Cleaving bonds in CH3OSO2CF3 with [1,2,4-(Me3C)3C5H2]2CeH; an experimental and computational study†‡
Evan L.
Werkema
a,
Ludovic
Castro
b,
Laurent
Maron
*b,
Odile
Eisenstein
*c and
Richard A.
Andersen
*a
aDepartment of Chemistry and Chemical Sciences, Division of Lawrence Berkeley National Laboratory, University of California, Berkeley, California 94720-1460, USA. E-mail: raandersen@lbl.gov
bLPCNO, Université de Toulouse, INSA, UPS, CNRS, LPCNO, 135 avenue de Rangueil, F-31077 Toulouse, France. E-mail: laurent.maron@irsamc.ups-tlse.fr
cInstitut Charles Gerhardt, UMR 5253 CNRS, Université Montpellier 2, cc 1501, Place E. Bataillon, F-34095 Montpellier, France. E-mail: Odile.eisenstein@univ-montp2.fr
First published on 3rd October 2012
Abstract
The reaction at 20 °C of the metallocenelanthanide hydride, [1,2,4-(Me3C)3C5H2]2CeH,  , and excess methyltrifluoromethanesulfonate, CH3OSO2CF3, results in formation of
, and excess methyltrifluoromethanesulfonate, CH3OSO2CF3, results in formation of 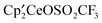 ,
,  ,
,  and the bimetallic complex
and the bimetallic complex  . The metallocenes
. The metallocenes  ,
, 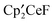 , and
, and  react with excess CH3OSO2CF3 to form
react with excess CH3OSO2CF3 to form  , CH3OCH3, CH3F, and (CH3O)2SO, respectively, at 20 °C. Thus, the net reaction is
, CH3OCH3, CH3F, and (CH3O)2SO, respectively, at 20 °C. Thus, the net reaction is  but the pathway is not a direct methyl transfer. Comparison of the reactivity of CH3OSO2CF3 and CH3OSO2CH3 (Werkema et al., Organometallics, 2012, 31, 870) is revealing since both form a similar set of products but the rates of reaction of CH3OSO2CF3 are faster. The bimetallic complex, in which the SO32− anion bridges two
but the pathway is not a direct methyl transfer. Comparison of the reactivity of CH3OSO2CF3 and CH3OSO2CH3 (Werkema et al., Organometallics, 2012, 31, 870) is revealing since both form a similar set of products but the rates of reaction of CH3OSO2CF3 are faster. The bimetallic complex, in which the SO32− anion bridges two  fragments, is unique in organometallic chemistry. The 1H NMR spectrum is fluxional at 20 °C and the low temperature spectrum is consistent with the geometry observed in the solid state. Density Functional Theory (DFT) calculations of the Gibbs energy profiles for the reaction of CH3OSO2CF3 with
fragments, is unique in organometallic chemistry. The 1H NMR spectrum is fluxional at 20 °C and the low temperature spectrum is consistent with the geometry observed in the solid state. Density Functional Theory (DFT) calculations of the Gibbs energy profiles for the reaction of CH3OSO2CF3 with  show that the CH-bond activation and direct CH3 group transfer have similar activation energy barriers. This contrasts with what is observed in the reaction of
show that the CH-bond activation and direct CH3 group transfer have similar activation energy barriers. This contrasts with what is observed in the reaction of  with CH3OSO2CH3, where CH-bond activation at the SCH3 group is preferred. Remarkably, the activation energy barriers for C–O-bond cleavage are similar in CH3OSO2CH3 and CH3OSO2CF3, which is traced to the calculated small exoergicity of −1.7 kcal mol−1 for the reaction of
with CH3OSO2CH3, where CH-bond activation at the SCH3 group is preferred. Remarkably, the activation energy barriers for C–O-bond cleavage are similar in CH3OSO2CH3 and CH3OSO2CF3, which is traced to the calculated small exoergicity of −1.7 kcal mol−1 for the reaction of 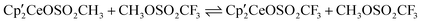 . This contrasts, perhaps, with conventional wisdom that overemphasizes the effect of the electron-withdrawing ability of the CF3 group on the chemical and physical properties of sulfonate esters.
. This contrasts, perhaps, with conventional wisdom that overemphasizes the effect of the electron-withdrawing ability of the CF3 group on the chemical and physical properties of sulfonate esters.
Introduction
The monomeric metallocenelanthanide hydride, [1,2,4-(Me3C)3C5H2]2CeH, abbreviated or [Ce]′H, was prepared in order to explore the intrinsic reactivity patterns of the Ce–H bond and to compare and contrast these patterns with those of the d-transition metal hydrides. The general reactivity pattern that was discovered in the experimental studies was that H for X metathesis occurred in reactions with CH3X, when X is a halide or an ether. The mechanism, shown by labeling studies guided by DFT computations, was not a direct, one-step process in which the CH3 group is transferred intact but rather a two-step pathway with a lower activation energy. The two-step pathway begins with CH-bond activation followed by elimination and trapping of the ejected carbene fragment, eqn (1).1–3
or [Ce]′H, was prepared in order to explore the intrinsic reactivity patterns of the Ce–H bond and to compare and contrast these patterns with those of the d-transition metal hydrides. The general reactivity pattern that was discovered in the experimental studies was that H for X metathesis occurred in reactions with CH3X, when X is a halide or an ether. The mechanism, shown by labeling studies guided by DFT computations, was not a direct, one-step process in which the CH3 group is transferred intact but rather a two-step pathway with a lower activation energy. The two-step pathway begins with CH-bond activation followed by elimination and trapping of the ejected carbene fragment, eqn (1).1–3 | (1) |
 formed.4,5 Thus, the reactivity pattern in the above reactions of
formed.4,5 Thus, the reactivity pattern in the above reactions of  is that intermolecular CH-bond breaking, a relatively low activation energy process, is the initial event, followed by subsequent reactions resulting in the products of the net metathetical exchange of H for X.
is that intermolecular CH-bond breaking, a relatively low activation energy process, is the initial event, followed by subsequent reactions resulting in the products of the net metathetical exchange of H for X.
This article is the culmination of the studies outlined above in which  is allowed to react with CH3OSO2CF3 in order to explore how the activation energy barriers and therefore the products and their distribution are manipulated by the electronegative fluorines in the OSO2CF3 group.
is allowed to react with CH3OSO2CF3 in order to explore how the activation energy barriers and therefore the products and their distribution are manipulated by the electronegative fluorines in the OSO2CF3 group.
Results
Experimental studies
 with an excess of methyltrifluoromethanesulfonate, CH3OSO2CF3, in C6D12 in an NMR tube at 20 °C results in a color change from purple to orange within 20 minutes. Monitoring the changes by 1H NMR spectroscopy shows that all of the resonances due to
with an excess of methyltrifluoromethanesulfonate, CH3OSO2CF3, in C6D12 in an NMR tube at 20 °C results in a color change from purple to orange within 20 minutes. Monitoring the changes by 1H NMR spectroscopy shows that all of the resonances due to  disappear and are replaced by four new sets of paramagnetic Me3C resonances due to metallocenes labeled A, B, C, and D, in the approximate ratio of 1.5
disappear and are replaced by four new sets of paramagnetic Me3C resonances due to metallocenes labeled A, B, C, and D, in the approximate ratio of 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2.5:1, respectively, eqn (2). After one day at 20 °C, the resonances due to C disappear and the ratio of A:B:D is 11:1:2, respectively. After five days at 20 °C the only paramagnetic resonances remaining are due to A, the known metallocene,
:1:2.5:1, respectively, eqn (2). After one day at 20 °C, the resonances due to C disappear and the ratio of A:B:D is 11:1:2, respectively. After five days at 20 °C the only paramagnetic resonances remaining are due to A, the known metallocene, 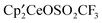 .4 Thus, the net reaction involves H for OSO2CF3 exchange. However, the mechanism is not a simple metathesis, since three intermediates are also observed, two of which are known metallocenes, C,
.4 Thus, the net reaction involves H for OSO2CF3 exchange. However, the mechanism is not a simple metathesis, since three intermediates are also observed, two of which are known metallocenes, C,  , and D,
, and D,  , and one, B, is new,
, and one, B, is new,  , abbreviated as
, abbreviated as  (see eqn (2)).
(see eqn (2)). | (2) |
In independent reactions,  and excess CH3OSO2CF3 form A and CH3F,
and excess CH3OSO2CF3 form A and CH3F,  and excess CH3OSO2CF3 form A and CH3OCH3; the details are in the Experimental section. The metallocene, labeled B, has chemical shifts in its 1H NMR spectrum that are identical to one of the products obtained upon mixing
and excess CH3OSO2CF3 form A and CH3OCH3; the details are in the Experimental section. The metallocene, labeled B, has chemical shifts in its 1H NMR spectrum that are identical to one of the products obtained upon mixing  and
and 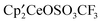 , A, as reported earlier.1 The metallocenes B and C are formed straightaway when
, A, as reported earlier.1 The metallocenes B and C are formed straightaway when  and A are mixed, and although misidentified as
and A are mixed, and although misidentified as 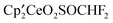 , B is now known to be a bridging sulfite, Fig. 2. Thus, the transformation of CH3OSO2CF3 into SO32− is a net reduction of sulfur from SVI to SIV. The metallocene products formed in the reaction of
, B is now known to be a bridging sulfite, Fig. 2. Thus, the transformation of CH3OSO2CF3 into SO32− is a net reduction of sulfur from SVI to SIV. The metallocene products formed in the reaction of  and CH3OSO2CF3 are illustrated in Scheme 1.
and CH3OSO2CF3 are illustrated in Scheme 1.
