2-Deoxystreptamine-containing aminoglycoside antibiotics: Recent advances in the characterization and manipulation of their biosynthetic pathways†
Sung Ryeol Park‡a, Je Won Park‡b, Yeon Hee Bana, Jae Kyung Sohngb and Yeo Joon Yoon*a
aDepartment of Chemistry and Nano Science, Ewha Womans University, Seoul 120-750, Republic of Korea. E-mail: joonyoon@ewha.ac.kr; Fax: +82-2-3277-3419; Tel: +82-2-3277-4082
bDepartment of Pharmaceutical Engineering, SunMoon University, Asan 336-708, Republic of Korea
First published on 26th November 2012
Abstract
Covering: 2007 to September 2012
The 2-deoxystreptamine-containing aminoglycosides, such as neomycin, kanamycin and gentamicin, are an important class of antibiotics. A detailed understanding of the complete biosynthetic pathway of aminoglycosides and their biosynthetic enzymes will allow us to not only generate more robust antibiotic agents or drugs with other altered biological activities, but also to produce clinically important semi-synthetic antibiotics by direct fermentation. This Highlight focuses on recent advances in the characterization of their biosynthetic enzymes and pathway as well as some chemo-enzymatic and metabolic engineering approaches for the biological production of natural, semi-synthetic, and novel aminoglycosides.
 Sung Ryeol Park | Sung Ryeol Park received her B.S. (chemistry) and M.S. (biochemistry) at Ewha Womans University. She obtained her Ph.D. (Biochemistry) in 2010 under the supervision of Yeo Joon Yoon at the same university. During her Ph.D. course, she worked on elucidating aminoglycoside biosynthetic pathways as well as generating bioactive natural compounds based on heterologous expression systems. She is currently a postdoctoral fellow at the University of Michigan in the laboratory of David H. Sherman where her research has focused on the molecular genetic and biochemical studies on the biosynthesis of secondary metabolites from terrestrial and marine microorganisms. |
 Je Won Park | Je Won Park (1971, Korea) obtained his Ph.D. in 2002 at Korea University under the supervision of the late Prof. Young Bae Kim. In 2003 he started as a postdoc at Health Canada, and then moved to Ewha Womans University as a research professor. Since 2011, he has been appointed as an assistant professor at the Department of Pharmaceutical Engineering in SunMoon University. Currently his research interests focus on the discovery and development of bioactive natural products, which are mainly obtained as microbial secondary metabolites, by using metabolomics, fluxomics, and biosynthetic pathway engineering. |
 Yeon Hee Ban | Yeon Hee Ban received her Master degree in microbiology from the Chungbuk National University, Korea, in 2007. She then joined the group of Prof. Yeo Joon Yoon at the Ewha Womans University for PhD studies on the biosynthesis of natural products. She is currently working on the investigation of the biosynthetic pathway for gentamicin and development of novel derivatives via pathway engineering. |
 Jae Kyung Sohng | Jae Kyung Sohng received his M.S. (1986) at Yonsei University and Ph.D. (1991) from Brown University under the supervision of Prof. David Cane. He worked as a postdoc in Prof. Heinz Floss Lab., University of Washington from 1991. In 1994, he was appointed as a full lecturer at the Department of Chemistry, SunMoon University. Now, he is a Professor at the Department of Pharmaceutical Engineering and Director at the Institute of Biomolecule Reconstruction, SunMoon University since 2004. His research interests are the biosynthesis of microbial secondary metabolites and glycosylation of small molecules for novel natural product by combination of plant aglycone and microbial glycone. |
 Yeo Joon Yoon | Yeo Joon Yoon is currently Professor of Chemistry and Nano Science at Ewha Womans University, Korea (since 2004). He received his B.S. degree (1992) and Ph.D. degree (2000) from Seoul National University, Korea. During his Ph.D. he studied the biosynthesis of rifamycin under the guidance of Prof. C. Richard Hutchinson at the University of Wisconsin-Madison, USA. As a post-doctoral researcher at the University of Minnesota, USA, with Prof. David H. Sherman he worked on pikromycin biosynthesis. His research interests are characterization of biosynthetic pathways for polyketides/aminoglycosides, and application of combinatorial biosynthesis/synthetic biology to the production of secondary metabolites. |
1 Introduction
Aminoglycosides are one of the classical antibiotics that act by binding to the 16S rRNA subunit of the 30S bacterial ribosome, which inhibits protein synthesis by generating errors in translation.1 Since the discovery of the first effective anti-tuberculosis agent, streptomycin, by Waksman in 1944,2 aminoglycoside antibiotics, such as neomycin (1949), kanamycin (1957), paromomycin (1959), gentamicin (1963), tobramycin (1967), sisomicin (1970), butirosin (1971), lividomycin (1971), and others have been widely used to treat bacterial infections, particularly against Gram-negative bacteria, for more than half a century (Fig. 1). However, as with other antibiotics, the emergence and rapid spread of aminoglycoside-resistant pathogens limit their continued intensive clinical use.3 Since the modification and deactivation of aminoglycosides by aminoglycoside-modifying enzymes (AME), for example N-acetylation, O-nucleotidylation, or O-phosphorylation, which are the major mechanisms of bacterial resistance to aminoglycosides,4 the resistance issue has been addressed through the chemical modification of natural aminoglycosides in order to avoid the above enzymatic deactivation.5 Such earlier chemical modification efforts have resulted in the development of semi-synthetic aminoglycosides, such as dibekacin, amikacin, netilmicin, and isepamicin, during the 1970s. However, since the latest approved semi-synthetic aminoglycoside arbekacin in 1990, no new aminoglycoside antibiotic has been launched (Fig. 1).5 Although untreatable multi-drug resistant (MDR) Gram-negative bacteria are on the increase, most recently introduced antibiotics after 2000 (i.e. oxazolidinones, lipopeptides, and mutilins) are exclusively against Gram-positive bacteria.6 Their urgent medical need, their well-known mechanism of action and resistance, recent advances in chemical/chemo-enzymatic modification as well as engineered biosynthesis of aminoglycosides, and their potential for the treatment of HIV7 and human genetic diseases8 have renewed interest for further investigation of this class of old drugs.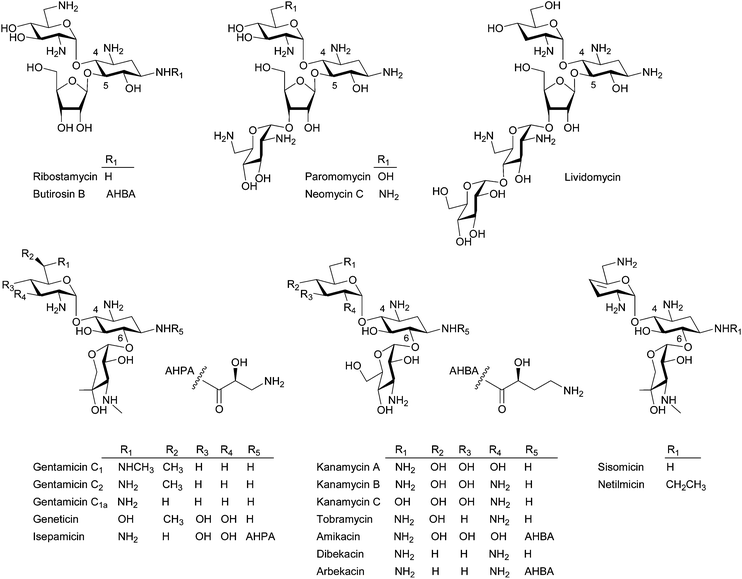 | ||
