Anion receptors based on ureidocalix[4]arenes immobilised in the partial cone conformation†
Oldřich
Hudeček
a,
Jan
Budka
a,
Hana
Dvořáková
b,
Petra
Cuřínová
c,
Ivana
Císařová
d and
Pavel
Lhoták
*a
aDepartment of Organic Chemistry, Institute of Chemical Technology Prague (ICT), Technicka 5, 166 28 Prague 6, Czech Republic. E-mail: lhotakp@vscht.cz; Fax: +420 220444288; Tel: +420 220445055
bLaboratory of NMR Spectroscopy, Institute of Chemical Technology Prague (ICT), Technicka 5, 166 28 Prague 6, Czech Republic
cInstitute of Chemical Process Fundamentals, v.v.i., Academy of Sciences of the Czech Republic, Rozvojová 135, 165 02 Prague 6, Czech Republic
dDepartment of Inorganic Chemistry, Charles University, Hlavova 8, 128 43 Prague 2, Czech Republic
First published on 22nd October 2012
Abstract
A novel method enabling the synthesis of receptors based on the calix[4]arenes immobilized in the partial cone conformation is reported. The application of a protection/deprotection strategy using nosyl groups enables the regioselective introduction of functional groups (NO2 and NH2) into the upper rim of calix[4]arenes, leading finally to the substitution pattern so far inaccessible to calixarene chemistry. The introduction of two ureido functions at the para positions of the partial cone conformer yields a novel type of receptor which can bind anions even in a highly competitive solvent (DMSO). This revealed that the partial cone conformation, so far rather ignored in supramolecular chemistry, can be also very useful in design of novel anion receptors.
Introduction
The importance of anions in various biological systems is well recognised. Hence, the study of the complexation of anionic species plays an important role in contemporary supramolecular chemistry as witnessed by many review articles1 and books2 published recently. Given the importance of anions in chemistry, medicine, environmental pollution or industrial processes, much effort has been focused on the design of anion binding ligands, receptors and sensors.3Not surprisingly, the main strategy for anion recognition relies on the application of positively charged species like polyammonium, guanidinium, or quaternary ammonium salts,4 which can bind anions mainly via electrostatic interactions. On the other hand, neutral organic receptors for anion recognition via hydrogen bonding interactions are especially useful in the design. These receptors can use highly directional hydrogen bonds from amide, sulfonamide, urea or thiourea moieties5 which can effectively interact with anions, especially if they are suitably preorganised. From this point of view, calix[4]arenes6 offer many possibilities due to their tuneable 3D shapes of the molecule leading to four different conformations (cone, partial cone, 1,3-alternate, and 1,2-alternate) (Fig. 1). This makes calix[4]arenes ideal candidates for the role of molecular scaffolds in the design of novel receptors allowing the elaborated introduction of various functional groups into precisely defined mutual positions.
![Four basic conformations of calix[4]arene: (a) cone, (b) partial cone, (c) 1,2-alternate and (d) 1,3-alternate.](/image/article/2013/NJ/c2nj40724h/c2nj40724h-f1.gif) | ||
| Fig. 1 Four basic conformations of calix[4]arene: (a) cone, (b) partial cone, (c) 1,2-alternate and (d) 1,3-alternate. | ||
Many neutral calixarene-based ligands for anion recognition have been reported in the literature so far.7,8 We have previously focused on a series of arylamide- or arylureido-substituted calix[4]arenes9 which can be used for anion recognition in solution. The systematic study of ureidocalix[4]arenes revealed that bis(phenylureido)-calix[4]arene immobilised in the cone conformation10 can bind benzoate with surprisingly high selectivity. On the other hand, receptors in the 1,3-alternate conformation bearing four ureido functions bind anions with a negative allosteric effect11 where only one site of the molecule is capable of forming a complex. Interestingly, albeit there are many references for the cone or 1,3-alternate conformations, there is almost no precedence9d,12 of using anion receptors in the remaining two conformations – partial cone or 1,2-alternate.
While the 1,2-alternate is scarcely used because of the lack of general synthetic methods leading to this conformation, preparation of the partial cone isomers is relatively easy. The problem lies in the wrong mutual positions of nitro groups used as the starting point for subsequent derivatization. Usually we start with distally disubstituted calix[4]arenes (Scheme 1, path a), where subsequent nitration and final alkylation of dinitro intermediate A leads to substitution pattern PC1. Unfortunately, this partial cone conformer bears nitro groups on the opposite sites of the cavity which is not suitable for the design of receptors. Recently we have reported a straightforward regioselective synthesis13 enabling the preparation of dinitro isomers B with the nitro groups on the unsubstituted aromatic rings (Scheme 1, path b). Here we report on the synthesis of novel type of anion receptors based on the calix[4]arenes immobilized in the partial cone conformation possessing a PC2 substitution pattern. These isomers have been so far inaccessible to calixarene chemistry and represent structures which are complementary to commonly used calixarene-based receptors (compare PC1 and PC2 in Scheme 1).
![Two different substitution patterns in dinitro substituted partial cone calix[4]arenes: (a) common nitration followed by alkylation and (b) nitration using nosyl protection/deprotection method followed by alkylation.](/image/article/2013/NJ/c2nj40724h/c2nj40724h-s1.gif) | ||
| Scheme 1 Two different substitution patterns in dinitro substituted partial cone calix[4]arenes: (a) common nitration followed by alkylation and (b) nitration using nosyl protection/deprotection method followed by alkylation. | ||
Results and discussion
The regioselective introduction of nitro groups into the calixarene skeleton was recently described by our group.13 The synthetic strategy is based on a direct nitration or ipso-nitration of the corresponding nosyl-substituted derivatives 1b–3b. As the reactivity of alkoxy substituted aromatic rings is higher compared to that of aromatic units bearing nosyl groups, one can selectively introduce nitro groups at the para positions of alkylated aromatic subunits. The whole strategy consists of three basic steps: (i) lower-rim protection (introduction of a nosyl moiety) leading to the formation of compounds 1b–3b, (ii) nitration or ipso-nitration (depending on the upper rim substitution) to form 1c–3c, and (iii) deprotection of the nosyl group by an alkaline hydrolysis to yield 1–3. This procedure straightforwardly (overall yield for the transformation of 1a to 1 is 75%) leads to the substitution pattern (Scheme 1) which is so far inaccessible to calix[4]arene chemistry and which is complementary to the commonly used isomers (Scheme 2).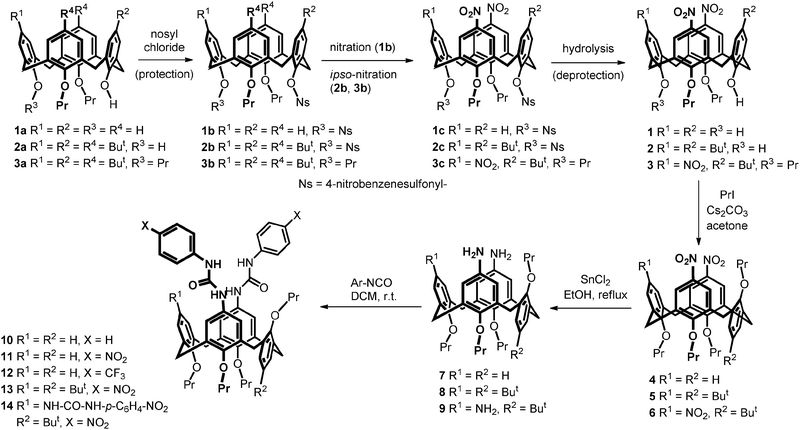 | ||
| Scheme 2 Synthesis of calixarene-based receptors in the partial cone conformation. | ||
The isomers 1–3 were then used for the formation of the partial cone conformers. The alkylation with propyl iodide using Cs2CO3 as a base led always to the mixture of the partial cone and cone isomers in approximately 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ratio. Unfortunately, these mixtures could not be separated by column chromatography either on silica gel or on alumina. On the other hand, a simple precipitation from CH2Cl2–MeOH system gave pure partial cone isomers 4–6 in medium yields (≈40%).
