Precursors for pyromellit-bridged silica sol–gel hybrid materials†
Stefan
Pfeifer
ab,
Anke
Schwarzer
a,
Dana
Schmidt
a,
Erica
Brendler
c,
Michael
Veith
b and
Edwin
Kroke
*a
aInstitute of Inorganic Chemistry, TU Bergakademie Freiberg, Leipziger Str. 29, D-09596 Freiberg, Germany. E-mail: edwin.kroke@chemie.tu-freiberg.de; Fax: +49 (0) 3731-39-4058
bLaboratory of Biophysics, Westphalian University of Applied Sciences, August-Schmidt-Ring 10, D-45665 Recklinghausen, Germany
cInstitute of Analytical Chemistry, TU Bergakademie Freiberg, Leipziger Str. 29, D-09596 Freiberg, Germany
First published on 31st August 2012
Abstract
Bridged bis(trialkoxysilylalkyl)pyromellitic diimides 3–6 were prepared as single-source precursors for sol–gel derived organic–inorganic hybrid materials. The synthesis route starts with the formation pyromellit diimide 1 from pyromellitic dianhydride and hexamethyldisilazane (HMDS), followed by metallation of the NH groups to give the dipotassium salt 2. The four molecular hybrid precursors 3–6 were obtained according to the first step of the Gabriel synthesis. The reaction rates were studied as a function of the alkyl chain length and the nature of the halide (Cl vs. I). All products 1–6 were comprehensively analysed using FT-IR, 1H and 13C NMR spectroscopy as well as elemental analysis, and – in the case of 3–6 – also with 29Si NMR spectroscopy. For compounds 3 (with propylene groups and methoxy substituents) and 6 (with methylene groups and ethoxy substituents) single crystal X-ray structures were determined and discussed. Hydrolysis and condensation of the alkoxides 3–6 were carefully monitored with solution 29Si and 1H NMR spectroscopy providing a basis for further studies on the formation of silica-pyromellit organic–inorganic hybrids from precursors 3–6. Finally the formation of flexible and transparent hydride films using precursors 4 and 6 was proved.
Introduction
Polyimides are typically formed by the reaction between aromatic acid dianhydrides (e.g. pyromellitic dianhydride) and primary diamines, e.g. ethylenediamine,1–3 as shown in Scheme 1. They are well known as high performance materials with excellent properties like oxidative stability at high temperatures, good mechanical properties and resistance against acids and solvents.4–8 These properties of polyimides make them interesting for a wide field of applications like coatings for electrical insulations9,10 or membranes.11 | ||
| Scheme 1 Synthesis and structure of imides. | ||
In many cases the low solubility of polyimides in organic solvents causes processing problems. In order to improve the solubility the imides were functionalized.12–14 Further investigations were carried out on the thermal stability of imides, which is generally high, ranging from 200 up to 450 °C. Aromatic anhydrides and diamines lead to higher thermal stabilities in comparison to aliphatic derivatives.
Another approach to increase the thermal stability of polyimides is to combine them with sol–gel materials to form organic–inorganic hybrid materials.15–20,27 These materials can be divided into two major classes. Class I hybrid materials consists of two phases with weak interactions (van-der-Waals) between both phases. Class II hybrids are materials with inorganic moieties covalently bonded to the organic moiety.21 A typical method to class II hybrid materials is the use of organically modified sol–gel precursors such as bridged molecules. A bridged precursor consists of an organic spacer and two terminal alkoxysilyl groups as depicted in Scheme 2. These hybrid precursors can easily be integrated in common sol–gel systems, which may be used for surface functionalization, fiber drawings or coatings.
 | ||
| Scheme 2 Schematic structure of bridged sol–gel precursors. | ||
Common polyimides are synthesized according to Scheme 1. This reaction results in the amidocarboxylic acid which can be converted under thermal influence to the corresponding imide. The conversion is possible in the solution and in the solid phase. A by-product of the imid formation is water. To serve as a bridged precursor for sol–gel chemistry the diamine must be replaced by aminoalkyltrialkoxysilane. These compounds are sensitive to moisture, and even small quantities of water which are formed during the imid formation can lead to hydrolysis of the alkoxysilane followed by condensation to siloxanes. To circumvent this problem the trialkoxysilyl group must be introduced after the completion of imidization and drying of the product.