![The metallocene products resulting from the reaction of , [Ce]′H, and CH3OSO2CF3.](/image/article/2013/NJ/c2nj40624a/c2nj40624a-s1.gif) | ||
Scheme 1 The metallocene products resulting from the reaction of  , [Ce]′H, and CH3OSO2CF3. , [Ce]′H, and CH3OSO2CF3. | ||
When the reaction shown in eqn (2) and Scheme 1 is complete, that is when the only metallocene in solution is  , A, hydrolysis (H2O) and analysis by GCMS shows the presence of (Me3C)3C5H3, Cp′H, (Me3C)2(Me2Et)C5H3, Cp′′H, 1,3,5-(Me3C)3C6H3, 1,2,4-(Me3C)3C6H3, and 1-F,2,4,6-(Me3C)3C6H2F. The presence of Cp′′H was observed in earlier studies1–3 in the reaction between
, A, hydrolysis (H2O) and analysis by GCMS shows the presence of (Me3C)3C5H3, Cp′H, (Me3C)2(Me2Et)C5H3, Cp′′H, 1,3,5-(Me3C)3C6H3, 1,2,4-(Me3C)3C6H3, and 1-F,2,4,6-(Me3C)3C6H2F. The presence of Cp′′H was observed in earlier studies1–3 in the reaction between  and CH3X and results from insertion of CH2 into the CH bond of a Me3C group; the CH2 fragment is derived from CH activation of CH3X. Thus, it is reasonable to attribute the presence of Cp′′H to CH-bond activation of the CH3O group in CH3OSO2CF3 generating
and CH3X and results from insertion of CH2 into the CH bond of a Me3C group; the CH2 fragment is derived from CH activation of CH3X. Thus, it is reasonable to attribute the presence of Cp′′H to CH-bond activation of the CH3O group in CH3OSO2CF3 generating  (and H2), which rapidly eliminates CH2, forming
(and H2), which rapidly eliminates CH2, forming  . The substituted benzene derivatives were also observed in previous studies in the reaction of
. The substituted benzene derivatives were also observed in previous studies in the reaction of  and CF3X.1 For example,
and CF3X.1 For example,  and CF3SiMe3 yield
and CF3SiMe3 yield 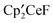 (and Me3SiH) along with the isomeric tri-tert-butyl benzenes and isomeric tri-tert-butylfluorobenzenes. The benzene derivatives are thought to result from the transient formation of
(and Me3SiH) along with the isomeric tri-tert-butyl benzenes and isomeric tri-tert-butylfluorobenzenes. The benzene derivatives are thought to result from the transient formation of  and
and  , which eliminate the CF2 and CHF fragments that are trapped by a Cp′ ring. By analogy, CH3OSO2CF3 can function as a source of CF3 and perhaps CHF2, generating
, which eliminate the CF2 and CHF fragments that are trapped by a Cp′ ring. By analogy, CH3OSO2CF3 can function as a source of CF3 and perhaps CHF2, generating  , CF2 and CHF, and the carbene fragments are trapped by the Cp′-ring. Similarly, the CH3O group in CH3OSO2CF3 is the source of
, CF2 and CHF, and the carbene fragments are trapped by the Cp′-ring. Similarly, the CH3O group in CH3OSO2CF3 is the source of  , as it is in the reaction between
, as it is in the reaction between  and CH3OSO2CH3.3 Identification of B as containing a bridging sulfite is important since it is formed from
and CH3OSO2CH3.3 Identification of B as containing a bridging sulfite is important since it is formed from  and either CH3OSO2CF3 or
and either CH3OSO2CF3 or  .
.
Since both 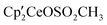 3 and
3 and  are known, a natural question arises as to the direction of the equilibrium in eqn (3).
are known, a natural question arises as to the direction of the equilibrium in eqn (3).
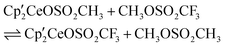 | (3) |
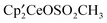 with excess CH3OSO2CF3 in an NMR tube at 19 °C and monitoring the C6D6 solution by 1H NMR spectroscopy as a function of time shows that the ratio of
with excess CH3OSO2CF3 in an NMR tube at 19 °C and monitoring the C6D6 solution by 1H NMR spectroscopy as a function of time shows that the ratio of  to
to  increases over time: 1:37 (10 min), 1:16 (2 days), 1:5 (6 days), 1:2 (30 days). Monitoring a mixture of
increases over time: 1:37 (10 min), 1:16 (2 days), 1:5 (6 days), 1:2 (30 days). Monitoring a mixture of  and CH3OSO2CH3 in a similar manner shows that
and CH3OSO2CH3 in a similar manner shows that 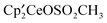 does not appear after 8 days at 19 °C. However, heating a solution at 60 °C for additional four days results in formation of some
does not appear after 8 days at 19 °C. However, heating a solution at 60 °C for additional four days results in formation of some  ; the ratio of
; the ratio of  to
to  is approximately 70:1 and the ratio changes to 34:1 after 15 days at 60 °C. These experiments show that the equilibrium illustrated by eqn (3) lies slightly to the right, which is perhaps unexpected given the large electronegativity difference between H and F. The computational studies presented below support these experimental results and provide a molecular level of understanding for the reactivity patterns.
is approximately 70:1 and the ratio changes to 34:1 after 15 days at 60 °C. These experiments show that the equilibrium illustrated by eqn (3) lies slightly to the right, which is perhaps unexpected given the large electronegativity difference between H and F. The computational studies presented below support these experimental results and provide a molecular level of understanding for the reactivity patterns.
 were obtained by sublimation in an evacuated and sealed ampoule at 170 °C. The crystal data are given in the ESI.‡ An ORTEP is shown in Fig. 1 and some bond distances and angles are listed in Table 1 together with the computed values that are discussed later.
were obtained by sublimation in an evacuated and sealed ampoule at 170 °C. The crystal data are given in the ESI.‡ An ORTEP is shown in Fig. 1 and some bond distances and angles are listed in Table 1 together with the computed values that are discussed later.
![ORTEP, 50% thermal ellipsoids, of [1,2,4-(Me3C)3C5H2]2CeOSO2CF3, all non-hydrogen atoms were refined anisotropically and the hydrogens (not shown) were placed in calculated positions and not refined.](/image/article/2013/NJ/c2nj40624a/c2nj40624a-f1.gif) | ||
| Fig. 1 ORTEP, 50% thermal ellipsoids, of [1,2,4-(Me3C)3C5H2]2CeOSO2CF3, all non-hydrogen atoms were refined anisotropically and the hydrogens (not shown) were placed in calculated positions and not refined. | ||
 from the X-ray data and the DFT computations
from the X-ray data and the DFT computations
| X-ray | DFT | |
|---|---|---|
| Ce–C(Cp′), ave | 2.83 ± 0.06 | 2.87 ± 004 |
| Ce–C(Cp′), range | 2.766(1) to 2.931(2) | 2.82 to 2.93 |
| Ce–Cp′centroid, ave | 2.54 | 2.60 |
| Ce–O, ave | 2.595 ± 0.003 | 2.595 |
| κ2-O–S, ave | 1.463 ± 0.001 | 1.531 |
| Terminal S–O | 1.421(2) | 1.477 |
| S–C | 1.832(2) | 1.893 |
| O–Ce–O | 54.30(5) | 56.4 |
| Cp′centroid–Ce–Cp′centroid | 144 | 142 |
As can be seen in Fig. 1, the triflate ligand is κ2-bound and the individual Ce–O bond lengths are 2.590(1) and 2.599(1) Å. These distances are much longer than the Ce–O bond length in the cis- and trans-enediolate derivatives of  , 2.171(1) and 2.118(3) Å, respectively,6 and in
, 2.171(1) and 2.118(3) Å, respectively,6 and in  , 2.113(3) Å,7 and even longer than the Ce–O bond distance of 2.443(1) Å in the bridging formaldehyde derivative of
, 2.113(3) Å,7 and even longer than the Ce–O bond distance of 2.443(1) Å in the bridging formaldehyde derivative of  ,6 and in
,6 and in  2 of 2.406(2) Å. The reason for the long Ce–O bond distance in the triflate is addressed in the computational studies that follow; it is not likely due to intramolecular steric effects since the bond angles and distances in the
2 of 2.406(2) Å. The reason for the long Ce–O bond distance in the triflate is addressed in the computational studies that follow; it is not likely due to intramolecular steric effects since the bond angles and distances in the  fragment are the same within the ESD's as found in all the metallocene derivatives that contain the
fragment are the same within the ESD's as found in all the metallocene derivatives that contain the  fragment.
fragment.
Single crystals of  were obtained by crystallization from a saturated solution of
were obtained by crystallization from a saturated solution of 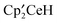 and
and  in C7D8. The crystal data are given in the ESI‡ and selected bond lengths and angles are given in Table 2.
in C7D8. The crystal data are given in the ESI‡ and selected bond lengths and angles are given in Table 2.

