestingly, only the N-terminal eight residues of the leader peptide are required for recognition of the precursor McjA by the maturation machinery.68 In particular, the Thr residue in the penultimate position of the leader peptide is proposed to be a recognition element for the maturation enzymes.314 The final product is believed to be transported from the cytoplasm by an ABC transporter.284,315 In contrast to microcin J25 and capistruin, which are encoded by four-gene clusters, lariatins A and B, produced by Rhodococcus jostii, result from a five-gene cluster (larABCDE).316 It is proposed that processing of the precursor LarA by LarB, LarC and LarD leads to the mature lasso peptide, which is then exported by LarF.316 Taking into account the sequence similarities between the larB and larD gene products and the modification enzymes identified for microcin J25 and capistruin, it is suggested that LarC, the function of which remains unclear, is a third enzyme essential for lariatin biosynthesis that would be specific to Gram-positive bacteria.Two systematic structure–activity relationship studies of microcin J25 have been performed, paving the way for future studies deciphering the roles of specific amino acids in the lasso structure and antibacterial activity.31,39 The most striking conclusion of these studies was that production and activity of the lasso peptide were tolerant to amino acid substitutions at most of the positions in the 21 amino acid core sequence. Another recent study demonstrated that the C-terminal residue, the bulky residue below the ring, and the size of the ring are critical for generating the lasso structure.317 Interestingly, some amino acid substitutions afforded branched-cyclic peptides instead of lasso peptides that were devoid of antibacterial activity. Moreover, the tail portion below the ring and the C-terminal glycine appeared critical for the antibacterial activity but could be modified without affecting lasso formation.
Using a heterologous capistruin production system in E. coli, a series of mutants of the precursor protein CapA have been generated, showing that only four residues of the lasso sequence are critical for maturation of the linear precursor into the mature lasso structure.41 Thus, the biosynthetic machineries of both microcin J25 and capistruin systems have low substrate specificity, which is a promising feature towards lasso peptide engineering.
10.3 Specific recommendations for the lasso peptide family
Similar to microcins, a common gene nomenclature should be adopted for lasso peptides in which the precursor and common biosynthetic genes are always given the same gene name: “A” for the genes encoding the precursor peptides, “B” for genes encoding the transglutaminase/cysteine protease homologs, “C” for genes coding for the Asn synthetase homologs, and “D” for genes encoding ABC transporters.11 Microviridins
Microviridins represent a family of cyclic N-acetylated trideca and tetradecapeptides that contain intramolecular ω-ester and/or ω-amide bonds. Like the lasso peptides, most microviridins contain lactams, but unlike the lasso peptides, these structures are not formed from the N-terminal amine but rather between ω-carboxy groups of glutamate or aspartate and the ε-amino group of lysine.318 Furthermore, microviridins also contain lactones generated by esterification of the ω-carboxy groups of glutamate or aspartate with the hydroxyl groups of serine and threonine. Lactone and lactam formation results in an unparalleled tricyclic architecture (Fig. 18). The crosslinking amino acids are part of a conserved TXKXPSDX(E/D)(D/E) central motif, whereas the N- and the C-termini of microviridin core peptides are highly variable.319 The related bicyclic dodecapeptide marinostatin (Fig. 18) contains only ω-ester bonds and a slightly altered microviridin-type central motif (TXRXPSDXDE).320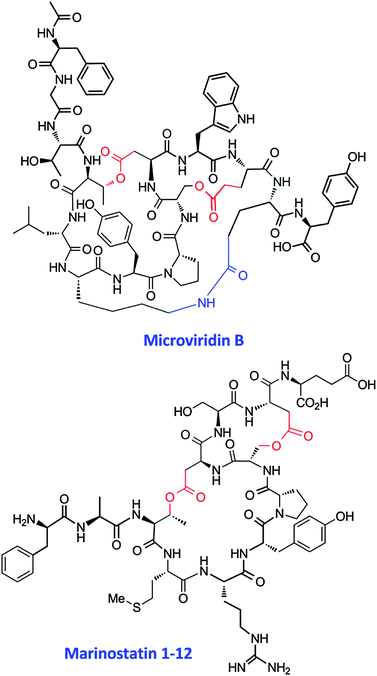 | ||
| Fig. 18 Structures of microviridin B from M. aeruginosa NIES298 and marinostatin 1-12 from Alteromonas sp. B-10-31. 322,325 Amino acid side chains forming ester and amide bonds are highlighted in red and blue, respectively. | ||
Microviridins have so far only been detected in bloom-forming freshwater cyanobacteria of the genera Microcystis and Planktothrix. The prototype of the family, microviridin A, was described for Microcystis viridis strain NIES 102 in 1990.318 Eleven further structures of related cyanobacterial metabolites, microviridin B–L, were elucidated in the past twenty years from laboratory strains and field samples.321–324 Bioinformatic analysis of complete microbial genomes, however, suggests a more widespread potential for microviridin biosynthesis than expected. Putative microviridin precursor genes were detected in a large number of cyanobacterial genomes, including strains of Cyanothece, Anabaena, Nodularia and Nostoc.323 In addition, the presence of related microviridin-type gene clusters in genomes of the sphingobacterium Microscilla marina ATCC 23134 and the myxobacterium Sorangium cellulosum Soce56 suggests an occurrence of microviridins beyond the cyanobacterial phylum,323 and the related bicyclic peptide marinostatin has been detected in the marine proteobacterial strain Alteromonas sp. B-10-31.320
11.1 Biological activities of microviridins
Microviridins were initially discovered through a bioactivity-guided screening program for inhibitors of serine type proteases in cyanobacteria.318 The different variants show distinct protease inhibition profiles with specificities for elastase, trypsin or chymotrypsin,321,322,324,326 and activities in the low nanomolar range.322,326 For example, microviridin B (Fig. 18) specifically inhibits elastase and has potential in the treatment of lung emphysema,322 and microviridin J inhibits a trypsin-type enzyme from Daphnia and disrupts the molting process of the animals.326 Microviridins may therefore play an important role in the trophic networks of freshwater lakes. Similarily, the bicyclic marinostatin (Fig. 18) has inhibitory activity against the protease subtilisin.32011.2 Biosynthesis of microviridins
Microviridin biosynthesis has been partially characterized in vivo and in vitro for the two cyanobacterial genera Microcystis and Planktothrix.323,327 The corresponding gene clusters have been designated mdnA–E and mvdA–E, respectively.323,327 The precursor peptides MdnA and MvdE contain a strictly conserved PFFARFL motif in their leader peptide that is essential for post-translational modification reactions.319 The lactone and lactam structures are introduced by two novel types of ATP grasp ligases, MdnC and MdnB, and MvdD and MvdC, respectively.323,327 The four enzymes show close similarity to each other and belong to the RimK family of enzymes that catalyze crosslinks of carboxyl groups with amine or thiol groups.328In vitro characterization revealed the activity of MvdD as the ω-ester ligase and MvdC as the ω-amide ligase.323 Further analysis of MvdD and mutational studies of the precursor peptide MvdE in Planktothrix suggested a strict order for the formation of the crosslinks, with the larger lactone forming first and the smaller lactone forming second.329 The size of both rings appears to be inflexible.329 Lactonization is then followed by lactam formation via MvdC.323 The cluster further encodes a GNAT-type N-acetyl transferase, designated MdnD and MvdB in Microcystis and Planktothrix, respectively.323,327 MvdB catalyzes the N-acetylation of the N-terminal amino acid of the lactonized and lactamized premature peptide.323 The microviridin gene cluster does not encode a protein with protease signatures that could be responsible for the cleavage of the leader peptide. However, the associated ABC transporter MdnE is crucial for correct processing in vivo and may play a scaffolding role thereby guaranteeing correct processing.319 Interestingly, the putative gene cluster in Anabaena PCC7120 encodes a precursor peptide that contains multiple putative core peptides,327 similarly to the multiple RiPPs generated from a single peptide that has been found for cyanobactins and cyclotides (sections 6 and 15).11.3 Specific recommendations for the microviridin family
The characteristic lactone and lactam structures are the signature motifs that define this group. Moreover, the three peptide modifying enzymes do not show close similarity to enzymes of other PRPS classes. The two enzymes catalyzing macrocyclization, MdnC/MvdD and MdnB/MvdC, can thus be taken as enzymatic basis for the classification of the microviridin group of peptides. Marinostatin and the corresponding biosynthesis gene cluster do not meet all criteria. However, as the two lactones and the ATP grasp ligase encoded by the marinostatin gene cluster resemble characteristic features of the microviridin class, marinostatins should be considered as a specific subgroup of microviridins. Two different gene nomenclatures (mdn, mvd) have been assigned to the related gene clusters in the two cyanobacterial genera.323,327 As both in vivo and in vitro characterization provided important insights into microviridin biosynthesis, standardization of this historical nomenclature is not possible without omitting important results. Future investigations into microviridins, however, should follow at least one of the existing gene designations.12 Sactipeptides: peptides with cysteine sulfur to α-carbon crosslinks
This section focuses on a small but growing class of ribosomally synthesized peptides that have undergone post-translational modification to introduce intramolecular linkages between cysteine sulfur and the α-carbon of another residue. Although some diketopiperazines (cyclodimers of amino acids) such as gliotoxin and aranotin have long been known to have sulfur to α-carbon linkages,330 those compounds are made by non-ribosomal peptide synthetase systems. At least for the case of gliotoxin, the sulfur is introduced by nucleophilic attack of glutathione on a diketopiperazine N-acyl imine.331 The first report of a ribosomally-made peptide with a sulfur to α-carbon linkage was the structural elucidation in 2003 of subtilosin A.332 In the meantime at least six such structures have been reported from Bacillus species. These include a naturally occurring hemolytic subtilosin A mutant (T6I)333 from B. subtilis, the anti-clostridial Trn-α and Trn-β from B. thuringiensis (together forming the two-component system thuricin CD),334 thurincin H from another strain of B. thuringiensis,335 and the cannibalistic sporulation killing factor (SKF) from B. subtilis (Fig. 19).336 The term sactibiotic (ulfur to lpha-arbon an) has been suggested to describe peptides in this group.337 The more general term sactipeptide may be appropriate as some compounds with such a structural feature but lacking antimicrobial activity may be discovered in the future. The sulfur to α-carbon linkage is also found in cyclothiazomycin, a thiopeptide from Streptomyces hygroscopicus that has a C-terminal cysteine sulfur to α-carbon link with an alanine.338–340
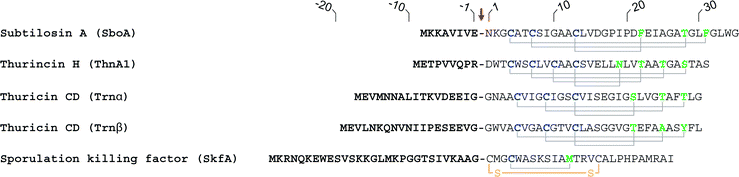 | ||
| Fig. 19 Sequences of sactipeptides based on structural genes. The arrow indicates the site of cleavage of the N-terminal leader peptide. Subtilosin A and SKF are then cyclized via an amide bond between the amino group of residue 1 and the C-terminal carboxyl. The other peptides are not cyclized in this fashion, but all have cysteine residues linked to α-carbons. | ||
All sactipeptides are relatively hydrophobic, and in the cases examined by NMR spectroscopy, have a 3D solution structure that resembles a hairpin with hydrophobic residues pointing to the exterior (Fig. 20). These include subtilosin A,332,341 Trn-α and Trn-β,342 and thurincin H.335 In subtilosin A and its T6I variant,333 as well as in SKF,336 the C- and N-termini at the ends of the hairpin are cyclized to form an amide bond. Both circular and open hairpin structures are conformationally restricted by the crosslinks of cysteine sulfur to the α-carbon of residues on the opposite side of the structure (Fig. 20). This unusual linkage is relatively stable under acidic conditions, but treatment with base and reducing agent rapidly cleaves it to form a free cysteine and a reactive N-acyl imine intermediate341 that can tautomerize to a more stable enamine form. An analogous process resulting in a related thiol elimination product occurs during mass spectrometric analysis, and can thereby give the initial impression that the cysteine residues are not modified and that the residues originally linked at the α-carbon are dehydroamino acids.334–336
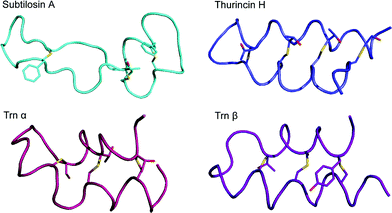 | ||
| Fig. 20 Three dimensional solution structures of sactipeptides. Subtilosin A has S to Cα crosslinks between C4 and F31, C7 and T28, and C13 and F22. Thurincin H has such links between C4 and S28, C7 and T25, C10 and T22, and C13 and N19. Trn-α has links between C5 and T28, C9 and T25, and C13 and S21. Trn-β has links between C5 and Y28, C9 and A25, and C13 and T21. SKF (not shown, 3D structure not reported) has a single S to Cα crosslink between C4 and M12. It also has a disulfide bond between C1 and C16. | ||
Most of the compounds isolated thus far having this modification show some antimicrobial activity, but the detailed mechanisms are not well understood. The high potency, and in some cases narrow specificity (e.g. Trn-α and Trn-β),343 of antimicrobial action suggests that many of these molecules require recognition of a receptor molecule in the cell membrane of target organisms. Thus far only the interaction of subtilosin A with lipid bilayers has been examined in detail.344,345 Subtilosin A adopts a partially buried orientation in such lipid bilayers. Interestingly, subtilosin A shows spermicidal activity although it is not haemolytic at low concentrations.346 However, replacement of its threonine-6 with isoleucine gives a variant that is potently haemolytic.333 As expected for a bacteriocin produced by a Gram-positive organism (B. subtilis), subtilosin A displays good activity against other Gram-positive species. Surprisingly, this effect is inhibited by EDTA through an unknown mechanism.347 However, at high concentrations in the presence of EDTA, subtilosin A can penetrate the outer membrane of some Gram-negative species to exert an antimicrobial effect.
The key sulfur to α-carbon crosslinks in subtilosin were proposed to be generated by a single radical S-adenosylmethionine (SAM) enzyme AlbA,348 a hypothesis that was recently confirmed by in vitro reconstitution of enzyme activity.349 AlbA was shown to sequentially install the crosslinks of all three cysteines with one threonine and two phenylalanine residues on the linear subtilosin precursor still bearing the leader peptide.349 The presence of the leader was essential for activity, and the enzyme distinguished between different residues to which the cysteine is coupled. Similarly, a single gene for a radical SAM enzyme is present in the cluster that is proposed to be responsible for generating all four crosslinks in thurincin H,350 and two such genes are found in the thuricin CD cluster, presumably one for crosslinking Trn-α and the other for modifying Trn-β.334 The requirement for such radical SAM enzymes has recently been used for genome mining to identify other possible peptides that may have analogous modifications.4,5 In addition to their involvement in sactipeptide biosynthesis, radical SAM proteins are also found near peptides that do not contain Cys and that therefore cannot make thioether linkages between Cys sulfurs and alpha carbons. Examples are found in the proteusins and PQQ biosynthetic pathways (sections 4 and 20). Hence, the presence of radical-SAM proteins must be evaluated in the context of the entire gene cluster and/or structure elucidation of the resulting RiPP before functional roles can be assigned.
12.1 Specific recommendations for sulfur to α-carbon linked peptides
To broaden the scope with respect to possible future biological activities that may be discovered, these compounds should be called sactipeptides rather than sactibiotics. A common gene nomenclature should be adopted for this group, at least for those cases where the genes are homologous. This nomenclature should be based on the thuricin CD genes,334 using the designation “A” for the precursor peptide, “C” and “D” for the radical-SAM protein(s), and “F” and “G” for ABC-transporters. The designation “P” should be used for currently unidentified proteases that may be encoded in biosynthetic gene clusters that await discovery.13 Bacterial head-to-tail cyclized peptides
Although many peptides are intramolecularly cyclized via amide bonds, this section focuses exclusively on relatively large bacterial peptides (35–70 residues) that are ribosomally synthesized and have a peptide bond between the C and N termini. As discussed below, these peptides are distinguished from other head-to-tail cyclized peptides discussed in this review (cyanobactins, amatoxins, orbitides, and cyclotides) by both their size and the biosynthetic machinery that achieves macrocyclization. The most well-studied and famous of these is enterocin AS-48, a potent antimicrobial agent isolated from Enterococcus,351 but over the last decade at least nine others have been purified and characterized (Fig. 21).200 All are bacteriocins from Gram-positive organisms. They include: butyrivibriocin AR10 (BviA),352 gassericin A (reutericin 6) (GaaA),353,354 circularin A (CirA),355 subtilosin A (SboA),332,341,348,356 uberolysin (UblA),357 carnocyclin A (CclA),358,359 lactocyclicin Q (LycQ),360 leucocyclicin Q (LcyQ),361 garvicin ML (GarML)362 and the cannibalistic sporulation killing factor (SKF) of Bacillus subtilis.336 Recent reviews have appeared on the structures, properties and genetics of production of this group,55,200 and more specifically, on enterocin AS-48.351 | ||
| Fig. 21 Comparison of the sequences of some cyclic bacteriocins based on the structural genes. The arrow indicates the cleavage site for the N-terminal leader, whose removal is followed by amide bond formation between the amino group of residue 1 and the carboxyl of the C-terminal residue. | ||
Although most of these peptides show little sequence homology, they tend to be resistant to high temperature (100 °C) treatment, to cleavage by many proteases, and to alterations in pH. They are usually quite hydrophobic and appear to exert their antimicrobial activity by targeting the cell membranes of other bacteria to create pores and cause ion permeability with consequent cell death. They have been classified into three groups.200,359 Type i are the AS-48 like peptides that also includes uberolysin, circularin A, lactocyclicin Q, and carnocyclin A. These peptides tend to be cationic at physiological pH and have relatively high isoelectric points (pI > 9). Type ii are the gassericin like peptides that includes butyrivibriocin AR10. These are anionic or neutral at physiological pH and have lower isoelectric points. Subtilosin A (35 residues) and its variants333 comprise an unrelated group that are not only much smaller, but also have three sactipeptide crosslinks as discussed in the previous section. The SKF peptide is also small (26 residues), has one sactipeptide link and one disulfide bridging the ring and is more closely related to subtilosin A than the larger circular peptides.336 The genetic determinants of a number of circular bacteriocins have been reported, and they can be located either on the chromosome or on plasmids. The structural genes all encode N-terminal leader peptides that are cleaved during maturation, but the length of these can vary from 2 residues (lactocyclicin Q) to 35 residues (AS-48). A significant number of other genes (up to 10 genes including the structural bacteriocin gene) are often necessary to obtain full production of the cyclic antimicrobial peptide. Many of these are predicted to code for membrane-bound proteins of uncertain function.
The three dimensional structures of enterocin AS-48 (X-ray crystallography),363 carnocyclin A (NMR spectroscopy),359 and subtilosin A (NMR spectroscopy)332,341 have been reported. Despite low sequence similarity, the three dimensional structures of the large bacteriocins AS-48 (70 residues) and carnocyclin A (60 residues) are remarkably similar and have a saposin fold of 5 and 4 α-helices, respectively (Fig. 22). Structural predictions based on the sequences of the other large type i and type ii cyclic bacteriocins suggests that they too may have saposin fold arrangements with four-helix bundles capped by either another α-helix as in AS-48 or with a less structured loop that replaces it for the shorter peptides of 60 residues.359 Despite similarities in three dimensional structures, the biological functions of these circular peptides appear to be quite different. For example, carnocyclin A is monomeric and shows anion binding and membrane transport properties.364 In contrast, AS-48 is dimeric and the dynamic reorganization of the interface between the two units is believed to play a key role in membrane pore formation.363 Subtilosin A is much smaller (35 residues) and has a quite different structure that does not belong to the saposin fold (see section 12).332,341 For all of these cyclic peptides, the N-to-C cyclizations appear to rigidify the structures in such a way that a significant number of hydrophobic residues are displayed to the outside, which is presumably important for their antimicrobial function. However, the detailed mechanisms of action of most of these head-to-tail cyclized peptides are not known.