In the literature hydrosilylation and transimidisation were reported as possible routes to triethoxysilylalkylimide.22–27 In the present study an approach based on the well-known Gabriel synthesis of amines is used for the preparation of bis(trialkoxysilylalkyl)imides.
Several trialkoxysilyl functionalized imides were prepared and characterized using different techniques including 1H, 13C, 29Si NMR and FT-IR spectroscopy. Selected monomers were comprehensively analyzed including single crystal X-ray structure determination, and further studied regarding their hydrolysis-condensation and film-formation behavior.
Results and discussion
Bis(trialkoxysilylalkyl)pyromellitic diimide may be prepared by various ways. The simplest variant would be the direct route using dianhydrides and aminosilanes as starting materials, which are both commercially available. This reaction leads to the respective amidocarboxylic acid as shown in Scheme 3. The water formed in the second reaction step leads very rapidly to hydrolysis and condensation of the alkoxysilane moieties. The silsesquioxanes which are formed during condensation are insoluble. Therefore, with this kind of precursor it is not possible to produce the corresponding molecular imide without hydrolysis. | ||
| Scheme 3 Formation of amidocarboxylic acid followed by formation of insoluble polyimide caused by hydrolysis and condensation. | ||
To illustrate the sensitivity to water, the gelation times of the amidocarboxylic acid, as presented in Scheme 3, with various molar ratios of alkoxy/water (A/W) in ethanol were studied. The reactions were performed at room temperature. It turned out that at an A/W-ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 after a few minutes a distinct turbidity of the solution occurred. After about 30 minutes, the samples were completely gelled. Further experiments showed that all reaction mixtures with A/W-ratios greater than 6:2 had gelled within 24 h. It is apparent from these attempts that the above-mentioned reaction is not suitable for synthesizing bis(trialkoxysilylalkyl)imides since the water liberated during imidization reaction directly leads to hydrolysis of Si–OR groups and condensation of the silanol groups, which again results in a further water elimination.
:6 after a few minutes a distinct turbidity of the solution occurred. After about 30 minutes, the samples were completely gelled. Further experiments showed that all reaction mixtures with A/W-ratios greater than 6:2 had gelled within 24 h. It is apparent from these attempts that the above-mentioned reaction is not suitable for synthesizing bis(trialkoxysilylalkyl)imides since the water liberated during imidization reaction directly leads to hydrolysis of Si–OR groups and condensation of the silanol groups, which again results in a further water elimination.
Methods reported so far for the synthesis of bis(trialkoxysilylalkyl)pyromellitic diimide are hydrosilylation22–24 and transimidation.25–27
An alternative route, which has not been reported until now, is the first step of the Gabriel synthesis, a well-known method for the preparation of primary amines.28,29 The Gabriel synthesis is based on the reaction of a potassium imide, generally potassium phthalimide, with alkyl halides to produce the alkylated imide and the potassium halide. In this synthetic route the final step is the hydrazinolysis to get the primary amine. However, this step is omitted in the present study. In Scheme 4 the complete synthesis route starting from pyromellitic diianhydride is shown.