| Ce(1)-κ2O2 | Ce(2)-κ1O | |
|---|---|---|
| Ce–C(Cp′), ave | 2.84 ± 0.06 | 2.82 ± 0.05 |
| Ce–C(Cp′), range | 2.769(5) to 2.921(4) | 2.769(4) to 2.891(4) |
| Ce–Cp′centroid | 2.57 | 2.54 |
| Ce–O, ave | 2.424 ± 0.009 | 2.264(3) |
| κn-O–S | 1.525 ± 0.001 | 1.537(3) |
| Cp′centroid–Ce–Cp′centroid, ave | 146 | 147 |
| O1–Ce–O2 | 58.65(1) | |
| O1–S–O2 | 102.2(2) | |
| O1–S–O3 | 105.1(2) | |
| O2–S–O3 | 104.9(2) |
Comparing the bond distances and angles in the sulfite structure at Ce(1) with those in 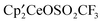 shows that the
shows that the  fragments are identical. The Ce–O bond distances are, however, rather different; the Ce(1)–O(1) and Ce(1)–O(2) distances are 2.415(3) and 2.432(3) Å, respectively, and significantly shorter than those in
fragments are identical. The Ce–O bond distances are, however, rather different; the Ce(1)–O(1) and Ce(1)–O(2) distances are 2.415(3) and 2.432(3) Å, respectively, and significantly shorter than those in  that average to 2.595 ± 0.001 Å. On the other hand, the average S–O bond lengths in the κ2-O2S fragment in the sulfite are longer than in the triflate, 1.525 ± 0.001 Å and 1.463 ± 0.001 Å, respectively. These bond distance trends reflect the higher coordination and oxidation number of sulfur in the triflate and its lower net negative charge. The most interesting feature in
that average to 2.595 ± 0.001 Å. On the other hand, the average S–O bond lengths in the κ2-O2S fragment in the sulfite are longer than in the triflate, 1.525 ± 0.001 Å and 1.463 ± 0.001 Å, respectively. These bond distance trends reflect the higher coordination and oxidation number of sulfur in the triflate and its lower net negative charge. The most interesting feature in  is the sulfite group that bridges the two
is the sulfite group that bridges the two  fragments. A search of the CCDC database did not generate any structure in which a single SO32− group bridges between two metal fragments. The geometry at sulfur is pyramidal since the O–S–O angles sum to 312°. The average S–O distance of 1.529 ± 0.005 Å and an O–S–O angle of 104.1 ± 1.0° are similar to those found in solid state structures that contain the SO32− ion. For example, the average S–O distance and the O–S–O angle in AgNaSO3·2H2O8 and [(NH4)9][(Fe(SO3)6]9 are 1.52 Å and 105°, respectively.
fragments. A search of the CCDC database did not generate any structure in which a single SO32− group bridges between two metal fragments. The geometry at sulfur is pyramidal since the O–S–O angles sum to 312°. The average S–O distance of 1.529 ± 0.005 Å and an O–S–O angle of 104.1 ± 1.0° are similar to those found in solid state structures that contain the SO32− ion. For example, the average S–O distance and the O–S–O angle in AgNaSO3·2H2O8 and [(NH4)9][(Fe(SO3)6]9 are 1.52 Å and 105°, respectively.
The pyramidal geometry of the SO32− group in  is anticipated by the Lewis structure and illuminated by the molecular orbital model originally developed by Walsh10 and described pedagogically by Gimarc.11 In the 26 electron dianion, a lone pair is located on sulfur, which disfavors a planar structure because the lone pair is in a pure 3p orbital. Upon lowering the symmetry from D3h to C3v, a symmetry allowed mixing of the sulfur 3p orbital with the empty a1 orbital, built from the in-phase combination of
is anticipated by the Lewis structure and illuminated by the molecular orbital model originally developed by Walsh10 and described pedagogically by Gimarc.11 In the 26 electron dianion, a lone pair is located on sulfur, which disfavors a planar structure because the lone pair is in a pure 3p orbital. Upon lowering the symmetry from D3h to C3v, a symmetry allowed mixing of the sulfur 3p orbital with the empty a1 orbital, built from the in-phase combination of  orbitals, stabilizes the sulfur lone pair. The tendency for pyramidalization is enhanced in SO32− by the antibonding interaction between the sulfur lone pair and the oxygen lone pairs present in the a2 orbital that is strongly S–O π antibonding in D3h symmetry. The pyramidalization decreases the S–O π overlap and consequently the antibonding interaction as shown qualitatively in the resulting orbital represented as a1(C3v).
orbitals, stabilizes the sulfur lone pair. The tendency for pyramidalization is enhanced in SO32− by the antibonding interaction between the sulfur lone pair and the oxygen lone pairs present in the a2 orbital that is strongly S–O π antibonding in D3h symmetry. The pyramidalization decreases the S–O π overlap and consequently the antibonding interaction as shown qualitatively in the resulting orbital represented as a1(C3v).

Solution structures
The 1H NMR spectra of three of the metallocenes, B, C, and D at 20 °C show the Me3C resonances in a 2:1 ratio, as expected if the metallocene has idealized C2v symmetry. In contrast, the Me3C resonances in the 1H NMR spectrum (20 °C) of A appear in a 1:1:1 pattern showing that the two cyclopentadienyl rings are chemically equivalent but the three Me3C groups on a given ring are chemically inequivalent.
The crystal structure of  , A, Fig. 1, shows that the molecule has C1 symmetry and therefore the 20 °C 1H NMR spectrum is due to fluxions that generate a metallocene with averaged Cs symmetry. Raising the temperature of a toluene-d8 solution results in coalescence of the 1:1:1 pattern into a 2:1 pattern around 355 K. The δ vs. 1/T plot is available in the ESI‡ and ΔG≠ (Tc = 355 K) is 14 kcal mol−1. If the solid state structure is maintained in solution the Me3C groups in the 1H NMR spectrum will appear as a pair of 1:1:1 resonances. A single set of 2:1 Me3C resonances may be observed by allowing the Cp′ rings to librate around their pseudo C5 axes, while the κ2-OSO2CF3 group rotates about the Ce⋯S vector or undergoes a κ2 ⇌ κ1 equilibrium. The 1:1:1 pattern of the Me3C resonances at lower temperatures is consistent with hindered ring rotation such that a horizontal mirror plane is present that interconverts the individual rings while the vertical mirror plane is absent resulting in the chemical inequivalence of all three Me3C groups on a given cyclopentadienyl ring. The horizontal mirror plane that interconverts the Cp′ rings also requires that the κ2-OSO2CF3 group lies in the plane, which requires that it too is fluxional.
, A, Fig. 1, shows that the molecule has C1 symmetry and therefore the 20 °C 1H NMR spectrum is due to fluxions that generate a metallocene with averaged Cs symmetry. Raising the temperature of a toluene-d8 solution results in coalescence of the 1:1:1 pattern into a 2:1 pattern around 355 K. The δ vs. 1/T plot is available in the ESI‡ and ΔG≠ (Tc = 355 K) is 14 kcal mol−1. If the solid state structure is maintained in solution the Me3C groups in the 1H NMR spectrum will appear as a pair of 1:1:1 resonances. A single set of 2:1 Me3C resonances may be observed by allowing the Cp′ rings to librate around their pseudo C5 axes, while the κ2-OSO2CF3 group rotates about the Ce⋯S vector or undergoes a κ2 ⇌ κ1 equilibrium. The 1:1:1 pattern of the Me3C resonances at lower temperatures is consistent with hindered ring rotation such that a horizontal mirror plane is present that interconverts the individual rings while the vertical mirror plane is absent resulting in the chemical inequivalence of all three Me3C groups on a given cyclopentadienyl ring. The horizontal mirror plane that interconverts the Cp′ rings also requires that the κ2-OSO2CF3 group lies in the plane, which requires that it too is fluxional.
The crystal structure of  , B, Fig. 2, shows that the molecule has Cs symmetry, but the two
, B, Fig. 2, shows that the molecule has Cs symmetry, but the two 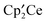 fragments are distinct and therefore the solution 1H NMR spectrum is an averaged one. Assuming that the Cp′ rings are undergoing libration about their pseudo-C5 axes, the two Cp′ ligands on Ce(1) are inequivalent and the Me3C groups will appear as a pair of 2:1 resonances, and those on Ce(2) are equivalent and the Me3C groups will appear as a single set of 1:1:1 resonances. Thus, seven Me3C resonances are expected if the molecule has Cs symmetry in solution as it does in the solid state. The δ vs. 1/T plot in Fig. 3 shows that the 2:1 pattern of Me3C groups at 93 K decoalesces around 263 K and emerges as seven resonances in the correct relative areas below 243 K. It is not possible to assign the individual resonances, but the NMR spectrum at low temperature is consistent with the solid state structure. A least motion path that accounts for the average symmetry at high temperature involves moving O(2) from Ce(1) to Ce(2), thereby exchanging their sites and orientation of their Cp′ ligands as the denticity of the μ3-SO3 group at Ce(1) changes from bidentate to unidentate.
fragments are distinct and therefore the solution 1H NMR spectrum is an averaged one. Assuming that the Cp′ rings are undergoing libration about their pseudo-C5 axes, the two Cp′ ligands on Ce(1) are inequivalent and the Me3C groups will appear as a pair of 2:1 resonances, and those on Ce(2) are equivalent and the Me3C groups will appear as a single set of 1:1:1 resonances. Thus, seven Me3C resonances are expected if the molecule has Cs symmetry in solution as it does in the solid state. The δ vs. 1/T plot in Fig. 3 shows that the 2:1 pattern of Me3C groups at 93 K decoalesces around 263 K and emerges as seven resonances in the correct relative areas below 243 K. It is not possible to assign the individual resonances, but the NMR spectrum at low temperature is consistent with the solid state structure. A least motion path that accounts for the average symmetry at high temperature involves moving O(2) from Ce(1) to Ce(2), thereby exchanging their sites and orientation of their Cp′ ligands as the denticity of the μ3-SO3 group at Ce(1) changes from bidentate to unidentate.