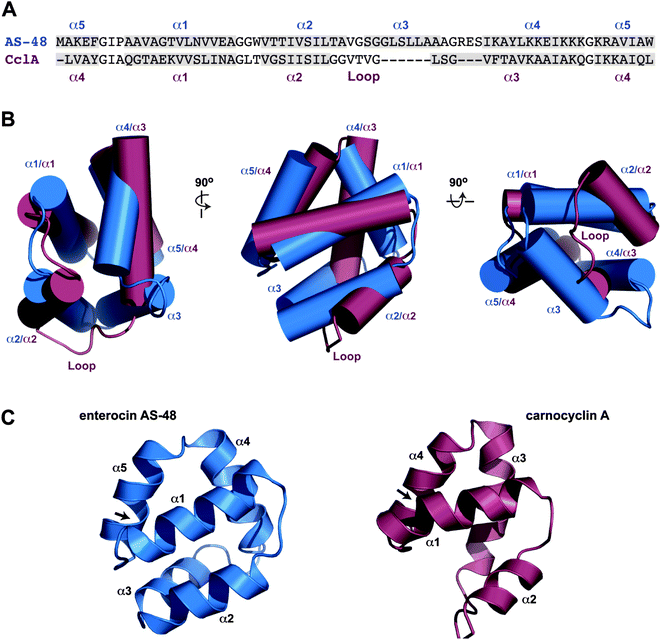 | ||
| Fig. 22 Comparison of the three dimensional structures of enterocin AS-48 (blue) and carnocyclin A (magenta). (A) Sequence comparison with helices and loop indicated. (B) Overlay of backbones of the two peptides showing the saposin fold and similarities in structure. (C) Ribbon diagrams for the two peptides. | ||
13.1 Biosynthesis of bacterial head-to-tail cyclized peptides
In terms of biosynthesis, the structural genes for these peptides indicate formation of a precursor with an N-terminal leader sequence that can vary greatly in length. The N-terminal leaders for leucocyclicin Q and lactocyclicin Q consist of only two residues.360,361 Circularin A and uberolysin are initially made with N-terminal leaders of three and six residues, respectively.355,357 In contrast, enterocin AS-48 has a 35 residue leader.351 In all cases discovered thus far, the structural gene shows no additional residues at the C-terminus, unlike the precursors for cyanobactins, amatoxins, cyclotides, and most orbitides (sections 6 and 14–16). This observation suggests that some type of activation of the C-terminal carboxyl group must be accomplished by another enzyme to assist amide bond formation with the amino group at the N-terminus after leader peptide cleavage. However, the identities of such activating enzymes have not been confirmed in the gene clusters for biosynthesis of these circular peptides (Fig. 23). Generally the head-to-tail ligation occurs between two hydrophobic residues, and membrane bound proteins may be involved. When either Met1 or Trp70, which are involved in the head-to-tail cyclization of enterocin AS-48, were substituted with alanine, the amide bond formation still proceeded albeit at lower production levels.365 Replacement of the last amino acid, histidine, of the leader peptide with isoleucine blocked formation of AS-48, suggesting that enzymatic recognition of that residue is essential for the leader peptide cleavage reaction.365 With the exception of the gene clusters for subtilosin A and SKF, the operons for production of circular peptide bacteriocins generally contain a membrane-bound protein of unknown function referred to as DUF95.55 This protein may be involved in the maturation process and/or cyclization.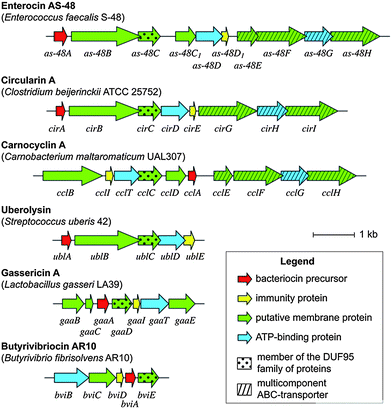 | ||
| Fig. 23 Comparison of gene clusters for biosynthesis of large ring bacteriocins. | ||
It is interesting to note that all the gene clusters for the circular bacterial peptides also contain ATP-binding proteins that could potentially activate the C-terminal carboxyl group for cyclization, although they may be responsible for other processes involving export. All the clusters also have a small immunity protein of unknown function that may at least partly protect the producing organism from its own cyclic antimicrobial peptide.55
13.2 Specific recommendations for bacterial N-to-C cyclized peptides
Historically, a common gene nomenclature has not been adopted for this group (Fig. 23), except for using the designation “A” for the precursor peptide. Since the “B” designation has been mostly, but not uniformly, used for the putative membrane protein, we recommend using this designation for new gene clusters that may be discovered in the future.14 Amatoxins and phallotoxins
The amatoxins and phallotoxins are produced by several species of mushrooms in the genera Amanita, Galerina, Lepiota, and Conocybe.54 They are N-to-C cyclized peptides of 7 and 8 residues, respectively, that also contain several other post-translational modifications including a tryptathionine formed by crosslinking a Cys to a Trp residue (Fig. 24A).366 The tryptathionine in α-amanitin is oxidized to the sulfoxide and hydroxylated on the indole in addition to other post-translational modifications such as a bishydroxylated Ile and cis-4-hydroxyPro. The phallotoxins are structurally similar but have one amino acid less. The amatoxins potently inhibit RNA polymerase II,367 whereas the phallotoxins such as phalloidin bind to actin368 and have been used to stain the cytoskeleton. The precursor peptides contain a 10-residue leader peptide, a 7- or 8-residue core peptide for phallotoxins and amatoxins, respectively, and a C-terminal recognition sequence (Fig. 24B).22 Based on genomic survey sequences and chemical characterization, the core peptide of this family of ribosomal peptides can range in size from 6 to 10 amino acids. Like the cyanobactins and cyclotides discussed elsewhere in this review, the leader and recognition sequences are highly conserved, but the core peptide is more variable. Interestingly, the P1 position for both proteolytic events that excise the core peptide from the precursor peptide is occupied by a proline. The proteins that carry out cyclization, hydroxylation of Leu, Ile and Pro, epimerization (for phallotoxins which contain one D-amino acid), and crosslinking of the Cys and Trp residues are currently still unknown, but a Pro oligopeptidase has been shown to cleave the precursor peptides after the Pro residues in an ordered manner,369 just as was found for the cyanobactins. α-Amanitin is also biosynthesized on ribosomes and initially processed by a Pro oligopeptidase in Galerina marginata, a mushroom not closely related to Amanita.370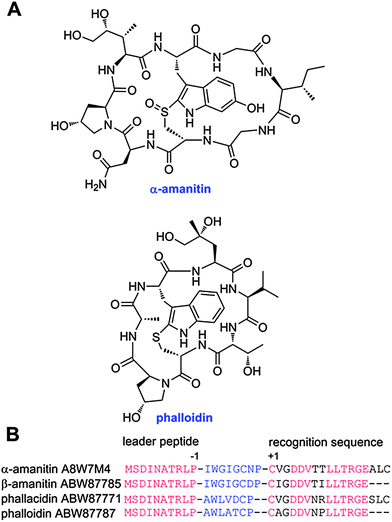 | ||
| Fig. 24 A. Chemical structures of a representative amatoxin and phallotoxin. B. Alignment of a select number of precursor peptides illustrating the highly conserved leader peptide and recognition sequences in red. The core peptide that is post-translationally modified, excised, and cyclized is shown in blue font. For beta-amanitin and phalloidin, the sequences are from genomic DNA whereas the other two sequences are from cDNA. Because of an intron in the genomic DNA, the last three amino acids of the recognition sequence are not known with certainty for beta-amanitin and phalloidin and are represented with dashes. | ||
15 Cyclotides
The name cyclotides was introduced for ribosomally-synthesized peptides from plants that are characterized by a head-to-tail cyclic peptide backbone and a cystine knot arrangement of three conserved disulfide bonds.371 They were originally discovered in plants of the Rubiaceae (coffee) and Violaceae (violet) families but have since also been found in the Cucurbitaceae (squash) and Fabaceae (legume) families. They are expressed in many plant tissues, including leaves, stems, flowers and roots. Dozens to hundreds of different cyclotides are expressed in an individual plant and there appears to be very little crossover of cyclotides between different plants, i.e., most plants have a unique set of cyclotides.The prototypic cyclotide, kalata B1, from the African herb Oldenlandia affinis (Rubiaceae) comprises 29 amino acids, including the six conserved Cys residues that form the signature cyclic cystine knot (CCK) motif of the family (Fig. 25).372 The backbone segments between successive Cys residues are referred to as loops and the sequence variations of cyclotides occur within these loops. Cyclotides have been classified into two main subfamilies, Möbius or bracelet, based on the presence or absence of a cis X-Pro peptide bond in loop 5 of the sequence. A smaller, third group is referred to as the trypsin inhibitor subfamily. This third subfamily has high sequence homology to some members of the knottin family of proteins from squash plants and its members are also referred to as cyclic knottins.373
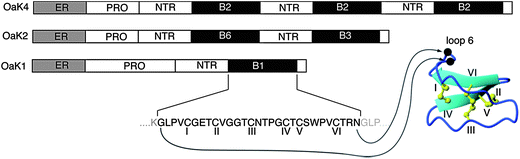 | ||
| Fig. 25 Structure of kalata B1 and cyclotide precursor proteins. A schematic representation of three precursor proteins from O. affinis is shown at the top of the figure. The proteins comprise an endoplasmic reticulum signal sequence labeled ER, a leader sequence (comprising a pro-region and a conserved repeated fragment labeled NTR) and either one or multiple copies of the core peptides. A short hydrophobic C-terminal recognition sequence is present in each precursor. At the bottom of the figure the amino acid sequence of kalata B1 is shown, with the cysteine residues labeled with Roman numerals. The cleavage sites for excision of a core cyclotide domain with an N-terminal Gly and a C-terminal Asn, which are thought to be subsequently linked by an asparaginyl endopeptidase, are indicated by arrows. The location of the ligation point, which forms loop 6 of the mature cyclic peptide, is shown on the right. Parts of the precursor protein flanking the mature domain are shown in lighter shading. Figure reprinted with permission from Daly et al.379 | ||
Thus far more than 200 sequences of cyclotides have been reported and they are documented in a database dedicated to circular proteins called CyBase (www.cybase.org.au).374 They range in size from 28–37 amino acids and are typically not highly charged peptides. Aside from their disulfide bonds375 and head-to-tail cyclized backbone, no other post-translational modifications have been found thus far in cyclotides. Cyclotides appear to be ubiquitous in the Violaceae but are more sparsely distributed in the Rubiaceae, occurring in about 5% of the hundreds of plants screened thus far. The reports of cyclotides in other plant families are more recent and there is limited information available on the distribution of cyclotides in these families. Nevertheless, it appears that cyclotides are a very large family of plant proteins, potentially numbering in the tens of thousands.376
Cyclotides are gene-encoded and are processed from larger precursor peptides. The precursors of Rubiaceae and Violaceae cyclotides are dedicated proteins, whose purpose appears to be only to produce cyclotides. By contrast, cyclotides in Clitoria ternatea, a member of the Fabaceae family, are produced from a chimeric precursor protein that also encodes an albumin,377,378 a situation similar to that recently reported for a small cyclic peptide trypsin inhibitor from sunflower seeds.377 Thus, there appear to be multiple types of precursors leading to circular proteins in plants. In the case of Rubiaceae and Violaceae cyclotides, the leader sequence is considered to comprise a pro-region and an N-terminal region (NTR) that is repeated in some genes along with the adjacent core peptide region, as illustrated in Fig. 25.
15.1 Biological activities of cyclotides
Cyclotides are thought to be plant defence molecules, given their potent insecticidal activity against Helicoverpa species.380–383 However, they also have a broad range of other biological activities, including anti-HIV,384 antimicrobial,385,386 cytotoxic,387 molluscicidal,388 anti-barnacle,389 nematocidal,390,391 and haemolytic activities.392 Some of these activities are of potential pharmaceutical interest, and because of their exceptional stability, cyclotides have also attracted attention as potential protein engineering or drug design templates.393,394The diverse range of activities of cyclotides seems to have a common mechanism that involves binding to and disruption of biological membranes.395 For example, electron micrographs of Helicoverpa larvae fed a diet containing cyclotides at a similar concentration to that which occurs naturally in plants show marked swelling and blebbing of mid-gut cells.380 Larvae that have ingested cyclotides are markedly stunted in their growth and development, presumably as a result of this disruption of their mid-gut membranes. A range of biophysical studies396–398 have confirmed membrane interactions for both Möbius and bracelet cyclotides, and detailed information is available on the residues involved in making contact with membrane surfaces. Furthermore, electrophysiological and vesicle leakage studies have confirmed the leakage of marker molecules through membranes treated with cyclotides. It appears that cyclotides are able to self-associate in the membrane environment to form large pores.
15.2 Cyclotide biosynthesis
As indicated in Fig. 25, cyclotides are derived from precursor proteins that encode one or more copies of the core peptide sequences, similar to the precursor peptides for cyanobactins (section 6) and some orbitides (section 16.3). For example the Oak1 (![[O with combining low line]](https://www.rsc.org/images/entities/i_char_004f_0332.gif) ldenlandia
ldenlandia ![[a with combining low line]](https://www.rsc.org/images/entities/i_char_0061_0332.gif) ffinis
ffinis ![[k with combining low line]](https://www.rsc.org/images/entities/char_006b_0332.gif) alata B
alata B![[1 with combining low line]](https://www.rsc.org/images/entities/char_0031_0332.gif) gene) encodes an 11 kDa precursor protein that contains an endoplasmic reticulum (ER) signal, leader peptide, kalata B1 core peptide, and a C-terminal peptide region, whereas the Oak4 gene encodes a precursor containing three copies of kalata B2 (Fig. 25).382 The single or multiple copies of the cyclotide domains are flanked in each case by N-terminal and C-terminal recognition sequences that are thought to be important for the processing reactions.
gene) encodes an 11 kDa precursor protein that contains an endoplasmic reticulum (ER) signal, leader peptide, kalata B1 core peptide, and a C-terminal peptide region, whereas the Oak4 gene encodes a precursor containing three copies of kalata B2 (Fig. 25).382 The single or multiple copies of the cyclotide domains are flanked in each case by N-terminal and C-terminal recognition sequences that are thought to be important for the processing reactions.
Because of the presence of the ER signal it is believed that cyclotide precursors are probably folded in the ER prior to processing (excision and cyclization) of the cyclotide domain. Protein disulfide isomerases (PDIs) are often involved in the folding of disulfide-rich proteins399 and in vitro experiments have shown increased yields of folded cyclotide kalata B1 in the presence of PDI,399 although so far there have been no definitive studies on the involvement of PDI for in planta cyclotide folding. A recent study has shown that cyclotides are targeted to vacuoles in plant cells and this is where the excision and cyclization processes are thought to occur.400
The N-terminal repeat (NTR) region of cyclotide precursors has been found to adopt an α-helical structure as an isolated synthetic peptide,62 but the significance, if any, of this is not yet known. It has been proposed that it might represent a recognition sequence for cyclotide folding by PDI, but, interestingly, cyclotide precursors from the Fabaceae plant Clitorea ternatea lack the NTR region seen in Violaceae or Rubiaceae precursors. The nature of the processing reaction(s) occurring at the N-terminal end of the cyclotide domain is still essentially unknown.
However, there is strong evidence that asparaginyl endoprotease (AEP) activity is responsible for processing at the C-terminal end of the cyclotide domain including involvement in the cyclization process.401,402 With just one exception (circulin F) all cyclotides have an Asn or Asp residue as the C-terminal end of the core peptide, and an AEP is in principle able to cleave after this residue. It has been proposed that this cleavage reaction occurs contemporaneously with ligation to the earlier-released N-terminus of the core peptide region. Mutagenesis experiments in transgenic tobacco and Arabidopsis plants have shown a vital role for the N-terminal Asn residue, as well as other key flanking residues.401 Similarly, gene silencing experiments in which AEP activity is knocked down show decreased production of cyclic peptide in transgenic plants.402 Overall, the processing of cyclotides can be summarized as involving excision and cyclization from a pre-folded precursor.
15.3 Specific recommendations for the cyclotide family
At present there is no common gene nomenclature for cyclotide precursors and given the limited knowledge of the full complement of associated biosynthetic genes encoding proteins involved in processing it seems premature to propose one. A naming scheme for the mature cyclic peptides that involves an indicative and pronounceable acronym based on the binomial name of the plant species in which the cyclotide was first discovered has been proposed,403 but has not been uniformly followed so far. We recommend that this suggestion be followed for individual peptide names; this does not exclude the additional use of ‘family’ names for subgroups of similar cyclotides, for example the cycloviolacins.40416 Orbitides: plant cyclic peptides lacking disulfide bonds
16.1 Perspective
In 1959, Kaufman and Tobschirble405 reported the discovery of a nonapeptide present in a slime that had precipitated from a container of flaxseed oil. Curiously, this peptide composed of proteinogenic amino acids was cyclized by an N-to-C terminal amide bond. Cyclolinopeptide A, cyclo-[ILVPPFFLI], was the first of more than 168 peptides discovered in plants with similar structures.198,406 Small plant cyclic peptides consisting entirely of alpha-amide linkages of natural amino acids were called Caryophyllaceae-like homomonocyclopeptides in a recent review.198 It is suggested here that the name orbitides be used to refer to all N-to-C cyclized peptides from plants that do not contain disulfides. Orbitides are made by at least nine individual plant families including the Annonaceae, Caryophyllaceae, Euphorbiaceae, Lamiaceae, Linaceae, Phytolaccaceae, Rutaceae, Schizandraceae, and Verbenaceae. The peptides range in size from five to twelve amino acids. It is notable that two-amino-acid cyclic peptides from plants have been observed but it is uncertain whether these arise from a ribosomal precursor.The 98 Caryophyllaceae-like non-redundant orbitides represented on cybase.org.au collectively contain 719 amino acids (Spring 2012). Based on simple statistics, many amino acids are highly underrepresented. Unlike most of the RiPPs discussed in this review, cysteine is the least abundant amino acid observed in orbitides as it appears just twice in a single core peptide predicted based on homology with a precursor EST sequence. Acidic residues (Asp, Glu) and their amides (Asn, Gln), basic residues (Lys, Arg), and His are also rare. Methionine, while rare overall, is well represented in the amino acids of currently known orbitides from Linum usitatissimum. Clearly, the amino acid composition of the orbitides described to date is significantly biased towards hydrophobic amino acids. Glycine and proline are found in most peptides, and serine, threonine and tyrosine occur 25, 31, and 54 times respectively.
Orbitides are composed largely of unmodified L-amino acids (e.g.Fig. 26) although there are several exceptions.198 Methionine oxidation is frequently observed in these peptides, but it has not been determined whether this is a true modification or an artifact of isolation. Determination of the stereochemistry of the sulfoxide by amino acid analysis may be able to address this question. Hydroxylated amino acids, including gamma-hydroxy isoleucine and delta-hydroxy-isoleucine,407,408 have also been observed as well as an unusual N-methyl-4-aminoproline present in cyclolinopeptide X.409 Additionally, D-tryptophan was observed in a peptide discovered in Schnabelia oligophylla.410
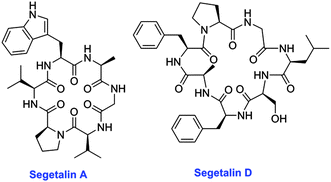 | ||
| Fig. 26 Structures of segetalins containing six and seven amino acids, isolated from Vaccaria segetalis (Caryophyllaceae).406 | ||
The rigid structure of orbitides and low amino acid complexity has enabled extensive structural studies.198 They have been crystallized from organic solvents, and solution structure information has been obtained using CD, NMR spectroscopy, and other methods. The published three-dimensional structures show a single stable conformation for most orbitides. Prolyl residues may be either in the cis or trans conformation depending on the peptide, and structures are typically stabilized by internal hydrogen bonds between amide backbone atoms.198
Only recently has it been shown that these peptides are derived from post-translational modification of ribosomally synthesized precursor peptides.411 Based on observations of the apparent segregation of segetalin peptides in closely related varieties of Vaccaria hispanica and the segregation of peptides in double haploid lines derived from crosses, it was reasoned that a RiPP origin was possible. A strategy for searching for the genes encoding these compounds was developed based on sequence tags obtained primarily by tandem mass spectrometry. The N- or C-terminus of the linear precursor peptide cannot be determined from the amino acid sequence analysis of the cyclized peptide, therefore, EST libraries were searched for all possible linear sequences of a peptide.411 Positive hits were found for cyclolinopeptide G, F and D (Fig. 27) as well as a number of segetalin orbitides. Previously these sequences had been disregarded due to their small size relative to other sequences. Expression of a cDNA encoding the precursor sequence of segetalin A in transformed S. vaccaria hairy roots from the variety White Beauty, which is unable to produce segetalin A, led to the production of the peptide.411 Furthermore, extracts of developing S. vaccaria seeds were shown to catalyze the production of segetalin A from a synthetic peptide precursor representing the gene product. Moreover, the presence of two segetalins, J [cyclo-(FGTHGLPAP)] and K [cyclo-(GRVKA)] were predicted by sequence analysis of S. vaccaria cDNA. A focused search by liquid chromatography/mass spectrometry confirmed the existence of these compounds. Sequence analysis also predicts the presence of similar precursor genes in Dianthus caryophyllus, Citrus spp., Linum usitatissimum, Gypsophila arrostil, and Jatropha spp.
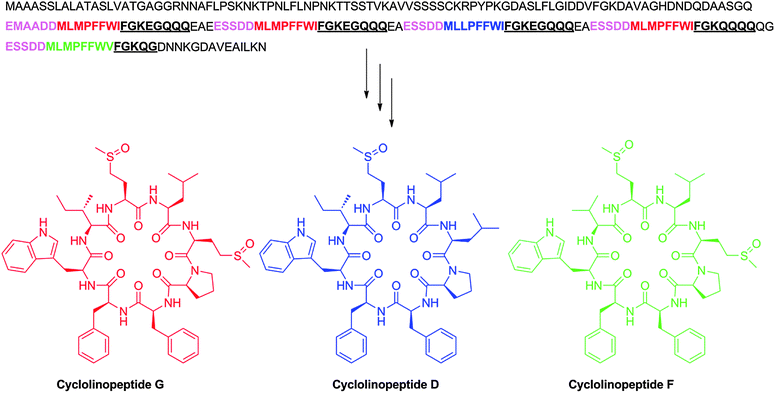 | ||
| Fig. 27 Amino acid sequences of cyclic peptides arising from Linaceae species Linum usitatissimum. The RiPP core peptides putatively encoding cyclolinopeptides D, F, and G is shown in blue, green, and red, respectively, with putative recognition sequences shown in pink and underlined font. | ||
16.2 Biosynthesis of orbitides
Searches of EST libraries derived from members of the family Caryophyllaceae has yielded gene sequences that appear to encode the precursor peptides.411 To date such sequences have only been observed in Caryophyllaceae mRNA as no genome databases of Caryophyllaceae species are available. The mRNA sequences are ubiquitously expressed in many plant tissues and the copy number of these sequences is high relative to other genes. The mechanism of processing of the precursor peptides is not known at this time. They typically contain a leader peptide, one core peptide, and a recognition sequence (Table 2). Caryophyllacaceae orbitide precursor sequences observed thus far exhibit a highly conserved recognition sequence with nine invariant residues. mRNA sequences of Dianthus caryophyllus and Gypsophila arrostil encode conservative recognition sequences that are highly similar, but identification of the cyclic peptides is necessary to confirm that the mRNAs lead to peptide products. Curiously, an EST sequence from Gypsophila arrostil contains stop codons and may be a pseudogene (Table 2). The leader sequences of all observed mRNA sequences that potentially encode RiPPs of caryophyllaceae orbitides contain 13 amino acids (Table 2).



| Rutaceae | |||
|---|---|---|---|
| a Products of bolded core peptides have been observed by MS/MS. b Amino acid X reflects a stop codon within the sequence. | |||

|

|

|
|
The sequences of Rutaceae precursor peptides are known from both mRNA and genomic DNA as genome sequences are available for Citrus sinensis412 and Citrus clementina.413 The observed DNA sequences are short and there are many more potential RiPP coding sequences than known peptides. The recognition sequence is highly conserved in both length and amino acid composition (Table 2), whereas the leader sequences are more variable.
16.3 Biosynthesis of orbitides in Linaceae
The genomic and mRNA sequences from the flaxseed variety Linum usitatissimum cv Bethune have been determined.414,415 Blast searches of EST sequences derived from this cultivar using a search string based on the known amino acids of cyclolinopeptide G revealed a sequence that potentially includes five core peptides in a single precursor peptide (Fig. 27).416 Three of these sequences corresponded to cyclolinopeptide G. The ability of a single precursor to be processed to multiple cyclic peptides is unique among known precursor peptides of orbitides but is also found in the precursors to cyclotides (section 15), cyanobactins (section 6), and possibly microviridins (section 11). A search of a genome database that comprised shotgun sequences of Bethune flax using the polyRiPP precursor sequence revealed both the DNA sequence that encodes cyclolinopeptides D, F and G (Fig. 27) as well as an additional sequence that encodes a polyRiPP precursor that may be processed to cyclolinopeptides A, B and E.41616.4 Biosynthesis of orbitides in Euphorbiaceae
The interest in Jatropha curcas as a potential biofuel crop has resulted in generation of a substantial database of EST sequences and a genomic sequence is also available.417 A search of EST libraries produced from Jatropha curcas conducted for this review identified two mRNA sequences that encode the two known cyclic peptides previously isolated from J. curcas, curcacyclines A and B.418 These sequences have no C-terminal recognition sequence as the gene terminates with the core peptide, thus requiring activation of the C-terminal carboxylate, similarly to the bacterial N-to-C cyclized peptides discussed in section 13. The leader sequences of both peptides contain 36 amino acids and are highly conserved (Table 3). Many other cyclic peptides have been isolated from Jatropha,419 and may have a RiPP origin.