| Fig. 1 Representative structures of the 4,5- and 4,6-disubstituted 2-DOS containing aminoglycosides. | ||
Structurally, aminoglycosides contain a core aminocyclitol moiety, streptamine, streptidine, or 2-deoxystreptamine (2-DOS) and a majority of aminoglycosides are composed of 2-DOS, which are decorated by sugar substituents at the C4, C5, and C6 positions (Fig. 1). The 4,5- and 4,6-disubstituted 2-DOS-containing aminoglycosides are the most clinically important antibiotics. The 4,5-disubstituted aminoglycosides represented by neomycin and paromomycin are used topically and against intestinal parasites, respectively, whereas the 4,6-disubstituted aminoglycosides represented by gentamicin, tobramycin, and amikacin have been used for the treatment of serious opportunistic infections by Gram-negative bacteria.8,9
A recent focused medicinal chemistry campaign by the biopharmaceutical company Achaogen yielded a collection of over 400 sisomicin analogs and ACHN-490, which showed promise as a “next-generation” aminoglycoside, was selected.10 ACHN-490 is the only novel aminoglycoside derivative currently under clinical trial to combat MDR Gram-negative pathogens.11 In addition, a novel aminoglycoside derivative NB54 exhibiting superior read-through efficiency and lower toxicity was developed and demonstrated its potential in the treatment of human genetic diseases.12 Despite the successful examples of historical and recent chemical modification approaches, biological approaches, including chemo-enzymatic methods and engineered biosynthesis, can be attractive alternatives to create more diverse novel aminoglycosides.
For biological approaches to be practical, a detailed understanding of the biosynthetic pathways of aminoglycosides is a prerequisite. Unfortunately, although the biosynthetic pathways of 4,5-disubstituted aminoglycosides have been well characterized in the context of butirosin and neomycin using the recombinant proteins,13 those of 4,6-disubstituted aminoglycosides still remain unclear. However, a recent discovery of the complete kanamycin biosynthetic pathway14,15 and the engineered biosynthesis of a novel kanamycin analogue, that is active against some kanamycin- and amikacin-resistant Gram-negative bacteria,14 as well as the successful chemo-enzymatic synthesis of N-acylated aminoglycosides16–18 demonstrated the potential of biological approaches for production of more robust aminoglycosides. Since there are several reviews covering the biosynthesis of aminoglycoside19–21 and the chemical modification of this class of molecule,4,8 herein we highlight the recent advances in understanding the biosynthesis of the 2-DOS-containing aminoglycosides as well as the current status and potential of the biological approaches for the heterologous production of the existing natural/semi-synthetic aminoglycosides and the creation of novel aminoglycosides. As many identical aminoglycoside biosynthetic genes are deposited by different names, a set of gene annotations, which is systematically coded by Piepersberg and co-workers21 will be mainly used in this review, except for the gene annotations for butirosin by Eguchi and co-worker20 (see ESI† for the complete list of aminoglycoside biosynthetic genes deposited to date).
2 Biosynthetic route to pseudodisaccharides
2.1 Biosynthesis of 2-DOS
Although most biosynthetic steps to the core moiety 2-DOS from D-glucose (Glc)-6-phosphate by 2-deoxy-scyllo-inosose (2-DOI) synthase, a dual-functional L-glutamine:2-DOI aminotransferase, and 2-deoxy-scyllo-inosamine (2-DOIA) dehydrogenase are well-established (Fig. 2; see the review19 for the detailed biosynthetic mechanism of 2-DOS), it has been revealed that an unusual radical S-adenosylmethionine (SAM) dehydrogenase BtrN catalyzes the oxidation of 2-DOIA to keto-2-DOIA under anaerobic conditions in the butirosin producer Bacillus circulans.22 In the neomycin-producing Streptomyces fradiae, an NAD+-dependent dehydrogenase NeoE catalyzes the same oxidation step.23 All other aminoglycoside gene clusters have NeoE homologs19,20 and some of them are functionally characterized. Therefore, it is clear that B. circulans has a unique 2-DOS biosynthetic system compared to the rest of the aminoglycoside producers, which is also supported by the presence of a 2-DOI synthase stabilizing protein found only in B. circulans. The product of the btrC2 gene, which is located outside of the butirosin cluster, is involved in the biosynthesis of vitamin B6 in the primary metabolism and also acts as a stabilizer of 2-DOI synthase by forming a heterodimer with BtrC.24 Interestingly, an NAD+-dependent dehydrogenase BtrE, which was believed to catalyze the oxidation of 2-DOIA, is also present in the butirosin cluster. However, BtrE did not demonstrate any 2-DOIA dehydrogenase activity, probably due to the lack of two important zinc motifs, and its function is presently unknown.22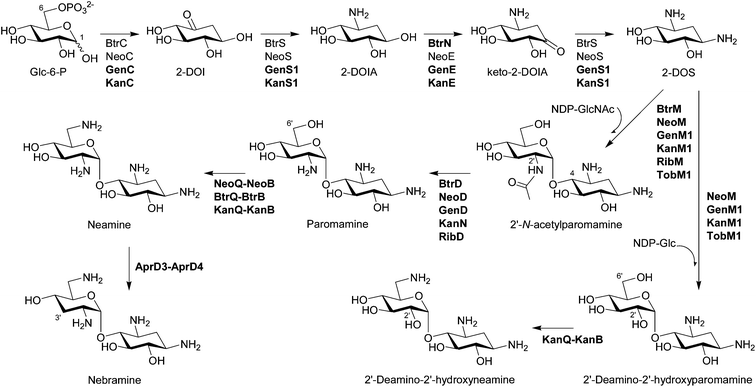 | ||
| Fig. 2 Biosynthesis of pseudodisaccharides. The biosynthetic enzymes whose functions have been characterized by in vivo and/or in vitro experiments are listed. The enzymes that have been functionally identified since 2007 are written in bold characters. | ||