 | ||
| Scheme 4 Synthesis of the diimide 1, its dipotassium salt 2 and the bis(trialkoxysilylalkyl)imides 3–6. | ||
Pyromellitic diimide (PMDI)
The synthesis and characteristics of pyromellitic diimide 1 are described in the literature.30–33 However, we used a different route to obtain 1. Pyromellitic diimide was prepared via a reaction of pyromellitic dianhydride (PMDA) with hexamethyldisilazane (HMDS). Reactions in dry acetone using molar ratios of PMDA: HMDS of 1:2 or 1:4 lead to the respective amidocarboxylic acid after hydrolysis of trimethylsilylgroups with water. The thermal imidization occurs by holding the temperature at 200 °C for two hours. Dimethylsulfoxide and dimethylformamide are suitable solvents to crystallize and purify the diimide 1.33 The purified product showed 1H NMR signals at 11.82 ppm and 8.02 ppm which agree with the literature.34 The FT-IR data are also in accordance with the reported spectra.35
Dipotassium pyromellitic diimide (K2PMDI)
Potassium diimide 2 is a starting material for the synthesis of the target compounds, i.e. bis(trialkoxysilylalkyl)pyromellitic diimides 3–6. It was synthesized from pyromellitic diimide 1. In the literature it is reported that the potassium salt is formed by reactions of ethanolic KOH solutions with imides.36 However, our attempts did not lead to the product. Much better results were obtained when DMSO was used as solvent. Due to the insolubility of 2 in DMSO a white precipitate formed immediately, which was examined with FT-IR, 1H and 13C NMR spectroscopy. 18-Crone-6 was used to dissolve the product for NMR investigations in d6-DMSO.Based on the absence of the vibration at 3200 cm−1 it can be concluded that a complete substitution of the hydrogen atom of the imide group against potassium occurred (see Fig. S1, ESI†). Furthermore the characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bands are shifted to smaller wave numbers. In contrast to the 1H NMR results of 1 the potassium salt 2 shows only one signal at 7.16 ppm. It can be assumed that the conversion is close to 100% because no 1H signals appear caused by imides or amides. Also, the 13C NMR data show that the signals for C2 and C3 are shifted downfield, while the signal for C1 is shifted upfield. All NMR data and assignments of compounds 1 and 2 are listed in the ESI.†
O bands are shifted to smaller wave numbers. In contrast to the 1H NMR results of 1 the potassium salt 2 shows only one signal at 7.16 ppm. It can be assumed that the conversion is close to 100% because no 1H signals appear caused by imides or amides. Also, the 13C NMR data show that the signals for C2 and C3 are shifted downfield, while the signal for C1 is shifted upfield. All NMR data and assignments of compounds 1 and 2 are listed in the ESI.†
Bis(trialkoxysilylalkyl)pyromellitic diimides 3–6
The bis(trialkoxysilylalkyl)pyromellitic diimides were prepared according to Scheme 4 by treatment of 2 with two molar equivalents of the respective trialkoxyhalogenoalkylsilane. The reactions were performed in dry dimethylformamide (DMF). The applied reaction temperature and time differed depending on the nature of the used silane. In the case of the iodine compounds the reaction was performed at 100 °C and the transformation was found to be completed after eight hours. For all chlorine compounds a temperature of 140 °C and a reaction time of several days were necessary. A change in colour of the reaction mixture from colourless to dark brown was noticed. In contrast to the iodine compound, no homogeneous solutions were obtained, because in contrast to potassium iodide potassium chloride is not soluble in DMF. In conclusion the reaction rate as a function of the studied silane is as follows:| iodopropyl ≫ chloromethyl > chloroethyl > chloropropyl |
The isolated imides 3–6 were examined by FT-IR spectroscopy. The results are shown in Fig. 1. The most important peaks are the CO vibration at approximately 1768–1765 cm−1 and 1712–1698 cm−1. This position of the CO bands is characteristic for five-membered cyclic imides. Specific bands due to the Si–O–C units are present at 1100–1065 cm−1 for Si–O–Me in the case of 3 and for Si–O–Et in the case of 4–6.