|
16.5 Relationship to other RiPP subfamilies
Orbitides have been observed in a range of plant tissues including roots, stems, leaves, developing seeds, mature seeds, fruits, and peels,198 and the corresponding mRNA has been detected in the various plant tissues. To date over one hundred unique peptides have been discovered, but only a small number of the species that are likely to produce them have been surveyed. Combined the families of Rutaceae, Linnaceae, Caryophyllaceae, Verbenacea, Annonaceae, Lamiaceae, Phytolaccaceae, Euphorbiaceae, and Schizandraceae have thousands of members.420 It is probable that a thorough study will reveal hundreds if not thousands of additional peptides.Although the cyclotides and orbitides appear to be quite different in size, structure, and primary composition there is a striking similarity in gene organization between the cyclotides and the orbitides from flax (cyclolinopeptides). Genes in both groups comprise, at the carboxy terminal end of the gene, multiple copies of the cyclic peptide sequences flanked by highly conserved regions. In contrast DNA sequences that comprise orbitides from Caryophyllacea genera such as Vaccaria and Rutaceae genera such as Citrus comprise the highly conserved flanking sequences but contain only a single cyclopeptide sequence.
This apparent difference in gene structure could result from the fact that the published Vaccaria and Citrus sequences were only derived from mRNA and as such may only represent pieces of pre-mRNA from a more complex gene. This possibility is supported by the fact that the majority of these DNA sequences are lacking a methionine amino terminus of the precursor peptide and the cDNAs of those that do have an intact amino terminal portion do not have features such as a tata-box expected in a promoter. The sequences flanking the precursor peptides have a high AT content and features typical of introns.
It is noteworthy that despite the relatively extensive size of the flax EST library and the abundance of cyclolinopeptides A, C and E, the mRNA from this gene has not been found and the gene was only uncovered when the genomic sequence of flax was determined. The unusual structure of the mRNAs from these genes apparently precludes discovery by standard cloning methodology. In consideration of this issue, and very few genomic sequences that are yet available, it is possible that the organization of the genes in other plant families that are known to make orbitide type cyclopeptides will be similar to that of the cyclotides and the orbitides from flax.
16.6 Biological activity
The innate biological role of orbitides is currently unknown. Generally the lipophilic composition of the orbitides is consistent with a membrane active role.421 Attention has largely been focused on the use of these compounds as drug candidates and in agriculture.198 Preparations containing orbitides have been tested using a number of bioassays that do not always relate to mechanism of action. For example, orbitides are classified as cytotoxic, antiplatelet, antimalarial, immunomodulating, and inhibiting T cell proliferation.198 These assays provide promising leads to understanding biologial activity and potential applications of the peptides, but a universal mechanism of action is unlikely. Analysis of the effects of peptides using in vitro assays has lead to a more precise understanding of the possible mechanism of action of some peptides. Curcacycline B enhances prolyl cis–trans isomerase activity of human cyclophilin B,422 and cycloleonuripeptide D inhibits cyclooxygenase.423 Structure activity studies of pseudostellarin A showed that γ- and β- turn structures were fixed by trans-annular hydrogen bonds between Gly and Leu.424 This rigid conformation was hypothesized to be important in tyrosinase activity exhibited by this compound. Similarly, the Trp-Ala-Gly-Val sequence in segetallin A and B has been suggested to play an important role in their estrogen-like activities.42516.7 Specific recommendations for the orbitide family
We propose that the name “orbitide” replace the longer designation “Caryophyllaceae like homomonocyclopeptides” and is used for all plant N-to-C cyclized peptides that do not contain disulfide crosslinks. It is too early to classify the various orbitides when so few confirmed examples are available.17 Conopeptides and other toxoglossan venom peptides
The venoms of predatory marine snails (such as species in the genus Conus, the cone snails) are a complex mixture of ribosomally-synthesized peptides that are post-translationally modified to various extents.426 Although the possibility that these venoms may also contain gene products from bacterial symbionts cannot be completely excluded, the vast majority of peptides are encoded in the genome of the marine snail itself, expressed in the epithelial cells lining the venom duct, and secreted into the lumen of the duct. Transcriptome analyses that provide quantitative estimates of the mRNAs expressed in the snail's venom duct typically reveal that a major fraction of all transcripts encode the secreted venom peptides (see for example ref. 10, 427, and 428).Biologically-active peptides from animal venoms are an increasingly important class of natural products.429 In total, these comprise many millions of different peptides, each presumably evolved to alter the function of a specific physiological target. Like the other RiPPs discussed in this review, venom peptides are generally post-translationally processed from larger precursors, to yield peptides that are mostly in a size range of 30–90 amino acids after processing.24 The venom peptides evolved by marine snails differ from most animal venom peptides in several distinct ways. The best characterized of the marine snail peptides, those of the cone snails (generally known as conopeptides or conotoxins) are mostly smaller (10–30 amino acids), with a significantly higher density of disulfide crosslinks than most animal venom peptides. Another feature is the larger diversity of post-translational modifications in these peptides compared to other venom peptides (e.g., scorpion or snake peptides).426 This diversity of post-translational modifications make them more like the RiPPs from bacterial and plant sources.
As detailed below, marine snail venom peptides have an entirely different set of post-translational modifications compared to other RiPPs. One likely fundamental cause for the lack of overlap in PTMs is that the evolution of post-translational modification in marine snail venom peptides has followed a very different path from post-translational modifications of peptidic natural products produced by bacteria. In the evolution and adaptive radiation of venomous animals, a delivery system must be evolved; thus, venoms are intrinsically injectable mixtures of natural products. Since a delivery route has already been established when the pharmacological agents in venoms are being evolved, peptides and polypeptides that suit the biological purposes of the venomous animal can immediately be selected for. In general, there are no major barriers to delivery of peptides and polypeptides once venom is injected into the targeted animal and most molecular targets are plasma membrane proteins. Thus, post-translational modifications are presumably evolved primarily to increase potency and selectivity, rather than to improve the efficiency of delivery, which may have been an evolutionary driving force for bacterial RiPPs.
As a class, marine snail peptides are structurally extremely diverse, and a comprehensive discussion of all of the structural and functional subclasses that have been described would require a much longer exposition than warranted in the present overview. Venom peptides of marine snails run the gamut from ribosomally-synthesized peptides that are quite minimally post-translationally modified, to peptidic natural products that have been subjected to more diverse post-translational modifications than most other RiPPs. The different post-translational modifications that have been reported in conopeptides to date are shown in Table 4. One feature that makes the literature on molluscan venom peptides particularly confusing compared to the other RiPPs discussed in this review is that the nomenclature in use names these peptides on the basis of their functional specificity and taxonomic origins, rather than their structural and/or chemical features.
| Modification | Amino acid residue | Final product |
|---|---|---|
| C-terminal amidation | Glycine | C-terminal –CO–NH2 |
| N-terminal cyclization | Glutamine | N-terminal pyroglutamate |
| Hydroxylation | Proline; Valine; Lysine | 4-Hydroxyproline; D-γ hydroxyvaline; 5-hydroxylysine |
| Carboxylation | Glutamate | γ-Carboxyglutamate |
| Sulfation | Tyrosine | Sulfotyrosine |
| Bromination | Tryptophan | 6-Bromotryptophan |
| Oxidation | Cysteine | Disulfide bridge |
| Glycosylation | Threonine | Various O-glycosyl amino acids |
| Epimerization | Valine; Leucine; Phenylalanine; Tryptophan | D-Valine; D-leucine; D-phenylalanine; D-tryptophan |
| Proteolysis | Various | Mature conopeptides |
17.1 Biological and toxicological perspectives
Venomous marine snails comprise a forbiddingly complex assemblage of >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 species.430 Taxonomists typically refer all venomous species to a single superfamily (“Conoidea”) or suborder (“Toxoglossa”), referred to as “conoideans” or “toxoglossans.” This is likely the most species-rich of all molluscan lineages with a large number of extremely small, deep-water forms that mostly remain undescribed. Venoms of all toxoglossans are produced in a specialized venom duct; the analysis of any individual venom has invariably demonstrated that it is a complex mixture with essentially all of the biologically-active components being peptides. Although, cone snails (genus Conus or family Conidae) comprise less than 5% of the total biodiversity of venomous molluscs, greater than 99% of all published papers in the scientific literature concern peptides derived from cone snails.
000 species.430 Taxonomists typically refer all venomous species to a single superfamily (“Conoidea”) or suborder (“Toxoglossa”), referred to as “conoideans” or “toxoglossans.” This is likely the most species-rich of all molluscan lineages with a large number of extremely small, deep-water forms that mostly remain undescribed. Venoms of all toxoglossans are produced in a specialized venom duct; the analysis of any individual venom has invariably demonstrated that it is a complex mixture with essentially all of the biologically-active components being peptides. Although, cone snails (genus Conus or family Conidae) comprise less than 5% of the total biodiversity of venomous molluscs, greater than 99% of all published papers in the scientific literature concern peptides derived from cone snails.
A nomenclature for peptides from other lineages of venomous marine snails has recently been suggested, based on family groups within the superfamily Conoidea. Thus, in addition to conopeptides from cone snails, venom peptides from species in the family Turridae are referred to as turripeptides, and from the family Clathurellidae as clathurellipeptides, etc. (12 other family groups have been proposed).431,432 Very few venom peptides from toxoglossans other than the cone snails have been characterized, and these mostly conform to the generalizations about conopeptides that we outline below. Because conopeptides have been much more extensively investigated, our focus will be on these peptides, but the reader should be aware of the vast trove of uncharacterized, post-translationally modified peptides from the other toxoglossan lineages.
One general feature of all marine snail venoms is their complexity both in the number of different peptides found, as well as in their structures. Furthermore, the genes encoding these venom peptides undergo accelerated evolution, so that there is essentially no molecular overlap in the modified core peptides found in venoms of even closely-related cone snail species. Most genes that encode these peptides have introns, and the initial transcripts have to undergo splicing before the mRNA can be exported from the nucleus. A small minority of modified core peptides are apparently encoded by genes without introns.
The translated precursor peptides have the canonical organization characteristic of RiPPs, with the leader peptide varying considerably in size; most conopeptide precursor peptides are 70–120 amino acids, and the majority of well-characterized modified core peptides found in cone snail venoms fall in the range of 12–30 amino acids. One unique feature of conopeptide superfamilies is the remarkable conservation of signal sequences within all members of the superfamily, providing an unequivocal marker for the gene superfamily to which a specific venom peptide belongs. Although there may be 50–200 different conopeptides found in the venom of an individual species, the vast majority of these typically belong to only 5–8 superfamilies.
17.2 Conopeptide structures and biological activities
Conopeptides comprise a more or less continuous spectrum of post-translational modification, with several widely distributed in all animal venom peptides: proteolytic cleavage, disulfide bond formation, C-terminal amidation, and cyclization of N-terminal Gln residues to pyroglutamate. Some small, disulfide-rich, endogenous peptides such as endothelin are similar in their general biochemical characteristics to many conopeptides. A superfamily of larger conopeptides (called conkunitzins) have clearly co-opted a well-characterized endogenous structural framework known as the Kunitz domain, which is widely found in protease inhibitors such as bovine plasma trypsin inhibitor (BPTI). Some venom peptides are expressed in other tissues of the cone snail, presumably with endogenous physiological functions (e.g., the conopressins). However, the origin of the majority of conopeptide superfamilies has not been established and in contrast to snake toxins and scorpion peptides that have 1–2 conserved structural frameworks, each gene superfamily represents at least one distinctive structural fold.Many conopeptides have a surprising array of diverse covalent modifications, each carried out by a different post-translational modification enzyme. One notable difference from RiPPs from bacteria and archaea is that they are encoded by eukaryotic genes that are not organized in a biosynthetic gene cluster. Adding to the complexity is that in a number of cases, peptides that are post-translationally modified at a particular locus are found in the venom along with the corresponding peptide with the identical amino acid sequence that has not been post-translationally modified. This trend is particularly observed for hydroxylation of proline to 4-hydroxyproline. The functional significance of the incomplete penetrance of some post-translational modifications has not been critically assessed.
For most conopeptides, the most important structural feature that confers the correct three-dimensional conformation to the biologically-active peptide are disulfide bonds. Conopeptides can have an unprecedented density of disulfide crosslinks; some conopeptides are only twelve amino acids in length, with three disulfide crosslinks. Since six Cys residues forming three disulfides can generate fifteen distinct isomers, the oxidative folding pathway of an unmodified core peptide to the specific biologically-active disulfide crosslinked isomer may well be enzyme catalyzed.433 This “conotoxin oxidative folding problem” is a challenging issue that has not been mechanistically elucidated, and it is likely that formation of the correct disulfide crosslinks in conotoxins requires special post-translational enzymes and accessory factors.
The pharmacological characterization of conopeptides to date has demonstrated great diversity of targets. A large majority of the molecular targets of conopeptides that have been elucidated are ion channels and receptors present on the plasma membrane; many are expressed in nervous systems. Thus, conopeptides have become a mainstay of ion channel pharmacology,434 and are widely used as basic research tools in neuroscience.
It is perhaps easiest to illustrate the range of conopeptide functional and structural diversity by focusing on their direct biomedical applications. At present, several conopeptides have been developed as lead compounds for therapeutic drugs, and one conopeptide is an approved commercial drug in the U.S. and European community (Prialt, for intractable pain). Some of the conopeptides that have reached human clinical trials are shown in Fig. 28.435 They demonstrate the structural and functional diversity in this compound family.
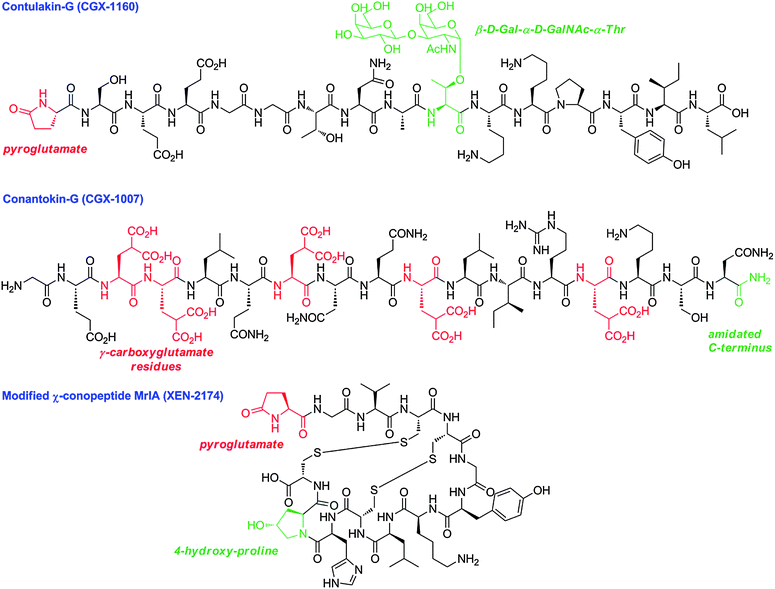 | ||
| Fig. 28 Structures of three post-translationally modified conopeptides that reached human clinical trials. | ||
Two of the peptides in Fig. 28, contulakin G and conotokin G, both characterized from the venom of Conus geographus, reached Phase I clinical trials for pain and epilepsy, respectively. Contulakin G has a glycosylated threonine residue that is essential for its potent analgesic activity. It is targeted to a G-protein coupled receptor (the neurotensin receptor) and has striking sequence homology at its C-terminal end to neurotensin. Interestingly, although the glycosylation makes the peptide less potent as an agonist at the neurotensin receptor, it renders the peptide a more effective analgesic. Fig. 28 also shows the structure of a modified conopeptide χ-MrIA that is in clinical development and inhibits the norepinephrine transporter; this analgesic peptide has been re-engineered by chemists to introduce a modification (pyroglutamate) in addition to the preexisting two disulfide bridges and 4-hydroxy-Pro found in the native peptide isolated from Conus marmoreus.
17.3 The conantokin superfamily and γ-carboxylation of glutamate
Although the three-dimensional conformation of most modified conopeptides is determined by the pattern of disulfide crosslinking, a notable exception are members of the conantokin superfamily, including conantokin G (Fig. 28).436 Conantokin G is an NMDA receptor antagonist, highly selective for one particular subtype of this receptor (i.e., receptors with NR2B subunits). For all conantokin peptides, an unusual post-translational modification, the carboxylation of glutamate residues to form γ-carboxyglutamate (γ or Gla) is required. When these modified amino acids are spaced every three or four residues apart in the primary amino acid sequence, the peptide folds into a stable helical conformation in the presence of Ca2+.437 In essence, the modification of glutamate to γ-carboxyglutamate creates a functional group for coordinating Ca2+; Two adjacent Gla residues on the same side of a helix chelate Ca2+ in a manner analogous to EDTA or EGTA. Thus, the post-translational modification of glutamate residues by the enzyme γ-glutamyl carboxylase is essential for the biologically-active conformation of conantokin G.A similar motif is found in many of the proteases comprising the mammalian blood-clotting cascade (such as prothrombin). The presence of a domain containing Gla residues is essential for the accumulation of blood-clotting factors on the membranes of activated platelets, thereby triggering the formation of blood clots. However, in mammals this modification is only found in large proteins. Among RiPPs, the γ-carboxylation of glutamate appears to be unique to toxoglossan venom peptides (the presence of this modification has been established both in conopeptides, and in certain turripeptides from venomous snails in the family Turridae).
17.4 Biosynthesis of conopeptides
The enzyme converting glutamate to Gla is a highly-conserved, widely-distributed enzyme.438,439 Not only do the enzymes that carry out this modification in cone snails and in the mammalian blood-clotting system have high sequence similarity, they are also functionally homologous—both require reduced vitamin K as a cofactor, and are stimulated by binding to a recognition sequence that determines which glutamate residue will be modified. An even more unexpected and striking piece of evidence for the common evolutionary origin of these enzymes was the discovery that the ten different introns in the human gene are completely conserved in their location in the Conus gene. Thus, if an intron is found between codons in the human gene, it is also found between the homologous codons in Conus. If an intron is between the second and third nucleotide of a codon in the human gene, it is in precisely the same location as the homologous codon in the Conus gene sequence. The introns themselves have no detectable sequence similarity, and the cone snail introns are longer than those in humans.Interestingly, the same enzyme was identified in Drosophila (where its function is unknown); in the Drosophila gene only three introns remain (all at precisely conserved locations), and they are all shorter than either the corresponding mammalian or Conus introns. Thus, in this instance, the post-translational modification enzyme that the cone snails have recruited for one gene superfamily of RiPPs is clearly an ancient enzyme with its origins early in metazoan evolution. It has been speculated that because the post-translational modification promotes the formation of short α-helices, that the carboxylation of glutamate is a relict of a pre-cellular, pre-ribosomal biotic world. This one case of a well-characterized post-translational enzyme that acts on conopeptides suggests that the PTMs used in marine snail venoms were already present early in evolution, and subsequently recruited in the snails venom duct for novel biological purposes.
18 Glycocins
The glycocins are a small group of glycosylated antimicrobial peptides produced by bacteria. The two currently characterized members, sublancin 168 produced by Bacillus subtilis 168 and glycocin F produced by Lactobacillus plantarum KW30, are both glycosylated on Cys residues,440,441 a very unusual form of glycosylation. In sublancin, a glucose is attached to one of five Cys residues in the precursor peptide (Cys22) with the other four Cys residues engaged in two disulfides (Fig. 29). Glycocin F has a very similar arrangement of disulfide bonds as well as a connecting loop that is glycosylated, but an N-acetylglucosamine is conjugated to a Ser in this loop rather than to a Cys residue. The S-linked glycosylation in glycocin F is located on a Cys that is the last residue of the peptide. The NMR structure of glycocin F has been reported showing two alpha helices held together by the two disulfide linkages.442 These two helices also appear to be the recognition elements for the S-glycosyltransferase SunS involved in sublancin formation.443 This glycosyltransferase has specificity for Cys because a Cys22Ser mutant of the SunA precursor peptide was not accepted as substrate.440 However, the enzyme was promiscuous with respect to its nucleotide-sugar donor and could add a variety of different hexoses to Cys22. Furthermore, SunS glucosylated mutant peptides in which the position of the Cys was varied along the loop connecting the two helices.443 Hence, it appears that as long as one or more Cys residues are present in this loop, SunS will carry out the S-linked glycosylation. Unlike most biosynthetic enzymes discussed in this review, SunS does not require the leader peptide for its activity.440 Whether one or more glycosyltransferases are responsible to install the two glycosylations in glycocin F is currently not known. Furthermore, the mode of action of sublancin 168 and glycocin F has yet to be established.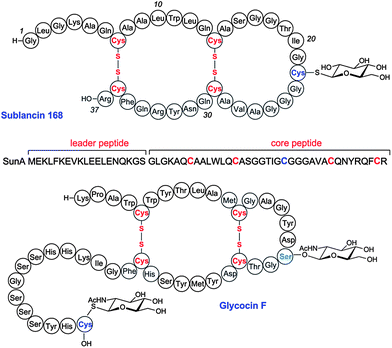 | ||
| Fig. 29 Structures of two glycocins. The precursor peptide that is transformed into sublancin by disulfide formation and S-glycosylation is shown below the sublancin 168 structure. | ||
18.1 Recommendations for the glycocin family
The glycocin family currently encompasses both O- and S-linked bacterial glycopeptides with antimicrobial activities. Although at present both members contain S-linked sugars, the name glycocin is not limited to S-linked glycopeptides as future studies may reveal new members that only contain O-linked and/or N-linked carbohydrates. The suffix –cin in glyco presently limits the family to antimicrobial peptides. It is possible that additional bacterial glycosylated peptides with other functions are awaiting discovery in which case a broader nomenclature may be required, similar to the extension of lantibiotics to lanthipeptides and sactibiotics to sactipeptides. The designation “A” is recommended for the precursor peptides and the designation “S” for the glycosyltransferase, “I” for self-resistance proteins, “T” for transporters, and “B” for thiol-disulfide isomerases.