The crystal structure of 2-DOI synthase BtrC from B. circulans and its complex with the substrate analog inhibitor carbaglucose-6-phosphate, NAD+, and Co2+ have been determined and suggest that the phosphate elimination mechanism is syn-elimination assisted by Glu 243 and the aldol condensation proceeds through a boat conformation, indicating the mechanical resemblance and difference between 2-DOI synthase and dehydoquinate synthase in the primary metabolism.25 Interestingly, 19 DNA fragments encoding 2-DOI synthase were identified from deep-sea environmental DNA, suggesting the possibility to expand potential aminoglycoside producers.26
2.2 Glycosylation in the formation of pseudodisaccharides
Bioinformatics analysis suggested that BtrM in the butirosin cluster and its homolog NeoM in the neomycin cluster transfer N-acetylglucosamine (GlcNAc) to DOS to give 2′-N-acetylparomamine.19 Indeed, it was confirmed that NeoM catalyzes the glycosylation of 2-DOS with UDP-GlcNAc using the cell-free extract of Escherichia coli expressing neoM.27 An independent in vivo study also confirmed the role of NeoM by an in-frame gene deletion.28 NeoM is the first characterized glycosyltransferase (GT) involved in the formation of pseudodisaccharide. In case of 4,6-disubstituted aminoglycosides, GenM1 in the gentamicin cluster of Micromonospora echinospora was found to catalyze the same glycosylation reaction by heterologous expression of minimal genes for the 2′-N-acetylparomamine biosynthesis in S. venezuelae and using its cell-free extract expressing GenM1.29 Following these studies, RibM of the ribostamycin producer S. ribosidificus and BtrM were also characterized as UDP-GlcNAc:2-DOS GTs through interspecies complementation of S. fradiae ΔneoM mutant with ribM and the heterologous expression of butirosin genes in E. coli to produce paromamine, respectively.30,31 Heterologous production of paromamine in S. lividans by expression of kanM1 with 2-DOS biosynthetic genes and deacetylase gene (see below) from the kanamycin producer S. kanamyceticus suggested that KanM1 is also the UDP-GlcNAc GT.32 Remarkably, it has been revealed that KanM1 accepts both UDP-Glc and UDP-GlcNAc as glycosyl donors but preferentially transfers Glc to 2-DOS, producing the previously undescribed pesudodisaccharide intermediate 2′-deamino-2′-hydroxyparomamine (Fig. 2) by the heterologous expression of kanamycin genes in S. venezuelae and kinetic analysis using the purified recombinant KanM1 expressed in S. venezuelae.14 In addition, the heterologous expression of neoM, tobM1 from the tobramycin gene cluster, and genM1 together with 2-DOS biosynthetic genes showed that these GTs are also flexible toward UDP-sugars but seem to use UDP-GlcNAc preferentially over UDP-Glc in contrast to KanM1.14 The discovery of an early branch point in the kanamycin biosynthetic pathway, governed by the substrate promiscuity of GT KanM1, established two independent parallel biosynthetic routes to kanamycins A and B (Fig. 3).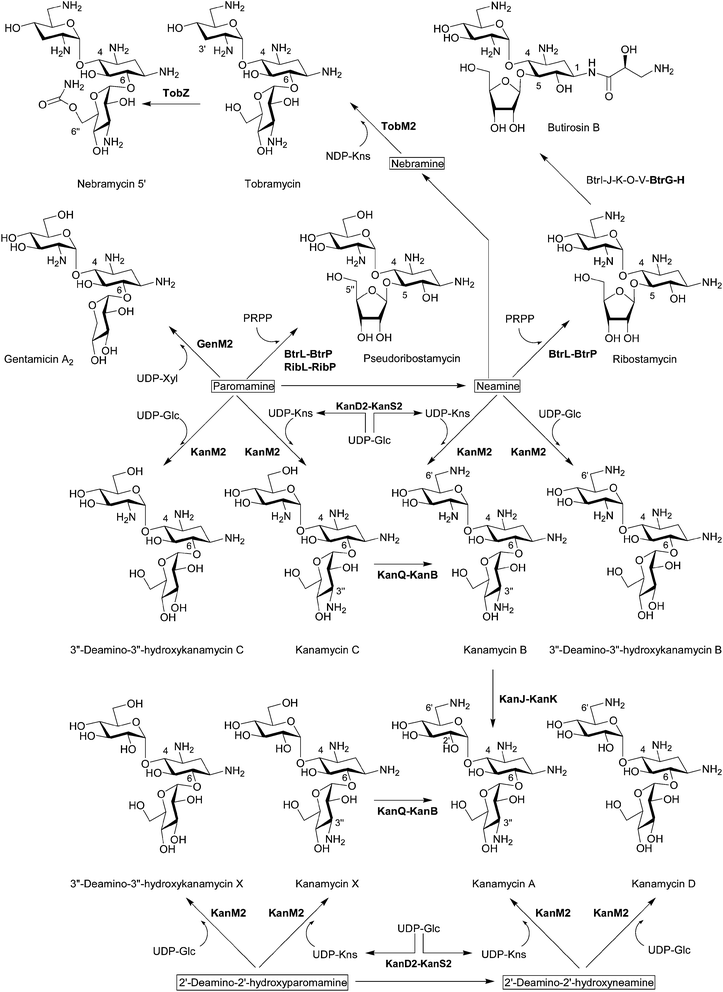 | ||
| Fig. 3 Biosynthesis of pseudotrisaccharides. The biosynthetic enzymes whose functions have been characterized by in vivo and/or in vitro experiments are listed. The enzymes that have been functionally identified since 2007 are written in bold characters. | ||
2.3 Deacetylation of 2′-N-acetylparomamine to paromamine
BtrD in the butirosin gene cluster was proposed to encode a nucleotidyltransferase generating the putative sugar donor dTDP-glucosamine (GlcN) for the GT BtrM, which was supposed to transfer GlcN onto 2-DOS to give paromamine.33 However, its function was reassigned as a 2′-N-acetylparomamine deacetylase using the recombinant BtrD and the enzymatically synthesized 2′-N-acetylparomamine (Fig. 2).34 This result suggested that UDP-GlcNAc is the true substrate for the GT BtrM, which was later confirmed by using the cell-free extracts containing BtrM-homologous GTs (NeoM27 and GenM129) and the purified recombinant KanM1.14 Again, it has also been shown that GenD,29 KanN,29 and RibD35 in the gentamicin, kanamycin, and the ribostamycin cluster, respectively, are also 2′-N-acetylparomamine deacetylases by the heterologous expression of genes for 2-DOS biosynthesis, deacetylase, and GT to produce paromamine in S. venezuelae. Paromamine can be further modified by C6′ amination and C3′ deoxygenation (Fig. 2) or can function as a sugar acceptor directly for the second glycosylation reaction to give pseudotrisaccharides (Fig. 3).2.4 C6′ amination of paromamine to neamine
The enzymes responsible for the conversion of paromamine to neamine through sequential dehydrogenation and transamination and their cofactors were first characterized using the recombinant proteins in the neomycin and butirosin gene cluster.36 NeoQ is FAD-dependent 6′-paromamine dehydrogenase, and NeoB and BtrB are pyridoxal-5′-phosphate (PLP)-dependent 6′-oxoparomamine:L-glutamate aminotransferases (Fig. 2).36 The NeoQ/NeoB-homologous proteins KanQ/KanB in the kanamycin cluster were found to be responsible for the C6′ amination of both paromamine and 2′-deamino-2′-hydroxyparomamine generating neamine and 2′-deamino-2′-hydroxyneamine, respectively (Fig. 2) using the cell-free extract of S. venezuelae expressing kanQ and kanB.14 An in-frame deletion of tobQ in the S. tenebrarius resulted in the production of 3′-deoxy-carbamoylkanamycin C, which lacks a C6 amino group compared to carbamoyltobramycin, also suggesting that tobQ encodes 6′-paromamine dehydrogenase in the tobramycin and apramycin biosynthetic pathway.372.5 C3′ deoxygenation of neamine to nebramine