 | ||
| Fig. 1 FT-IR spectra of bis(trialkoxysilylalkyl)pyromellitic diimides 3–6. | ||
Compounds 3–6 were analyzed by means of 1H, 13C and 29Si NMR spectroscopy. The 1H NMR results show that the resonance of N-CH2-CH2-R shifts downfield from 3.60 ppm for both bis(trimethoxysilylpropyl)-pyromellitic diimide 3 and bis(triethoxysilylpropyl)pyromellitic diimide 4 to 3.69 ppm for bis(triethoxysilylethyl)pyromellitic diimide 5. In contrast, compound 6 (bis(triethoxysilylmethyl)-pyromellitic diimide) comprises an upfield shift to 3.21 ppm. The aromatic protons, which were found at 7.16 ppm for 1 and 8.15 ppm for the potassium salt 2, appear again at 8.15 ppm for compounds 3–4 and at 8.13 ppm for 5. For compound 6 the signal can be observed at 8.22 ppm. The shift of the signal can be explained as follows. The shortening of the alkyl group from compound 4 to 6 causes a decreased +I-effect. This is reflected by the downfield shift of the signal to 3.69 ppm. By further shortening of the alkyl group from ethylene 5 to methylene 6 the protons of the methylene bridge appear upfield shifted at 3.22 ppm, due to the electron-donating properties of silicon.
The 29Si NMR data of compounds 3–6 are shown in Fig. 2. An upfield shift is observed with shortening of the alkylene bridge from propylene to methylene.

Single crystal structures of compounds 3 and 6
Single crystals of 3 were obtained from methanol at room temperature while suitable crystals of 6 were grown at 3 °C in hexane solution. Both crystal structures show disordered moieties.37 In 3, lying on an inversion centre, the whole Si(OMe3) group occurs in two different positions (89:11). In contrast, compound 6 shows three ethoxy groups being disordered in two positions independent of each other (92:8, 73:29 and 28:72). All bond lengths and angles are within the range of the expected values (Fig. 3). The bond lengths of the imide unit indicate a conjugated system, while Calkyl–N bond to the silyl group is a typical single bond (mean value: 1.47 Å).
 | ||
| Fig. 3 Molecular structures of compounds 3 and 6. The thermal displacement ellipsoids of the non-hydrogen atoms are drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. Compound 3 shows a disordered Si(OMe)3 group (89:11), while in 6 three ethoxy groups are disordered in two positions (92:8, 73:29 and 28:72). | ||
In both crystal structures (3 and 6), the minimal distance between adjacent aromatic fragments is larger than 5 Å indicating no π–π stacking interactions. On the other hand, in 3 the disordered ethoxy group O9-C22-C21 shows a C–H⋯π interaction (d = 2.97 Å, 147°) for the lower occupied position. In fact, the crystal structure of 3 is dominated by C–H⋯O contacts, which are rather weak (H–O: 2.6 Å). Only one contact C11-H11⋯O5 (H–O: 2.42 Å, C–H–O: 162°) forms molecular strands along the crystallographic a axis. In the case of 6, C16-H16⋯O10 (H–O: 2.48 Å, C–H–O: 149°) forms a zigzag chain along the crystallographic b axis. Other C–H⋯O contacts are in the range of 2.5–2.6 Å indicating a rather weak influence on the crystal structure.
Hydrolysis and condensation of bis(trialkoxysilylalkyl)pyromellitic diimides 3–6
In sol–gel chemistry the hydrolysis and condensation behaviour of hydrolysable compounds are of high interest. There are many studies of pure molecular precursors or mixtures of various alkoxy silanes and other molecular compounds.38–44 The most important parameters which control the hydrolysis and condensation, such as pH, temperature, solvents and nature of catalysts as well as the amount of water which is available for the reaction were studied.45–50 Most spectroscopic investigations regarding alkoxy silanes rely on 29Si NMR studies.40,41,51–541H NMR spectroscopy52,55 was less frequently used. Investigations which mainly use FT-IR spectroscopy38,39,42 as analysis method have also been published.Especially 29Si NMR spectroscopy allows us to examine the chemical environments of the individual silicon atoms and to analyse them. The most frequently investigated sol–gel precursors are of the type RxSi(OR)3−x where R is an alkyl group which may be functionalized and OR stands for methoxy or ethoxy groups.