:5 ratio. Unfortunately, these mixtures could not be separated by column chromatography either on silica gel or on alumina. On the other hand, a simple precipitation from CH2Cl2–MeOH system gave pure partial cone isomers 4–6 in medium yields (≈40%).
The conformation of 4–6 was confirmed using 1H NMR spectroscopy. The partial cone conformer can be imaginary bisected into two halves – one having the cone-like arrangement with the syn orientation of the aromatic units, and the second one possessing the 1,3-alternate-like features (anti oriented phenolic rings). Both halves retained their typical splitting pattern and signal multiplicity known from 1H NMR spectra of common conformers. Thus, dinitro derivative 4 (Fig. 2) showed one doublet at 3.12 ppm for equatorial CH bond of the bridging CH2 moiety, and another doublet at 4.06 ppm for the axial CH bond, both with typical geminal interaction constants (J = 13.8 Hz). On the other hand, signals of bridging units in the 1,3-alternate-like part of the molecule possess almost identical chemical shifts (≈3.72 ppm). While the propyl group in the 1,3-alternate-like half of the molecule exhibits typical shifts (Fig. 2), the analogous signals of the opposite propyl group in the cone-like part are distinctly shifted towards the stronger field (upfield shift). This indicates that protons Ha–Hc are located close to the distal inverted aromatic unit which leads to substantial shielding of all aliphatic signals due to the magnetic anisotropy of the aromatic system (for total structure assignment see ESI†).
 | ||
| Fig. 2 Typical features of the 1H NMR spectra of derivative 4 (CDCl3, 298 K, 300 MHz). | ||
Thus, the comparison of the corresponding chemical shifts (Fig. 2) clearly shows the substantial upfield shift (approx. 0.60 ppm) for all protons (compare 3.84 (Ha′)/3.25 (Ha) ppm for –OCH2– moiety, or 1.15 (Hc′)/0.69 (Hc) ppm for terminal CH3 group). Similar features and trends can be observed in all newly prepared derivatives 4–14 possessing the partial cone conformation (for complete structure assignment of compound 4, see ESI†).
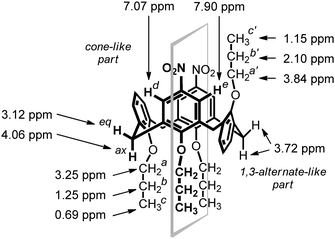
The final unambiguous structural evidence for compound 5 was obtained by X-ray crystallographic analysis of monocrystals grown from the methanol–CH2Cl2 mixture. The molecule adopts a partial cone conformation (Fig. 3) where the aromatic units bearing NO2 groups are pointing towards each other into the cavity, and the third aromatic unit with the syn orientation is almost ideally perpendicular (89.06(8)°) to the main plane formed by the four carbon bridging atoms. The remaining anti-oriented aromatic unit is noticeably tilted from the cavity with the interplanar angle = 151.10(8)° (with the main plane). Interestingly, both nitro groups are slightly turned out from the ideal coplanar arrangement with attached aromatic units by 10.1(3)° and 15.9(3)°.
 | ||
| Fig. 3 X-Ray structure of derivative 5. | ||
Nitro derivatives 4–6 were then smoothly reduced with SnCl2·2H2O in refluxing ethanol to give amino derivatives 7, 8 and 9 in 95, 97, and 98% yields, respectively. Target receptors 10–14 were obtained by the reaction of amines 7–9 with appropriate isocyanates (phenyl, p-trifluoromethylphenyl or p-nitrophenyl) in dichloromethane at room temperature and they were isolated in 63–78% yields (for 10–13) and 31% yield (for 14).
The complexation ability of ligands 10–13 was studied by standard 1H NMR titration experiments using an increasing concentration of appropriate anions to obtain different calixarene:anion ratios (1:0.3–10). To avoid possible unwanted interactions of receptors with counter-cations all anions studied were used in the form of tetrabutylammonium salts. In the beginning, the 1H NMR titration experiments were performed in the mixed solvent (CDCl3/CD3CN = 4:1, v/v) to ensure the solubility of both organic and inorganic parts. Under these conditions, the addition of anions to the receptor led to the remarkable down-field shifts of ureido NH signals thus indicating the complexation phenomenon under fast exchange conditions. Very high complexation induced chemical shifts (CIS) were observed for chloride and bromide (>3 ppm). Unfortunately, the binding constants were too high (K ≫ 104) to be reliably measured by the NMR technique.14
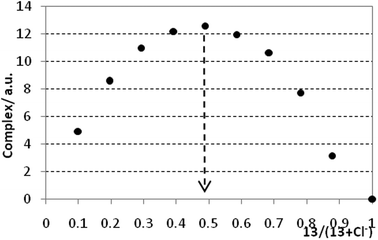
Based on these preliminary results, we decided to change the solvent system for more polar DMSO-d6. Despite the usage of this highly competitive solvent towards hydrogen bonding interactions, reasonable CIS values for NH protons were observed for all anions measured. Thus, CIS value for the 11/AcO− system was 1.50 ppm and similar values were obtained for all receptors with benzoate and acetate, while other anions usually showed lower values (compare CIS = 0.65 for the 11/Cl− system). The binding constants were determined from the CIS values of NH protons, Fig. 4 shows a typical binding isotherm constructed for 12/Cl− system. All titration curves (with exception of receptor 14 bearing three ureido moieties) best fit with the formation of 1:1 complexes (calix/anion), the stoichiometry was also proved by the independent Job plot analysis15 for selected anions (Fig. 5). The calculated binding constants are summarised in Table 1.