Since aminoglycosides, which lack the C3′ hydroxyl group, such as tobramycin, are less prone to deactivation by the most well-studied AME aminoglycoside phosphotransferases (APHs) and the chemically modified 3′-deoxykanamycins showed excellent activity against resistant bacteria producing APH(3′),5,18 C3′ deoxygenation has long been of interest. Bioinformatics analysis suggested a radical SAM oxidoreductase AprD3 and a dehydrogenase AprD4 from S. tenebrarius, which produces a small amount of tobramycin, are responsible for C3′ deoxygenation.20 The cell-free extract of S. venezuelae containing AprD3 and AprD4 indeed eliminated a 3′-hydroxyl group in neamine to produce nebramine (Fig. 2) but did not transform kanamycin B to tobramycin, indicating that these enzymes are active only for pseudodisaccharides.143 Biosynthetic route to pseudotrisaccharides
3.1 Second glycosylation in 4,5-disubstituted aminoglycosides
The first functionally characterized glycosylation step from aminoglycoside gene clusters is the O-ribosylation of neamine for the formation of ribostamycin, a common intermediate for neomycins and butirosin, during butirosin biosynthesis (Fig. 3). Using recombinant proteins, it has been revealed that BtrL catalyzes phosphoribosylation of neamine to form 5′′-phosphoribostamycin using 5-phosphoribosyl-1-pyrophosphate (PRPP) as a ribosyl donor and BtrP dephosphorylates 5′′-phosphoribostamycin to generate ribostamycin.38 BtrL and BtrP also catalyzed the two-step ribosylation of paromamine to give 6′-deamino-6′-hydroxyribostamycin (pseudoribostamycin).38 The production of pseudoribostamycin in the S. venezuelae mutant by heterologous expression of ribostamycin genes for direct ribosylation of paromamine also showed that RibL and RibP in the ribostamycin producer S. ribosidificus play the same role as BtrL and BtrP, respectively.353.2 Second glycosylation in 4,6-disubstituted aminoglycosides
In contrast to 4,5-disubstituted aminoglycosides, it has been believed that the gentamicin pseudotrisaccharide scaffold is derived from the addition of nucleotide-activated D-xylose (Xyl) to paromamine.19 Using the cell-free extract of S. venezuelae expressing the minimal genes for the formation of gentamicin A2, it has been proved that GenM2 catalyzes the transfer of UDP-Xyl directly to paromamine to give gentamicin A2 (Fig. 3).29 This result excluded the possibility that the xylose moiety of gentamicin A2 is generated from glucose or ribose attached to paromamine.Remarkably, it has been discovered that the GT KanM2 in the kanamycin gene cluster is able to use four different pseudodisaccharides, including paromamine, neamine, 2′-deamino-2′-hydroxyparomamine, and 2′-deamino-2′-hydroxyneamine as glycosyl acceptors and two different glycosyl donors such as UDP-Glc and UDP-kanosamine (UDP-Kns, UDP-D-3-glucosamine) (Fig. 3).14,39 Comparative kinetic analysis using the recombinant KanM2 expressed in S. venezuelae together with the UDP-Glc and UDP-Kns, which was enzymatically synthesized from UDP-Glc by the joint activity of KanD2 and KanS2 (see below), revealed that KanM2's preferred sugar donor is UDP-Kns.14
Despite this remarkable substrate flexibility of KanM2, this enzyme seems to selectively transfer Kns to paromamine and 2′-deamino-2′-hydroxyparomamine but not to nebramine based on the metabolite profile of the S. venezuelae expressing kanamycin B minimal biosynthetic genes with aprD3 and aprD4.14 However, it turned out that TobM2 from the S. tenebrarius is able to use nebramine as a glycosyl acceptor of UDP-Kns and synthesize tobramycin when expressed with kanamycin B minimal biosynthetic genes with aprD3 and aprD4 in S. venezuelae (Fig. 3).14
3.3 1-N-acylation of pseudotrisaccharide
The unique (S)-4-amino-2-hydroxybutyrate (AHBA) side chain of butirosin improves pharmacological properties and protects the aminoglycoside from several common resistance mechanisms.4,8 The AHBA or analogous (S)-4-amino-2-hydroxypropionate (AHPA) are incorporated to the natural aminoglycoside scaffolds giving the semi-synthetic amikacin, arbekacin, and isepamicin (Fig. 1). The comparison of butirosin and neomycin gene clusters suggested that seven butirosin genes that lack homologs in the neomycin cluster could be involved in the biosynthesis and transfer of the AHBA side chain, and it has been revealed that five of these gene products (BtrI, BtrJ, BtrK, BtrO, and BtrV) synthesize γ-L-Glu-AHBA-S-BtrI (acyl carrier protein, ACP) via the BtrI-mediated pathway involving a rare protective chemistry in biosynthesis (i.e. γ-L-glutamylation of a potentially nucleophilic primary amine on the ACP-bound intermediate).19,40 Later, it was shown that the γ-glutamylated dipeptide is transferred from BtrI to ribostamycin by the 1-N-acyltransferase BtrH, resulting in the formation of γ-L-Glu-butirosin, and the protective γ-glutamyl group is cleaved by the γ-glutamyl cyclotransferase BtrG (Fig. 3).163.4 C6′ amination of pseudotrisaccharides
The cell-free extract of S. venezuelae expressing kanQ and kanB catalyzes C6′ amination of kanamycins C and X to generate kanamycins B and A, respectively, revealing their substrate flexibility accepting both pseudodisaccharides and pseudotrisaccharides (Fig. 3).14 The substrate flexibility of KanQ-KanB and GTs (KanM1 and KanM2) led to the unexpected architecture of two parallel kanamycin biosynthetic pathways (Fig. 2 and Fig. 3).3.5 C2′ deamination of pseudotrisaccharides
Although kanamycin A can be biosynthesized independently from kanamycin B via 2′-deamino-2′-hydroxyparomamine and kanamycin X,14 a recent study demonstrated that kanamycin B can also be converted to kanamycin A by two enzymes, which are exclusively present in the kanamycin cluster.15 Synthesis of 2′-oxokanamycin and the release of ammonia from kanamycin B is catalysed by a FeII/α-ketoglutarate (KG)-dependent dioxygenase KanJ, and an NADPH-dependent ketoreductase KanK stereoselectively reduces 2′-oxokanamycin to give kanamycin A (Fig. 3). Moreover, it has been revealed that KanJ catalyzes the hydroxylation of C2′ to afford a hemiaminal intermediate, which is simultaneously converted into 2′-oxokanamycin, as in the case of the usual α-KG-dependent hydroxylase.153.6 C6′′-O-carbamoylation of pseudotrisaccharides