19 Autoinducing peptides, ComX, methanobactin, and N-formylated peptides
The autoinducing peptides (AIPs) are quorum sensing peptides produced by most members of the low G + C Gram-positive bacteria (Firmicutes).444–446 In all of these AIP signals, the presence of a cyclic ester or thioester is a conserved feature, which differentiates the AIP structures from linear regulatory peptides. The Staphylococcus aureus AIPs have received the most attention, and in producing strains the extracellular AIP binds to a receptor on a cognate AgrC histidine kinase, inducing a regulatory cascade that increases expression of virulence factors.447 The S. aureus thiolactone-containing peptide is generated from a larger precursor peptide AgrD (Fig. 30A) by AgrB,448,449 a unique transmembrane protein that has endopeptidase activity.449–451 The molecular details of the thiolactonization process are currently not known but the general process of excision of a cyclic peptide from a linear precursor has similarities with the cyclization processes described for the cyanobactins, amatoxins, cyclotides, and some orbitides. The ArgD peptides are about 45 amino acids in length and contain three different regions: an N-terminal amphiphatic helical region,452 a core peptide that encodes the residues that will form the mature AIP,448 and a C-terminal, highly negatively charged region.449 The N-terminal helix is thought to be important for interaction of the peptide with the membrane and with AgrB. Overall, there is considerable variation in the sequences of the three regions in different species with only the Cys required for thiolactone formation (or Ser for lactone formation) conserved in the core peptide. The negatively charged C-terminal putative helix is more conserved than the N-terminal helix.449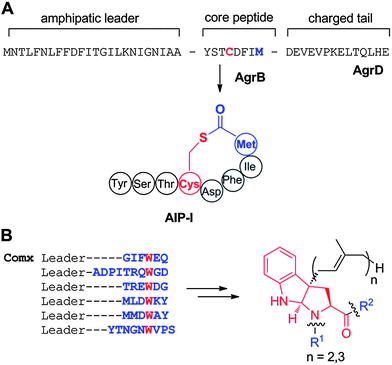 | ||
| Fig. 30 A. Structure of AIP-I and the ribosomally synthesized precursor peptide from which it is produced. B. Structures of ComX and its derivatives and the ribosomally synthesized precursor peptides from which they are produced. The red font Trp is converted to the structure shown by cyclization of the amide nitrogen of Trp onto C2 of the indole and prenylation at C3. After leader peptide removal, the modified Trp is present in short peptides with the sequences of R1 and R2 shown in blue font. | ||
Another example of ribosomally synthesized quorum sensing compounds are ComX and its derivatives produced by bacilli. These compounds are biosynthesized from a much longer precursor peptide that contains a leader peptide and a variable core peptide (Fig. 30B). After unusual prenylation and cyclization steps involving a fully conserved Trp residue (Fig. 30B),453 the leader peptide is released and the mature compound is produced.454
Methanobactins are copper chelators, or chalkophores (cf. siderophores for iron chelators) that are used by methanotrophic bacteria that use methane as their sole carbon source. Copper acquisition is critical for these Gram-negative bacteria as methane oxidation is carried out by a copper-dependent monooxygenase.455 The initially reported structure of the methanobactin isolated from Methylosinus trichosporium OB3b determined by X-ray crystallography456 has been revised on the basis of NMR studies and more recent high resolution X-ray structures (Fig. 31).457,458 It contains two oxazolones and a disulfide. When the genome of M. trichosporium was sequenced, a small gene was identified that encodes for a peptide that could serve as the precursor for methanobactin (Fig. 31). The peptide contains a putative 19-amino acid leader peptide and a 11-amino acid core peptide.459 If this is indeed the precursor to methanobactin, post-translational modifications must involve transformation of two of the Cys residues to the oxazolones with an adjacent thioamide, deamination of an N-terminal Leu, and formation of the disulfide. Interestingly, homologous precursor peptide genes and flanking genes occur outside the methanotrophs, such as in the rice endophyte Azospirillum sp. B510.459 Further studies may provide insights into this fascinating system.
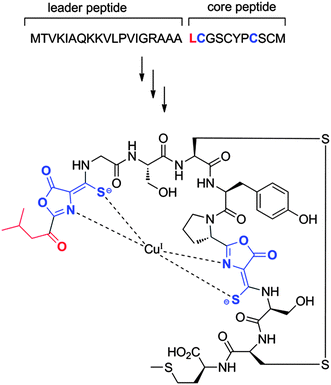 | ||
| Fig. 31 Currently accepted structure of methanobactin from M. trichosporium OB3b and the precursor peptide from which it may be derived. | ||
Peptidogenomic approaches have uncovered several short N-formylated linear peptides.6 Bacterial proteins are typically formylated on the N-terminal Met residue, because translation initiates with formyl-methionine. The formyl group is usually removed by a peptide deformylase, but in some short peptides, the deformylation step appears to be incomplete or omitted altogether. In most cases, the functional relevance is unclear and the lack of deformylation may simply be the result of translation termination before the short peptide emerges from the ribosome to be deformylated. But in the case of the formylated δ-toxin of Staphylococcus aureus, deformylation appears to be inhibited by depletion of iron during stationary phase.460 Furthermore, the formylated toxin, as well as other N-formylated peptides,461 serves as a chemoattractant for neutrophils.460 N-Terminal formylation is also found in microcin C discussed above. It is possible that N-formylation helps protect the peptides from aminopeptidases, similarly to other modifications of the N-terminus found in other RiPPs (e.g. acetyl,462 lactyl,140,146 methyl,150,180,181,463 2-oxobutyryl,163 and 2-oxopropionyl464). We note that N-formylated peptides are not RiPPs since they are not post-translationally modified, but they are included here as they are often detected when searching for RiPPs by mass spectrometry.
20 PQQ, pantocin, and thyroid hormones
A number of other molecules that are smaller than the compounds discussed thus far are generated from a ribosomally synthesized peptide. Pyrroloquinoline quinone (PQQ, Fig. 32A) is a cofactor for several bacterial dehydrogenases.465 In a series of studies in the late 1980s and early 1990s, PQQ was first shown to be generated from a ribosomally synthesized precursor peptide.466–468 Currently, the biosynthetic genes have been found in over 100 bacteria.8 The precursor peptide is PqqA, a 22 amino acid peptide containing a conserved glutamate and tyrosine that are oxidatively morphed into the final structure and excised from the peptide by a series of enzymatic reactions (Fig. 32A).466,469 Genetic studies have implicated PqqACDE to be required for PQQ biosynthesis,8,470,471 but the details of the PTM process are largely unknown. PqqC has been shown to catalyze the final oxidation step in an oxygen dependent mechanism.472 PqqB and PqqE, a putative non-heme iron oxygenase and a radical-SAM protein respecutively, carry out currently unknown transformations.8,473 PqqE and PqqD form a protein complex that may be important for the overall timing of the reactions and/or the protection of reactive intermediates.8,474 Although PqqF is not absolutely required, its sequence places it in the zinc-dependent protease family and it may be involved in the peptide excision process. At present the role of the flanking amino acids in the PqqA precursor peptide is not known, but it is likely that they serve a similar function as the recognition sequences discussed for cyanobactin, amatoxin, and cyclotide biosynthesis. With only two, non-sequential amino acids of the precursor peptide ending up in the final product, the term core peptide is not pertinent. As mentioned before, PQQ is made by an overall process that is similar to the other RiPPs discussed here, but the final product is no longer a peptide. | ||
| Fig. 32 Structures of (A) PQQ, (B) coelenterazine, (C) the pantocins, and (D) the thyroid hormones T3 and T4. For PQQ and the pantocins, the ribosomally synthesized precursor peptides from which they are produced are also depicted. | ||
Another potential small molecule excised from a larger protein is coelenterazine (also called coelenterate-type luciferin, Fig. 32B). This molecule is used as a light emitting substrate by various luciferases and Ca2+-binding photoproteins in marine organisms. It is not well defined what organisms produce coelenterazine and its biosynthesis is currently unresolved. However, two observations suggest it may be ribosomally synthesized. First, feeding experiments indicate the compound is made from L-Tyr and L-Phe.475 Second, and more intriguing, expression in E. coli of a mutant of the green fluorescent protein from Aequorea victoria in which the conserved sequence FSYG leading to the GFP chromophore was replaced with FYYG resulted in the production of coelenterazine.476 At present, no details are available regarding this remarkable observation, but it suggests that coelenterazine can be made from a ribosomally synthesized protein.
Another example of a small molecule produced from a much larger ribosomally synthesized precursor is pantocin A produced by the bacterium Pantoea agglomerans (Fig. 32C).477 The gene paaP in its biosynthetic cluster encodes a 30-residue peptide with the sequence -Glu-Glu-Asn- near its center.478 Mutagenesis and heterologous expression studies demonstrated that the final product is formed from PaaP by two enzymes, PaaA and PaaB, and unknown proteases in the host.478 PaaA has significant sequence similarity to the ThiF/MoeB family of proteins that adenylate carboxyl groups, hinting that it may be involved in cyclization of the side chain of the first Glu with the amide nitrogen of the second Glu residue. PaaB has a domain with homology to the Fe(II)-α-ketoglutarate dependent family of enzymes, suggesting it may catalyze an oxidation step that will be required for formation of the final product. As with the PQQ pathway, the roles of the flanking residues in the precursor peptide is not clear but they may be recognition sequences for PaaA and PaaB, and like many leader peptides, they may keep the biosynthetic intermediates inactive.
Perhaps the most remarkable example of construction of a biologically active small molecule from a ribosomally synthesized precursor is the assembly mechanism of the thyroid hormones 3,3′,5,5′-tetraiodothyronine (T4) and 3,30,5-triiodothyronine (T3) (Fig. 32D). These compounds are not made from free Tyr but rather from Tyr imbedded in thyroglobulin, a very large glycoprotein comprised of two identical subunits of 330000 Da each.479 The aromatic rings of proximal Tyr residues are diiodinated by thyroid peroxidase, and this enzyme subsequently carries out the oxidatively coupling reaction of two iodinated Tyr residues. Hydrolysis then releases T4 and a small amount of T3 from incompletely iodinated Tyr. Since T3 is the major active form, T4 is converted to T3 by iodothyronine deiodinases.479 Although clearly this biosynthetic pathway is very different from that of the other compounds discussed in this review, the thyroid hormone examples provide yet another demonstration of the diverse ways that nature makes use of ribosomal products to generate small molecules.
21 Shorthand linear notations for RiPPs
As a consequence of their generally large molecular structures, shorthand nomenclature has been introduced for several of the subclasses of RiPPs (e.g. for lanthipeptides in Fig. 4). Here, we suggest a more general shorthand notation that can represent structures of RiPPs in linear format, which is useful for description of structures in abstracts of manuscripts and conference proceedings. The proposed notation uses a prefix code of 3–4 characters used to specify the type of modification for each modified amino acid, which are identified by their residue number in the core peptide sequence. For instance, 1-NαAc refers to acetylation of the N-terminus of residue 1 (e.g. in microviridin B, Fig. 33), 22-ΔCO2 refers to decarboxylation of residue 22 (e.g. gallidermin, Fig. 33), and 1-CγOH refers to hydroxylation of the γ-carbon of residue 1 (e.g. α-amanitin, Fig. 34). For post-translational modifications that introduce crosslinks, the amino acids that are crosslinked are indicated first by residue number followed by the type of crosslink. For instance, 3R-7-CβS refers to a crosslink between the β-carbon of amino acid 3 and the sulfur of residue 7, with the stereochemistry at Cβ being R (e.g. the A-ring of nisin, Fig. 33), 1-8-NαC refers to N-to-C cyclization between the N-terminal amino group of residue 1 and the C-terminal carboxylate of residue 8 (e.g. patellamide C, Fig. 33), 5-19-SS refers to a disulfide between residues 5 and 19 (e.g. kalata B1, Fig. 34), and 4-31-SCα refers to a crosslink between the sulfur of residue 4 to the α-carbon of residue 31 (e.g. in subtilosin, Fig. 34). All modifications in a given peptide are provided in front of the linear sequence of the core peptide in square brackets starting with the lowest residue numbers and working towards the higher residues. When multiple identical modifications are present, they are grouped together as illustrated for the five (methyl)lanthionine crosslinks in nisin (Fig. 33). The pre-fixes for a select number of PTMs discussed in this review are listed in Table 5, and examples of select RiPPs are shown in Fig. 33 and 34. Using the same reasoning, new pre-fixes can be readily introduced for the many PTMs that are not covered in Table 5.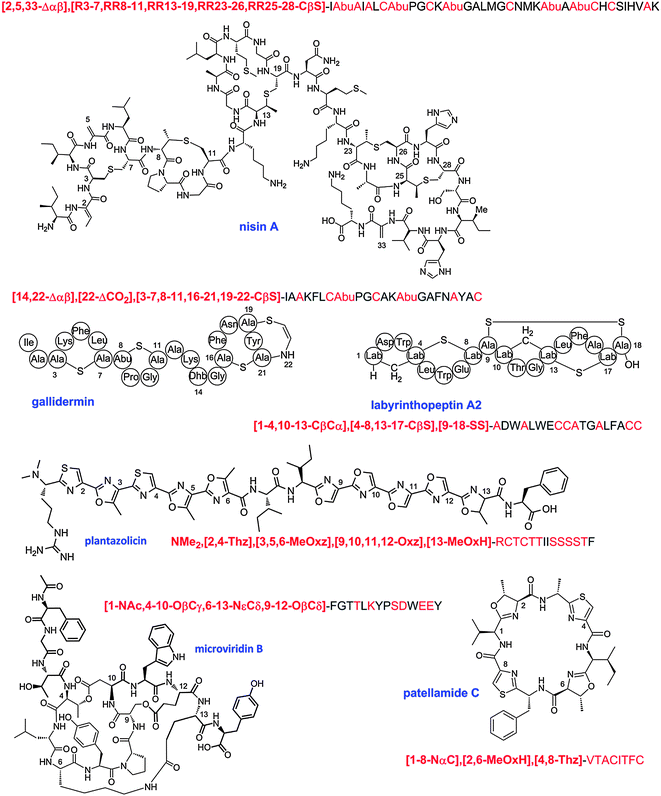 | ||
| Fig. 33 Select examples of the linear description of RiPPs. The linear description is shown above each chemical structure. The 3–4 character descriptors are shown in bold red font, the amino acids to which they refer are shown in non-bold red font. Abu, 2-aminobutyric acid. | ||
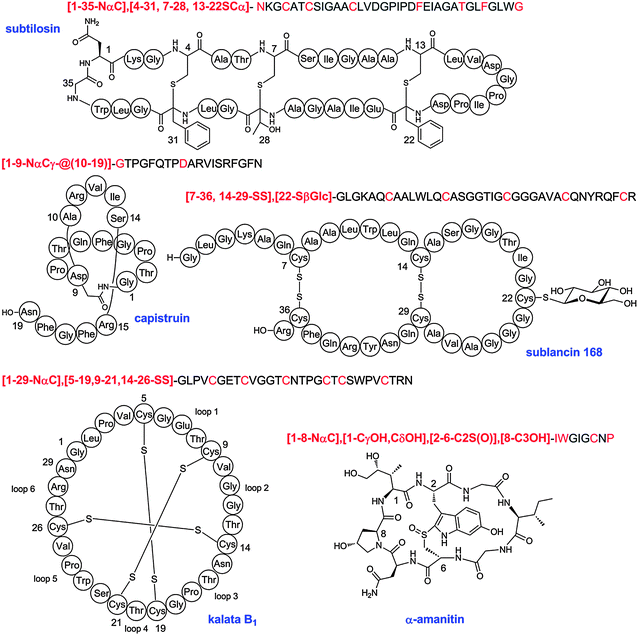 | ||
| Fig. 34 Additional examples of linear shorthand notation for RiPPs. | ||
| PTM | Shorthand notation | Structures and examples from this review | Figure | Occurrence |
|---|---|---|---|---|
| a In many cases, amidation of the C-terminus may be better described as cleavage of the bond between the amide nitrogen and the α-carbon of what was the C-terminal amino acid. b The peptide sequence determines which of the two possible notations should be used. For instance, a lanthionine formed from Cys1 and Ser2 would be SCβ whereas a lanthionine formed from Ser1 and Cys2 would be CβS. | ||||
| Single residue modifications | ||||
| 2,3-Dehydroamino acid | Δαβ | dehydroalanine | Lanthipeptides, LAPs, thiopeptides | |
| dehydrobutyrine | ||||
| nisin | 2/33 | |||
| lacticin 3147 | 4 | |||
| goadsporin | 9 | |||
| thiostrepton A | 12 | |||
| nosiheptide | 12 | |||
| thiomuracin | 12 | |||
| O-Phosphorylation of Ser/Thr | OβP | pSer, pThr | 2 | Lanthipeptides |
| Descarboxy | ΔCO2 | decarboxylated C-terminal residue | Lanthipeptides, linaridins, thiopeptides | |
| gallidermin | 33 | |||
| epidermin | 5 | |||
| cypemycin | 5 | |||
| micrococcin P1 | 12 | |||
| Cβ-hydroxylation | CβOH | e.g. β-hydroxy-Asp | Lanthipeptides, proteusins | |
| duramycin | 4 | |||
| e.g. β-hydroxy-Val, β-hydroxy-Asn | ||||
| polytheonamide A | 6 | |||
| Cγ-hydroxylation | CγOH | e.g. γ-hydroxy-Pro | Lanthipeptides, conopeptides, phallotoxins, amatoxins | |
| α-amanitin | 24/34 | |||
| MrIA | 28 | |||
| e.g. γ-hydroxy-Leu/Ile | ||||
| phalloidin | 24 | Phallotoxins, orbitides | ||
| Cδ-hydroxylation | CδOH | e.g. δ-hydroxy-Ile | Amatoxins, orbitides, conopeptides | |
| α-amanitin | 24/34 | |||
| Aromatic ring hydroxylation | C6OH | e.g. C6 hydroxy-Trp | Amatoxins | |
| α-amanitin | 24/34 | |||
| Cγ-carboxylation | CγCO2 | Glu carboxylation | Conopeptides | |
| conantokin-G | 28 | |||
| Cβ-methylation | CβMe | e.g. β-methyl-Val polytheonamides | 6 | Proteusins, bottromycins |
| bottromycin A2 | 14 | |||
| e.g. β-methyl-Pro, β-methyl-Phe | ||||
| bottromycin A2 | 14 | |||
| e.g. β-methyl-Met | ||||
| polytheonamide | 6 | |||
| N-terminal acetylation | NαAc | goadsporin | 9 | Lanthipeptides, microviridins |
| microviridin B | 18/33 | |||
| N-terminal mono or dimethylation | NαMe | Linaridins, LAPs, orbitides | ||
| NαMe2 | cypemycin | 5 | ||
| plantazolicin | 9 | |||
| N-terminal pyroglutamate formation | NαCδanhydro | contulakin-G | 28 | |
| MrIA | 28 | |||
| Glycosylation | Cys, Thr and Ser glycosylation | Glycocins, conopeptides | ||
| SβGlc | sublancin | 29/34 | ||
| SβGlcNAc | glycocin F | 29/34 | ||
| OβGlcNAc | glycocin F | 29 | ||
| OαGalNAc | GalNAc-Thr, contulakin-G | 28 | ||
| Epimerization | 2R | D-Ala in lacticin 3147 | 4 | Nearly all compound classes discussed in this review |
| polytheonamides | 6 | |||
| C-terminal amidationa | CONH2 | conantokin-G | 28 | Conopeptides, thiopeptides |
| nosiheptide | 12 | |||
| thiostrepton A | 12 | |||
| Crosslink of two residues | ||||
| Disulfide | SS | labyrinthopeptin | 4/33 | Lanthipeptides, cyanobactins, cyclotides, lasso peptides, conopeptides, glycocins, sactipeptides |
| ulithiacyclamide | 10 | |||
| siamycins | 17 | |||
| kalata B1 | 25/34 | |||
| MrIA | 28 | |||
| sublancin | 29/34 | |||
| glycocin F | 29 | |||
| Lanthionine | SCβ or CβSb | thioether | Lanthipeptides | |
| nisin | 2 | |||
| actagardine | 4 | |||
| duramycin | 4 | |||
| lacticin 3147 | 4 | |||
| SapT | 4 | |||
| Lanthionine sulfoxide | S(O)Cβ | actagardine | 4 | Lanthipeptides |
| Lysinoalanine | NεCβ | secondary amine | Lanthipeptides | |
| duramycin | 4 | |||
| α-carbon to β-carbon | CαCβ | labionin | Lanthipeptides | |
| labyrinthopeptin | 4/33 | |||
| Oxazole | Oxz | microcin B17 | 9 | LAPs, cyanobactins, thiopeptides |
| Oxazoline | OxH | patellamide A | 10 | LAPs, cyanobactins, thiopeptides |
| Methyloxazole | MeOxz | plantazolicin | 9/33 | LAPs, cyanobactins |
| goadsporin | 9 | |||
| microcyclamide | 10 | |||
| Methyloxazoline | MeOxH | plantazolicin | 9/33 | LAPs, cyanobactins |
| patellamide A | 10 | |||
| patellamide C | 33 | |||
| Thiazole | Thz | microcin B17 | 9 | LAPs, cyanobactins, thiopeptides, bottromycins |
| goadsporin | 9 | |||
| plantazolicin | 9/33 | |||
| patellamide A | 10 | |||
| patellamide C | 33 | |||
| microcyclamide | 10 | |||
| trichamide | 10 | |||
| bottromycin A2 | 14 | |||
| Thiazoline | ThH | trunkamide | 10 | LAPs, cyanobactins, thiopeptides |
| Tryptathionine | C2S or SC2b | thioether | Phallotoxins | |
| phalloidin | 24 | |||
| Tryptathionine sulfoxide | C2S(O) | α-amanitin | 24/34 | Amatoxins |
| α-carbon linked to sulfur | SCα (or CαS)b | thioether | Sactipeptides, thiopeptides | |
| subtilosin | 20/34 | |||
| thuricin CD | 20 | |||
| C- to N-terminal cyclization | NαC | patellamide A | 10 | Cyanobactins, amanitins, cyclotides, orbitides, sactipeptides |
| patellamide C | 33 | |||
| subtilosin | 20/34 | |||
| enterocin AS-48 | 22 | |||
| carnocyclin A | 22 | |||
| α-amanitin | 24/34 | |||
| kalata B1 | 34 | |||
| cyclolinopeptides | 27 | |||
| segetalins | 26 | |||
| N-terminal amine and Glu/Asp side chain | NαCδ | isopeptide | ||
| NαCγ | ||||
| N-terminal amine and Glu/Asp side chain peptide tail goes through the lactam | NαCδ@ | siamycin | 17 | Lasso peptides |
| NαCγ@ | capistruin | 17/34 | ||
| microcin J25 | 16 | |||
| Indication of size of peptide tail through lactam | NαCδ−@[9–21] | microcin J25 | 17 | Lasso peptides |
| NαCγ−@[10–19] | capistruin | 34 | ||
| Lys side chain with Glu side chain | CδNε or NεCδb | isopeptide | Microviridins | |
| microviridin B | 18/33 | |||
| Lys side chain with Asp side chain | NεCγ or CγNεb | isopeptide | Microviridins | |
| Asp side chain with Ser/Thr: | CγOβ or OβCγb | ester | Microviridins | |
| microviridin B | 18/33 | |||
| marinostatin 1–12 | 18 | |||
| Glu side chain with Ser/Thr: | CδOβ or OβCδb | ester | Microviridins | |
| microviridin B | 18/33 | |||
| Cys side chain and C-terminal carboxylate | SβCO | thioester | ||
| AIP-I | 30 | AIPs | ||
It is likely that the future will see more and more engineered, non-natural RiPP analogs having one or more amino acid substitutions, insertions, or deletions. Such molecules should be denoted by standard mutagenesis nomenclature. Analogs containing additional modified residues by in vitro synthesis or by synthetic biology approaches can in most cases also be designated by the standard nomenclature, e.g. a Met residue that is replaced by a chlorinated Trp residue at postion 17 in nisin would be designated as Met17ClTrp. In cases where modifications from different RiPP classes are combined into one molecule (e.g. a classical lantibiotic attached to a lasso-peptide-like ring), the new molecule should get a descriptive name (e.g. nisin[1–18]-GSG linker-capistruin[1–9]), accompanied by a description of the structure at the amino acid level according to the proposed nomenclature rules described in this paper. Use of newly invented names, such as lassonisin or micronisin to describe hybrids are discouraged as they would cause possible confusion, especially if many such analogs are prepared.