![ORTEP, 50% thermal ellipsoids, of [1,2,4-(Me3C)3C5H2]2Ce (μ3-O2SO–Ce(1)κ2O–Ce(2)κ1O)Ce[1,2,4-(Me3C)3C5H2]2. All non-hydrogen atoms were refined anisotropically and the hydrogens (not shown) were placed in calculated positions and not refined.](/image/article/2013/NJ/c2nj40624a/c2nj40624a-f2.gif) | ||
| Fig. 2 ORTEP, 50% thermal ellipsoids, of [1,2,4-(Me3C)3C5H2]2Ce (μ3-O2SO–Ce(1)κ2O–Ce(2)κ1O)Ce[1,2,4-(Me3C)3C5H2]2. All non-hydrogen atoms were refined anisotropically and the hydrogens (not shown) were placed in calculated positions and not refined. | ||
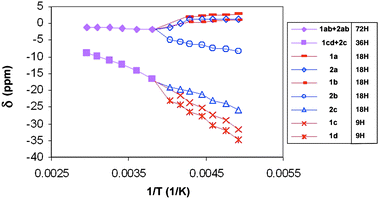 | ||
Fig. 3
1H NMR chemical shifts, δ, vs. 1/T plot of CMe3 resonances in 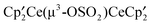 in C7D8. Numerals refer to the cerium center, Ce(1) or Ce(2) as labeled in Fig. 2 and letters refer to individual CMe3 groups along with the number of hydrogen atoms in each set of Me3C resonances. in C7D8. Numerals refer to the cerium center, Ce(1) or Ce(2) as labeled in Fig. 2 and letters refer to individual CMe3 groups along with the number of hydrogen atoms in each set of Me3C resonances. | ||
Computational studies
The computational study evolves in the following way. (i) The excellent agreement between the calculated and experimental structures of 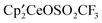 suggests that the calculated structure of
suggests that the calculated structure of  is close to that of the experimental complex whose crystal structure is not available. Comparison between these two calculated structures defines the structural effect of CF3 relative to CH3. (ii) The Gibbs energies for the CH-bond activation and the methyl transfer in the reaction of the CH3OSO2CF3 with
is close to that of the experimental complex whose crystal structure is not available. Comparison between these two calculated structures defines the structural effect of CF3 relative to CH3. (ii) The Gibbs energies for the CH-bond activation and the methyl transfer in the reaction of the CH3OSO2CF3 with  are compared. The same processes have been studied for the reaction with the metallacycle, [(1,2,4-(Me3C)3C5H2)(1,2-(Me3C)2-4-(Me2CCH2)C5H2)]Ce, for completeness; these data are available in the ESI.‡ (iii) The Gibbs energies of the reactions of CH3OSO2CF3 and CH3OSO2CH33 with
are compared. The same processes have been studied for the reaction with the metallacycle, [(1,2,4-(Me3C)3C5H2)(1,2-(Me3C)2-4-(Me2CCH2)C5H2)]Ce, for completeness; these data are available in the ESI.‡ (iii) The Gibbs energies of the reactions of CH3OSO2CF3 and CH3OSO2CH33 with  are compared in order to define the effect of CF3 on the kinetic and thermodynamic values. To keep the comparison meaningful only the reactions at the OCH3 group of CH3OSO2CH3 are considered. The CF-bond activation pathways have not been studied. (iv) The thermodynamics of X (X = OCH3, F) for OSO2CH3 or OSO2CF3 exchange on
are compared in order to define the effect of CF3 on the kinetic and thermodynamic values. To keep the comparison meaningful only the reactions at the OCH3 group of CH3OSO2CH3 are considered. The CF-bond activation pathways have not been studied. (iv) The thermodynamics of X (X = OCH3, F) for OSO2CH3 or OSO2CF3 exchange on 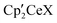 are compared in order to define the effect of CF3 on the affinity of
are compared in order to define the effect of CF3 on the affinity of  for OSO2R (R = CH3, CF3).
for OSO2R (R = CH3, CF3).
 .
As illustrated by the Newman projections in the previous study on CH3OSO2CH3, a gauche and a trans conformation are obtained as minima in the geometry optimization of CH3OSO2CF3.3 In both molecules, these two conformations are close in energy but the preferred isomer is not the same; for CH3OSO2CF3 the gauche isomer is preferred by 1.5 kcal mol−1 over the trans isomer, while for CH3OSO2CH3 the preference is reversed with a difference in energy of 1.4 kcal mol−1. The bond distances in the two molecules are similar, for example, the SO bonds in CH3OSO2CF3 are shorter by only 0.007 Å relative to the equivalent bonds in CH3OSO2CH3.
.
As illustrated by the Newman projections in the previous study on CH3OSO2CH3, a gauche and a trans conformation are obtained as minima in the geometry optimization of CH3OSO2CF3.3 In both molecules, these two conformations are close in energy but the preferred isomer is not the same; for CH3OSO2CF3 the gauche isomer is preferred by 1.5 kcal mol−1 over the trans isomer, while for CH3OSO2CH3 the preference is reversed with a difference in energy of 1.4 kcal mol−1. The bond distances in the two molecules are similar, for example, the SO bonds in CH3OSO2CF3 are shorter by only 0.007 Å relative to the equivalent bonds in CH3OSO2CH3.
The optimized structure of [1,2,4-(Me3C)3C5H2]2CeOSO2CF3 is represented in Fig. 4. The calculated bond distances in  are given in Table 1 where they are compared to the values obtained in the X-ray structure. The orientation of the cyclopentadienyl rings in the calculated metallocene is identical to that found in the crystal structure, as shown by comparing Fig. 1 and 4. The triflate is κ2-coordinated to the cerium fragment with average Ce–O distances of 2.595 Å. Additional distances and angles are compared in Table 1. The calculated Ce–O bond distance is identical to that observed in the crystal structure but all bond distances to sulfur are about 0.05 Å longer in the calculated structure. Comparing the calculated bond distances in
are given in Table 1 where they are compared to the values obtained in the X-ray structure. The orientation of the cyclopentadienyl rings in the calculated metallocene is identical to that found in the crystal structure, as shown by comparing Fig. 1 and 4. The triflate is κ2-coordinated to the cerium fragment with average Ce–O distances of 2.595 Å. Additional distances and angles are compared in Table 1. The calculated Ce–O bond distance is identical to that observed in the crystal structure but all bond distances to sulfur are about 0.05 Å longer in the calculated structure. Comparing the calculated bond distances in  with those in
with those in  , all of the distances in the OSO2CF3 group are slightly shorter but the Ce–O(ave) distance is slightly longer (0.06Å).
, all of the distances in the OSO2CF3 group are slightly shorter but the Ce–O(ave) distance is slightly longer (0.06Å).
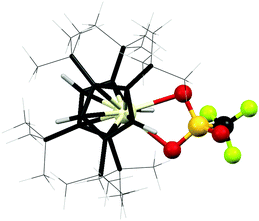 | ||
Fig. 4 Optimized structure of 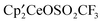 . The color codes for atoms are off white for Ce, red for O, gold yellow for S; black for C, yellow green for F, white for H. Selected distances are given in Table 1. . The color codes for atoms are off white for Ce, red for O, gold yellow for S; black for C, yellow green for F, white for H. Selected distances are given in Table 1. | ||
The adducts, transition states, and expected products associated with the CH-bond activation of the hydride are shown in Fig. 5. The CH-bond activation is a proton-transfer from the OCH3 group to the hydride. The reaction begins by coordination of the substrate to the cerium fragment by the oxygen lone pair on SO. A transition state is located for coordination either by the oxygen lone pairs on SO or on OCH3. The CH-bond activation path is different, depending upon which oxygen lone pair is used in the transition state: CH/O when the substrate coordinates with the terminal oxygen and CH/OCH3 when the oxygen of the OCH3 group is used.
![Adducts, transition states, intermediates, and products for the CH-bond activation of the OCH3 group with labels indicating the type of oxygen coordinated to the [Ce]′ fragment at the transition state.](/image/article/2013/NJ/c2nj40624a/c2nj40624a-f5.gif) | ||
| Fig. 5 Adducts, transition states, intermediates, and products for the CH-bond activation of the OCH3 group with labels indicating the type of oxygen coordinated to the [Ce]′ fragment at the transition state. | ||
The methyl transfer reaction between [Ce]′X and CH3OSO2CF3 yields [Ce]′OSO2CF3 and CH3X as products. The adducts, the transition states, and the expected products for X = H, OCH3, and F are shown in Fig. 6. For this reaction, CH3OSO2CF3 coordinates exclusively by way of the SO lone pair on oxygen. The methyl transfer reaction occurs with inversion of configuration at carbon in an SN2 transition state.