It has been confirmed that a previously proposed O-carbamoyltransferase TobZ indeed catalyzes the ATP-dependent 6′′-O-carbamoylation of tobramycin to form nebramycin 5′ using the recombinant TobZ from Streptoalloteichus tenebrarius (recently reclassified from Streptomyces tenebrarius)41 expressed in S. lividans (Fig. 3).42 In addition, the structural and biochemical data suggested a reaction sequence for TobZ, which possesses two major domains, a larger Kae 1-like domain and a smaller YrdC-like domain. The YrdC-like domain catalyzes the first half-reaction, namely the hydrolysis of carbamoylphosphate and subsequent adenylation by ATP to form carbamoyladenylate, which is channelled to the Kae 1-like domain where the transfer of the carbamoyl moiety to the 6′′-hydroxyl group of tobramycin occurs. TobZ is also able to carbamoylate kanamycin A to form a novel carbamoyl-kanamycin A exhibiting a certain degree of substrate flexibility.423.7 C3′′ amination of pseudotrisaccharides
4,6-disubstituted aminoglycosides, such as gentamicins, kanamycins and sisomicin, contain a 3′′-amino group (Fig. 1). In the case of kanamycin, it was found that UDP-Kns is synthesized by a dehydrogenase KanD2 and an aminotransferase KanS2 and that the C3′′ amination activity of KanD2–KanS2 precedes that of GT KanM2 by in vitro reactions using cell-free extracts (Fig. 3).14 These results indicate that this enzyme pair acts only on the nucleotide-activated sugar, as in the biosynthesis of free Kns in rifamycin-producing Amycolatopsis mediterranei.43A M. echinospora mutant, in which the genD1 gene encoding a putative radical SAM C-methyltransferase was inactivated, accumulated gentamicin A2, suggesting that GenD1 might participate in the pseudotrisaccharide modification steps after gentamicin A2 formation.44 The first modification step of gentamicin A2 is presumably the oxidation of the hydroxyl group on C3′′ and amination. Therefore, it was hypothesized that the putative radical SAM domain of GenD1 might be involved in the C3′′ oxidation.44 However, previously it was proposed that GenD1 catalyzes the C-methylation at C6′′ of gentamicins.20 Therefore, it is more plausible that the replacement of genD1 with a thiostrepton resistance gene might have a polar effect on the expression of the downstream gene genS2, which is homologous to kanS2 and encodes a putative aminotransferase probably involved the oxidative amination of 3′′-hydroxyl group, and thus gentamicin A2 accumulated. However, further enzymatic analysis of GenD1 and GenS2 is required to determine their exact functions.
4 Biosynthetic route to pseudotetrasaccharides
4.1 Glycosylation in the formation of pseudotetrasaccharides
Bioinformatics analysis suggested NeoF as a potential GT involved in the attachment of the fourth ring in neomycin and it has been confirmed that NeoF is a GT that transfers UDP-GlcNAc onto ribostamycin to generate 2′′′-N-acetyl-6′′′-deamino-6′′′-hydroxyneomycin C (N-acetylglucosaminylribostamycin) using the recombinant purified NeoF (Fig. 4).27 An in vivo in-frame gene deletion of neoF in S. fradiae resulting in the accumulation of ribostamycin also confirmed the role of NeoF.28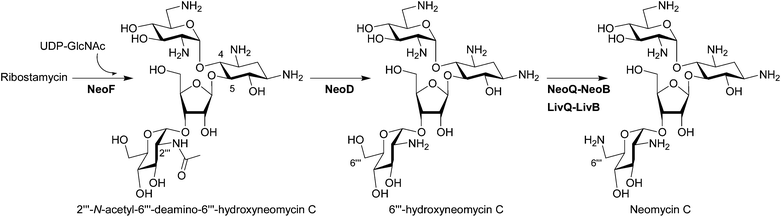 | ||
| Fig. 4 Biosynthesis of pseudotetrasaccharides. The biosynthetic enzymes whose functions have been characterized by in vivo and/or in vitro experiments are listed. The enzymes which have been functionally identified since 2007 are written in bold characters. | ||
4.2 Deacetylation of pseudotetrasaccharides
Interestingly, although two deacetylation steps are needed in the biosynthesis of neomycin (see Fig. 2 and Fig. 4) based on the functional characterization of NeoM and NeoF as N-acetylglucosaminyltransferases,27 only one BtrD-homologous deacetylase, NeoD, is present in the neomycin gene cluster. The in vitro experiment using recombinant NeoD proved that this enzyme recognizes both acetylated pseudodisaccharide 2′-N-acetylparomamine and acetylated pseudotetrasacchride N-acetylglucosaminylribostamycin (Fig. 4).27 However, the ΔneoD S. fradiae mutant supplemented with neamine still produced neomycin, suggesting the presence of a second deacetylase in S. fradiae capable of the deacetylation of N-acetylglucosaminylribostamycin.284.3 C6′′′ amination of pseudotetrasaccharides
NeoB, which is responsible for the amination of 6′-oxoparomamine to neamine (Fig. 2) was also suggested to be involved in the C6′′′-amination based on its deamination activity at C6′′′ of neomycin (Fig. 4).36 Recently, it has indeed been shown that NeoQ and NeoB can recognize both pseudodisaccharide paromamine and pseudotetrasaccharide 6′′′-hydroxyneomycin C.45 Interestingly the NeoQ homolog LivQ in the lividomycin producer S. lividus oxidized only 6′′′-hydroxyneomycin C, whereas the corresponding aminotransferase LivB showed the same enzymatic property as NeoB capable of transaminating both C6′ and C6′′′ positions.45 These results demonstrated that the different modification pattern at the C6′-position in neomycin and lividomycin (Fig. 1) is only due to the substrate specificities of the oxidoreductase NeoQ and LivQ.5 Heterologous production of natural and semi-synthetic aminoglycosides