22 Outlook
The ribosomal origin of some of the classes of RiPPs discussed in this review has been known for quite some time, but for most classes this realization has been reached more recently as a result of the rapid advancement of DNA sequencing techniques. The availability of genome sequences first allowed the identification of putative precursor genes for known natural products. For compounds of bacterial, archaeal, and fungal sources, the convenient clustering of genes also provided immediate insights into the biosynthetic pathways, but deciphering biosynthetic pathways has been more challenging for compounds from higher organisms. In recent years, the use of genome information has been shifting slowly from identification of the genes for the biosynthesis of known compounds to genome mining for new compounds.1 RiPPs are particularly suited for genome mining because of the direct link between precursor gene and final product and because of the relatively short biosynthetic pathways, which lend them well for heterologous expression. This approach has allowed both isolation of new compounds from producing organisms,6,26,47,48,64,133,134,144,179,190,192,308,327,480,481 and heterologous production of compounds that are not produced under laboratory conditions by the organism containing the biosynthetic genes (“silent or cryptic clusters”).89 These studies either used genes encoding related precursor peptides as query for searching genomes, resulting in analogs of known compounds, or used the genes encoding key biosynthetic enzymes, often resulting in more distant analogs of known natural products.Although they have been quite successful, these genome mining strategies currently have two disadvantages. Unlike phenotypic screens for discovery of new natural products with a known bioactivity, the genome mining approach typically does not provide any clues about the biological activities of the products. A second current shortcoming of the genome mining approach is that compound classes that fall into new structural scaffolds are difficult to discover because neither their biosynthetic genes nor their precursor genes are known. The large number of short open reading frames in genomes that are not annotated during genome sequencing exercises suggests that such classes may be quite common. Even with these current limitations, genome mining is likely to become an even more active area of research that potentially may uncover compounds with exciting bioactivities because analysis of the currently available genomes for known classes illustrates the ubiquitous nature of RiPPs. Going forward, it will be important to use the most powerful bioinformatics methods to analyze the explosion of sequence information. In this respect, Hidden Markov Models (HMMs) can identify distantly related protein families when other methods do not.482–484 The recently developed, freely available genome mining tool Bagel2 (http://bagel2.molgenrug.nl/) also offers many features to screen for putative (modified) antimicrobial peptides based on a large set of search criteria.485 Another useful site in the context of posttranslational modifications is the RESID Database of Protein Modifications (http://www.ebi.ac.uk/RESID/index.html),486 which provides a comprehensive compilation of naturally occurring modifications annotated in the UniProt Protein Knowledgebase.
Caution is warranted with respect to drawing conclusions from bioinformatics alone as several common expectations about RiPP biosynthesis systems are not completely reliable. In particular, sequence similarity of precursor peptides does not guarantee that the final products belong to the same family of RiPPs. For example, lanthionine synthetases, cyclodehydratases, and radical SAM enzymes all act on members of the nitrile hydratase-like leader peptide family, as well as the nif11-like leader peptide family.3,26,45 Meanwhile, proximity between genes for a RiPP precursor and a maturase does not guarantee a target/substrate relationship. The genes encoding the precursor peptide, its maturation enzyme(s), and its export transporter can all be far apart in a genome.26,487
This review has attempted to bring together groups of scientists working on different classes of RiPPs to work out a common nomenclature within the class and across classes. We hope that similar concerted efforts will be used to classify new RiPP classes and their biosynthetic machinery that are yet to be discovered. In particular, we recommend in general that the gene encoding the precursor gene is always given the designation A. If surrounding genes have homology with enzymes used by the RiPP classes discussed in this review, we recommend trying to use a similar gene designation as that used previously to illustrate the connection. For all genes encoding proteins with unknown function, we recommend alphabetically naming the genes in the order they appear in the operon in the first disclosure of the gene cluster. All studies following the initial report of a new RiPP are strongly encouraged to use the same letter designations of the initial study such that a common nomenclature arises.
23 Acknowledgements
The recommendations in this review were compiled from the answers to two sets of successive questionnaires. Several discussion groups then made recommendations to the entire group of authors. The discussion groups were led by David Craik (cyclotides), Elke Dittmann (microviridins), Baldomero Olivera (conopeptides), Sylvie Rebuffat (microcins), Eric Schmidt (cyanobactins and LAPs), Wilfred van der Donk (lanthipeptides and glycocins), John Vederas (sactipeptides and N-to-C cyclized peptides), and Christopher Walsh (thiopeptides). The final write-ups for the several classes of compounds were provided by David Craik (cyclotides), Elke Dittmann (microviridins), Wendy Kelly (thiopeptides), Douglas Mitchell (LAPs), Joern Piel (proteusins), Baldomero Olivera (conopeptides), Martin Reaney (orbitides), Sylvie Rebuffat (microcins and lasso peptides), Eric Schmidt (cyanobactins), Wilfred van der Donk (introduction, lanthipeptides, linaridins, bottromycins, glycocins, quorum sensing peptides, PQQ, methanobactin, linear nomenclature, and outlook) and John Vederas (sactipeptides, N-to-C cyclized peptides, linear nomenclature). Coordination of the discussions and assembly and editing of the final review was done by Wilfred van der Donk. We thank Dr Qi Zhang (UIUC) for help with illustrations.24 References
- J. E. Velásquez and W. A. van der Donk, Curr. Opin. Chem. Biol., 2011, 15, 11 CrossRef.
- H. Wang, D. P. Fewer and K. Sivonen, PLoS One, 2011, 6, e22384 CAS.
- D. H. Haft, M. K. Basu and D. A. Mitchell, BMC Biol., 2010, 8, 70 CrossRef.
- K. Murphy, O. O'Sullivan, M. C. Rea, P. D. Cotter, R. P. Ross and C. Hill, PLoS One, 2011, 6, e20852 CAS.
- D. H. Haft and M. K. Basu, J. Bacteriol., 2011, 193, 2745 CrossRef CAS.
- R. D. Kersten, Y.-L. Yang, Y. Xu, P. Cimermancic, S.-J. Nam, W. Fenical, M. A. Fischbach, B. S. Moore and P. C. Dorrestein, Nat. Chem. Biol., 2011, 7, 794 CrossRef CAS.
- S. W. Lee, D. A. Mitchell, A. L. Markley, M. E. Hensler, D. Gonzalez, A. Wohlrab, P. C. Dorrestein, V. Nizet and J. E. Dixon, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5879 CrossRef CAS.
- Y. Q. Shen, F. Bonnot, E. M. Imsand, J. M. Rosefigura, K. Sjölander and J. P. Klinman, Biochemistry, 2012, 51, 2265 CrossRef CAS.
- Q. Qin, E. J. McCallum, Q. Kaas, J. Suda, I. Saska, D. J. Craik and J. S. Mylne, BMC Genomics, 2010, 11, 111 CrossRef.
- A. O. Lluisma, B. A. Milash, B. Moore, B. M. Olivera and P. Bandyopadhyay, Marine Genomics, 2012, 43 CrossRef.
- J. A. McIntosh, M. S. Donia and E. W. Schmidt, Nat. Prod. Rep., 2009, 26, 537 RSC.
- S. Duquesne, D. Destoumieux-Garzon, J. Peduzzi and S. Rebuffat, Nat. Prod. Rep., 2007, 24, 708 RSC.
- T. R. Klaenhammer, FEMS Microbiol. Rev., 1993, 12, 39 CAS.
- R. Finking and M. A. Marahiel, Annu. Rev. Microbiol., 2004, 58, 453 CrossRef CAS.
- M. A. Fischbach and C. T. Walsh, Chem. Rev., 2006, 106, 3468 CrossRef CAS.
- T. J. Oman and W. A. van der Donk, Nat. Chem. Biol., 2010, 6, 9 CrossRef CAS.
- L. Huo, S. Rachid, M. Stadler, S. C. Wenzel and R. Müller, Chem. Biol., 2012, 19, 1278 CrossRef CAS.
- W. J. K. Crone, F. J. Leeper and A. W. Truman, Chem. Sci., 2012, 3, 3516 RSC.
- J. P. Gomez-Escribano, L. Song, M. J. Bibb and G. L. Challis, Chem. Sci., 2012, 3, 3522 RSC.
- Y. Hou, M. D. Tianero, J. C. Kwan, T. P. Wyche, C. R. Michel, G. A. Ellis, E. Vazquez-Rivera, D. R. Braun, W. E. Rose, E. W. Schmidt and T. S. Bugni, Org. Lett., 2012, 14, 5050 CrossRef CAS.
- E. W. Schmidt, J. T. Nelson, D. A. Rasko, S. Sudek, J. A. Eisen, M. G. Haygood and J. Ravel, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7315 CrossRef CAS.
- H. E. Hallen, H. Luo, J. S. Scott-Craig and J. D. Walton, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19097 CrossRef CAS.
- M. Trabi, J. S. Mylne, L. Sando and D. J. Craik, Org. Biomol. Chem., 2009, 7, 2378 CAS.
- S. R. Woodward, L. J. Cruz, B. M. Olivera and D. R. Hillyard, EMBO J., 1990, 9, 1015 CAS.
- M. S. Donia, B. J. Hathaway, S. Sudek, M. G. Haygood, M. J. Rosovitz, J. Ravel and E. W. Schmidt, Nat. Chem. Biol., 2006, 2, 729 CrossRef CAS.
- B. Li, D. Sher, L. Kelly, Y. Shi, K. Huang, P. J. Knerr, I. Joewono, D. Rusch, S. W. Chisholm and W. A. van der Donk, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10430 CrossRef CAS.
- O. P. Kuipers, G. Bierbaum, B. Ottenwälder, H. M. Dodd, N. Horn, J. Metzger, T. Kupke, V. Gnau, R. Bongers, P. van den Bogaard, H. Kosters, H. S. Rollema, W. M. de Vos, R. J. Siezen, G. Jung, F. Götz, H. G. Sahl and M. J. Gasson, Antonie van Leeuwenhoek, 1996, 69, 161 CrossRef CAS.
- P. D. Cotter, L. H. Deegan, E. M. Lawton, L. A. Draper, P. M. O'Connor, C. Hill and R. P. Ross, Mol. Microbiol., 2006, 62, 735 CrossRef CAS.
- C. Chatterjee, G. C. Patton, L. Cooper, M. Paul and W. A. van der Donk, Chem. Biol., 2006, 13, 1109 CrossRef CAS.
- R. Rink, J. Wierenga, A. Kuipers, L. D. Kluskens, A. J. M. Driessen, O. P. Kuipers and G. N. Moll, Appl. Environ. Microbiol., 2007, 73, 5809 CrossRef CAS.
- O. Pavlova, J. Mukhopadhyay, E. Sineva, R. H. Ebright and K. Severinov, J. Biol. Chem., 2008, 283, 25589 CrossRef CAS.
- M. S. Donia, J. Ravel and E. W. Schmidt, Nat. Chem. Biol., 2008, 4, 341 CrossRef CAS.
- A. N. Appleyard, S. Choi, D. M. Read, A. Lightfoot, S. Boakes, A. Hoffmann, I. Chopra, G. Bierbaum, B. A. Rudd, M. J. Dawson and J. Cortés, Chem. Biol., 2009, 16, 490 CrossRef CAS.
- M. R. Levengood, P. J. Knerr, T. J. Oman and W. A. van der Donk, J. Am. Chem. Soc., 2009, 131, 12024 CrossRef CAS.
- M. R. Islam, K. Shioya, J. Nagao, M. Nishie, H. Jikuya, T. Zendo, J. Nakayama and K. Sonomoto, Mol. Microbiol., 2009, 72, 1438 CrossRef CAS.
- M. G. Acker, A. A. Bowers and C. T. Walsh, J. Am. Chem. Soc., 2009, 131, 17563 CrossRef CAS.
- D. A. Mitchell, S. W. Lee, M. A. Pence, A. L. Markley, J. D. Limm, V. Nizet and J. E. Dixon, J. Biol. Chem., 2009, 284, 13004 CrossRef CAS.
- A. A. Bowers, M. G. Acker, A. Koglin and C. T. Walsh, J. Am. Chem. Soc., 2010, 132, 7519 CrossRef CAS.
- S. J. Pan and A. J. Link, J. Am. Chem. Soc., 2011, 133, 5016 CrossRef CAS.
- T. A. Knappe, F. Manzenrieder, C. Mas-Moruno, U. Linne, F. Sasse, H. Kessler, X. Xie and M. A. Marahiel, Angew. Chem., Int. Ed., 2011, 50, 8714 CrossRef CAS.
- T. A. Knappe, U. Linne, L. Robbel and M. A. Marahiel, Chem. Biol., 2009, 16, 1290 CrossRef CAS.
- C. Li, F. Zhang and W. L. Kelly, Chem. Commun., 2012, 48, 558 RSC.
- C. Li, F. Zhang and W. L. Kelly, Mol. BioSyst., 2011, 7, 82 RSC.
- H. Onaka, M. Nakaho, K. Hayashi, Y. Igarashi and T. Furumai, Microbiology, 2005, 151, 3923 CrossRef CAS.
- M. F. Freeman, C. Gurgui, M. J. Helf, B. I. Morinaka, A. R. Uria, N. J. Oldham, H. G. Sahl, S. Matsunaga and J. Piel, Science, 2012, 338, 387 CrossRef CAS.
- W. L. Kelly, L. Pan and C. Li, J. Am. Chem. Soc., 2009, 131, 4327 CrossRef CAS.
- L. C. Wieland Brown, M. G. Acker, J. Clardy, C. T. Walsh and M. A. Fischbach, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2549 CrossRef CAS.
- R. Liao, L. Duan, C. Lei, H. Pan, Y. Ding, Q. Zhang, D. Chen, B. Shen, Y. Yu and W. Liu, Chem. Biol., 2009, 16, 141 CrossRef CAS.
- R. P. Morris, J. A. Leeds, H. U. Naegeli, L. Oberer, K. Memmert, E. Weber, M. J. Lamarche, C. N. Parker, N. Burrer, S. Esterow, A. E. Hein, E. K. Schmitt and P. Krastel, J. Am. Chem. Soc., 2009, 131, 5946 CrossRef CAS.
- C. T. Walsh, S. J. Malcolmson and T. S. Young, ACS Chem. Biol., 2012, 7, 429 CrossRef CAS.
- Y. M. Li, J. C. Milne, L. L. Madison, R. Kolter and C. T. Walsh, Science, 1996, 274, 1188 CrossRef CAS.
- J. O. Melby, N. J. Nard and D. A. Mitchell, Curr. Opin. Chem. Biol., 2011, 15, 369 CrossRef CAS.
- J. Lee, J. McIntosh, B. J. Hathaway and E. W. Schmidt, J. Am. Chem. Soc., 2009, 131, 2122 CrossRef CAS.
- J. D. Walton, H. E. Hallen-Adams and H. Luo, Biopolymers, 2010, 94, 659 CrossRef CAS.
- M. J. van Belkum, L. A. Martin-Visscher and J. C. Vederas, Trends Microbiol., 2011, 19, 411 CrossRef CAS.
- S. Gunasekera, N. L. Daly, M. A. Anderson and D. J. Craik, IUBMB Life, 2006, 58, 515 CrossRef CAS.
- P. J. Knerr and W. A. van der Donk, Annu. Rev. Biochem., 2012, 81, 479 CrossRef CAS.
- R. S. Roy, S. Kim, J. D. Baleja and C. T. Walsh, Chem. Biol., 1998, 5, 217 CrossRef CAS.
- G. C. Patton, M. Paul, L. E. Cooper, C. Chatterjee and W. A. van der Donk, Biochemistry, 2008, 47, 7342 CrossRef CAS.
- W. E. Houssen, S. H. Wright, A. P. Kalverda, G. S. Thompson, S. M. Kelly and M. Jaspars, ChemBioChem, 2011, 11, 1867 CrossRef.
- T. Sprules, K. E. Kawulka, A. C. Gibbs, D. S. Wishart and J. C. Vederas, Eur. J. Biochem., 2004, 271, 1748 CrossRef CAS.
- J. L. Dutton, R. F. Renda, C. Waine, R. J. Clark, N. L. Daly, C. V. Jennings, M. A. Anderson and D. J. Craik, J. Biol. Chem., 2004, 279, 46858 CrossRef CAS.
- W. M. Müller, P. Ensle, B. Krawczyk and R. D. Süssmuth, Biochemistry, 2011, 50, 8362 CrossRef.
- M. S. Donia and E. W. Schmidt, Chem. Biol., 2011, 18, 508 CrossRef CAS.
- Y. Kotake, S. Ishii, T. Yano, Y. Katsuoka and H. Hayashi, Biochemistry, 2008, 47, 2531 CrossRef CAS.
- S. Ishii, T. Yano, A. Ebihara, A. Okamoto, M. Manzoku and H. Hayashi, J. Biol. Chem., 2010, 285, 10777 CrossRef CAS.
- L. A. Furgerson Ihnken, C. Chatterjee and W. A. van der Donk, Biochemistry, 2008, 47, 7352 CrossRef CAS.
- W. L. Cheung, S. J. Pan and A. J. Link, J. Am. Chem. Soc., 2010, 132, 2514 CrossRef CAS.
- F. Garcia-Olmedo, A. Molina, J. M. Alamillo and P. Rodriguez-Palenzuela, Biopolymers, 1998, 47, 479 CrossRef CAS.
- R. E. Hancock and G. Diamond, Trends Microbiol., 2000, 8, 402 CrossRef CAS.
- L. Padovan, M. Scocchi and A. Tossi, Curr. Protein Pept. Sci., 2010, 11, 210 CrossRef CAS.
- A. A. Vassilevski, S. A. Kozlov and E. V. Grishin, Biochemistry (Moscow), 2009, 74, 1505 CrossRef CAS.
- S. Dutertre and R. J. Lewis, J. Biol. Chem., 2010, 285, 13315 CrossRef CAS.
- R. M. Kini and R. Doley, Toxicon, 2010, 56, 855 CrossRef CAS.
- D. C. Koh, A. Armugam and K. Jeyaseelan, Cell. Mol. Life Sci., 2006, 63, 3030 CrossRef CAS.
- L. A. Rogers, J. Bacteriol., 1928, 16, 321 CAS.
- E. Gross and J. L. Morell, J. Am. Chem. Soc., 1971, 93, 4634 CrossRef CAS.
- N. Schnell, K.-D. Entian, U. Schneider, F. Götz, H. Zahner, R. Kellner and G. Jung, Nature, 1988, 333, 276 CrossRef CAS.
- L. Ingram, Biochim. Biophys. Acta, 1970, 224, 263 CrossRef CAS.
- G. W. Buchman, S. Banerjee and J. N. Hansen, J. Biol. Chem., 1988, 263, 16260 CAS.
- C. Kaletta and K. D. Entian, J. Bacteriol., 1989, 171, 1597 CAS.
- S. Banerjee and J. N. Hansen, J. Biol. Chem., 1988, 262, 9508 Search PubMed.
- C. Kaletta, K. D. Entian, R. Kellner, G. Jung, M. Reis and H. G. Sahl, Arch. Microbiol., 1989, 152, 16 CrossRef CAS.
- S. Boakes, J. Cortés, A. N. Appleyard, B. A. Rudd and M. J. Dawson, Mol. Microbiol., 2009, 72, 1126 CrossRef CAS.
- G. Bierbaum, H. Brötz, K. P. Koller and H. G. Sahl, FEMS Microbiol. Lett., 1995, 127, 121 CrossRef CAS.
- C. Kaletta, K. D. Entian and G. Jung, Eur. J. Biochem., 1991, 199, 411 CrossRef CAS.
- C. Chatterjee, L. M. Miller, Y. L. Leung, L. Xie, M. Yi, N. L. Kelleher and W. A. van der Donk, J. Am. Chem. Soc., 2005, 127, 15332 CrossRef CAS.
- W. M. Müller, T. Schmiederer, P. Ensle and R. D. Süssmuth, Angew. Chem., Int. Ed., 2010, 49, 2436 CrossRef.
- Y. Goto, B. Li, J. Claesen, Y. Shi, M. J. Bibb and W. A. van der Donk, PLoS Biol., 2010, 8, e1000339 Search PubMed.
- A. K. Sen, A. Narbad, N. Horn, H. M. Dodd, A. J. Parr, I. Colquhoun and M. J. Gasson, Eur. J. Biochem., 1999, 261, 524 CrossRef CAS.
- O. Koponen, M. Tolonen, M. Qiao, G. Wahlstrom, J. Helin and P. E. J. Saris, Microbiology, 2002, 148, 3561 CAS.
- L. D. Kluskens, A. Kuipers, R. Rink, E. de Boef, S. Fekken, A. J. Driessen, O. P. Kuipers and G. N. Moll, Biochemistry, 2005, 44, 12827 CrossRef CAS.
- H. P. Weil, A. G. Beck-Sickinger, J. Metzger, S. Stevanovic, G. Jung, M. Josten and H. G. Sahl, Eur. J. Biochem., 1990, 194, 217 CrossRef CAS.
- C. Meyer, G. Bierbaum, C. Heidrich, M. Reis, J. Süling, M. I. Iglesias-Wind, C. Kempter, E. Molitor and H.-G. Sahl, Eur. J. Biochem., 1995, 232, 478 CrossRef CAS.
- B. Li, J. P. Yu, J. S. Brunzelle, G. N. Moll, W. A. van der Donk and S. K. Nair, Science, 2006, 311, 1464 CrossRef CAS.
- M. Helfrich, K. D. Entian and T. Stein, Biochemistry, 2007, 46, 3224 CrossRef CAS.
- K. Siegers, S. Heinzmann and K.-D. Entian, J. Biol. Chem., 1996, 271, 12294 CrossRef CAS.
- R. J. Siezen, O. P. Kuipers and W. M. de Vos, Antonie van Leeuwenhoek, 1996, 69, 171 CrossRef CAS.
- L. Xie, L. M. Miller, C. Chatterjee, O. Averin, N. L. Kelleher and W. A. van der Donk, Science, 2004, 303, 679 CrossRef CAS.
- Y. Goto, A. Ökesli and W. A. van der Donk, Biochemistry, 2011, 50, 891 CrossRef CAS.
- S. Kodani, M. E. Hudson, M. C. Durrant, M. J. Buttner, J. R. Nodwell and J. M. Willey, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11448 CrossRef CAS.
- N. M. Okeley, M. Paul, J. P. Stasser, N. Blackburn and W. A. van der Donk, Biochemistry, 2003, 42, 13613 CrossRef CAS.