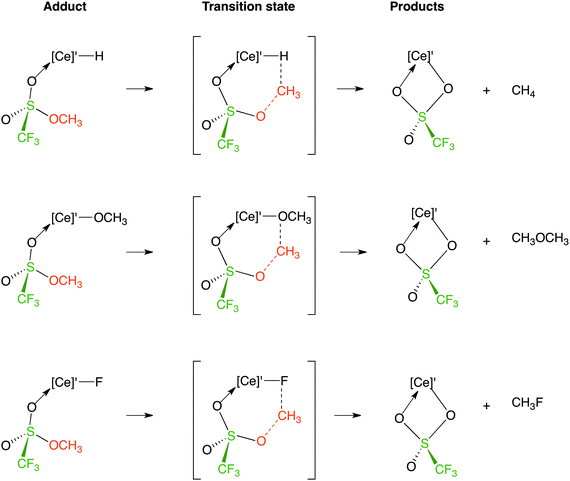 | ||
| Fig. 6 Adducts, transition states, and products for the methyl transfer reaction. | ||
The Gibbs energy profiles for the CH-bond activation and methyl transfer in the reaction of CH3OSO2CF3 with [Ce]′H are shown in Fig. 7 as histograms. The full Gibbs energy profiles are given in the ESI.‡ The Gibbs energy profiles obtained for the corresponding reactions with CH3OSO2CH3, presented in detail in an earlier paper, are shown for a comparison.3
![Gibbs energy profiles, in kcal mol−1, for CH-bond activation and methyl transfer for the reaction of CH3OSO2CF3 and [Ce]′H. The results of the corresponding reactions with CH3OSO2CH3 are included for comparison.3 The Gibbs energies of the adducts are in blue, the transition states are in red, and the products are in green.](/image/article/2013/NJ/c2nj40624a/c2nj40624a-f7.gif) | ||
| Fig. 7 Gibbs energy profiles, in kcal mol−1, for CH-bond activation and methyl transfer for the reaction of CH3OSO2CF3 and [Ce]′H. The results of the corresponding reactions with CH3OSO2CH3 are included for comparison.3 The Gibbs energies of the adducts are in blue, the transition states are in red, and the products are in green. | ||
Adduct formation with either the SO or OCH3 oxygen lone pairs is endoergic by 7 kcal mol−1. The calculated activation energy barriers for CH-bond activation and methyl transfer are also close in energy although the calculated energies of the product forming reactions are very different. The CH-bond activation forms an intermediate with η2-CH2OSO2CF3 bonded to [Ce]′ via the carbon and one of the oxygen atoms. This intermediate evolves into product by ejection of a CH2 fragment forming [Ce]′OSO2CF3, the most stable of all possible products. Methyl transfer forms [Ce]′OSO2CF3 and CH4 in a single step and is accordingly much more exoergic. The key message that emerges from Fig. 7 is that CH-bond activation and methyl transfer have similar activation barriers.
The Gibbs energy profiles for the methyl transfer between [Ce]′X, (X = H, OCH3, and F) with CH3OSO2CF3 are illustrated using histograms in Fig. 8. The full Gibbs energy profiles are given in the ESI.‡ The activation energy barriers are lower for the hydride than for the methoxide and fluoride by more than 15 kcal mol−1. This agrees with the observation that the OCH3/OSO2CF3 and F/OSO2CF3 exchange reactions occur more slowly, which is why C and D are observed in the 1H NMR spectrum in the presence of CH3OSO2CF3 on short reaction times. In all cases, the formation of [Ce]′OSO2CF3 is exoergic and the extent of exoergicity lies in the order of F < OCH3 ≪ H.
![Gibbs energy profiles, in kcal mol−1, for the methyl transfer reaction between [Ce]′X, (X = H, OCH3, and F) and CH3OSO2CF3. The Gibbs energies of the adducts are in blue, the transition states are in red, and the products are in green.](/image/article/2013/NJ/c2nj40624a/c2nj40624a-f8.gif) | ||
| Fig. 8 Gibbs energy profiles, in kcal mol−1, for the methyl transfer reaction between [Ce]′X, (X = H, OCH3, and F) and CH3OSO2CF3. The Gibbs energies of the adducts are in blue, the transition states are in red, and the products are in green. | ||
Discussion
Both reactions of CH3OSO2CH33 or CH3OSO2CF3 with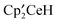 result in the net exchange of H for OSO2R (R = CH3 or CF3). This provides the opportunity to determine the effect of the CF3 group on the reaction energy profiles. The equilibrium reaction illustrated by eqn (3) shows a slight thermodynamic preference for
result in the net exchange of H for OSO2R (R = CH3 or CF3). This provides the opportunity to determine the effect of the CF3 group on the reaction energy profiles. The equilibrium reaction illustrated by eqn (3) shows a slight thermodynamic preference for 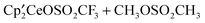 over
over  . This is confirmed by the calculated value of ΔG0 of −1.7 kcal mol−1 for this reaction. This small Gibbs energy favoring
. This is confirmed by the calculated value of ΔG0 of −1.7 kcal mol−1 for this reaction. This small Gibbs energy favoring 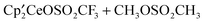 is somewhat surprising since the notion that OSO2CF3 is an excellent leaving group (a kinetic concept) is implicitly associated with a thermochemical meaning attributed to the electron-withdrawing ability of CF3 relative to CH3. This implicit connection is only weakly supported by the small value of ΔG0, showing that the difference in energy between the two Ce–O bonds in the cerium complexes is similar to that between the CH3–O bonds in the two sulfonate esters.
is somewhat surprising since the notion that OSO2CF3 is an excellent leaving group (a kinetic concept) is implicitly associated with a thermochemical meaning attributed to the electron-withdrawing ability of CF3 relative to CH3. This implicit connection is only weakly supported by the small value of ΔG0, showing that the difference in energy between the two Ce–O bonds in the cerium complexes is similar to that between the CH3–O bonds in the two sulfonate esters.
The products formed in the reaction of CH3OSO2CH3 or CH3OSO2CF3 with 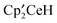 are
are  , where R is CH3 or CF3. However, the intermediates that are spectroscopically observed are rather different and this difference addresses the question of mechanism. In both reactions,
, where R is CH3 or CF3. However, the intermediates that are spectroscopically observed are rather different and this difference addresses the question of mechanism. In both reactions,  , D, is observed as an intermediate that evolves into
, D, is observed as an intermediate that evolves into  in the presence of excess CH3OSO2R. When R is CF3, the rate is faster than when R is CH3, and in both reactions the CH3O group is derived by cleaving the O–S bond. The only source of fluoride is the CF3 group, and
in the presence of excess CH3OSO2R. When R is CF3, the rate is faster than when R is CH3, and in both reactions the CH3O group is derived by cleaving the O–S bond. The only source of fluoride is the CF3 group, and  is presumably derived from CF-bond activation of CF3 or CH3−xFx; trapping of the CHF and CF2 fragments by Cp′H is responsible for the formation of the tri-tert-butyl benzene and tri-tert-butylfluorobenzene isomers, as previously reported.1 The most interesting intermediate is the bridging sulfite, B, that is generated by two independent pathways, (i)
is presumably derived from CF-bond activation of CF3 or CH3−xFx; trapping of the CHF and CF2 fragments by Cp′H is responsible for the formation of the tri-tert-butyl benzene and tri-tert-butylfluorobenzene isomers, as previously reported.1 The most interesting intermediate is the bridging sulfite, B, that is generated by two independent pathways, (i)  and CH3OSO2CF3 and (ii)
and CH3OSO2CF3 and (ii) 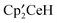 and
and  . Since both reactions are rapid at room temperature it is reasonable to suggest that when
. Since both reactions are rapid at room temperature it is reasonable to suggest that when  , A, is formed in the reaction of
, A, is formed in the reaction of  and CH3OSO2CF3, it is converted into
and CH3OSO2CF3, it is converted into 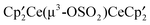 , B, along with
, B, along with  , C; in the presence of CH3OSO2CF3, both B and C are converted into the net product
, C; in the presence of CH3OSO2CF3, both B and C are converted into the net product  , A, along with (CH3O)2SO and CH3F. Thus the sulfite is an intermediate to and from
, A, along with (CH3O)2SO and CH3F. Thus the sulfite is an intermediate to and from 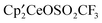 , A, depending on the reagents present in solution. The reaction of CH3OSO2CF3 and
, A, depending on the reagents present in solution. The reaction of CH3OSO2CF3 and  is rather complex since the pathways forming A, B, C, and D, eqn (2), must have similar rates and the intermediates must have similar lifetimes. However, regardless of the pathway, ultimately
is rather complex since the pathways forming A, B, C, and D, eqn (2), must have similar rates and the intermediates must have similar lifetimes. However, regardless of the pathway, ultimately  is consumed and
is consumed and  is produced, a net H for X exchange reaction like those discussed earlier.1–4,6,7
is produced, a net H for X exchange reaction like those discussed earlier.1–4,6,7
The Gibbs energy profiles for the CH-bond activation and methyl transfer processes show that the activation energy barriers are similar in CH3OSO2CF3 and CH3OSO2CH3 (considering only the CH-bond activation of the OCH3 group), Fig. 7. Thus, replacing the CH3 group by a CF3 group does not influence the reactivity of the OCH3 group. In CH3OSO2CH3, CH-bond activation of the SCH3 group is more favorable and the CH-bond activation of the OCH3 group is considerably more difficult.3 The calculations show that the CF3 group does not lower the energy barrier of CH-bond activation at OCH3 and it is rather surprising that the CH3OSO2CH3 and CH3OSO2CF3 are equally good Me+ transfer reagents. The small difference in ΔG0 for the reaction shown in eqn (3) is a ramification of this unexpected result.