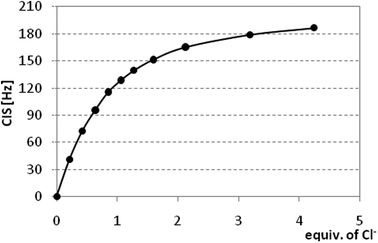 | ||
| Fig. 4 binding isotherm of receptor 12 with a chloride anion (1H NMR titration, 300 MHz, 298 K, DMSO-d6). | ||
| Anion | K/M−1 | |||
|---|---|---|---|---|
| 10 | 11 | 12 | 13 | |
| a Too small changes in chemical shifts upon addition of anions. b Titration experiments were carried out in pure CDCl3. c The complexation constant for the cone analogue of receptor 11 (measured for the comparison): KCl = 173 ± 24 M−1. | ||||
| Cl− | 870 ± 200 | 1540 ± 370c | 450 ± 50 | 800 ± 180 |
| Br− | 120 ± 40 | 920 ± 100 | 60 ± 10 | 210 ± 80 |
| I− | a | 30 ± 6 | a | 175 ± 40 |
| AcO− | 6400 ± 560 | 2250 ± 300 | 13000 ± 3000 |
2700 ± 650 |
| BzO− | 2750 ± 390 | 16500 ± 3500 |
16000 ± 2800 |
6200 ± 1200 |
| CN− | 50 ± 13 | 730 ± 120 | 240 ± 50 | 680 ± 150 |
| NO3− | 120 ± 20 | 380 ± 70 | 28 ± 7 | a |
| DMSO-d6b | 8 ± 2 | 12 ± 3 | 11 ± 1 | 7 ± 2 |
All ureido or amidic receptors for anions are known to operate via the hydrogen bonds between the N–H moiety and negatively charged species (N–H⋯A−). Albeit DMSO is known as a highly competitive solvent towards HB interactions, the binding constants of receptors 10–13 are surprisingly high. In all cases, the carboxylates possess the highest binding constants, with selectivity for benzoate over acetate (for 11–13). Thus, both receptors 11 and 12 bearing electron withdrawing substituents (NO2 and CF3) possess almost the same complexation ability towards benzoate (KBz ≈ 16000 M−1), while the complexation of acetate is reasonably different KAc = 13000 M−1 for 12 and KAc = 2250 M−1 for 11. This leads to selectivity factor (KBz/KAc) 1.23 for 12 and 7.33 for 11. The only exception is represented by the unsubstituted receptor 10 showing higher selectivity for acetate (KBz = 2750 M−1, KAc = 6400 M−1).
As follows from Table 1, the binding site created by two preorganised ureido functions can bind also other anions possessing various geometries. Thus, a linear cyanide anion is efficiently bound by 11 (KCN = 730 M−1) or by 13 (KCN = 680 M−1). The binding constants for spherical halides are inversely related to their basicity as can be documented by the results for compound 11: KCl = 1540 M−1, KBr = 920 M−1, and KI = 30 M−1.
Receptor 14 bearing three ureido functions on the upper rim was inefficient towards NO3− and CN− anions. On the other hand, halogens or carboxylates showed rather complicated behaviour which did not correspond to the formation of complexes possessing 1:1 stoichiometry. We have also studied the interaction of receptors with solvent used – DMSO. The 1H NMR titrations of receptors 10–13 with DMSO-d6 was carried out in CDCl3 and the corresponding complexation constants are shown in Table 1. As the K values are very small (<12 M−1), these interactions were neglected in experiments carried out using DMSO-d6 as solvent.
Conclusions
A novel method enabling the synthesis of receptors based on the calix[4]arenes immobilized in the partial cone conformation is reported. The application of a protection/deprotection strategy using nosyl groups enables the regioselective introduction of functional groups (NO2 and NH2) into the upper rim of calix[4]arenes, leading finally to the substitution pattern so far inaccessible to calixarene chemistry, and structurally complementary to commonly used calixarene-based receptors The introduction of two ureido functions at the para positions of calix[4]arenes immobilised in the partial cone conformation gave a novel type of receptor used successfully for the complexation of anions.18 Albeit based on the hydrogen-bonding interactions, these receptors can bind anions even in a highly competitive solvent like DMSO. The complexation study revealed that the partial cone conformation, so far rather ignored in supramolecular chemistry, can be very useful in design of novel anion receptors.Experimental
Materials and instrumentations
All chemicals were purchased from commercial sources and used without further purification. Solvents were dried and distilled using conventional methods. Melting points were measured on a Heiztisch Mikroskop-Polytherm A (Wagner & Munz, Germany). NMR spectra were performed on Varian Gemini 300 (1H: 300 MHz, 13C: 75 MHz) and on Bruker Avance DRX 500 (1H: 500 MHz, 13C: 125 MHz) spectrometers. Deuterated solvents used are indicated in each case. Chemical shifts (δ) are expressed in ppm and are referred to the TMS as an internal standard; coupling constants (J) are in Hz. The signal assignment was supported by 1H–1H COSY, 1H–13C HMQC or 1H–13C HMBC 2D NMR and 1D 1H–DPFGSE NOE experiments using the standard pulse