The progress to characterize biosynthetic routes to 4,6-disubstitued aminoglycosides has been slow mainly because of difficulties in genetically manipulating the aminoglycoside-producing actinomycetes. For example, the gentamicin producer M. echinospora and kanamycin producer S. kanamyceticus have innate resistance to most antibiotics precluding the use of standard Streptomyces cloning vectors and show low transformation efficiency. Moreover, obtaining soluble functional recombinant enzymes from the 4,6-disubstitued aminoglycoside producers by expression in E. coli is often very challenging. Some of the above mentioned key enzymes involved in 4,6-disubstituted aminoglycoside biosynthesis, such as KanM1, KanM2, and TobZ, were obtained by expression in Streptomyces.14,42 Therefore, an efficient heterologous expression platform of aminoglycoside biosynthetic genes based on genetically amenable streptomycetes has been developed to facilitate the deciphering of biosynthetic pathways to this class of molecules, biosynthetic engineering to generate more robust aminoglycoside derivatives, or direct fermentative production of clinically important semi-synthetic antibiotics.5.1 Streptomyces sp.
The potential for the heterologous production of aminoglycosides in a readily manipulable Streptomyces species was first demonstrated by the heterologous production of kanamycin or spectinomycin, which is an aminocyclitol-containing aminoglycoside, through expression of the cosmid containing the entire gene cluster of each compound in a mutant strain of S. venezuelae YJ003, which lacks the genes for biosynthesis of its native deoxysugar (dTDP-D-desosamine).46,47 Among Streptomyces, the pikromycin-producing S. venezuelae has certain advantages as a heterologous host for the production of aminoglycosides as well as polyketides and hybrid polyketide-nonribosomal peptides due to its rapid growth and relative ease of genetic manipulation.48 The heterologous expression system based on S. venezuelae has also been used successfully to dissect the biosynthetic route to gentamicin A2,29 pseudoribostamycin,35 and kanamycin14 by expression of their minimal gene sets as described above. Importantly, the microbial production of amikacin by co-expression of a set of seven genes for the biosynthesis and incorporation of the AHBA side chain with kanamycin A biosynthetic genes in S. venezuelae YJ003 demonstrated the possibility of the direct fermentative production of semi-synthetic aminoglycoside for the first time.14 In addition to S. venezuelae, another non-aminoglycoside-producing strain S. lividans has been used to produce paromamine by expression of genes from S. kanamyceticus.32 A continued challenge of this Streptomyces-based heterologous expression approach is the relatively low productivity of the target compounds.5.2 Escherichia coli
E. coli offers obvious advantages as a heterologous host in terms of rapid growth and well-developed genetic tools and has been exploited for the production of natural products, such as polyketides, nonribosomal peptides, and hybrid polyketide-nonribosomal peptides.49E. coli can also serve as an alternate platform for the production of aminoglycosides. The first heterologous production of aminoglycoside biosynthetic intermediate in E. coli has been demonstrated by the production of relatively large amounts of 2-DOI (29.5 mg L−1), which can be a valuable starting material for the benzene-free synthesis of catechol or other benzenoids, by the expression of 2-DOI synthase gene btrC in an engineered E. coli, in which genes encoding enzymes involved in the primary metabolism of Glc-6-phosphate and competing for Glc-6-phosphate with 2-DOI synthase were disrupted.50 By adding genes for 2-DOS biosynthesis (btrC, btrS, and btrN) and GT gene (btrM) in a similarly engineered E. coli, approximately 5 mg L−1 of pseudodisaccharide paromamine has been heterologously produced.31 The production of pseudoribostamycin genes in the same E. coli strain by expression of the recombinant biosynthetic pathway composed of butirosin, ribostamycin, and kanamycin genes demonstrated the potential of E. coli for the production of pseudotrisaccharides although the production level seemed to be very low compared to pseudodisaccharide.51 However, significant challenges remain in the heterologous production of complete aminoglycosides in E. coli because some of the aminoglycoside biosynthetic genes could not be functionally expressed in E. coli. Synthesis and expression of codon-optimized genes corresponding to the E. coli codon bias would likely improve expression and production.6 Biosynthesis of new aminoglycosides
6.1 Chemo-enzymatic synthesis
The tolerance of BtrH and BtrG for non-native aminoglycoside substrates were exploited to synthesize AHBA-paromomycin and AHBA-neomycin by using γ-L-Glu-AHBA-S-BtrI as the side chain donor, suggesting the possibility for the engineered biosynthesis of novel AHBA-aminoglycosides. However, BtrH could not transfer the γ-L-Glu-AHBA chain from BtrI to the 4,6-disubstituted aminoglycosides.16 Whereas, kanamycin A and gentamicin C are acylated with modest yield using a synthetic N-acetylcysteamine thioester (SNAC) as the side chain donor, γ-L-Glu-AHBA-SNAC, instead of the ACP acyl donor.17 The in vitro synthesis of AHBA-aminoglycoside by this method requires two steps: the acyltransferase BtrH first transfers γ-L-Glu-AHBA-SNAC onto the aminoglycosides and the resulting acyl-aminoglycoside products are treated with the γ-glutamyl cyclotransferase BtrG to give AHBA-aminoglycosides because γ-L-Glu-AHBA-SNAC is also a good substrate for BtrG.17 It is at present unclear why using SNAC as the acyl donor expanded the substrate range of BtrH.Another chemo-enzymatic method to generate N-acylated aminoglycosides using aminoglycoside N-aceytyltransferases (AAC) and unnatural acyl-CoAs was reported.18 Two AACs (AAC(6′)-APH(2′′) and AAC(3)-IV) were used and found to be tolerant towards a number of aminoglycosides as well as a diverse acyl-CoAs, allowing for the generation of single-N-acylated as well as homo- and hetero-di-N-acylated aminoglycosides in quantities sufficient for a qualitative screen for antibacterial activity.18 These two innovative examples of chemo-enzymatic synthesis afford quick and efficient access to unnatural acylated aminoglycosides, although further investigation is needed to increase production capacity.