To describe the degree of hydrolysis and condensation of alkoxy silanes the well-known M, D, T and Q notation is used.56 In Scheme 5 a T group is shown schematically with its possibilities of hydrolysis and condensation states. The subscript number indicates the number of silanol groups. The superscript number indicates how far the condensation has progressed.
 | ||
| Scheme 5 Assignment of notation for hydrolyzed T-groups (T00–T03) and condensed/hydrolyzed T-groups (T12–T30) to molecular structures. | ||
The hydrolysis and condensation behaviour of imides 4–6 were examined with 1H and 29Si NMR spectroscopy. The reactions were performed in d6-DMSO. Aqueous HCl was used for catalysis to provide an acidic medium. The reaction has been monitored for 4 hours. In Fig. 4 the time dependent measurements of substance 4 are shown.
 | ||
| Fig. 4 Hydrolysis of 4 observed by 29Si NMR spectroscopy (solvent: d6-DMSO, for further details see experimental part). | ||
The 29Si NMR signal for compound 4 is located at a chemical shift of −46.08 ppm. After 25 minutes signals for T01 (−44.58 ppm), T02 (−43.60 ppm) and T03 (−42.87 ppm) appear downfield shifted from the original signal of 4. The structure assignments to these signals are shown in Scheme 5. T00 completely disappeared after about 50 minutes. Simultaneous to the hydrolysis products, upfield shifted signals occur for the condensation products T10 (−52.58 ppm) and T11 (−51.60 ppm). T12 (−50.53 ppm) appears first after 50 minutes. The signal intensity for hydrolysis products T01–T03 has a maximum intensity after 20 min. The increase in the signal intensity with reaction time is nearly similar for T02 and T03. Even after 4 hours both can be noticed but with very low intensity. T01 disappears after 100 minutes. Considering the signals assigned to the condensation products it can be observed that the relatively broad bands consist of three signals. This splitting can be explained with the fact that for example a T01 species can condensate with a T01, T02 or a T03 group. Therefore three possibilities exist for a T01 group to form condensation products. The options for condensation products just mentioned are shown in Scheme 6. Similar to T10 various condensation products can occur for T11 and T12.
 | ||
| Scheme 6 Possibilities for T01 group to form condensation products and signal assignment. | ||
For compound 5 the time dependent spectra of the hydrolysis-condensation reactions are shown in Fig. 5. The initial 29Si NMR signal for 5 appears at −48.6 ppm. This signal is shifted 2.52 ppm upfield compared to the signal of compound 4. The signals for hydrolysis products are as observed for 4 downfield shifted (T01 = − 52.58 ppm, T02 = − 51.60 ppm and T03 = − 50.53 ppm). Both compounds 4 and 5 show similar shifts from the initial signal to T01 of the hydrolysis products. For compound 4 the signal appears 1.5 ppm downfield and for 5 1.44 ppm. From T01 to T02 the shift is 1 ppm in both cases. Except from T02 to T03 the different shifts vary more. In the case of 4 the value is 0.72 ppm and for 5 0.44 ppm. The formation of the hydrolysis product T02 and T03 seems to be preferred in contrast to T01 which is not detectable over the measured time. Hydrolysis products are still detectable even after four hours. It is particularly noticeable that the intensity of T02 and T03 is decreasing over the time but nearly no condensation products occur. The signal for T11 appears at a chemical shift of −55.14 ppm and in the case of T12 at 54.22 ppm.