sequences provided by Bruker. The mass analyses were performed using ESI technique on a Q-TOF (Micromass) spectrometer. Elemental analyses were done on Perkin-Elmer 240, Elementar vario EL (Elementar, Germany) or Mitsubishi TOX-100 instruments. All samples were dried in the desiccator over P2O5 under vacuum (1 Torr) at 80 °C for 8 hours. The IR spectra were measured on an FT-IR spectrometer Nicolet 740 or Bruker IFS66 spectrometers equipped with a heatable Golden Gate Diamante ATR-Unit (SPECAC) in KBr. 100 Scans for one spectrum were co-added at a spectral resolution of 4 cm−1. The courses of the reactions were monitored by TLC using TLC aluminum sheets with Silica gel 60 F254 (Merck). The column chromatography was performed using Silica gel 60 (Merck).Synthesis
All intermediates 1a–1c, 2a–2c, 3a–3c and 1–3 were prepared according to the recently published procedures.13General procedure: synthesis of 25,26,27,28-tetrapropoxy derivatives 4–7 in the partial cone conformation
The corresponding calix[4]arene derivatives 1–3 were dissolved in anhydrous acetone (200 ml for 1 g), then powdered Cs2CO3 (2 equiv. for each hydroxyl group) and 1-iodopropane (3 equiv. for each hydroxyl group) were added. The mixture was heated to reflux for 3 days (monitored by TLC), and the solvent was removed under reduced pressure. The residue was acidified with 1 M hydrochloric acid and extracted twice with CH2Cl2. The combined organic layers were washed with water, then with brine and dried over MgSO4. Organic solvent was evaporated to dryness. The further purification is described below for each particular derivative.5,17-Dinitro-25,26,27,28-tetrapropoxycalix[4]arene (partial cone, syn-dinitro) 4
A general procedure was applied to 5,17-dinitro-26,28-dipropoxycalix[4]arene-25,27-diol 1 (0.50 g, 0.84 mmol), Cs2CO3 (1.09 g, 3.35 mmol) and 1-iodopropane (0.5 ml, 5.13 mmol). The residue after evaporation was dissolved in 2 ml of CH2Cl2 and precipitated by addition of 50 ml of methanol. The precipitate was filtered off, dissolved in 2 ml of CH2Cl2, and then the whole procedure was repeated two more times, the third precipitate was air dried to give 0.23 g of product 4 as a white powder in 40% yield, mp: 197–198 °C. 1H NMR (300 MHz, CDCl3, 298 K) δ: 7.87 (2H, d, J = 2.9 Hz, ArH), 7.23 (2H, d, J = 7.6 Hz, ArH), 7.17 (2H, d, J=7.6 Hz, ArH), 7.09 (2H, d, J = 2.9 Hz, ArH), 7.02 (1H, t, J = 7.5 Hz, ArH), 6.92 (1H, t, J = 7.5 Hz, ArH), 4.06 (2H, d, J = 13.8 Hz, ArCH2Ar ax), 3.86–3.59 (10H, m, 3 × OCH2 + 2 × ArCH2Ar), 3.29–3.21 (2H, m, OCH2), 3.12 (2H, d, J = 14.1 Hz, ArCH2Ar eq), 2.14–2.06 (2H, m, OCH2CH2), 1.95–1.83 (4H, m, OCH2CH2), 1.29–1.21 (2H, m, OCH2CH2), 1.17–1.08 (9H, m, CH3), 0.69 (3H, t, J = 7.6 Hz, CH3). 13C{1H} NMR (75 MHz, CDCl3, 298 K): δ 161.1, 157.4, 156.5, 142.3, 136.1, 135.5, 133.3, 133.2, 131.0, 130.0, 125.1, 123.8, 122.6, 76.9, 75.8, 75.5, 35.7, 30.9, 24.1, 24.0, 22.5, 11.0, 10.9, 9.4; TOF HRMS (ESI+): calcd for [C40H46N2O8 + Na]+: 705.3146. Found: m/z (rel. int%) 705.3146 (100) [M + Na]+. For complete structure assignment using various NMR techniques, see ESI.†5,17-Di-tert-butyl-11,23-dinitro-25,26,27,28-tetrapropoxycalix[4]arene (partial cone, syn-dinitro) 5
A general procedure was applied to 11,23-di-tert-butyl-5,17-dinitro-26,28-dipropoxycalix[4]arene-25,27-diol 2 (1.00 g, 1.41 mmol), Cs2CO3 (1.83 g, 5.62 mmol) and 1-iodopropane (0.82 ml, 8.41 mmol). The residue after evaporation was dissolved in 2 ml of CH2Cl2, then 40 ml of methanol was added and the resulting solution was stored at 0 °C overnight. The crystals formed were filtered off, quickly washed with cold methanol and air dried to give 0.50 g of product 5 as a yellowish powder in 45% yield, mp: 237–240 °C. 1H NMR (300 MHz, CDCl3, 298 K) δ: 7.86 (2H, d, J = 2.9 Hz, ArH), 7.21 (2H, s, ArH), 7.15–7.13 (4H, m, ArH), 4.09 (2H, d, J = 13.5 Hz, ArCH2Ar ax), 3.80–3.57 (10H, m, 3 × OCH2 + 2 × ArCH2Ar), 3.33–3.27 (2H, m, OCH2), 3.10 (2H, d, J = 13.8 Hz, ArCH2Ar eq), 2.06–1.98 (2H, m, OCH2CH2), 1.96–1.84 (4H, m, OCH2CH2), 1.41–1.32 (20H, m, OCH2CH2 + 2 × But), 1.12–1.05 (9H, m, CH3), 0.65 (3H, t, J = 7.5 Hz, CH3). 13C{1H} NMR (75 MHz, CDCl3, 296 K) δ: 161.3, 155.2, 153.4, 146.5, 144.5, 142.2, 135.4, 135.4, 133.5, 132.2, 127.7, 126.6, 125.3, 123.9, 76.9, 74.9, 74.8, 36.5, 34.5, 34.3, 31.8, 31.5, 24.1, 23.8, 21.6, 11.0, 10.8, 9.9; TOF HRMS (ESI+): calcd for [C48H62N2O8 + Na]+: 817.4398. Found: m/z (rel. int. %) 817.4397 (100) [M + Na]+.5-tert-Butyl-11,17,23-trinitro-25,26,27,28-tetrapropoxycalix[4]arene (partial cone) 6