The thermodynamics, ΔG0, of X/Y ligand exchange between [Ce]′X (X = H, OCH3, F, OSO2CF3, OSO2CH3) and CH3Y (Y = OCH3, F, OSO2CF3, OSO2CH3) are summarized in Fig. 9. The diagram is organized such that values associated with OSO2CH3 are on the left side and those associated with OSO2CF3 are on the right side. The bottom reaction, which is the X/Y (OSO2CF3/OSO2CH3) exchange reaction between 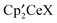 and CH3Y, closes several thermodynamical cycles, resulting in a difference in ΔG0 for the reactions on the left and on the right of 1.7 kcal mol−1.
and CH3Y, closes several thermodynamical cycles, resulting in a difference in ΔG0 for the reactions on the left and on the right of 1.7 kcal mol−1.
 | ||
Fig. 9 Gibbs energies, ΔG0, in kcal mol−1 for X/Y ligand exchanges in the reaction of  with CH3Y where the products are Cp′2CeY and CH3X. with CH3Y where the products are Cp′2CeY and CH3X.  . . | ||
The exchange of a hydride with methoxide, fluoride, mesylate, or triflate is strongly exoergic as is expected for a reaction in which hydride is replaced by an electronegative anion. The exoergicity increases in the order of OCH3 < F < OSO2CH3 < OSO2CF3. The metallocenes 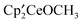 ,
,  , and
, and  have in common a Ce–O bond, which might be thought to determine the thermochemical behavior of these compounds. However, the exchange reaction between OCH3 and either OSO2CH3 or OSO2CF3 indicates a strong preference of the [Ce]′ fragment for OSO2CH3 or OSO2CF3 over OCH3, even though the Ce–O bond in the sulfonate esters is about 0.5 Å longer than in the alkoxides as mentioned above. Furthermore, the similar affinity of OSO2CH3 and OSO2CF3 for the [Ce]′ fragment indicates that the CF3 group has a marginal influence on the thermodynamic properties of the OSO2R group, implying that the difference between OSO2R and OR′ is associated with the SO bonds. The S–O bond has low-lying
have in common a Ce–O bond, which might be thought to determine the thermochemical behavior of these compounds. However, the exchange reaction between OCH3 and either OSO2CH3 or OSO2CF3 indicates a strong preference of the [Ce]′ fragment for OSO2CH3 or OSO2CF3 over OCH3, even though the Ce–O bond in the sulfonate esters is about 0.5 Å longer than in the alkoxides as mentioned above. Furthermore, the similar affinity of OSO2CH3 and OSO2CF3 for the [Ce]′ fragment indicates that the CF3 group has a marginal influence on the thermodynamic properties of the OSO2R group, implying that the difference between OSO2R and OR′ is associated with the SO bonds. The S–O bond has low-lying  orbitals, which can delocalize the lone pair of any atom (here an oxygen) bound to sulfur, and this effect is the origin of the stability of the OSO2R anion and the elongated Ce–O bonds in
orbitals, which can delocalize the lone pair of any atom (here an oxygen) bound to sulfur, and this effect is the origin of the stability of the OSO2R anion and the elongated Ce–O bonds in 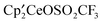 . The S–CF3 bond is less efficient in delocalizing density because the
. The S–CF3 bond is less efficient in delocalizing density because the 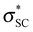 lies higher in energy than the
lies higher in energy than the  orbital, Scheme 2. The participation of
orbital, Scheme 2. The participation of  in the lowest unoccupied orbital of the sulfonate has been shown to be important in the computational study at the MP2 level of alkynyl sulfonate.12
in the lowest unoccupied orbital of the sulfonate has been shown to be important in the computational study at the MP2 level of alkynyl sulfonate.12
 | ||
Scheme 2 Schematic representation as a Newman projection along the S–O bond of the delocalization of an oxygen lone pair into the empty orbitals of the SO2CF3 group, mostly located on the  orbitals. orbitals. | ||
The NBO charge analysis of the neutral species and the cerium compounds given in Fig. 10 are consistent with the postulate advocated in the previous paragraph. The values show that the CF3 group slightly modifies the charge on the sulfur atom but has little effect on the other atoms. Thus, the NBO charges are consistent with the notion presented that the electron density on these sulfonate ligands is manipulated by the low-lying  orbitals, which are only marginally modified when CH3 is replaced by CF3.
orbitals, which are only marginally modified when CH3 is replaced by CF3.
 | ||
Fig. 10 NBO charges on CH3OSO2CH3, CH3OSO2CF3,  , ,  . CH3OSO2CH3 and CH3OSO2CF3 are shown in their optimal conformation. . CH3OSO2CH3 and CH3OSO2CF3 are shown in their optimal conformation. | ||
Conclusion
The reaction of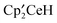 with an excess of CH3OSO2CF3 forms three monometallic metallocenes,
with an excess of CH3OSO2CF3 forms three monometallic metallocenes,  ,
,  , and
, and 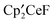 , along with the dimetallic metallocene
, along with the dimetallic metallocene  at 20 °C. In the dimetallic complex, variable temperature 1H NMR spectrum shows that the Cp′ rings are fluxional at 20 °C but the low temperature spectrum is consistent with the solid state structure. Over a period of 2–5 days, all metallocenes are transformed into
at 20 °C. In the dimetallic complex, variable temperature 1H NMR spectrum shows that the Cp′ rings are fluxional at 20 °C but the low temperature spectrum is consistent with the solid state structure. Over a period of 2–5 days, all metallocenes are transformed into  . DFT calculations exploring whether the initial step in the reaction is a CH-bond activation or a direct methyl transfer show that the activation barriers for these two pathways are essentially the same. This result deviates from all other computational results on
. DFT calculations exploring whether the initial step in the reaction is a CH-bond activation or a direct methyl transfer show that the activation barriers for these two pathways are essentially the same. This result deviates from all other computational results on  in which the CH-bond activation step is always lower in energy than a direct methyl transfer step. The initial rationalization for the preferred methyl transfer with CH3OSO2CF3 is that the electron withdrawing ability of the CF3 group stabilizes the OSO2CF3 anion and facilitates the CH3–OSO2CF3 cleavage. The DFT results do not support this interpretation since the effect of replacing a CH3 group with a CF3 group is small. Rather, methyl group transfer from CH3OSO2CF3 and CH3OSO2CH3 is controlled by the low-lying
in which the CH-bond activation step is always lower in energy than a direct methyl transfer step. The initial rationalization for the preferred methyl transfer with CH3OSO2CF3 is that the electron withdrawing ability of the CF3 group stabilizes the OSO2CF3 anion and facilitates the CH3–OSO2CF3 cleavage. The DFT results do not support this interpretation since the effect of replacing a CH3 group with a CF3 group is small. Rather, methyl group transfer from CH3OSO2CF3 and CH3OSO2CH3 is controlled by the low-lying  orbitals that are marginally influenced by the nature of the other groups at sulfur.
orbitals that are marginally influenced by the nature of the other groups at sulfur.
Experimental procedures
General
All manipulations were performed under an inert atmosphere using standard Schlenk and dry box techniques. All solvents were dried and distilled from sodium or sodium benzophenoneketyl. Methyltrifluoromethanesulfonate, CH3OSO2CF3, and dimethylsulfite, (CH3O)2SO, were obtained commercially and purified by distillation. NMR spectra were recorded on Bruker AV-300, AV-400, and AV600 spectrometers at 20 °C in the solvent specified. J. Young NMR tubes were used for all NMR tube experiments. Electron impact mass spectrometry was performed by the microanalytical facility at the University of California, Berkeley. The abbreviation Cp′ is used for the 1,2,4-tri-tert-butylcyclopentadienyl ligand. Unless otherwise specified, samples for GC-MS were prepared by adding a drop of nitrogen-purged H2O, agitating, and allowing the samples to stand closed for 10 min. The samples were then dried over magnesium sulfate, filtered, and diluted ten-fold with pentane. A 1 μL sample was injected into an HP6890 GC system with a J&W DB-XLB universal non-polar column, attached to an HP5973 Mass Selective Detector. and
and  in C6D6.
in C6D6.