- K. Meindl, T. Schmiederer, K. Schneider, A. Reicke, D. Butz, S. Keller, H. Guhring, L. Vertesy, J. Wink, H. Hoffmann, M. Bronstrup, G. M. Sheldrick and R. D. Süssmuth, Angew. Chem., Int. Ed., 2010, 49, 1151 CrossRef CAS.
- H. Wang and W. A. van der Donk, ACS Chem. Biol., 2012, 7, 1529 CrossRef CAS.
- O. P. Kuipers, H. S. Rollema, W. M. Yap, H. J. Boot, R. J. Siezen and W. M. de Vos, J. Biol. Chem., 1992, 267, 24340 CAS.
- W. Liu and J. N. Hansen, J. Biol. Chem., 1992, 267, 25078 CAS.
- H. S. Rollema, O. P. Kuipers, P. Both, W. M. de Vos and R. J. Siezen, Appl. Environ. Microbiol., 1995, 61, 2873 CAS.
- G. Bierbaum, C. Szekat, M. Josten, C. Heidrich, C. Kempter, G. Jung and H. G. Sahl, Appl. Environ. Microbiol., 1996, 62, 385 CAS.
- P. Chen, J. Novak, M. Kirk, S. Barnes, F. Qi and P. W. Caufield, Appl. Environ. Microbiol., 1998, 64, 2335 CAS.
- Y. Shi, X. Yang, N. Garg and W. A. van der Donk, J. Am. Chem. Soc., 2011, 133, 2338 CrossRef CAS.
- F. Oldach, R. Al Toma, A. Kuthning, T. Caetano, S. Mendo, N. Budisa and R. D. Süssmuth, Angew. Chem., Int. Ed., 2012, 51, 415 CrossRef CAS.
- A. Chakicherla and J. N. Hansen, J. Biol. Chem., 1995, 270, 23533 CrossRef CAS.
- O. P. Kuipers, H. S. Rollema, W. M. de Vos and R. J. Siezen, FEBS Lett., 1993, 330, 23 CrossRef CAS.
- J. R. van der Meer, H. S. Rollema, R. J. Siezen, M. M. Beerthuyzen, O. P. Kuipers and W. M. de Vos, J. Biol. Chem., 1994, 269, 3555 CAS.
- T. Stein and K. D. Entian, Rapid Commun. Mass Spectrom., 2002, 16, 103 CrossRef CAS.
- T. J. Oman, P. J. Knerr, N. A. Bindman, J. E. Velasquez and W. A. van der Donk, J. Am. Chem. Soc., 2012, 134, 6952 CrossRef CAS.
- T. Kupke, C. Kempter, G. Jung and F. Götz, J. Biol. Chem., 1995, 270, 11282 CrossRef CAS.
- A. Ökesli, L. E. Cooper, E. J. Fogle and W. A. van der Donk, J. Am. Chem. Soc., 2011, 133, 13753 CrossRef.
- Y. Shi, A. Bueno and W. A. van der Donk, Chem. Commun., 2012, 48, 10966 RSC.
- R. Rink, A. Kuipers, E. de Boef, K. J. Leenhouts, A. J. Driessen, G. N. Moll and O. P. Kuipers, Biochemistry, 2005, 44, 8873 CrossRef CAS.
- R. Rink, J. Wierenga, A. Kuipers, L. D. Kluskens, A. J. M. Driessen, O. P. Kuipers and G. N. Moll, Appl. Environ. Microbiol., 2007, 73, 1792 CrossRef CAS.
- M. R. Levengood and W. A. van der Donk, Bioorg. Med. Chem. Lett., 2008, 18, 3025 CrossRef CAS.
- G. N. Moll, A. Kuipers and R. Rink, Antonie van Leeuwenhoek, 2010, 97, 319 CrossRef CAS.
- M. R. Levengood, G. C. Patton and W. A. van der Donk, J. Am. Chem. Soc., 2007, 129, 10314 CrossRef CAS.
- R. Khusainov and O. P. Kuipers, ChemBioChem, 2012, 13, 2433 CrossRef CAS.
- S. Kodani, M. A. Lodato, M. C. Durrant, F. Picart and J. M. Willey, Mol. Microbiol., 2005, 58, 1368 CrossRef CAS.
- J. Lubelski, R. Rink, R. Khusainov, G. N. Moll and O. P. Kuipers, Cell. Mol. Life Sci., 2008, 65, 455 CrossRef CAS.
- G. Bierbaum and H. G. Sahl, Curr. Pharm. Biotechnol., 2009, 10, 2 CAS.
- E. Breukink and B. de Kruijff, Nat. Rev. Drug Discovery, 2006, 5, 321 CrossRef CAS.
- A. M. Jones and J. M. Helm, Drugs, 2009, 69, 1903 CrossRef CAS.
- M. P. Ryan, R. W. Jack, M. Josten, H. G. Sahl, G. Jung, R. P. Ross and C. Hill, J. Biol. Chem., 1999, 274, 37544 CrossRef CAS.
- N. I. Martin, T. Sprules, M. R. Carpenter, P. D. Cotter, C. Hill, R. P. Ross and J. C. Vederas, Biochemistry, 2004, 43, 3049 CrossRef CAS.
- A. L. McClerren, L. E. Cooper, C. Quan, P. M. Thomas, N. L. Kelleher and W. A. van der Donk, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17243 CrossRef CAS.
- E. M. Lawton, P. D. Cotter, C. Hill and R. P. Ross, FEMS Microbiol. Lett., 2007, 267, 64 CrossRef CAS.
- I. Wiedemann, T. Bottiger, R. R. Bonelli, A. Wiese, S. O. Hagge, T. Gutsmann, U. Seydel, L. Deegan, C. Hill, P. Ross and H. G. Sahl, Mol. Microbiol., 2006, 61, 285 CrossRef CAS.
- T. J. Oman, T. J. Lupoli, T. S. Wang, D. Kahne, S. Walker and W. A. van der Donk, J. Am. Chem. Soc., 2011, 133, 17544 CrossRef CAS.
- S. M. Morgan, M. P. O'Connor, P. D. Cotter, R. P. Ross and C. Hill, Antimicrob. Agents Chemother., 2005, 49, 2606 CrossRef CAS.
- J. M. Willey and W. A. van der Donk, Annu. Rev. Microbiol., 2007, 61, 477 CrossRef CAS.
- J. M. Willey, A. Willems, S. Kodani and J. R. Nodwell, Mol. Microbiol., 2006, 59, 731 CrossRef CAS.
- J. E. Velásquez, X. Zhang and W. A. van der Donk, Chem. Biol., 2011, 18, 857 CrossRef.
- M. Begley, P. D. Cotter, C. Hill and R. P. Ross, Appl. Environ. Microbiol., 2009, 75, 5451 CrossRef CAS.
- A. J. Marsh, O. O'Sullivan, R. P. Ross, P. D. Cotter and C. Hill, BMC Genomics, 2010, 11, 679 CrossRef CAS.
- Q. Zhang, Y. Yu, J. E. Velasquez and W. A. van der Donk, Proc. Natl. Acad. Sci. U. S. A., 2012 DOI:10.1073/pnas.1210393109.
- G. H. Völler, J. M. Krawczyk, A. Pesic, B. Krawczyk, J. Nachtigall and R. D. Süssmuth, ChemBioChem, 2012, 13, 1174 CrossRef.
- T. Kupke, C. Kempter, V. Gnau, G. Jung and F. Götz, J. Biol. Chem., 1994, 269, 5653 CAS.
- C. Heidrich, U. Pag, M. Josten, J. Metzger, R. W. Jack, G. Bierbaum, G. Jung and H. G. Sahl, Appl. Environ. Microbiol., 1998, 64, 3140 CAS.
- D. A. Widdick, H. M. Dodd, P. Barraille, J. White, T. H. Stein, K. F. Chater, M. J. Gasson and M. J. Bibb, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4316 CrossRef CAS.
- P. D. Cotter, P. M. O'Connor, L. A. Draper, E. M. Lawton, L. H. Deegan, C. Hill and R. P. Ross, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18584 CrossRef CAS.
- C. Chatterjee, M. Paul, L. Xie and W. A. van der Donk, Chem. Rev., 2005, 105, 633 CrossRef CAS.
- Y. Minami, K. Yoshida, R. Azuma, A. Urakawa, T. Kawauchi, T. Otani, K. Komiyama and S. Omura, Tetrahedron Lett., 1994, 35, 8001 Search PubMed.
- C. Kempter, T. Kupke, D. Kaiser, J. W. Metzger and G. Jung, Angew. Chem., Int. Ed. Engl., 1996, 35, 2104 CrossRef CAS.
- J. Claesen and M. J. Bibb, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16297 CrossRef CAS.
- J. Claesen and M. J. Bibb, J. Bacteriol., 2011, 193, 2510 CrossRef CAS.
- T. Hamada, T. Sugawara, S. Matsunaga and N. Fusetani, Tetrahedron Lett., 1994, 35, 719 CrossRef CAS.
- U. Hentschel, J. Hopke, M. Horn, A. B. Friedrich, M. Wagner, J. Hacker and B. S. Moore, Appl. Environ. Microbiol., 2002, 68, 4431 CrossRef CAS.
- T. Hamada, S. Matsunaga, G. Yano and N. Fusetani, J. Am. Chem. Soc., 2005, 127, 110 CrossRef CAS.
- N. Kessler, H. Schuhmann, S. Morneweg, U. Linne and M. A. Marahiel, J. Biol. Chem., 2004, 279, 7413 CrossRef CAS.
- T. Hamada, S. Matsunaga, M. Fujiwara, K. Fujita, H. Hirota, R. Schmucki, P. Guntert and N. Fusetani, J. Am. Chem. Soc., 2010, 132, 12941 CrossRef CAS.
- M. Iwamoto, H. Shimizu, I. Muramatsu and S. Oiki, FEBS Lett., 2010, 584, 3995 CrossRef CAS.
- P. A. Frey, A. D. Hegeman and F. J. Ruzicka, Crit. Rev. Biochem. Mol. Biol., 2008, 43, 63 CrossRef CAS.
- A. Jilek and G. Kreil, Monatsh. Chem., 2008, 139, 1 CrossRef CAS.
- Q. Zhang, W. A. van der Donk and W. Liu, Acc. Chem. Res., 2012, 45, 555 CrossRef CAS.
- R. Kellner, G. Jung, M. Josten, C. Kaletta, K. D. Entian and H. G. Sahl, Angew. Chem., 1989, 101, 618 CrossRef CAS.
- Besredka, Ann. Inst. Pasteur, 1901, 15, 880 Search PubMed.
- J. H. Brown, Monograph No. 9 Rockefeller Institute for Medical Research, New York, 1919 Search PubMed.
- J. T. Weld, J. Exp. Med., 1934, 59, 83 CrossRef CAS.
- E. W. Todd, J. Pathol. Bacteriol., 1938, 47, 423 CrossRef CAS.
- V. Nizet, B. Beall, D. J. Bast, V. Datta, L. Kilburn, D. E. Low and J. C. De Azavedo, Infect. Immun., 2000, 68, 4245 CrossRef CAS.
- V. Datta, S. M. Myskowski, L. A. Kwinn, D. N. Chiem, N. Varki, R. G. Kansal, M. Kotb and V. Nizet, Mol. Microbiol., 2005, 56, 681 CrossRef CAS.
- E. M. Molloy, P. D. Cotter, C. Hill, D. A. Mitchell and R. P. Ross, Nat. Rev. Microbiol., 2011, 9, 670 CrossRef CAS.
- P. D. Cotter, L. A. Draper, E. M. Lawton, K. M. Daly, D. S. Groeger, P. G. Casey, R. P. Ross and C. Hill, PLoS Pathog., 2008, 4, e1000144 Search PubMed.
- D. J. Gonzalez, S. W. Lee, M. E. Hensler, A. L. Markley, S. Dahesh, D. A. Mitchell, N. Bandeira, V. Nizet, J. E. Dixon and P. C. Dorrestein, J. Biol. Chem., 2010, 285, 28220 CrossRef CAS.
- K. L. Dunbar, J. O. Melby and D. A. Mitchell, Nat. Chem. Biol., 2012, 8, 569 CrossRef CAS.
- J. C. Milne, R. S. Roy, A. C. Eliot, N. L. Kelleher, A. Wokhlu, B. Nickels and C. T. Walsh, Biochemistry, 1999, 38, 4768 CrossRef CAS.
- E. W. Triplett and T. M. Barta, Plant Physiol., 1987, 85, 335 CrossRef CAS.
- A. J. Scupham and E. W. Triplett, J. Appl. Microbiol., 2006, 100, 500 CrossRef CAS.
- Y. Igarashi, Y. Kan, K. Fujii, T. Fujita, K. Harada, H. Naoki, H. Tabata, H. Onaka and T. Furumai, J. Antibiot., 2001, 54, 1045 CrossRef CAS.
- H. Onaka, H. Tabata, Y. Igarashi, Y. Sato and T. Furumai, J. Antibiot., 2001, 54, 1036 CrossRef CAS.
- R. Scholz, K. J. Molohon, J. Nachtigall, J. Vater, A. L. Markley, R. D. Süssmuth, D. A. Mitchell and R. Borriss, J. Bacteriol., 2011, 193, 215 CrossRef CAS.
- B. Kalyon, S. E. Helaly, R. Scholz, J. Nachtigall, J. Vater, R. Borriss and R. D. Süssmuth, Org. Lett., 2011, 13, 2996 CrossRef CAS.
- K. J. Molohon, J. O. Melby, J. Lee, B. S. Evans, K. L. Dunbar, S. B. Bumpus, N. L. Kelleher and D. A. Mitchell, ACS Chem. Biol., 2011, 6, 1307 CrossRef CAS.
- J. C. Milne, R. S. Roy, A. C. Eliot, N. L. Kelleher, A. Wokhlu, B. Nickels and C. T. Walsh, Biochemistry, 1999, 38, 4768 CrossRef CAS.
- J. A. McIntosh and E. W. Schmidt, ChemBioChem, 2010, 11, 1413 CrossRef CAS.
- J. A. McIntosh, M. S. Donia and E. W. Schmidt, J. Am. Chem. Soc., 2010, 132, 4089 CrossRef CAS.
- M. S. Donia and E. W. Schmidt, in Comprehensive Natural Products II Chemistry and Biology, ed. B. S. Moore and P. Crews, Elsevier: Oxford, 2010, p. 539 Search PubMed.
- C. Ireland and P. J. Scheuer, J. Am. Chem. Soc., 1980, 102, 5688 CrossRef CAS.
- T. W. Hambley, C. J. Hawkins, M. F. Lavin, A. Van den Brenk and D. J. Watters, Tetrahedron, 1992, 48, 341 CrossRef CAS.
- M. R. Prinsep, R. E. Moore, I. A. Levine and G. M. L. Patterson, J. Nat. Prod., 1992, 55, 140 CrossRef CAS.
- P. F. Long, W. C. Dunlap, C. N. Battershill and M. Jaspars, ChemBioChem, 2005, 6, 1760 CrossRef CAS.
- S. Sudek, M. G. Haygood, D. T. Youssef and E. W. Schmidt, Appl. Environ. Microbiol., 2006, 72, 4382 CrossRef CAS.
- N. Leikoski, D. P. Fewer and K. Sivonen, Appl. Environ. Microbiol., 2009, 75, 853 CrossRef CAS.
- N. Leikoski, D. P. Fewer, J. Jokela, M. Wahlsten, L. Rouhiainen and K. Sivonen, Appl. Environ. Microbiol., 2010, 76, 701 CrossRef CAS.
- M. S. Donia, W. F. Fricke, F. Partensky, J. Cox, S. I. Elshahawi, J. R. White, A. M. Phillippy, M. C. Schatz, J. Piel, M. G. Haygood, J. Ravel and E. W. Schmidt, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, E1423 CrossRef CAS.
- M. S. Donia, W. F. Fricke, J. Ravel and E. W. Schmidt, PLoS One, 2011, 6, e17897 CAS.
- M. S. Donia, D. E. Ruffner, S. Cao and E. W. Schmidt, ChemBioChem, 2011, 12, 1230 CrossRef CAS.
- K. Sivonen, N. Leikoski, D. P. Fewer and J. Jokela, Appl. Microbiol. Biotechnol., 2010, 86, 1213 CrossRef CAS.
- L. Cascales and D. J. Craik, Org. Biomol. Chem., 2010, 8, 5035 CAS.
- N. H. Tan and J. Zhou, Chem. Rev., 2006, 106, 840 CrossRef CAS.
- H. Luo, H. E. Hallen-Adams, J. S. Scott-Craig and J. D. Walton, Eukaryotic Cell, 2010, 9, 1891 CrossRef CAS.
- L. A. Martin-Visscher, M. J. van Belkum and J. C. Vederas, in Prokaryotic Antimicrobial Peptides: From Genes to Applications, ed. D. Drider and S. Rebuffat, Springer Science, 2011, p. 213 Search PubMed.
- X. H. Jian, H. X. Pan, T. T. Ning, Y. Y. Shi, Y. S. Chen, Y. Li, X. W. Zeng, J. Xu and G. L. Tang, ACS Chem. Biol., 2012, 646 CrossRef CAS.
- J. A. McIntosh, C. R. Robertson, V. Agarwal, S. K. Nair, G. W. Bulaj and E. W. Schmidt, J. Am. Chem. Soc., 2010, 132, 15499 CrossRef CAS.
- J. Koehnke, A. Bent, W. E. Houssen, D. Zollman, F. Morawitz, S. Shirran, J. Vendome, A. F. Nneoyiegbe, L. Trembleau, C. H. Botting, M. C. M. Smith, M. Jaspars and J. H. Naismith, Nat. Struct. Mol. Biol., 2012, 19, 767 CAS.
- J. A. McIntosh, M. S. Donia, S. K. Nair and E. W. Schmidt, J. Am. Chem. Soc., 2011, 133, 13698 CrossRef CAS.
- B. F. Milne, P. F. Long, A. Starcevic, D. Hranueli and M. Jaspars, Org. Biomol. Chem., 2006, 4, 631 CAS.
- M. D. Tianero, M. S. Donia, T. S. Young, P. G. Schultz and E. W. Schmidt, J. Am. Chem. Soc., 2012, 134, 418 CrossRef CAS.
- W. E. Houssen and M. Jaspars, ChemBioChem, 2010, 11, 1803 CrossRef CAS.
- A. R. Carroll, J. C. Coll, D. J. Bourne, J. K. MacLeod, T. M. Zabriskie, C. M. Ireland and B. F. Bowden, Aust. J. Chem., 1996, 49, 659 CAS.
- A. B. Williams and R. S. Jacobs, Cancer Lett., 1993, 71, 97 CrossRef CAS.
- S. G. Aller, J. Yu, A. Ward, Y. Weng, S. Chittaboina, R. Zhuo, P. M. Harrell, Y. T. Trinh, Q. Zhang, I. L. Urbatsch and G. Chang, Science, 2009, 323, 1718 CrossRef CAS.
- P. Wipf, S. Venkatraman, C. P. Miller and S. J. Geib, Angew. Chem., 1994, 106, 1554 CrossRef CAS.
- A. Bertram and G. Pattenden, Nat. Prod. Rep., 2007, 24, 18 RSC.
- T. L. Su, Br. J. Exp. Pathol., 1948, 29, 473 CAS.
- M. C. Bagley, J. W. Dale, E. A. Merritt and X. Xiong, Chem. Rev., 2005, 105, 685 CrossRef CAS.
- M. C. Carnio, A. Holtzel, M. Rudolf, T. Henle, G. Jung and S. Scherer, Appl. Environ. Microbiol., 2000, 66, 2378 CrossRef CAS.
- J. Shoji, H. Hinoo, Y. Wakisaka, K. Koizumi, M. Mayama, S. Matsura and K. Matsumoto, J. Antibiot., 1976, 29, 366 CrossRef CAS.
- F. Benazet, M. Cartier, J. Florent, C. Godard, G. Jung, J. Lunel, D. Mancy, C. Pascal, J. Renaut, P. Tarridec, J. Theilleux, R. Tissier, M. Dubost and L. Ninet, Experientia, 1980, 36, 414 CrossRef CAS.
- J. Vandeputte and J. D. Dutcher, Antibiot. Annu., 1955, 3, 560 Search PubMed.
- O. D. Hensens and G. Albers-Schonberg, Tetrahedron Lett., 1978, 39, 3649 CrossRef.
- O. D. Hensens and G. Albers-Schonberg, J. Antibiot., 1983, 36, 814 CrossRef CAS.
- B. Anderson, D. C. Hodgkin and M. A. Viswamitra, Nature, 1970, 225, 233 CrossRef CAS.
- M. S. Puar, T. M. Chan, V. Hegde, M. Patel, P. Bartner, K. J. Ng, B. N. Pramanik and R. D. MacFarlane, J. Antibiot., 1998, 51, 221 CrossRef CAS.
- B. W. Bycroft and M. S. Gowland, J. Chem. Soc., Chem. Commun., 1978, 256 RSC.
- M. C. Bagley and E. A. Merritt, J. Antibiot., 2004, 57, 829 CrossRef CAS.
- C. Pascard, A. Ducruix, J. Lunel and T. Prange, J. Am. Chem. Soc., 1977, 99, 6418 CrossRef CAS.
- T. Prange, A. Ducruix and C. Pascard, Nature, 1977, 265 Search PubMed.
- M. V. Rodnina, A. Savelsbergh, N. B. Matassova, V. I. Katunin, Y. P. Semenkov and W. Wintermeyer, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9586 CrossRef CAS.
- P. H. Anborgh and A. Parmeggiani, EMBO J., 1991, 10, 779 CAS.
- A. Mikolajka, H. Liu, Y. Chen, A. L. Starosta, V. Marquez, M. Ivanova, B. S. Cooperman and D. N. Wilson, Chem. Biol., 2011, 18, 589 CrossRef CAS.
- A. L. Starosta, H. Qin, A. Mikolajka, G. Y. Leung, K. Schwinghammer, K. C. Nicolaou, D. Y. Chen, B. S. Cooperman and D. N. Wilson, Chem. Biol., 2009, 16, 1087 CrossRef CAS.
- G. Lentzen, R. Klinck, N. Matassova, F. Aboul-ela and A. I. H. Murchie, Chem. Biol., 2003, 10, 769 CrossRef CAS.
- B. T. Porse, E. Cundliffe and R. A. Garrett, J. Mol. Biol., 1999, 287, 33 CrossRef CAS.
- B. T. Porse, I. Leviev, A. S. Mankin and R. A. Garrett, J. Mol. Biol., 1998, 276, 391 CrossRef CAS.
- D. M. Cameron, J. Thompson, P. E. March and A. E. Dahlberg, J. Mol. Biol., 2002, 319, 27 CrossRef CAS.
- R. L. Gonzalez, Jr., S. Chu and J. D. Puglisi, RNA, 2007, 13, 2091 CrossRef.
- J. Modolell, B. Cabrer, A. Parmeggiani and D. Vazquez, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 1796 CrossRef CAS.