A general procedure was applied to 5-tert-butyl-11,17,23-trinitro-26,27,28-tripropoxycalix[4]arene-25-ol 3 (0.80 g, 1.08 mmol), Cs2CO3 (0.70 g, 2.15 mmol) and 1-iodopropane (0.30 ml, 3.08 mmol). The residue after evaporation was dissolved in 2 ml of CH2Cl2, then 40 ml of methanol was added and the resulting solution was stored at 0 °C overnight. The crystals formed were filtered off, quickly washed with cold methanol and air dried to give 0.30 g of product 6 as a white powder in 35% yield, mp: 222–224 °C. 1H NMR (300 MHz, CDCl3, 298 K) δ: 8.11 (2H, s, ArH), 7.92 (2H, d, J = 2.6 Hz, ArH), 7.24 (2H, s, ArH), 7.12 (2H, d, J = 2.6 Hz, ArH), 4.16 (2H, d, J = 13.8 Hz, ArCH2Ar ax), 3.85–3.58 (10H, m, 3 × OCH2 + 2 × ArCH2Ar), 3.44–3.39 (2H, m, OCH2), 3.28 (2H, d, J = 14.1 Hz, ArCH2Ar eq), 2.09–1.86 (6H, m, OCH2CH2), 1.37–1.30 (11H, m, OCH2CH2 + But), 1.13–1.06 (9H, m, CH3), 0.67 (3H, t, J = 7.5 Hz, CH3). 13C{1H} NMR (75 MHz, CDCl3, 296 K) δ: 161.7, 161.3, 155.05, 144.7, 143.0, 142.4, 137.4, 134.0, 133.6, 132.0, 127.8, 126.0, 125.2, 123.4, 77.2, 75.2, 75.0, 36.4, 34.3, 31.8, 31.2, 24.0, 23.9, 21.8, 11.0, 10.7, 10.0; TOF HRMS (ESI+): calcd for [C44H53N3O10 + Na]+: 806.3623. Found: m/z (rel. int. %) 806.3627 (100) [M + Na]+.General procedure: reduction of nitro derivatives 4–6
The corresponding nitro derivatives 4–6 were dispersed in ethanol (10 ml for 100 mg) and SnCl2·2H2O (5 equiv. for each nitro group) was added. The mixture was heated to reflux overnight and then poured into 1 M NH3 (aq.). Resulting suspension was repeatedly extracted with CH2Cl2, combined organic layers were washed with water, then with brine and dried over MgSO4. Organic solvent was removed under reduced pressure to give the pure products 7–9.5,17-Diamino-25,26,27,28-tetrapropoxycalix[4]arene (partial cone, syn-diamino) 7
A general procedure was applied to dinitro derivative 4 (0.43 g, 0.63 mmol), SnCl2·2H2O (1.42 g, 6.30 mmol). The product 7 was isolated as a brown powder (0.37 g) in 95% yield, mp: 176–179 °C. 1H NMR (300 MHz, CDCl3, 298 K) δ: 7.20 (2H, d, J = 7.6 Hz, ArH), 7.05 (2H, d J = 7.3 Hz, ArH), 6.87 (1H, t, J = 7.0 Hz, ArH), 6.83 (1H, t, J = 7.5 Hz, ArH), 6.45 (2H, d, J = 2.6 Hz, ArH), 5.61 (2H, d, J = 2.6 Hz, ArH), 4.04 (2H, d, J = 13.5 Hz, ArCH2Ar ax), 3.75–3.45 (10H, m, 3 × OCH2 + 2 × ArCH2Ar), 3.29–3.23 (2H, m, OCH2), 2.96 (2H, d, J = 13.5 Hz, ArCH2Ar eq), 2.00–1.93 (2H, m, OCH2CH2), 1.89–1.77 (4H, m, OCH2CH2), 1.42–1.34 (2H, m, OCH2CH2), 1.14–1.07 (9H, m, 3 × CH3), 0.68 (3H, t, J = 7.6 Hz, CH3). 13C{1H} NMR (75 MHz, CDCl3, 296 K) δ: 157.7, 149.5, 140.3, 137.4, 134.3, 134.1, 132.9, 130.5, 129.1, 122.0, 121.9, 118.1, 116.1, 76.4, 75.6, 74.8, 35.9, 31.0, 24.6, 24.0, 22.6, 11.2, 11.1, 9.6; TOF HRMS (ESI+): calcd for [C40H50N2O4 + H]+: 623.3843. Found: m/z (rel. int. %) 623.3847 (100) [M + H]+.5,17-Diamino-11,23-di-tert-butyl-25,26,27,28-tetrapropoxycalix[4]arene (partial cone, syn-diamino) 8
A general procedure was applied to dinitro derivative 5 (0.30 g, 0.38 mmol), SnCl2·2H2O (0.85 g, 3.77 mmol). The product 8 was isolated as a brown powder (0.27 g) in 97% yield, mp: 104–105 °C. 1H NMR (300 MHz, CDCl3, 298 K) δ: 7.17 (2H, s, ArH), 7.01 (2H, s, ArH), 6.42 (2H, d, J = 2.6 Hz, ArH), 5.70 (2H, d, J = 2.9 Hz, ArH), 4.07 (2H, d, J = 13.2 Hz, ArCH2Ar ax), 3.75–3.63 (2H, m, OCH2), 3.58 (4H, d, J = 4.7 Hz, ArCH2Ar), 3.52–3.44 (2H, m, OCH2), 3.36–3.30 (4H, m, OCH2), 2.94 (2H, d, J = 13.5 Hz, ArCH2Ar eq), 1.89–1.75 (6H, m, OCH2CH2), 1.41–1.32 (20H, m, OCH2CH2 + 2 × But), 1.12–1.02 (6H, m, CH3), 0.91 (3H, t, J = 7.5 Hz, CH3), 0.63 (3H, t, J = 7.6 Hz, CH3). 13C{1H} NMR (75 MHz, CDCl3, 296 K) δ: 155.4, 154.2, 149.9, 144.7, 143.5, 140.0, 136.4, 134.3, 133.3, 133.1, 127.4, 125.8, 118.3, 116.2, 76.5, 74.7, 73.9, 37.0, 36.5, 34.3, 34.2, 31.9, 24.2, 24.0, 21.6, 11.2, 10.6, 9.9; TOF HRMS (ESI+): calcd for [C48H66N2O4 + H]+: 735.5095. Found: m/z (rel. int. %) 735.5095 (100) [M + H]+.5,11,17-Triamino-23-tert-butyl-25,26,27,28-tetrapropoxycalix[4]arene (partial cone) 9
A general procedure was applied to trinitro derivative 6 (0.25 g, 0.32 mmol), SnCl2·2H2O (1.08 g, 4.79 mmol). The product 9 was isolated as a dark brown powder (0.22 g) in 98% yield, mp: 124–126 °C. 1H NMR (300 MHz, CDCl3, 296 K) δ: 7.16 (2H, s, ArH), 6.44 (2H, s, ArH), 6.41 (2H, d, J = 2.9 Hz, ArH), 5.79 (2H, d, J = 2.6 Hz, ArH), 3.99 (2H, d, J = 12.9 Hz, ArCH2Ar ax), 3.72–3.42 (10H, m, 3 × OCH2 + 2 × ArCH2Ar), 3.30–3.25 (2H, m, OCH2), 2.83 (2H, d, J = 12.9 Hz, ArCH2Ar eq), 1.89–1.77 (6H, m, OCH2CH2), 1.37–1.32 (11H, m, OCH2CH2 + But), 1.08–0.98 (9H, m, CH3), 0.63 (3H, t, J = 7.5 Hz, CH3). 13C{1H} NMR (75 MHz, CDCl3, 298 K) δ: 155.5, 149.7, 149.6, 143.4, 140.5, 140.0, 137.9, 133.9, 133.1, 133.0, 127.3, 118.3, 116.2, 116.0, 76.5, 74.9, 73.9, 36.9, 31.9, 31.2, 29.9, 24.3, 24.0, 21.5, 11.1, 10.8, 10.0; TOF HRMS (ESI+): calcd for [C44H59N3O4 + H]+: 694.4578. Found: m/z (rel. int. %) 694.4582 (100) [M + H]+.General procedure: synthesis of calixarene-based receptors 10–14