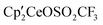 4 was dissolved in C6D6 in an NMR tube and
4 was dissolved in C6D6 in an NMR tube and 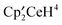 was added. After 20 minutes, the 1H NMR spectrum contained resonances due to
was added. After 20 minutes, the 1H NMR spectrum contained resonances due to  ,
,  ,
,  , and a pair of CMe3 resonances in a 2:1 area ratio corresponding to a new species, δ −1.38 (36H, ν1/2 = 250 Hz), −12.66 (18H, ν1/2 = 120 Hz). After one day at 19 °C, resonances due to
, and a pair of CMe3 resonances in a 2:1 area ratio corresponding to a new species, δ −1.38 (36H, ν1/2 = 250 Hz), −12.66 (18H, ν1/2 = 120 Hz). After one day at 19 °C, resonances due to 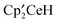 had disappeared from the 1H NMR spectrum and diamagnetic resonances corresponding to isomers of tri-tert-butylbenzene13 had appeared. Additional
had disappeared from the 1H NMR spectrum and diamagnetic resonances corresponding to isomers of tri-tert-butylbenzene13 had appeared. Additional  was added to the sample until all
was added to the sample until all  resonances had disappeared from the spectrum, by which point a yellow precipitate had formed. The sample was filtered and the precipitate was suspended in C7D8. The 1H NMR spectrum showed the new species to be the principal component. The sample was heated to 60 °C for two weeks, the tube was carefully inverted to separate the solution from the remaining solid, and the sample was allowed to stand at 19 °C for five days, after which small yellow crystals suitable for X-ray diffraction studies had formed. Successful solution of the crystal structure showed the new substance to be
resonances had disappeared from the spectrum, by which point a yellow precipitate had formed. The sample was filtered and the precipitate was suspended in C7D8. The 1H NMR spectrum showed the new species to be the principal component. The sample was heated to 60 °C for two weeks, the tube was carefully inverted to separate the solution from the remaining solid, and the sample was allowed to stand at 19 °C for five days, after which small yellow crystals suitable for X-ray diffraction studies had formed. Successful solution of the crystal structure showed the new substance to be  ; full crystallographic details are included in the ESI.‡ Monoclinic cell space group P21/c, a = 20.6275(8) Å, b = 10.5366(4) Å, c = 30.6641(12) Å, β = 99.5900(10)°, V = 6541.5(4) Å3. MS (M – Cp′)+m/z (calc., found) 1060 (100, 100) 1061 (57, 57) 1062 (46, 47) 1063 (20, 20) 1064 (8, 8), 1065 (3, 4).
; full crystallographic details are included in the ESI.‡ Monoclinic cell space group P21/c, a = 20.6275(8) Å, b = 10.5366(4) Å, c = 30.6641(12) Å, β = 99.5900(10)°, V = 6541.5(4) Å3. MS (M – Cp′)+m/z (calc., found) 1060 (100, 100) 1061 (57, 57) 1062 (46, 47) 1063 (20, 20) 1064 (8, 8), 1065 (3, 4).
 in C6D12.
in C6D12.
 was dissolved in C6D12 in an NMR tube. The tube was cooled in a liquid nitrogen isopropanol bath, the head space was evacuated, and an excess of CH3OSO2CF3 was added by vacuum transfer. The tube was warmed to 19 °C, the headspace was refilled with N2 (1 atm), and the sample was allowed to stand. After 20 minutes, the solution had turned orange, and resonances due to
was dissolved in C6D12 in an NMR tube. The tube was cooled in a liquid nitrogen isopropanol bath, the head space was evacuated, and an excess of CH3OSO2CF3 was added by vacuum transfer. The tube was warmed to 19 °C, the headspace was refilled with N2 (1 atm), and the sample was allowed to stand. After 20 minutes, the solution had turned orange, and resonances due to  had disappeared from the 1H NMR spectrum. Paramagnetic resonances due to
had disappeared from the 1H NMR spectrum. Paramagnetic resonances due to  ,
,  ,
,  ,4 and
,4 and  2,6 had appeared in the spectrum; the ratio of the four components was approximately 1.5:1:2.5:2. Diamagnetic resonances due to isomers of tri-tert-butylbenzene had also appeared. The 19F NMR spectrum contained resonances due to
2,6 had appeared in the spectrum; the ratio of the four components was approximately 1.5:1:2.5:2. Diamagnetic resonances due to isomers of tri-tert-butylbenzene had also appeared. The 19F NMR spectrum contained resonances due to  , CH3F, and three other resonances at −81.22, −89.11, and −90.80 ppm. After 50 minutes, the ratio of the paramagnetic species in the 1H NMR spectrum was 5:1:2:2.5. After 1 day, only resonances due to
, CH3F, and three other resonances at −81.22, −89.11, and −90.80 ppm. After 50 minutes, the ratio of the paramagnetic species in the 1H NMR spectrum was 5:1:2:2.5. After 1 day, only resonances due to  ,
,  , and
, and  remained in a 11:1:2 ratio, and after 5 days, only resonances due to
remained in a 11:1:2 ratio, and after 5 days, only resonances due to 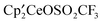 remained. The 19F NMR spectrum after five days contained resonances due to CH3OSO2CF3,
remained. The 19F NMR spectrum after five days contained resonances due to CH3OSO2CF3,  , CH3F, and two other resonances at −78.49 and −80.85. The GCMS analysis showed four principle components in addition to Cp′H, with (M)+m/z 246 (two isomers of tri-tert-butylbenzene), 264 (tri-tert-butylfluorobenzene) and 248 (Cp′H + CH2) in an approximate ratio of 1:1:1.5:60.
, CH3F, and two other resonances at −78.49 and −80.85. The GCMS analysis showed four principle components in addition to Cp′H, with (M)+m/z 246 (two isomers of tri-tert-butylbenzene), 264 (tri-tert-butylfluorobenzene) and 248 (Cp′H + CH2) in an approximate ratio of 1:1:1.5:60.
 in C6D12.
in C6D12.
 was dissolved in C6D12 in an NMR tube. The tube was cooled in a liquid nitrogen isopropanol bath, the head space was evacuated, and an excess of CH3OSO2CF3 was added by vacuum transfer. The tube was warmed to 19 °C, the headspace was refilled with N2 (1 atm), and the sample was allowed to stand. After one hour, the Cp′-ring tert-butyl resonances due to
was dissolved in C6D12 in an NMR tube. The tube was cooled in a liquid nitrogen isopropanol bath, the head space was evacuated, and an excess of CH3OSO2CF3 was added by vacuum transfer. The tube was warmed to 19 °C, the headspace was refilled with N2 (1 atm), and the sample was allowed to stand. After one hour, the Cp′-ring tert-butyl resonances due to  in the 1H NMR spectrum had shifted from −2.59 and −5.97 ppm to −2.35 and −4.64 ppm. After two hours, resonances due to
in the 1H NMR spectrum had shifted from −2.59 and −5.97 ppm to −2.35 and −4.64 ppm. After two hours, resonances due to 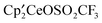 had appeared; the ratio of
had appeared; the ratio of  to
to  was approximately 1:8. The 19F NMR spectrum contained resonances due to
was approximately 1:8. The 19F NMR spectrum contained resonances due to  and three other resonances at −90.76, −130.74, and −131.35 ppm in a 10:4:1:2 area ratio. After 1 day, the ratio of
and three other resonances at −90.76, −130.74, and −131.35 ppm in a 10:4:1:2 area ratio. After 1 day, the ratio of  to
to 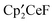 in the 1H NMR spectrum was 10:1. The 19F NMR spectrum contained resonances due to CH3F as well as
in the 1H NMR spectrum was 10:1. The 19F NMR spectrum contained resonances due to CH3F as well as  and two other resonances at −90.76 and −130.74 ppm in a 10:50:3:1 area ratio. After 5 days, only resonances due to
and two other resonances at −90.76 and −130.74 ppm in a 10:50:3:1 area ratio. After 5 days, only resonances due to  remained in the 1H NMR spectrum. The 19F NMR spectrum contained the same set of four resonances in a 6:45:2:1 area ratio.
remained in the 1H NMR spectrum. The 19F NMR spectrum contained the same set of four resonances in a 6:45:2:1 area ratio.
 in C6D12.
in C6D12.
 was synthesized from
was synthesized from  , C18O, and H2 in an NMR tube as previously described.2,6 An excess of CH316OSO2CF3 was added and the sample was allowed to stand at 19 °C for 5 days, after which time only resonances due to
, C18O, and H2 in an NMR tube as previously described.2,6 An excess of CH316OSO2CF3 was added and the sample was allowed to stand at 19 °C for 5 days, after which time only resonances due to  remained in the 1H NMR spectrum. The solvent was removed under reduced pressure, yielding a light red powder. EI MS analysis indicated the presence of
remained in the 1H NMR spectrum. The solvent was removed under reduced pressure, yielding a light red powder. EI MS analysis indicated the presence of  with no 18O enrichment. MS (M)+m/z (calc., found) 755 (100, 100) 756 (39, 32) 757 (25, 15).
with no 18O enrichment. MS (M)+m/z (calc., found) 755 (100, 100) 756 (39, 32) 757 (25, 15).
 in C6D6.
in C6D6.