- J. M. Harms, D. N. Wilson, F. Schluenzen, S. R. Connell, T. Stachelhaus, Z. Zaborowska, C. M. T. Spahn and P. Fucini, Mol. Cell, 2008, 30, 26 CrossRef CAS.
- S. E. Heffron and F. Jurnak, Biochemistry, 2000, 39, 37 CrossRef CAS.
- A. Parmeggiani, I. M. Krab, S. Okamura, R. C. Nielsen, J. Nyborg and P. Nissen, Biochemistry, 2006, 45, 6846 CrossRef CAS.
- G. A. McConkey, M. J. Rogers and T. F. McCutchan, J. Biol. Chem., 1997, 272, 2046 CrossRef CAS.
- M. J. Rogers, E. Cundliffe and T. F. McCutchan, Antimicrob. Agents Chemother., 1998, 42, 715 CAS.
- M. N. Aminake, S. Schoof, L. Sologub, M. Leubner, M. Kirschner, H. D. Arndt and G. Pradel, Antimicrob. Agents Chemother., 2011, 55, 1338 CrossRef CAS.
- S. Schoof, G. Pradel, M. N. Aminake, B. Ellinger, S. Baumann, M. Potowski, Y. Najajreh, M. Kirschner and H. D. Arndt, Angew. Chem., Int. Ed., 2010, 49, 3317 CrossRef CAS.
- U. G. Bhat, M. Halasi and A. L. Gartel, PLoS One, 2009, 4, e6593 Search PubMed.
- B. Pandit and A. L. Gartel, PLoS One, 2011, 6, e17110 CAS.
- N. S. Hegde, D. A. Sanders, R. Rodriguez and S. Balasubramanian, Nat. Chem., 2011, 3, 725 CrossRef CAS.
- M. Aoki, T. Ohtsuka, M. Yamada, Y. Ohba, H. Yoshizaki, H. Yasuno, T. Sano, J. Watanabe, K. Yokose and H. Seto, J. Antibiot., 1991, 44, 582 CrossRef CAS.
- M. Hashimoto, T. Murakami, K. Funahashi, T. Tokunaga, K. Nihei, T. Okuno, T. Kimura, H. Naoki and H. Himeno, Bioorg. Med. Chem., 2006, 14, 8259 CrossRef CAS.
- N. Mizuhara, M. Kuroda, A. Ogita, T. Tanaka, Y. Usuki and K. Fujita, Bioorg. Med. Chem., 2011, 19, 5300 CrossRef CAS.
- D. J. Holmes, J. L. Caso and C. J. Thompson, EMBO J., 1993, 12, 3183 CAS.
- Y. Ding, Y. Yu, H. Pan, H. Guo, Y. Li and W. Liu, Mol. BioSyst., 2010, 6, 1180 RSC.
- K. Engelhardt, K. F. Degnes and S. B. Zotchev, Appl. Environ. Microbiol., 2010, 76, 7093 CrossRef CAS.
- J. Wang, Y. Yu, K. Tang, W. Liu, X. He, X. Huang and Z. Deng, Appl. Environ. Microbiol., 2010, 76, 2335 CrossRef CAS.
- T. S. Young and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13053 CrossRef CAS.
- Y. Yu, L. Duan, Q. Zhang, R. Liao, Y. Ding, H. Pan, E. Wendt-Pienkowski, G. Tang, B. Shen and W. Liu, ACS Chem. Biol., 2009, 4, 855 CrossRef CAS.
- A. A. Bowers, C. T. Walsh and M. G. Acker, J. Am. Chem. Soc., 2010, 132, 12182 CrossRef CAS.
- Q. Zhang, Y. Li, D. Chen, Y. Yu, L. Duan, B. Shen and W. Liu, Nat. Chem. Biol., 2011, 7, 154 CrossRef CAS.
- J. M. Waisvisz, M. G. van der Hoeven, J. van Peppen and W. C. M. Zwennis, J. Am. Chem. Soc., 1957, 79, 4520 CrossRef CAS.
- H. Shimamura, H. Gouda, K. Nagai, T. Hirose, M. Ichioka, Y. Furuya, Y. Kobayashi, S. Hirono, T. Sunazuka and S. Omura, Angew. Chem., Int. Ed., 2009, 48, 914 CrossRef CAS.
- Y. Kobayashi, M. Ichioka, T. Hirose, K. Nagai, A. Matsumoto, H. Matsui, H. Hanaki, R. Masuma, Y. Takahashi, S. Omura and T. Sunazuka, Bioorg. Med. Chem. Lett., 2010, 20, 6116 CrossRef CAS.
- T. Otaka and A. Kaji, J. Biol. Chem., 1976, 251, 2299 CAS.
- T. Otaka and A. Kaji, FEBS Lett., 1981, 123, 173 CrossRef CAS.
- T. Otaka and A. Kaji, FEBS Lett., 1983, 153, 53 CrossRef CAS.
- J. M. Waisvisz, M. G. van der Hoeven and B. te Nijnhius, J. Am. Chem. Soc., 1957, 79, 4524 CrossRef CAS.
- M. Kaneda, J. Antibiot., 2002, 55, 924 CrossRef CAS.
- M. Kaneda, J. Antibiot., 1992, 45, 792 CrossRef CAS.
- H.-G. Lerchen, G. Schiffer, H. Brötz-Österheld, A. Mayer-Bartschmid, S. Eckermann, C. Freiberg, R. Endermann, J. Schuhmann, H. Meier, N. Svenstrup, S. Seip, M. Gehling and D. Häbich, Cyclic iminopeptide derivatives, WO 2006/103010, 2006.
- F. Baquero and F. Moreno, FEMS Microbiol. Lett., 1984, 23, 117 CrossRef CAS.
- K. Severinov, E. Semenova, A. Kazakov, T. Kazakov and M. S. Gelfand, Mol. Microbiol., 2007, 189, 8772 Search PubMed.
- S. Rebuffat, in Prokaryotic antimicrobial peptides: from genes to applications, ed. D. Drider and S. Rebuffat, Springer, New York, 2011, p. 55 Search PubMed.
- A. Bayer, S. Freund and G. Jung, Angew. Chem., Int. Ed. Engl., 1993, 32, 1336 CrossRef.
- J. I. Guijarro, J. E. Gonzalez-Pastor, F. Baleux, J. L. San Millan, M. A. Castilla, M. Rico, F. Moreno and M. Delepierre, J. Biol. Chem., 1995, 270, 23520 CrossRef CAS.
- S. Rebuffat, A. Blond, D. Destoumieux-Garzon, C. Goulard and J. Peduzzi, Curr. Protein Pept. Sci., 2004, 5, 383 CrossRef CAS.
- G. Bierbaum and A. Jansen, Chem. Biol., 2007, 14, 734 CrossRef CAS.
- K. J. Rosengren and D. J. Craik, Chem. Biol., 2009, 16, 1211 CrossRef CAS.
- X. Thomas, D. Destoumieux-Garzon, J. Peduzzi, C. Afonso, A. Blond, N. Birlirakis, C. Goulard, L. Dubost, R. Thai, J. C. Tabet and S. Rebuffat, J. Biol. Chem., 2004, 279, 28233 CrossRef CAS.
- G. Vassiliadis, D. Destoumieux-Garzon, C. Lombard, S. Rebuffat and J. Peduzzi, Antimicrob. Agents Chemother., 2010, 54, 288 CrossRef CAS.
- K. Severinov, E. Semenova and T. Kazakov, in Prokaryotic antimicrobial peptides: from genes to applications, ed. D. Drider and S. Rebuffat, Springer, New York, 2011, p. 289 Search PubMed.
- M. J. Bayro, J. Mukhopadhyay, G. V. Swapna, J. Y. Huang, L. C. Ma, E. Sineva, P. E. Dawson, G. T. Montelione and R. H. Ebright, J. Am. Chem. Soc., 2003, 125, 12382 CrossRef CAS.
- K. J. Rosengren, R. J. Clark, N. L. Daly, U. Goransson, A. Jones and D. J. Craik, J. Am. Chem. Soc., 2003, 125, 12464 CrossRef CAS.
- K. A. Wilson, M. Kalkum, J. Ottesen, J. Yuzenkova, B. T. Chait, R. Landick, T. Muir, K. Severinov and S. A. Darst, J. Am. Chem. Soc., 2003, 125, 12475 CrossRef CAS.
- G. H. M. Vondenhoff, S. Dubiley, K. Severinov, E. Lescrinier, J. Rozenski and A. Van Aerschot, Bioorg. Med. Chem., 2011, 19, 5462 CrossRef CAS.
- L. S. Håvarstein, D. B. Diep and I. F. Nes, Mol. Microbiol., 1995, 16, 229 CrossRef.
- S. Duquesne, D. Destoumieux-Garzon, S. Zirah, C. Goulard, J. Peduzzi and S. Rebuffat, Chem. Biol., 2007, 14, 793 CrossRef CAS.
- D. J. Clarke and D. J. Campopiano, Org. Biomol. Chem., 2007, 5, 2564 CAS.
- J. L. San Millan, R. Kolter and F. Moreno, J. Bacteriol., 1985, 163, 1016 CAS.
- N. Allali, H. Afif, M. Couturier and L. Van Melderen, J. Bacteriol., 2002, 184, 3224 CrossRef CAS.
- R. F. Roush, E. M. Nolan, F. Lohr and C. T. Walsh, J. Am. Chem. Soc., 2008, 130, 3603 CrossRef CAS.
- E. M. Nolan, M. A. Fischbach, A. Koglin and C. T. Walsh, J. Am. Chem. Soc., 2007, 129, 14336 CrossRef CAS.
- E. M. Nolan and C. T. Walsh, Biochemistry, 2008, 47, 9289 CrossRef CAS.
- G. Vassiliadis, J. Peduzzi, S. Zirah, X. Thomas, S. Rebuffat and D. Destoumieux-Garzon, Antimicrob. Agents Chemother., 2007, 51, 3546 CrossRef CAS.
- M. O. Maksimov, S. J. Pan and A. J. Link, Nat. Prod. Rep., 2012, 29, 996 RSC.
- K. Yano, S. Toki, S. Nakanishi, K. Ochiai, K. Ando, M. Yoshida, Y. Matsuda and M. Yamasaki, Bioorg. Med. Chem., 1996, 4, 115 CrossRef CAS.
- P. F. Lin, H. Samanta, C. M. Bechtold, C. A. Deminie, A. K. Patick, M. Alam, K. Riccardi, R. E. Rose, R. J. White and R. J. Colonno, Antimicrob. Agents Chemother., 1996, 40, 133 CAS.
- M. Tsunakawa, S. L. Hu, Y. Hoshino, D. J. Detlefson, S. E. Hill, T. Furumai, R. J. White, M. Nishio, K. Kawano, S. Yamamoto, Y. Fukagawa and T. Oki, J. Antibiot., 1995, 48, 433 CrossRef CAS.
- G. Helynck, C. Dubertret, J. F. Mayaux and J. Leboul, J. Antibiot., 1993, 46, 1756 CrossRef CAS.
- Y. Morishita, S. Chiba, E. Tsukuda, T. Tanaka, T. Ogawa, M. Yamasaki, M. Yoshida, I. Kawamoto and Y. Matsuda, J. Antibiot., 1994, 47, 269 CrossRef CAS.
- M. Yamasaki, K. Yano, M. Yoshida, Y. Matsuda and K. Yamaguchi, J. Antibiot., 1994, 47, 276 CrossRef CAS.
- K. Yano, M. Yamasaki, M. Yoshida, Y. Matsuda and K. Yamaguchi, J. Antibiot., 1995, 48, 1368 CrossRef CAS.
- T. Ogawa, K. Ochiai, T. Tanaka, E. Tsukuda, S. Chiba, K. Yano, M. Yamasaki, M. Yoshida and Y. Matsuda, J. Antibiot., 1995, 48, 1213 CrossRef CAS.
- K. I. Kimura, F. Kanou, H. Takahashi, Y. Esumi, M. Uramoto and M. Yoshihama, J. Antibiot., 1997, 50, 373 CrossRef CAS.
- Y. Esumi, Y. Suzuki, Y. Itoh, M. Uramoto, K. Kimura, M. Goto, M. Yoshihama and T. Ichikawa, J. Antibiot., 2002, 55, 296 CrossRef CAS.
- W. Weber, W. Fischli, E. Hochuli, E. Kupfer and E. K. Weibel, J. Antibiot., 1991, 44, 164 CrossRef CAS.
- D. F. Wyss, H. W. Lahm, M. Manneberg and A. M. Labhardt, J. Antibiot., 1991, 44, 172 CrossRef CAS.
- M. Iwatsuki, R. Uchida, Y. Takakusagi, A. Matsumoto, C.-L. Jiang, Y. Takahashi, M. Arai, S. Kobayashi, M. Matsumoto, J. Inokoshi, H. Tomoda and S. Omura, J. Antibiot., 2007, 60, 357 CrossRef CAS.
- R. A. Salomon and R. N. Farias, J. Bacteriol., 1992, 174, 7428 CAS.
- M. A. Delgado, M. R. Rintoul, R. N. Farias and R. A. Salomon, J. Bacteriol., 2001, 183, 4543 CrossRef CAS.
- T. A. Knappe, U. Linne, S. Zirah, S. Rebuffat, X. Xie and M. A. Marahiel, J. Am. Chem. Soc., 2008, 130, 11446 CrossRef CAS.
- K. Kuznedelov, E. Semenova, T. A. Knappe, D. Mukhamedyarov, A. Srivastava, S. Chatterjee, R. H. Ebright, M. A. Marahiel and K. Severinov, J. Mol. Biol., 2011, 412, 842 CrossRef CAS.
- T. A. Knappe, U. Linne, X. Xie and M. A. Marahiel, FEBS Lett., 2010, 584, 785 CrossRef CAS.
- M. O. Maksimov, I. Pelczer and A. J. Link, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15223 CrossRef CAS.
- S. J. Pan, J. Rajniak, W. L. Cheung and A. J. Link, ChemBioChem, 2012, 13, 367 CrossRef CAS.
- K.-P. Yan, Y. Li, S. Zirah, C. Goulard, T. Knappe, M. Marahiel and S. Rebuffat, ChemBioChem, 2012, 13, 1046 CrossRef CAS.
- S. J. Pan, J. Rajniak, M. O. Maksimov and A. J. Link, Chem. Commun., 2012, 48, 1880 RSC.
- J. O. Solbiati, M. Ciaccio, R. N. Farías, J. E. González-Pastor, F. Moreno and R. A. Salomón, J. Bacteriol., 1999, 181, 2659 CAS.
- J. Inokoshi, M. Matsuhama, M. Miyake, H. Ikeda and H. Tomoda, Appl. Microbiol. Biotechnol., 2012, 95, 451 CrossRef CAS.
- R. Ducasse, K. P. Yan, C. Goulard, A. Blond, Y. Li, E. Lescop, E. Guittet, S. Rebuffat and S. Zirah, ChemBioChem, 2012, 13, 371 CrossRef CAS.
- M. O. Ishitsuka, T. Kusumi, H. Kakisawa, K. Kaya and M. M. Watanabe, J. Am. Chem. Soc., 1990, 112, 8180 CrossRef CAS.
- A. R. Weiz, K. Ishida, K. Makower, N. Ziemert, C. Hertweck and E. Dittmann, Chem. Biol., 2011, 18, 1413 CrossRef CAS.
- K. Miyamoto, H. Tsujibo, Y. Hikita, K. Tanaka, S. Miyamoto, M. Hishimoto, C. Imada, K. Kamei, S. Hara and Y. Inamori, Biosci., Biotechnol., Biochem., 1998, 62, 2446 CrossRef CAS.
- M. Murakami, Q. Sun, K. Ishida, H. Matsuda, T. Okino and K. Yamaguchi, Phytochemistry, 1997, 45, 1197 CrossRef CAS.
- T. Okino, H. Matsuda, M. Murakami and K. Yamaguchi, Tetrahedron, 1995, 51, 10679 CrossRef CAS.
- B. Philmus, G. Christiansen, W. Y. Yoshida and T. K. Hemscheidt, ChemBioChem, 2008, 9, 3066 CrossRef CAS.
- H. J. Shin, M. Murakami, H. Matsuda and K. Yamaguchi, Tetrahedron, 1996, 52, 8159 CrossRef CAS.
- K. Kanaori, K. Kamei, M. Taniguchi, T. Koyama, T. Yasui, R. Takano, C. Imada, K. Tajima and S. Hara, Biochemistry, 2005, 44, 2462 CrossRef CAS.
- T. Rohrlack, K. Christoffersen, M. Kaebernick and B. A. Neilan, Appl. Environ. Microbiol., 2004, 70, 5047 CrossRef CAS.
- N. Ziemert, K. Ishida, A. Liaimer, C. Hertweck and E. Dittmann, Angew. Chem., Int. Ed., 2008, 47, 7756 CrossRef CAS.
- M. Y. Galperin and E. V. Koonin, Protein Sci., 1997, 6, 2639 CrossRef CAS.
- B. Philmus, J. P. Guerrette and T. K. Hemscheidt, ACS Chem. Biol., 2009, 4, 429 CrossRef CAS.
- D. M. Gardiner, P. Waring and B. J. Howlett, Microbiology, 2005, 151, 1021 CrossRef CAS.
- D. H. Scharf, N. Remme, A. Habel, P. Chankhamjon, K. Scherlach, T. Heinekamp, P. Hortschansky, A. A. Brakhage and C. Hertweck, J. Am. Chem. Soc., 2011, 133, 12322 CrossRef CAS.
- K. Kawulka, T. Sprules, R. T. McKay, P. Mercier, C. M. Diaper, P. Zuber and J. C. Vederas, J. Am. Chem. Soc., 2003, 125, 4726 CrossRef CAS.
- T. Huang, H. Geng, V. R. Miyyapuram, C. S. Sit, J. C. Vederas and M. M. Nakano, J. Bacteriol., 2009, 191, 5690 CrossRef CAS.
- M. C. Rea, C. S. Sit, E. Clayton, P. M. O'Connor, R. M. Whittal, J. Zheng, J. C. Vederas, R. P. Ross and C. Hill, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9352 CrossRef CAS.
- C. S. Sit, M. J. van Belkum, R. T. McKay, R. W. Worobo and J. C. Vederas, Angew. Chem., Int. Ed., 2011, 50, 8718 CrossRef CAS.
- W. T. Liu, Y. L. Yang, Y. Xu, A. Lamsa, N. M. Haste, J. Y. Yang, J. Ng, D. Gonzalez, C. D. Ellermeier, P. D. Straight, P. A. Pevzner, J. Pogliano, V. Nizet, K. Pogliano and P. C. Dorrestein, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16286 CrossRef CAS.
- M. C. Rea, R. P. Ross, P. D. Cotter and C. Hill, in Prokaryotic Antimicrobial Peptides: From Genes to Applications, ed. D. Drider and S. Rebuffat, Springer Science, 2011, pp. 29–53 Search PubMed.
- J. Wang, Y. Yu, K. Tang, W. Liu, X. He, X. Huang and Z. Deng, Appl. Environ. Microbiol., 2010, 76, 2335 CrossRef CAS.
- M. Hashimoto, T. Murakami, K. Funahashi, T. Tokunaga, K. Nihei, T. Okuno, T. Kimura, H. Naoki and H. Himeno, Bioorg. Med. Chem., 2006, 14, 8259 CrossRef CAS.
- M. Aoki, T. Ohtsuka, M. Yamada, Y. Ohba, H. Yoshizaki, H. Yasuno, T. Sano, J. Watanabe, K. Yokose and H. Seto, J. Antibiot., 1991, 44, 582 CrossRef CAS.
- K. E. Kawulka, T. Sprules, C. M. Diaper, R. M. Whittal, R. T. McKay, P. Mercier, P. Zuber and J. C. Vederas, Biochemistry, 2004, 43, 3385 CrossRef CAS.
- C. S. Sit, R. T. McKay, C. Hill, R. P. Ross and J. C. Vederas, J. Am. Chem. Soc., 2011, 133, 7680 CrossRef CAS.
- M. C. Rea, A. Dobson, O. O'Sullivan, F. Crispie, F. Fouhy, P. D. Cotter, F. Shanahan, B. Kiely, C. Hill and R. P. Ross, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(Suppl 1), 4639 CrossRef CAS.
- S. Thennarasu, D. K. Lee, A. Poon, K. E. Kawulka, J. C. Vederas and A. Ramamoorthy, Chem. Phys. Lipids, 2005, 137, 38 CrossRef CAS.
- K. Yamamoto, J. Xu, K. E. Kawulka, J. C. Vederas and A. Ramamoorthy, J. Am. Chem. Soc., 2010, 132, 6929 CrossRef CAS.
- L. Silkin, S. Hamza, S. Kaufman, S. L. Cobb and J. C. Vederas, Bioorg. Med. Chem. Lett., 2007, 18, 3103 CrossRef.
- L. A. Martin-Visscher, S. Yoganathan, C. S. Sit, C. T. Lohans and J. C. Vederas, FEMS Microbiol. Lett., 2011, 317, 152 CrossRef CAS.
- G. Zheng, L. Z. Yan, J. C. Vederas and P. Zuber, J. Bacteriol., 1999, 181, 7346 CAS.
- L. Flühe, T. A. Knappe, M. J. Gattner, A. Schäfer, O. Burghaus, U. Linne and M. A. Marahiel, Nat. Chem. Biol., 2012, 8, 350 CrossRef.
- H. Lee, J. J. Churey and R. W. Worobo, FEMS Microbiol. Lett., 2009, 299, 205 CrossRef CAS.
- M. Sánchez-Hidalgo, M. Montalbán-Lopez, R. Cebrián, E. Valdivia, M. Martínez-Bueno and M. Maqueda, Cell. Mol. Life Sci., 2011, 68, 2845 CrossRef.
- M. L. Kalmokoff, T. D. Cyr, M. A. Hefford, M. F. Whitford and R. M. Teather, Can. J. Microbiol., 2003, 49, 763 CrossRef CAS.
- Y. Kawai, T. Saito, H. Kitazawa and T. Itoh, Biosci., Biotechnol., Biochem., 1998, 62, 2438 CrossRef CAS.
- K. Arakawa, Y. Kawai, Y. Ito, K. Nakamura, T. Chujo, J. Nishimura, H. Kitazawa and T. Saito, Lett. Appl. Microbiol., 2010, 50, 406 CrossRef CAS.
- R. Kemperman, M. Jonker, A. Nauta, O. P. Kuipers and J. Kok, Appl. Environ. Microbiol., 2003, 69, 5839 CrossRef CAS.
- K. Babasaki, T. Takao, Y. Shimonishi and K. Kurahashi, J. Biochem., 1985, 98, 585 CAS.
- R. E. Wirawan, K. M. Swanson, T. Kleffmann, R. W. Jack and J. R. Tagg, Microbiology, 2007, 153, 1619 CrossRef CAS.