The amino derivatives 7–9 were dissolved in anhydrous CH2Cl2 (30 ml for 0.100 g) under the atmosphere of dry nitrogen. The corresponding isocyanate (2.50 equiv. for each amino group) was added and the mixture was stirred overnight at room temperature. The reaction was quenched by the addition of methanol, then organic solvents were removed under reduced pressure and the residue was purified. The purification method is described below for each particular ligand.5,17-Bis[N′-(phenyl)ureido]-25,26,27,28-tetrapropoxycalix[4]arene (partial cone) 10
A general procedure was applied to amino derivative 7 (0.15 g, 0.24 mmol) and phenyl isocyanate (143 mg, 0.13 ml, 1.20 mmol). The product 10 was isolated by column chromatography (30 g of Al2O3, eluent CH2Cl2/MeOH = 50:1, then CH2Cl2/MeOH = 1:1, RF = 0.11) as a white powder (0.14 g) in 66% yield, mp: 163–166 °C. 1H NMR (300 MHz, DMSO-d6, 298 K) δ: 8.36 (2H, s, NH), 8.05 (2H, s, NH), 7.32 (4H, d, J = 7.9 Hz, ArH), 7.22–7.12 (8H, m, ArH), 7.06 (2H, d, J = 2.1 Hz, ArH), 6.91–6.82 (4H, m, ArH), 6.47 (2H, d, J = 2.3 Hz, ArH), 4.00 (2H, d, J = 12.6 Hz, ArCH2Ar ax), 3.76–3.57 (8H, m, OCH2 + ArCH2Ar), 3.44–3.30 (4H, m, OCH2), 3.08 (2H, d, J = 13.5 Hz, ArCH2Ar eq), 1.80–1.64 (6H, m, OCH2CH2), 1.43–1.23 (2H, m, OCH2CH2), 1.03 (6H, t, J = 7.3 Hz, CH3), 0.74 (3H, t, J = 7.2 Hz, CH3), 0.66 (3H, t, J = 7.6 Hz, CH3). 13C NMR (75 MHz, CDCl3, 296 K) δ: 157.4, 154.2, 153.4, 139.0, 137.0, 135.3, 133.8, 133.5, 131.0, 130.5, 129.5, 129.1, 124.0, 123.2, 122.7, 122.6, 122.2, 119.6, 76.4, 75.6, 35.5, 31.0, 24.4, 24.0, 22.6, 11.2, 10.9, 9.5; TOF HRMS (ESI+): calcd for [C54H60N4O6 + Na]+: 883.4405. Found: m/z (rel. int. %) 883.4402 (100) [M + Na]+.
5,17-Bis[N′-(4-nitrophenyl)ureido]-25,26,27,28-tetrapropoxycalix[4]arene (partial cone) 11
A general procedure was applied to amino derivative 7 (0.10 g, 0.16 mmol) and 4-nitrophenyl isocyanate (97% purity, 136 mg, 0.80 mmol). The product 11 was isolated by column chromatography (30 g of Al2O3, eluent CH2Cl2/MeOH = 160:1, RF = 0.14) as a red powder (0.12 g) in 78% yield, mp: 227–230 °C. 1H NMR (300 MHz, DMSO-d6, 298 K) δ: 8.97 (2H, s, NH), 8.10 (2H, s, NH), 8.01 (4H, d, J = 9.4 Hz, ArH), 7.43 (4H, d, J = 9.4 Hz, ArH), 7.19–7.13 (6H, m, ArH), 6.91 (1H, t, J = 7.5 Hz, ArH), 6.84 (1H, t, J = 7.5 Hz, ArH), 6.37 (2H, d, J = 2.6 Hz, ArH), 3.99 (2H, d, J = 12.9 Hz, ArCH2Ar ax), 3.77–3.58 (6H, m, OCH2 + ArCH2Ar), 3.51–3.39 (4H, m, OCH2), 3.26 (2H, t, J = 8.2 Hz, OCH2), 3.08 (2H, d, J = 13.2 Hz, ArCH2Ar eq), 1.85–1.72 (6H, m, OCH2CH2), 1.40–1.32 (2H, m, OCH2CH2), 1.05 (6H, t, J = 7.3 Hz, CH3), 0.87 (3H, t, J = 7.3 Hz, CH3), 0.66 (3H, t, J = 7.5 Hz, CH3). 13C{1H} NMR (75 MHz, DMSO-d6, 298 K) δ: 157.4, 156.6, 151.6, 151.3, 146.7, 140.6, 136.5, 133.6, 133.2, 132.4, 130.6, 129.1, 125.1, 122.7, 121.3, 120.4, 118.4, 117.1, 75.9, 75.1, 73.8, 36.2, 30.1, 23.4, 23.4, 21.4, 11.0, 10.4, 9.4; TOF HRMS (ESI+): calcd for [C54H58N6O10 + Na]+: 973.4107. Found: m/z (rel. int. %) 973.4101 (100) [M + Na]+. EA found (%) C, 68.14; H, 6.12; N, 8.83 (C54H58N6O10 requires C 68.20; H 6.15; N 8.84).
5,17-Bis{N′-[(4-trifluoromethyl)phenyl]ureido}-25,26,27,28-tetrapropoxycalix[4]arene (partial cone) 12
A general procedure was applied to amino derivative 7 (0.10 g, 0.16 mmol) and 4-(trifluoromethyl)phenyl isocyanate (150 mg, 0.11 ml, 0.80 mmol). The first purification was used to remove the majority of carbamate by column chromatography (50 g of Al2O3, eluent pure CH2Cl2 – carbamate fractions; second eluent pure MeOH – calix[4]arene fractions). The product 12 was finally isolated by preparative GPC (isocratic elution of CHCl3, column PLgel 10 μm, 100 Å, 600 × 25 μm, fl. rate 5 ml min−1, detector UV 254 nm) as a white powder (0.10 g) in 63% yield, mp: 197–199 °C. 1H NMR (300 MHz, DMSO-d6, 298 K) δ: 8.68 (2H, s, NH), 8.05 (2H, s, NH), 7.47 (8H, brs, ArH), 7.20–7.12 (6H, m, ArH), 6.93–6.82 (2H, m, ArH), 6.41 (2H, s, ArH), 4.00 (2H, d, J = 12.9 Hz, ArCH2Ar ax), 3.75–3.58 (8H, m, OCH2 + ArCH2Ar), 3.43–3.24 (4H, m, OCH2), 3.08 (2H, d, J = 12.9 Hz, ArCH2Ar eq), 1.81–1.74 (6H, m, OCH2CH2), 1.41–1.33 (2H, m, OCH2CH2), 1.04 (6H, t, J = 7.0 Hz, CH3), 0.82 (3H, t, J = 7.3 Hz, CH3), 0.66 (3H, t, J = 7.0 Hz, CH3). 19F NMR (282 MHz, DMSO-d6, 296 K) δ: −60.49 (6F, s, CF3). 13C{1H} NMR (75 MHz, CDCl3, 296 K) δ: 157.4, 156.99, 153.6, 153.5, 141.9, 136.7, 135.5, 133.5, 133.4, 130.3, 130.1, 129.3, 126.2 (q, J = 3.8 Hz), 124.7 (q, J = 32.0 Hz), 124.0 (q, J = 271.0 Hz), 123.6, 122.4, 122.1, 118.5, 76.3, 75.4, 75.2, 35.1, 30.3, 24.2, 23.8, 22.5, 10.9, 10.8, 9.3; TOF HRMS (ESI+): calcd for [C56H58F6N4O6 + Na]+: 1019.4159. Found: m/z (rel. int. %) 1019.4153 (100) [M + Na]+.5,17-Di-tert-butyl-11,23-bis[N′-(4-nitrophenyl)ureido]-25,26,27,28-tetrapropoxycalix[4]arene (partial cone) 13