 3 was dissolved in C6D6 in an NMR tube and a drop of CH3OSO2CF3 was added. After 10 minutes, resonances due to
3 was dissolved in C6D6 in an NMR tube and a drop of CH3OSO2CF3 was added. After 10 minutes, resonances due to  had appeared in the 1H NMR spectrum; the ratio of
had appeared in the 1H NMR spectrum; the ratio of  to
to  was 37:1. After 2 days at 19 °C, the ratio was 16:1. After six days, a set of two resonances in a 2:1 area ratio due to an unknown species had appeared in the 1H NMR spectrum [δ −4.368 (ν1/2 = 20 Hz), −7.981 (ν1/2 = 20 Hz)]; the ratio of
was 37:1. After 2 days at 19 °C, the ratio was 16:1. After six days, a set of two resonances in a 2:1 area ratio due to an unknown species had appeared in the 1H NMR spectrum [δ −4.368 (ν1/2 = 20 Hz), −7.981 (ν1/2 = 20 Hz)]; the ratio of  ,
,  , and the new species was 53:12:1. After 15 days, the ratio was 20:6:1; after 30 days, 10:5:1.
, and the new species was 53:12:1. After 15 days, the ratio was 20:6:1; after 30 days, 10:5:1.
 in C6D6.
in C6D6.
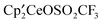 was dissolved in C6D6 in an NMR tube and a drop of CH3OSO2CH3 was added. After 20 minutes, the 1H NMR resonances due to the CMe3 groups of
was dissolved in C6D6 in an NMR tube and a drop of CH3OSO2CH3 was added. After 20 minutes, the 1H NMR resonances due to the CMe3 groups of 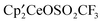 had coalesced from three resonances in a 1:1:1 area ratio to two resonances in a 2:1 area ratio [δ −0.83 (ν1/2 = 1850 Hz), −13.36 (ν1/2 = 60 Hz)]. After 8 days at 19 °C, the spectrum was unchanged. The sample was heated to 60 °C, and after 4 days, resonances due to
had coalesced from three resonances in a 1:1:1 area ratio to two resonances in a 2:1 area ratio [δ −0.83 (ν1/2 = 1850 Hz), −13.36 (ν1/2 = 60 Hz)]. After 8 days at 19 °C, the spectrum was unchanged. The sample was heated to 60 °C, and after 4 days, resonances due to  and the new species observed in the previous experiment had appeared; the ratio of
and the new species observed in the previous experiment had appeared; the ratio of  ,
,  , and the new species was 140:2:1. After 15 days, the ratio was 34:1:1; after 28 days, the ratio was 17:1:1.
, and the new species was 140:2:1. After 15 days, the ratio was 34:1:1; after 28 days, the ratio was 17:1:1.
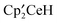 in C6D12.
in C6D12.
 was dissolved in C6D12 in an NMR tube and an excess of (CH3O)2SO was added. After 20 minutes, the solution had turned light red, resonances due to
was dissolved in C6D12 in an NMR tube and an excess of (CH3O)2SO was added. After 20 minutes, the solution had turned light red, resonances due to  were absent from the 1H NMR spectrum, and resonances due to
were absent from the 1H NMR spectrum, and resonances due to  had appeared. The spectrum was unchanged after 1 hour at 19 °C. Integration of the CMe3 resonances relative to the residual solvent 1H resonance showed approximately 63% conversion of
had appeared. The spectrum was unchanged after 1 hour at 19 °C. Integration of the CMe3 resonances relative to the residual solvent 1H resonance showed approximately 63% conversion of  to
to  .
.
Computational details
All molecules were calculated in full with no simplification of the ligands. The Stuttgart–Dresden–Bonn Relativistic large Effective Core Potential (RECP) was used to represent the inner shells of Ce.14 The associated basis set14 augmented by an f-polarization function (α = 1.000) was used to represent the valence orbitals.15 Sulfur has also been represented by an RECP,16 with the associated basis set augmented by a d-polarization Gaussian function (α = 0.421). The atoms C, O, and H were represented by an all-electron 6-31G(d,p) basis set.17 F was also represented by an RECP18 with the associated basis set of the type (4s5p/2s3p)18 augmented by two contracted d-polarization Gaussian functions (α1 = 3.3505(0.357851), α2 = 0.9924(0.795561)).19 Calculations were carried out at the DFT(B3PW91) level20 with Gaussian 03.21 The nature of the extrema (minimum or transition state) was established with analytical frequencies calculations and the intrinsic reaction coordinate (IRC) was followed to confirm that the transition states connect to reactants and products. The zero point energy (ZPE) and entropic contribution have been estimated within the harmonic potential approximation. The Gibbs free energy, G, was calculated at T = 298.15 K and 1 atm. The NBO analysis22 was carried out replacing Ce by La because of the technical requirement to have even number of f electrons for the calculations.Acknowledgements
This work was supported by the Director, Office of Science, Office of Basic Energy Sciences (OBES), of the U.S. Department of Energy (DOE) under Contract No. DE-AC02-05CH11231. L.M. and O.E. thank the CNRS and le Ministère de l'Enseignement Supérieure et de la Recherche (MESR) for fundings. L.C. thanks the Computer Centers, CINES and CALMIP, for a generous donation of computational time. L.M. is a junior member of the Institut Universitaire de France (IUF).References
- E. L. Werkema, E. Messines, L. Perrin, L. Maron, O. Eisenstein and R. A. Andersen, J. Am. Chem. Soc., 2005, 127, 7781 CrossRef CAS.
- E. L. Werkema, R. A. Andersen, A. Yahia, L. Maron and O. Eisenstein, Organometallics, 2009, 28, 3173 CrossRef CAS.
- E. L. Werkema, L. Castro, L. Maron, O. Eisenstein and R. A. Andersen, Organometallics, 2012, 31, 870 CrossRef CAS.
- L. Maron, E. Werkema, L. Perrin, O. Eisenstein and R. A. Andersen, J. Am. Chem. Soc., 2005, 127, 279 CrossRef CAS.
- E. L. Werkema and R. A. Andersen, J. Am. Chem. Soc., 2008, 130, 7153 CrossRef CAS.
- E. L. Werkema, L. Maron, O. Eisenstein and R. A. Andersen, J. Am. Chem. Soc., 2007, 129, 2529 CrossRef CAS . Correction p. 6662.
- E. L. Werkema, A. Yahia, L. Maron, O. Eisenstein and R. A. Andersen, Organometallics, 2010, 29, 5103 CrossRef CAS.
- L. Niinisto and L. O. Larsson, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1973, 29, 623 CrossRef CAS.
- L. O. Larsson and L. Niinisto, Acta Chem. Scand., 1973, 27, 859 CrossRef CAS.
- A. D. Walsh, J. Chem. Soc., 1953, 2301 RSC.
- B. H. Gimarc, Molecular Structure and Bonding, Academic Press, New York, 1979, p. 169 Search PubMed.
- P. J. Stang, C. M. Crittell, A. M. Arif, M. Karni and Y. Apeloig, J. Am. Chem. Soc., 1991, 113, 7461 CrossRef CAS.
- H. Kuenzer and S. Berger, J. Org. Chem., 1985, 50, 3222 CrossRef CAS.
- (a) M. Dolg, H. Stoll, A. Savin and H. Preuss, Theor. Chim. Acta, 1989, 75, 173 CrossRef CAS; (b) M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1993, 85, 441 CrossRef CAS.
- L. Maron and O. Eisenstein, J. Phys. Chem. A, 2000, 104, 7140 CrossRef CAS.
- A. Bergner, M. Dolg, W. Küchle, H. Stoll and H. Preuß, Mol. Phys., 1993, 80, 1431 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS.
- H. Igel-Mann, H. Stoll and H. Preuß, Mol. Phys., 1988, 65, 1321 CrossRef.
- L. Maron and C. Teichteil, Chem. Phys., 1998, 237, 105 CrossRef CAS.
- J. J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244 CrossRef CAS; A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef; K. Burke, J. P. Perdew and W. Yang, in Electronic Density Functional Theory: Recent Progress and New Directions, ed. J. F. Dobson, G. Vignale and M. P. Das, Plenum, New York, NY, 1998 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03, Revision C.02, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
Footnotes |
| † This article is included in the All Aboard 2013 themed issue. |
‡ Electronic supplementary information (ESI) available: Crystallographic information for 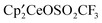 , A, and , A, and 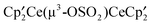 , B, and a δ vs. 1/T plot for A. Full Gibbs energy profiles corresponding to all paths studied. List of coordinates, energy E and Gibbs energy, G in a.u. for all calculated species. The results for the reaction of CH3OSO2CF3 with the metallacycle have been included for completeness. CCDC 901437 for [1,2,4-(Me3C)3C5H2]2CeOSO2CF3 and 889316 for [1,2,4-(Me3C)3C5H2]2Ce(μ3-OSO2)Ce[1,2,4-(Me3C)3C5H2]2. For ESI and crystallographic data in CIF or other format see DOI: 10.1039/c2nj40624a , B, and a δ vs. 1/T plot for A. Full Gibbs energy profiles corresponding to all paths studied. List of coordinates, energy E and Gibbs energy, G in a.u. for all calculated species. The results for the reaction of CH3OSO2CF3 with the metallacycle have been included for completeness. CCDC 901437 for [1,2,4-(Me3C)3C5H2]2CeOSO2CF3 and 889316 for [1,2,4-(Me3C)3C5H2]2Ce(μ3-OSO2)Ce[1,2,4-(Me3C)3C5H2]2. For ESI and crystallographic data in CIF or other format see DOI: 10.1039/c2nj40624a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2013 |