6.2 Combinatorial biosynthesis and metabolic engineering
The introduction of the above-mentioned seven btr genes for the biosynthesis and incorporation of the AHBA side chain in S. venezuelae engineered to biosynthesize kanamycin X (6′-deamino-6′-hydroxykanamycin A) resulted in the in vivo generation of a new 1-N-AHBA-kanamycin X that is highly active against Gram-negative bacteria including an amikacin-resistant clinical isolate.14 These results demonstrate the potential of combinatorial biosynthesis (or pathway engineering) toward more robust next generation aminoglycosides.Gene inactivation to elucidate the functions of genD1 and tobQ produced unnatural aminoglycosides, a methylated derivative of gentamicin A2 probably on the GlcN moiety and 3′-deoxy-carbamoylkanamycin C, respectively.37,44 Although they are less bioactive than their parent compounds, such as gentamicin Cs and tobramycin, these results indicate that the enzymes involved in the late-stage modifications of aminoglycosides are flexible towards the altered substrates and demonstrated the possibility to generate structurally diverse new aminoglycoside derivatives by simple metabolic engineering.
6.3 Engineering glycosyltransferases
In an effort to expand the substrate specificity of GT and synthesize a variety of aminoglycoside derivatives, a chimeric gene library encoding for a random length of the N-terminus (an acceptor recognition domain of GT-B superfamily) of KanM1 and C-terminus (a donor-recognition domain) of GtfE (vancomycin GT) was constructed by the time-dependent incremental truncation method.52 A plate-based whole cell color assay, which selects colonies based on the color changes of the pH indicator according to the proton release during glycosidic bond formation between the sugar donor and the acceptor, was employed for the selection of functional chimeric enzymes. The most active mutant (N-kanM1-669 bp and 753 bp-gtfE-C) displayed significantly improved activity for the transfer of dTDP-D-Glc, GDP-D-mannose, and UDP-D-galactose onto 2-DOS as compared to wild-type KanM1. In addition, it acquired new donor specificities for dTDP-L-rhamnose and dTDP-4-keto-6-D-deoxyglucose.52 This domain swapping method revealed the potential for the synthesis of a variety of aminoglycoside derivatives.7 Conclusions
Although total or semi-synthesis of new aminoglycosides will continue to play their roles in combating resistant pathogens, we saw the potential of the chemo-enzymatic synthesis or engineered biosynthesis to generate novel aminoglycosides. Even though remarkable advances have been made in the characterization of the biosynthetic pathways for the 2-DOS-containing aminoglycosides and the understanding of the enzyme functions involved in these biosynthetic pathways during the last half decade, there are still many aminoglycoside biosynthetic pathways and enzymes remaining to be characterized. Generally, aminoglycoside biosynthesis involves a set of repeated reactions, such as oxidation/transamination, glycosylation, deacetylation, dehydration/reduction, methylation and epimerization. However, it has now been revealed that even highly homologous enzymes from similar pathways may have significantly different biochemical properties. Therefore, the detailed understanding of such enzymes and the complex aminoglycoside biosynthetic networks will improve our understanding of the unknown enzymatic catalysis and eventually allow us to generate novel aminoglycoside derivatives with more robust antibiotic activities or other therapeutic potentials.8 Acknowledgements
Research in the authors' laboratories has been supported by the National Research Foundation of Korea (NRF) grant funded by the Korea Ministry of Education, Science and Technology (MEST) (20120006243, 20120000650 and 20122039424), the Intelligent Synthetic Biology Center of Global Frontier Project funded by the MEST (2011-0031961), and the Ewha Global Top5 Grant 2011 of Ewha Womans University. J.K.S gratefully acknowledges a grant (PJ008013) from the Next-Generation BioGreen 21 program, Rural Development Administration.9 References
- D. Moazed and H. F. Noller, Nature, 1987, 327, 389 CrossRef CAS.
- A. Schatz, E. Bugie and S. A. Waksman, Proc. Soc. Exp. Med. Biol., 1944, 55, 66 CAS.
- S. Magnet and J. S. Blanchard, Chem. Rev., 2005, 105, 477 CrossRef CAS.
- J. L. Houghton, K. D. Green, W. Chen and S. Garneau-Tsodikova, ChemBioChem, 2010, 11, 880 CrossRef CAS.
- S. Kondo and K. Hotta, J. Infect. Chemother., 1999, 5, 1 CrossRef CAS.
- M. A. Fischbach and C. T. Walsh, Science, 2009, 325, 1089 CrossRef CAS.
- A. Lapidot, A. Berchanski and G. Borkow, FEBS J., 2008, 275, 5236 CrossRef CAS.
- M. Hainrichson, I. Nudelman and T. Baasov, Org. Biomol. Chem., 2008, 6, 227 CAS; V. Pokrovskaya, I. Nudelman, J. Kandasamy and T. Bassov, Methods Enzymol., 2010, 478, 437 Search PubMed.
- S. Jana and J. K. Deb, Appl. Microbiol. Biotechnol., 2006, 70, 140 CrossRef CAS.
- J. B. Aggen, E. S. Armstrong, A. A. Goldblum, P. Dozzo, M. S. Linsell, M. J. Gliedt, D. J. Hildebrandt, L. A. Feeney, A. Kubo, R. D. Matias, S. Lopez, M. Gomez, K. B. Wlasichuk, R. Diokno, G. H. Miller and H. E. Moser, Antimicrob. Agents Chemother., 2010, 54, 4636 CrossRef CAS.
- P. Dozzo and H. E. Moser, Expert Opin. Ther. Pat., 2010, 20, 1321 CrossRef CAS.
- I. Nudelman, A. Rebibo-Sabbah, M. Cherniavsky, V. Belakhov, M. Hainrichson, F. Chen, J. Schacht, D. S. Pilch, T. Ben-Yosef and T. Baasov, J. Med. Chem., 2009, 52, 2836 CrossRef CAS.
- F. Kudo and T. Eguchi, Methods Enzymol., 2009, 459, 493 CAS.
- J. W. Park, S. R. Park, K. K. Nepal, A. R. Han, Y. H. Ban, Y. J. Yoo, E. J. Kim, E. M. Kim, D. Kim, J. K. Sohng and Y. J. Yoon, Nat. Chem. Biol., 2011, 7, 843 CrossRef CAS.