A general procedure was applied to amino derivative 8 (0.10 g, 0.14 mmol) and 4-nitrophenyl isocyanate (97% purity, 115 mg, 0.68 mmol). The product 13 was isolated by column chromatography (30 g of Al2O3, eluent CH2Cl2/MeOH = 160:1, RF = 0.15) as a red powder (0.10 g) in 75% yield, mp: 248–250 °C. 1H NMR (300 MHz, DMSO-d6, 286 K) δ: 9.09 (2H, s, NH), 8.30 (2H, s, NH), 8.09 (4H, d, J = 9.4 Hz, ArH), 7.54 (4H, d, J = 9.1 Hz, ArH), 7.22 (2H, d, J = 2.3 Hz, ArH), 7.20 (2H, s, ArH), 7.15 (2H, s, ArH), 6.58 (2H, d, J = 2.6 Hz, ArH), 4.04 (2H, d, J = 12.3 Hz, ArCH2Ar ax), 3.78–3.60 (6H, m, OCH2 + ArCH2Ar), 3.46–3.32 (4H, m, OCH2), 3.10 (2H, d, J = 12.9 Hz, ArCH2Ar eq), 2.95 (2H, t, J = 7.5 Hz, OCH2), 1.83–1.72 (4H, m, OCH2CH2), 1.50–1.31 (22H, m, OCH2CH2 + 2 × But), 0.99 (6H, t, J = 7.5 Hz, CH3), 0.63 (3H, t, J = 7.5 Hz, CH3), 0.54 (3H, t, J = 7.3 Hz, CH3). 13C{1H} NMR (75 MHz, DMSO-d6, 298 K) δ: 154.9, 153.8, 151.7, 146.7, 144.9, 142.8, 140.8, 135.2, 133.3, 133.2, 133.0, 132.5, 127.5, 125.8, 125.2, 120.2, 118.6, 117.2, 76.0, 74.8, 72.3, 37.8, 34.0, 33.8, 31.7, 31.7, 30.6, 30.0, 23.2, 22.8, 20.6, 10.9,9.6; TOF HRMS (ESI+): calcd for [C62H74N6O10 + Na]+: 1085.5359. Found: m/z (rel. int. %) 1085.5354 (100) [M + Na]+.
5-tert-Butyl-11,17,23-tris[N′-(4-nitrophenyl)ureido]-25,26,27,28-tetrapropoxycalix[4]arene (partial cone) 14
A general procedure was applied to amino derivative 9 (0.10 g, 0.14 mmol) and 4-nitrophenyl isocyanate (97% purity, 183 mg, 1.08 mmol). The product 14 was isolated by column chromatography (30 g of Al2O3, eluent CH2Cl2/MeOH = 160:1, then CH2Cl2/MeOH = 1:1, RF = 0.15) as a yellow powder (0.04 g) in 31% yield, mp: 232–235 °C. 1H NMR (300 MHz, DMSO-d6, 298 K) δ: 9.36 (1H, s, NH), 9.00 (2H, s, NH), 8.78 (1H, s, NH), 8.25 (2H, s, NH), 8.20 (2H, d, J = 9.1 Hz, ArH), 8.10 (4H, d, J = 9.1 Hz, ArH), 7.72 (2H, d, J = 9.1 Hz, ArH), 7.49 (4H, d, J = 9.1 Hz, ArH), 7.27 (2H, s, ArH), 7.20 (2H, s, ArH), 7.18 (2H, d, J = 2.6 Hz, ArH), 6.53 (2H, d, J = 2.3 Hz, ArH), 4.03 (2H, d, J = 12.9 Hz, ArCH2Ar ax), 3.77–3.60 (6H, m, OCH2 + ArCH2Ar), 3.46–3.32 (6H, m, OCH2), 3.06 (2H, d, J = 12.9 Hz, ArCH2Ar eq), 1.85–1.73 (4H, m, OCH2CH2), 1.70–1.62 (2H, m, OCH2CH2), 1.46–1.36 (11H, m, OCH2CH2 + But) 1.02 (6H, t, J = 7.5 Hz, CH3), 0.77 (3H, t, J = 7.3 Hz, CH3), 0.64 (3H, t, J = 7.5 Hz, CH3). 13C NMR (75 MHz, DMSO-d6, 298 K) δ: 155.6, 152.7, 152.2, 152.1, 152.0, 147.3, 147.2, 143.2, 141.5, 141.2, 137.1, 134.1, 133.4, 133.3, 133.2, 132.8, 125.9, 125.7, 121.2, 119.8, 119.0, 118.0, 117.6, 76.5, 75.2, 73.4, 37.7, 34.4, 32.1, 31.0, 23.9, 23.6, 21.2, 11.4, 10.7, 10.2; TOF HRMS (ESI+): calcd for [C65H71N9O13 + Na]+: 1208.5064. Found: m/z (rel. int. %) 1208.5073 (100) [M + Na]+.
Crystallographic measurements
233 total of measured reflections (θmax = 27.5°), from which 10398 were unique (Rint = 0.033) and 6859 observed (I > 2σ(I)). The structure was solved by direct methods16 and refined by full-matrix least squares based on F2 (SHELXL97).17 The hydrogen atoms were fixed into idealised positions (riding model) and assigned temperature factors either Hiso(H) = 1.2 Ueq(pivot atom) or Hiso(H) = 1.5 Ueq(pivot atom) for –CH3. The refinement converged (Δ/σmax = 0.001) to R = 0.061 for observed reflections and wR(F2) = 0.196, GOF = 1.00 for 533 parameters and all 10398 reflections. The final difference map displayed no peaks of chemical significance (Δρmax = 0.68, Δρmin −0.49 e Å−3). The displacement parameters of atoms of tert-butyl groups are wittnesing disorder of these moieties, however the attempt to split positions of these atoms did not improve the model.
Acknowledgements
This research was supported by the Czech Science Foundation (No. 203/09/0691), and by the Grant Agency of the Academy of Sciences of the Czech Republic (No. IAAX08240901).References
- (a) P. A. Gale, Chem. Commun., 2008, 4525–4540 RSC; (b) R. Vilar, Eur. J. Inorg. Chem., 2008, 357–367 CrossRef CAS; (c) I. Ravikumar and P. Ghosh, Chem. Soc. Rev., 2012, 41, 3077–3098 RSC; (d) M. Wenzel, J. R. Hiscock and P. A. Gale, Chem. Soc. Rev., 2012, 41, 480–520 RSC; (e) P. A. Gale, Chem. Commun., 2011, 47, 82–86 RSC; (f) P. S. Dieng and C. Sirlin, Int. J. Mol. Sci., 2010, 11, 3334–3348 CrossRef CAS.