- L. A. Martin-Visscher, M. J. van Belkum, S. Garneau-Tsodikova, R. M. Whittal, J. Zheng, L. M. McMullen and J. C. Vederas, Appl. Environ. Microbiol., 2008, 74, 4756 CrossRef CAS.
- L. A. Martin-Visscher, X. Gong, M. Duszyk and J. C. Vederas, J. Biol. Chem., 2009, 284, 28674 CrossRef CAS.
- N. Sawa, T. Zendo, J. Kiyofuji, K. Fujita, K. Himeno, J. Nakayama and K. Sonomoto, Appl. Environ. Microbiol., 2009, 75, 1552 CrossRef CAS.
- Y. Masuda, H. Ono, H. Kitagawa, H. Ito, F. Mu, N. Sawa, T. Zendo and K. Sonomoto, Appl. Environ. Microbiol., 2011, 77, 8164 CrossRef CAS.
- J. Borrero, D. A. Brede, M. Skaugen, D. B. Diep, C. Herranz, I. F. Nes, L. M. Cintas and P. E. Hernandez, Appl. Environ. Microbiol., 2011, 77, 369 CrossRef CAS.
- M. J. Sánchez-Barrena, M. Martínez-Ripoll, A. Gálvez, E. Valdivia, M. Maqueda, V. Cruz and A. Albert, J. Mol. Biol., 2003, 334, 541 CrossRef.
- X. Gong, L. A. Martin-Visscher, D. Nahirney, J. C. Vederas and M. Duszyk, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 1797 CrossRef CAS.
- R. Cebrián, M. Maqueda, J. L. Neira, E. Valdivia, M. Martínez-Bueno and M. Montalbán-López, Appl. Environ. Microbiol., 2010, 76, 7268 CrossRef.
- T. Wieland and H. Faulstich, Crit. Rev. Biochem. Mol. Biol., 1978, 5, 185 CrossRef CAS.
- D. A. Bushnell, P. Cramer and R. D. Kornberg, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 1218 CrossRef CAS.
- T. Wieland and V. M. Govindan, FEBS Lett., 1974, 46, 351 CrossRef CAS.
- H. Luo, H. E. Hallen-Adams and J. D. Walton, J. Biol. Chem., 2009, 284, 18070 CrossRef CAS.
- H. Luo, H. E. Hallen-Adams, J. S. Scott-Craig and J. D. Walton, Fungal Genet. Biol., 2012, 49, 123 CrossRef CAS.
- D. J. Craik, N. L. Daly, T. Bond and C. Waine, J. Mol. Biol., 1999, 294, 1327 CrossRef CAS.
- O. Saether, D. J. Craik, I. D. Campbell, K. Sletten, J. Juul and D. G. Norman, Biochemistry, 1995, 34, 4147 CrossRef CAS.
- L. Chiche, A. Heitz, J. C. Gelly, J. Gracy, P. T. Chau, P. T. Ha, J. F. Hernandez and D. Le-Nguyen, Curr. Protein Pept. Sci., 2004, 5, 341 CrossRef CAS.
- C. K. Wang, Q. Kaas, L. Chiche and D. J. Craik, Nucleic Acids Res., 2008, 36, D206 CrossRef CAS.
- U. Göransson and D. J. Craik, J. Biol. Chem., 2003, 278, 48188 CrossRef.
- C. W. Gruber, A. G. Elliott, D. C. Ireland, P. G. Delprete, S. Dessein, U. Goransson, M. Trabi, C. K. Wang, A. B. Kinghorn, E. Robbrecht and D. J. Craik, Plant Cell, 2008, 20, 2471 CrossRef CAS.
- G. K. Nguyen, S. Zhang, N. T. Nguyen, P. Q. Nguyen, M. S. Chiu, A. Hardjojo and J. P. Tam, J. Biol. Chem., 2011, 286, 24275 CrossRef CAS.
- A. G. Poth, M. L. Colgrave, R. E. Lyons, N. L. Daly and D. J. Craik, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10127 CrossRef CAS.
- N. L. Daly, K. J. Rosengren and D. J. Craik, Adv. Drug Delivery Rev., 2009, 61, 918 CrossRef CAS.
- B. L. Barbeta, A. T. Marshall, A. D. Gillon, D. J. Craik and M. A. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1221 CrossRef CAS.
- C. W. Gruber, M. Cemazar, M. A. Anderson and D. J. Craik, Toxicon, 2007, 49, 561 CrossRef CAS.
- C. Jennings, J. West, C. Waine, D. Craik and M. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10614 CrossRef CAS.
- C. V. Jennings, K. J. Rosengren, N. L. Daly, M. Plan, J. Stevens, M. J. Scanlon, C. Waine, D. G. Norman, M. A. Anderson and D. J. Craik, Biochemistry, 2005, 44, 851 CrossRef CAS.
- K. R. Gustafson, T. C. McKee and H. R. Bokesch, Curr. Protein Pept. Sci., 2004, 5, 331 CrossRef CAS.
- J. P. Tam, Y. A. Lu, J. L. Yang and K. W. Chiu, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 8913 CrossRef CAS.
- M. Pränting, C. Lööv, R. Burman, U. Göransson and D. I. Andersson, J. Antimicrob. Chemother., 2010, 65, 1964 CrossRef.
- E. Svangard, U. Göransson, Z. Hocaoglu, J. Gullbo, R. Larsson, P. Claeson and L. Bohlin, J. Nat. Prod., 2004, 67, 144 CrossRef.
- M. R. Plan, I. Saska, A. G. Cagauan and D. J. Craik, J. Agric. Food Chem., 2008, 56, 5237 CrossRef CAS.
- U. Göransson, M. Sjogren, E. Svangard, P. Claeson and L. Bohlin, J. Nat. Prod., 2004, 67, 1287 CrossRef.
- M. L. Colgrave, A. C. Kotze, Y. H. Huang, J. O'Grady, S. M. Simonsen and D. J. Craik, Biochemistry, 2008, 47, 5581 CrossRef CAS.
- M. L. Colgrave, A. C. Kotze, D. C. Ireland, C. K. Wang and D. J. Craik, ChemBioChem, 2008, 9, 1939 CrossRef CAS.
- D. G. Barry, N. L. Daly, R. J. Clark, L. Sando and D. J. Craik, Biochemistry, 2003, 42, 6688 CrossRef CAS.
- D. J. Craik, M. Cemazar, C. K. Wang and N. L. Daly, Biopolymers, 2006, 84, 250 CrossRef CAS.
- K. Jagadish and J. A. Camarero, Biopolymers, 2010, 94, 611 CrossRef CAS.
- Y. H. Huang, M. L. Colgrave, N. L. Daly, A. Keleshian, B. Martinac and D. J. Craik, J. Biol. Chem., 2009, 284, 20699 CrossRef CAS.
- H. Kamimori, K. Hall, D. J. Craik and M. I. Aguilar, Anal. Biochem., 2005, 337, 149 CrossRef CAS.
- Z. O. Shenkarev, K. D. Nadezhdin, E. N. Lyukmanova, V. A. Sobol, L. Skjeldal and A. S. Arseniev, J. Inorg. Biochem., 2008, 102, 1246 CrossRef CAS.
- Z. O. Shenkarev, K. D. Nadezhdin, V. A. Sobol, A. G. Sobol, L. Skjeldal and A. S. Arseniev, FEBS J., 2006, 273, 2658 CrossRef CAS.
- C. W. Gruber, M. Cemazar, R. J. Clark, T. Horibe, R. F. Renda, M. A. Anderson and D. J. Craik, J. Biol. Chem., 2007, 282, 20435 CrossRef CAS.
- B. F. Conlan, A. D. Gillon, B. L. Barbeta and M. A. Anderson, Am. J. Bot., 2011, 98, 2018 CrossRef CAS.
- A. D. Gillon, I. Saska, C. V. Jennings, R. F. Guarino, D. J. Craik and M. A. Anderson, Plant J., 2008, 53, 505 CrossRef CAS.
- I. Saska, A. D. Gillon, N. Hatsugai, R. G. Dietzgen, I. Hara-Nishimura, M. A. Anderson and D. J. Craik, J. Biol. Chem., 2007, 282, 29721 CrossRef CAS.
- A. M. Broussalis, U. Göransson, J. D. Coussio, G. Ferraro, V. Martino and P. Claeson, Phytochemistry, 2001, 58, 47 CrossRef CAS.
- B. Chen, M. L. Colgrave, C. Wang and D. J. Craik, J. Nat. Prod., 2006, 69, 23 CrossRef CAS.
- H. P. Kaufmann and A. Tobschirbel, Chem. Ber., 1959, 92, 2805 CrossRef CAS.
- H. Morita and K. Takeya, Heterocycles, 2010, 80, 739 CrossRef CAS.
- H. Morita, A. Shishido, T. Kayashita, M. Shimomura, K. Takeya and H. Itokawa, Chem. Lett., 1994, 12, 2415 CrossRef.
- H. Morita, T. Kayashita, A. Uchida, K. Takeya and H. Itokawa, J. Nat. Prod., 1997, 60, 212 CrossRef CAS.
- B. Picur, M. Lisowski and I. Z. Siemion, Lett. Pept. Sci., 1998, 5, 183 CAS.
- Y. Zhou, M.-K. Wang, S.-L. Peng and L.-S. Ding, Acta Bot. Sin, 2001, 43, 431 CAS.
- J. A. Condie, G. Nowak, D. W. Reed, J. J. Balsevich, M. J. Reaney, P. G. Arnison and P. S. Covello, Plant J., 2011, 67, 682 CrossRef CAS.
- Sweet Orange Genome, http://www.citrusgenomedb.org/species/sinensis.
- Haploid Clementine Genome, International Citrus Genome Consortium, 2011, http://int-citrusgenomics.org, http://www.phytozome.net/clementine.
- Z. Wang, N. Hobson, L. Galindo, J. McDill, L. Yang, S. Hawkins, G. Neutelings, R. Datla, G. Lambert, D. W. Galbraith, C. Cullis, G. K. S. Wong, J. Wang and M. K. Deyholos, 2011, GenBank accession number AFSQ00000000.
- P. Venglat, D. Xiang, S. Qiu, S. L. Stone, C. Tibiche, D. Cram, M. Alting-Mees, J. Nowak, S. Cloutier, M. Deyholos, F. Bekkaoui, A. Sharpe, E. Wang, G. Rowland, G. Selvaraj and R. Datla, BMC Plant Biol., 2011, 11, 74 CrossRef CAS.
- P. S. Covello, R. S. S. Datla, S. L. Stone, J. J. Balsevich, M. J. T. Reaney, P. G. Arnison and J. A. Condie, WO/2010/130030, 2010.
- S. Sato, H. Hirakawa, S. Isobe, E. Fukai, A. Watanabe, M. Kato, K. Kawashima, C. Minami, A. Muraki, N. Nakazaki, C. Takahashi, S. Nakayama, Y. Kishida, M. Kohara, M. Yamada, H. Tsuruoka, S. Sasamoto, S. Tabata, T. Aizu, A. Toyoda, T. Shin-i, Y. Minakuchi, Y. Kohara, A. Fujiyama, S. Tsuchimoto, S. Kajiyama, E. Makigano, N. Ohmido, N. Shibagaki, J. A. Cartagena, N. Wada, T. Kohinata, A. Atefeh, S. Yuasa, S. Matsunaga and K. Fukui, DNA Res., 2012, 18, 65 CrossRef.
- A. J. J. van den Berg, S. F. A. Horsten, J. J. Kettenes-van den Bosch, B. H. Kroes, C. J. Beukelman, B. R. Leeflang and R. P. Labadie, FEBS Lett., 1995, 358, 215 CrossRef CAS.
- R. K. Devappa, H. P. Makkar and K. Becker, J. Agric. Food Chem., 2010, 58, 6543 CrossRef CAS.
- http://www.ars-grin.gov/cgi-bin/npgs/html/taxfam.pl .
- A. Arouri, V. Kiessling, L. Tamm, M. Dathe and A. Blume, J. Phys. Chem. B, 2011, 115, 158 CrossRef CAS.
- C. Auvin-Guette, C. Baraguey, A. Blond, F. Lezenven, J.-L. Pousset and B. Bodo, Tetrahedron Lett., 1997, 38, 2845 CrossRef.
- H. Morita, A. Gonda, K. Takeya, H. Itokawa and Y. Iitaka, Tetrahedron, 1997, 53, 1617 CrossRef CAS.
- H. Morita, T. Kayashita, K. Takeya and H. Itokawa, Tetrahedron, 1994, 50, 12599 CrossRef CAS.
- H. Morita, Y. S. Yun, K. Takeya, H. Itokawa and K. Yamada, Tetrahedron, 1995, 51, 6003 CrossRef CAS.
- O. Buczek, G. Bulaj and B. M. Olivera, Cell. Mol. Life Sci., 2005, 62, 3067 CrossRef CAS.
- H. Hu, P. Bandyopadhyay, B. M. Olivera and M. Yandell, BMC Genomics, 2011, 60 CrossRef CAS.
- Y. Terrat, D. Biass, S. Dutertre, P. Favreau, M. Remmd, R. Stocklin, D. Piquemal and F. Ducancel, Toxicon, 2012, 34 CrossRef CAS.
- B. M. Olivera, in Handbook of Biologically Active Peptides, ed. A. J. Kastin, Elsevier Inc, 2006, p. 381 Search PubMed.
- R. A. Seronay, A. E. Fedosov, M. A. Astilla, M. Watkins, N. Saguil, F. M. Heralde, S. Tagaro, G. T. Poppe, P. M. Alino, M. Oliverio, Y. I. Kantor, G. P. Concepcion and B. M. Olivera, Toxicon, 2010, 56, 1257 CrossRef CAS.
- B. M. Olivera, J. Imperial and G. P. Concepcion, in Handbook of Biologically Active Peptides, ed. A. J. Kastin, Elsevier, Inc., 2013 Search PubMed.
- M. Watkins, D. R. Hillyard and B. M. Olivera, J. Mol. Evol., 2006, 62, 247 CrossRef CAS.
- G. Bulaj and B. M. Olivera, Antioxid. Redox Signaling, 2008, 10, 141 CrossRef CAS.
- H. Terlau and B. M. Olivera, Physiol. Rev., 2004, 84, 41 CrossRef CAS.
- T. S. Han, R. W. Teichert, B. M. Olivera and G. Bulaj, Curr. Pharm. Des., 2008, 14, 2462 CrossRef CAS.
- V. D. Twede, G. Miljanich, B. M. Olivera and G. Bulaj, Curr. Opin. Drug Discov. Devel., 2009, 12, 231 CAS.
- M. Prorok, S. E. Warder, T. Blandl and F. J. Castellino, Biochemistry, 1996, 35, 16528 CrossRef CAS.
- P. K. Bandyopadhyay, C. J. Colledge, C. S. Walker, L.-M. Zhou, D. R. Hillyard and B. M. Olivera, J. Biol. Chem., 1998, 273, 5447 CrossRef CAS.
- P. K. Bandyopadhyay, J. E. Garrett, R. P. Shetty, T. Keate, C. S. Walker and B. M. Olivera, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 1264 CrossRef CAS.
- T. J. Oman, J. M. Boettcher, H. Wang, X. N. Okalibe and W. A. van der Donk, Nat. Chem. Biol., 2011, 7, 78 CrossRef CAS.
- J. Stepper, S. Shastri, T. S. Loo, J. C. Preston, P. Novak, P. Man, C. H. Moore, V. Havlicek, M. L. Patchett and G. E. Norris, FEBS Lett., 2011, 585, 645 CrossRef CAS.
- H. Venugopal, P. J. Edwards, M. Schwalbe, J. K. Claridge, D. S. Libich, J. Stepper, T. Loo, M. L. Patchett, G. E. Norris and S. M. Pascal, Biochemistry, 2011, 50, 2748 CrossRef CAS.
- H. Wang and W. A. van der Donk, J. Am. Chem. Soc., 2011, 133, 16394 CrossRef CAS.
- G. Ji, R. Beavis and R. P. Novick, Science, 1997, 276, 2027 CrossRef CAS.
- M. Otto, R. Süssmuth, G. Jung and F. Götz, FEBS Lett., 1998, 424, 89 CrossRef CAS.
- A. Wuster and M. M. Babu, J. Bacteriol., 2008, 190, 743 CrossRef CAS.
- M. Thoendel, J. S. Kavanaugh, C. E. Flack and A. R. Horswill, Chem. Rev., 2011, 111, 117 CrossRef CAS.
- G. Ji, R. C. Beavis and R. P. Novick, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 12055 CrossRef CAS.
- M. Thoendel and A. R. Horswill, J. Biol. Chem., 2009, 284, 21828 CrossRef CAS.
- L. Zhang, L. Gray, R. P. Novick and G. Ji, J. Biol. Chem., 2002, 277, 34736 CrossRef CAS.
- R. Qiu, W. Pei, L. Zhang, J. Lin and G. Ji, J. Biol. Chem., 2005, 280, 16695 CrossRef CAS.
- L. Zhang, J. Lin and G. Ji, J. Biol. Chem., 2004, 279, 19448 CrossRef CAS.
- M. Okada, I. Sato, S. J. Cho, H. Iwata, T. Nishio, D. Dubnau and Y. Sakagami, Nat. Chem. Biol., 2005, 1, 23 CrossRef CAS.
- M. Okada, Biosci., Biotechnol., Biochem., 2011, 75, 1413 CrossRef CAS.
- G. E. Kenney and A. C. Rosenzweig, ACS Chem. Biol., 2011, 7, 260 CrossRef.
- H. J. Kim, D. W. Graham, A. A. DiSpirito, M. A. Alterman, N. Galeva, C. K. Larive, D. Asunskis and P. M. Sherwood, Science, 2004, 305, 1612 CrossRef CAS.
- L. A. Behling, S. C. Hartsel, D. E. Lewis, A. A. DiSpirito, D. W. Choi, L. R. Masterson, G. Veglia and W. H. Gallagher, J. Am. Chem. Soc., 2008, 130, 12604 CrossRef CAS.
- A. El Ghazouani, A. Basle, S. J. Firbank, C. W. Knapp, J. Gray, D. W. Graham and C. Dennison, Inorg. Chem., 2011, 50, 1378 CrossRef CAS.
- B. D. Krentz, H. J. Mulheron, J. D. Semrau, A. A. Dispirito, N. L. Bandow, D. H. Haft, S. Vuilleumier, J. C. Murrell, M. T. McEllistrem, S. C. Hartsel and W. H. Gallagher, Biochemistry, 2010, 49, 10117 CrossRef CAS.
- G. A. Somerville, A. Cockayne, M. Durr, A. Peschel, M. Otto and J. M. Musser, J. Bacteriol., 2003, 185, 6686 CrossRef CAS.
- E. Schiffmann, B. A. Corcoran and S. M. Wahl, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 1059 CrossRef CAS.
- Z. He, C. Yuan, L. Zhang and A. E. Yousef, FEBS Lett., 2008, 582, 2787 CrossRef CAS.
- Q. Zhang and W. A. van der Donk, FEBS Lett., 2012, 586, 3391 CrossRef CAS.
- M. Skaugen, J. Nissenmeyer, G. Jung, S. Stevanovic, K. Sletten, C. I. M. Abildgaard and I. F. Nes, J. Biol. Chem., 1994, 269, 27183 CAS.
- C. Anthony, Biochem Soc Trans, 1998, 26, 413 CAS.
- P. Mazodier, F. Biville, E. Turlin and F. Gasser, J. Gen. Microbiol., 1988, 134, 2513 CAS.
- N. Goosen, H. P. Horsman, R. G. Huinen and P. van de Putte, J. Bacteriol., 1989, 171, 447 CAS.
- N. Goosen, R. G. Huinen and P. van de Putte, J. Bacteriol., 1992, 174, 1426 CAS.
- D. R. Houck, J. L. Hanners, C. J. Unkefer, M. A. van Kleef and J. A. Duine, Antonie van Leeuwenhoek, 1989, 56, 93 CrossRef CAS.
- J. J. Meulenberg, E. Sellink, N. H. Riegman and P. W. Postma, Mol. Gen. Genet., 1992, 232, 284 CAS.
- J. S. Velterop, E. Sellink, J. J. Meulenberg, S. David, I. Bulder and P. W. Postma, J. Bacteriol., 1995, 177, 5088 CAS.
- O. T. Magnusson, H. Toyama, M. Saeki, A. Rojas, J. C. Reed, R. C. Liddington, J. P. Klinman and R. Schwarzenbacher, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7913 CrossRef CAS.
- S. R. Wecksler, S. Stoll, H. Tran, O. T. Magnusson, S. P. Wu, D. King, R. D. Britt and J. P. Klinman, Biochemistry, 2009, 48, 10151 CrossRef CAS.
- S. R. Wecksler, S. Stoll, A. T. Iavarone, E. M. Imsand, H. Tran, R. D. Britt and J. P. Klinman, Chem. Commun., 2010, 46, 7031 RSC.
- Y. Oba, S. Kato, M. Ojika and S. Inouye, Biochem. Biophys. Res. Commun., 2009, 390, 684 CrossRef CAS.
-
W. W. Ward and M. Chalfie, Expression of a gene for a modified green-fluorescent protein, US 5741668, 1998.
- M. Jin, L. Liu, S. A. Wright, S. V. Beer and J. Clardy, Angew. Chem., Int. Ed., 2003, 42, 2898 CrossRef CAS.
- M. Jin, S. A. Wright, S. V. Beer and J. Clardy, Angew. Chem., Int. Ed., 2003, 42, 2902 CrossRef CAS.
- S. E. Rokita, J. M. Adler, P. M. McTamney and J. A. Watson, Jr, Biochimie, 2010, 92, 1227 CrossRef CAS.
- N. Ziemert, K. Ishida, P. Quillardet, C. Bouchier, C. Hertweck, N. T. de Marsac and E. Dittmann, Appl. Environ. Microbiol., 2008, 74, 1791 CrossRef CAS.
- N. Garg, W. Tang, Y. Goto and W. A. van der Donk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5241 CrossRef CAS.
- S. R. Eddy, PLoS Comput. Biol., 2011, 7, e1002195 CAS.
- J. D. Selengut, D. H. Haft, T. Davidsen, A. Ganapathy, M. Gwinn-Giglio, W. C. Nelson, A. R. Richter and O. White, Nucleic Acids Res., 2007, 35, D260 CrossRef CAS.
- M. Punta, P. C. Coggill, R. Y. Eberhardt, J. Mistry, J. Tate, C. Boursnell, N. Pang, K. Forslund, G. Ceric, J. Clements, A. Heger, L. Holm, E. L. Sonnhammer, S. R. Eddy, A. Bateman and R. D. Finn, Nucleic Acids Res., 2012, 40, D290 CrossRef CAS.
- A. de Jong, A. J. van Heel, J. Kok and O. P. Kuipers, Nucleic Acids Res., 2010, 38, W647 CrossRef CAS.
- J. S. Garavelli, Proteomics, 2004, 4, 1527 CrossRef CAS.
- D. H. Haft, Biol. Direct, 2009, 4, 15 CrossRef.
| This journal is © The Royal Society of Chemistry 2013 |