- H. Sucipto, F. Kudo and T. Eguchi, Angew. Chem., Int. Ed., 2012, 51, 3428 CrossRef CAS.
- N. M. Llewellyn, Y. Li and J. B. Spencer, Chem. Biol., 2007, 14, 379 CrossRef CAS.
- N. M. Llewellyn and J. B. Spencer, Chem. Commun., 2008, 32, 3786 RSC.
- K. D. Green, W. Chen, J. L. Houghton, M. Fridman and S. Garneau-Tsodikova, ChemBioChem, 2010, 11, 119 CrossRef CAS.
- N. M. Llewellyn and J. B. Spencer, Nat. Prod. Rep., 2006, 23, 864 RSC.
- F. Kudo and T. Eguchi, J. Antibiot., 2009, 62, 471 CrossRef CAS.
- U. F. Wehmeier and W. Piepersberg, Methods Enzymol., 2009, 459, 459 CAS.
- K. Yokoyama, M. Numakura, F. Kudo, D. Ohmori and T. Eguchi, J. Am. Chem. Soc., 2007, 129, 15147 CrossRef CAS.
- F. Kudo, Y. Yamamoto, K. Yokoyama, T. Eguchi and K. Kakinuma, J. Antibiot., 2005, 58, 766 CrossRef CAS.
- H. Tamegai, H. Sawada, E. Nango, R. Aoki, H. Hirakawa, T. Iino and T. Eguchi, Biosci., Biotechnol., Biochem., 2010, 74, 1215 CrossRef CAS.
- E. Nango, T. Kumasaka, T. Hirayama, N. Tanaka and T. Eguchi, Proteins: Struct., Funct., Bioinf., 2008, 70, 517 CrossRef CAS.
- H. Tamegai, Y. Kanda and C. Kato, Biosci., Biotechnol., Biochem., 2010, 74, 1102 CrossRef CAS.
- K. Yokoyama, Y. Yamamoto, F. Kudo and T. Eguchi, ChemBioChem, 2008, 9, 865 CrossRef CAS.
- Q. Fan, F. Huang, P. F. Leadlay and J. B. Spencer, Org. Biomol. Chem., 2008, 6, 3306 CAS.
- J. W. Park, J. S. J. Hong, N. Parajuli, W. S. Jung, S. R. Park, S.-K. Lim, J. K. Sohng and Y. J. Yoon, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8399 CrossRef CAS.
- K. K. Nepal, T.-J. Oh, B. Subba, J. C. Yoo and J. K. Sohng, Mol. Cells, 2009, 27, 83 CrossRef CAS.
- N. P. Kurumbang, K. Liou and J. K. Sohng, J. Appl. Microbiol., 2010, 108, 1780 CrossRef CAS.
- K. K. Nepal, T.-J. Oh and J. K. Sohng, Mol. Cells, 2009, 27, 601 CrossRef CAS.
- F. Kudo, K. Kawabe, H. Kuriki, T. Eguchi and K. Kakinuma, J. Am. Chem. Soc., 2005, 127, 1711 CrossRef CAS.
- A. W. Truman, F. Huang, N. M. Llewellyn and J. B. Spencer, Angew. Chem., Int. Ed., 2007, 46, 1462 CrossRef CAS.
- N. P. Kurumbang, K. Liou and J. K. Sohng, Appl. Biochem. Biotechnol., 2011, 163, 373 CrossRef CAS.
- F. Huang, D. Spiteller, N. A. Koorbanally, Y. Li, N. M. Llewellyn and J. B. Spencer, ChemBioChem, 2007, 8, 283 CrossRef CAS.
- Y. Yu, X. Hou, X. Ni and H. Xia, J. Antibiot., 2008, 61, 63 CrossRef CAS.
- F. Kudo, T. Fujii, S. Kinoshita and T. Eguchi, Bioorg. Med. Chem., 2007, 15, 4360 CrossRef CAS.
- F. Kudo, H. Sucipto and T. Eguchi, J. Antibiot., 2009, 62, 707 CrossRef CAS.
- Y. Li, N. M. Llewellyn, R. Giri, F. Huang and J. B. Spencer, Chem. Biol., 2005, 12, 665 CrossRef CAS.
- T. Tamura, Y. Ishida, M. Otoguro, K. Hatano and K. Suzuki, Int. J. Syst. Evol. Microbiol., 2008, 58, 688 CrossRef CAS.
- C. Parthier, S. Görlich, F. Jaenecke, C. Breithaupt, U. Bräuer, U. Fandrich, D. Clausnitzer, U. F. Wehmeier, C. Böttcher, D. Scheel and M. T. Stubbs, Angew. Chem., Int. Ed., 2012, 51, 4046 CrossRef CAS.
- J. Guo and J. W. Frost, J. Am. Chem. Soc., 2002, 124, 10642 CrossRef CAS.
- J.-Y. Kim, J.-W. Suh, S.-H. Kang, T. H. Phan, S.-H. Park and H.-J. Kwon, Biochem. Biophys. Res. Commun., 2008, 372, 730 CrossRef CAS.
- D. Clausnitzer, W. Piepersberg and U. F. Wehmeier, J. Appl. Microbiol., 2011, 111, 642 CrossRef CAS.
- L. P. Thapa, T.-J. Oh, H. C. Lee, K. Liou, J. W. Park, Y. J. Yoon and J. K. Sohng, Appl. Microbiol. Biotechnol., 2007, 76, 1357 CrossRef CAS.
- L. P. Thapa, T.-J. Oh, K. Liou and J. K. Sohng, J. Appl. Microbiol., 2008, 105, 300 CrossRef CAS.
- S. R. Park, A. R. Han, Y. H. Ban, Y. J. Yoo, E. J. Kim and Y. J. Yoon, Appl. Microbiol. Biotechnol., 2010, 85, 1227 CrossRef CAS.
- H. Zhang, B. A. Boghigian, J. Armando and B. A. Pfeifer, Nat. Prod. Rep., 2011, 28, 125 RSC.
- T. Kogure, N. Wakisaka, H. Takaku and M. Takagi, J. Biotechnol., 2007, 129, 502 CrossRef CAS.
- N. P. Kurumbang, J. W. Park, Y. J. Yoon, K. Liou and J. K. Sohng, Res. Microbiol., 2010, 161, 526 CrossRef CAS.
- A. W. Truman, M. V. Dias, S. Wu, T. L. Blundell, F. Huang and J. B. Spencer, Chem. Biol., 2009, 16, 676 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of enzymes involved in the 4,5- and 4,6-disubstituted 2-DOS-containing aminoglycosides. See DOI: 10.1039/c2np20092a |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2013 |