- (a) Anion Coordination Chemistry, ed. K. Bowman-James, A. Bianchi and E. Garcia-Espana, Wiley-VCH Verlag, 2012 Search PubMed; (b) Anion Recognition in Supramolecular Chemistry, in Topics in Heterocyclic Chemistry, ed. P. A. Gale and W. Dehaen, Springer Verlag, 2010, vol. 24 Search PubMed; (c) J. L. Sessler, P. A. Gale and W. S. Cho, Anion Receptor Chemistry, The Royal Society of Chemistry, Cambridge, 2006 Search PubMed; (d) Recognition of Anions, in Structure and Bonding, ed. R. Vilar, Springer Verlag, 2008, vol. 129 Search PubMed; (e) Anion Sensing, Topics in Current Chemistry, ed. I. Stibor, Springer, Berlin, 2005, vol. 255 Search PubMed.
- (a) H. T. Ngo, X. Liu and K. A. Jolliffe, Chem. Soc. Rev., 2012, 41, 4928–4965 RSC; (b) X. Wu, Microchim. Acta, 2012, 176, 23–47 CrossRef CAS; (c) S. Kubik, Chem. Soc. Rev., 2010, 39, 3648–3663 RSC.
- (a) J. M. Llinares, D. Powell and K. Bowman-James, Coord. Chem. Rev., 2003, 240, 57–75 CrossRef CAS; (b) C. Rether and C. Schmuck, Eur. J. Org. Chem., 2011, 1459–1466 CrossRef CAS; (c) M. P. Coles, Chem. Commun., 2009, 3659–3676 RSC.
- (a) V. Amendola, L. Fabbrizzi and L. Mosca, Chem. Soc. Rev., 2010, 39, 3889–3915 RSC; (b) P. Dydio, D. Lichosyt and J. Jurczak, Chem. Soc. Rev., 2011, 40, 2971–2985 RSC; (c) M. O. Odago, A. E. Hoffman, R. L. Carpenter, D. C. T. Tse, S.-S. Sun and A. J. Lees, Inorg. Chim. Acta, 2011, 374, 558–565 CrossRef CAS.
- For books on calixarenes, see: (a) C. D. Gutsche, Calixarenes: An introduction, The Royal Society of Chemistry, Thomas Graham House, Cambridge, 2nd edn 2008 Search PubMed; (b) Calixarenes in the Nanoworld, ed. J. Vicens, J. Harrowfield and L. Backlouti, Springer, Dordrecht, 2007 Search PubMed; (c) Calixarenes 2001, ed. Z. Asfari, V. Böhmer, J. Harrowfield and J. Vicens, Kluwer Academic Publishers, Dordrecht, 2001 Search PubMed; (d) L. Mandolini and R. Ungaro, Calixarenes in Action, Imperial College Press, London, 2000 Search PubMed.
- For review on calixarene-based receptors for anions, see: (a) E. A. Shokova and V. V. Kovalev, Russ. J. Org. Chem., 2009, 45, 1275–1314 CrossRef CAS; (b) S. E. Matthews and P. D. Beer, Supramol. Chem., 2005, 17, 411–435 CrossRef CAS; (c) P. Lhotak, Top. Curr. Chem., 2005, 255, 65–95 CAS.
- For some recent examples on anion recognition using neutral calixarenes, see: (a) E. K. Bullough, C. A. Kilner, M. A. Little and C. E. Willans, Org. Biomol. Chem., 2012, 10, 2824–2829 RSC; (b) A. J. McConnell, C. J. Serpell and P. D. Beer, New J. Chem., 2012, 36, 102–112 RSC; (c) E. Quinlan, S. E. Matthews and T. Gunnlaugsson, J. Org. Chem., 2007, 72, 7497–7503 CrossRef CAS; (d) E. Metay, M. C. Duclos, S. Pellet-Rostaing, M. Lemaire, J. Schulz, R. Kannappan, C. Bucher, E. Saint-Aman and C. Chaix, Eur. J. Org. Chem., 2008, 4304–4312 CrossRef CAS.
- (a) K. Flidrova, M. Tkadlecova, K. Lang and P. Lhotak, Dyes Pigm., 2012, 92, 668–673 CrossRef CAS; (b) K. Flidrova, M. Tkadlecova, K. Lang and P. Lhoták, Tetrahedron Lett., 2012, 53, 678–680 CrossRef CAS; (c) J. Kroupa, I. Stibor, M. Pojarová, M. Tkadlecova and P. Lhotak, Tetrahedron, 2008, 64, 10075–10079 CrossRef CAS; (d) P. Curinova, I. Stibor, J. Budka, J. Sykora, K. Lang and P. Lhotak, New J. Chem., 2009, 33, 612–619 RSC; (e) A. Krajewska, P. Lhotak and H. Radecka, Sensors, 2007, 7, 1655–1666 CrossRef CAS.
- I. Stibor, J. Budka, V. Michlova, M. Tkadlecova, M. Pojarova, P. Curinová and P. Lhotak, New J. Chem., 2008, 32, 1597–1607 RSC.
- J. Budka, P. Lhotak, V. Michlova and I. Stibor, Tetrahedron Lett., 2001, 42, 1583–1586 CrossRef CAS.
- M. Kumar, A. Dhir and V. Bhalla, Chem. Commun., 2010, 46, 6744–6746 RSC.
- (a) O. Hudecek, P. Curinova, J. Budka and P. Lhoták, Tetrahedron, 2011, 67, 5213–5218 CrossRef CAS; (b) O. Hudecek, J. Budka, V. Eigner and P. Lhotak, Tetrahedron, 2012, 68, 4187–4193 CrossRef CAS.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- Z. D. Hill and P. MacCarthy, J. Chem. Educ., 1986, 63, 162–167 CrossRef CAS.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef.
- As suggested by one of the referees we have carried out the additional complexation study using the cone analogue of receptor 11 to directly compare the complexation abilities of both conformers. As one can see from ESI† (page 14), the complexation of Cl− occurred with a 1:1 stoichiometry (Job plot) and the corresponding complexation constant is 173 ± 24 M−1 which is much lower than that of the partial cone derivative 11 (1540 ± 370). This indicates that the partial cone conformation can be much better choice than the cone conformers used so far in the design of novel anion receptors.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, COSY, HMBC, HMQC and NOE spectra of compound 4, including complete structure assignment of this compound. CCDC 894549. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2nj40724h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2013 |