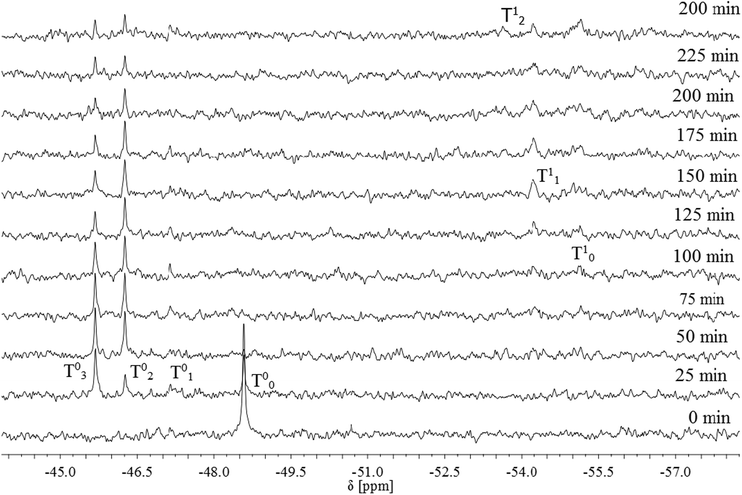 | ||
| Fig. 5 Hydrolysis of 5 as observed by 29Si NMR spectroscopy (solvent: d6-DMSO, for further details see experimental part). | ||
The 29Si NMR signal for bis(trialkoxyalkyl)imid 6 appears at 56.87 ppm (Fig. 6). After 25 min all hydrolysis products and condensation products are formed. In contrast to 4 and 5 where the shift between T00 and T01 is about 1.5 ppm downfield, the signal is observed at 2.06 ppm. Almost all hydrolysis products disappeared after 4 hours due to ongoing condensation reactions. The preferred hydrolysis products are T01 and T02. The latter shows the highest signal intensity during hydrolysis. Considering the signal for condensation grades T11 and T10 it can be recognized that these two are the major condensation products. The signals for T12 is very low over the whole investigated time but not absent. As mentioned for 4 (Scheme 6) the different kinds of hydrolysis products of T10 are also found for compound 6.
 | ||
| Fig. 6 Hydrolysis of 6 observed by 29Si NMR spectroscopy (solvent: d6-DMSO, further details see experimental part). | ||
As already pointed out above, there are a few studies which make use of 1H NMR spectroscopy to study the hydrolysis and condensation of alkoxy silanes. In this work, the hydrolysis of 3 was additionally examined by 1H NMR measurements. As in the previous experimental procedure d6-DMSO was used as solvent and 0.001 N HCl as catalyst. The amount of water was calculated that a maximum hydrolysis of 50% can occur (Rw = 2). The obtained result is shown in Fig. 7.
 | ||
| Fig. 7 Hydrolysis of 3 observed by 1H NMR spectroscopy (solvent: d6-DMSO, further details see experimental part). | ||
The spectrum marked with 0 min is the spectrum of pure 3 in d6-DMSO. The important signals for the reaction progress are located around 0.5 ppm, 3.2 ppm, 4.1 ppm and in the range of 5.8 to 6.8 ppm. In the following spectrum (10 min) the signal for the added water is located at 3.3 ppm. After just ten minutes of reaction time, structural changes can be detected. The signal for the protons of the methyl group of methanol with a chemical shift of 3.17 ppm appears. The protons of the hydroxyl groups can be detected first after 20 min reaction time at 4.14 ppm. During the hydrolysis progress an increase of both methanol signals can be observed while the area for the methoxy group is decreasing. Moreover, there is an alteration of the signals for the protons at the alpha carbon atoms. In addition to the signal of compound 3 at 0.63 ppm appears a new downfield shifted signal at 0.52 ppm. The intensity increases with ongoing reaction time. This signal belongs to the trimethoxysilyl group which is singly hydrolysed (T01). With increasing reaction time two additional signals appear further downfield. The signal at 0.44 ppm is assigned to T02 species and the signal for the protons on the alpha carbon of a completely hydrolysed silicon atom appears at 0.38 ppm. After about 10 minutes, three new signals appear. These are located at 6.78 ppm (T01), 6.32 ppm (T02) and 5.92 ppm (T03) and can be assigned to the hydroxyl groups of the different degrees of hydrolysis.
After 60 min 50% hydrolysis with regard to the Hα signal occurred. With prolonged reaction time the Hα signal and the signals 6.78 ppm and 5.92 ppm (T01–T03) should disappear due to complete hydrolysis and condensation, accompanied by an increase in the signal caused by liberated water. The other peaks which were assigned to hydrolysis products raised from the Hα signal show decreasing intensities.
All compounds formed transparent gels after a few days. No precipitation was observed. This observation encouraged us to prepare coatings and films using the novel hybrid precursors 3–6.
Film preparation of bis(trialkoxysilylalkyl)pyromellitic diimides 3–6
The precursors 3–6 can be used for producing flexible and transparent hybrid materials. Therefore, the ability of film formation of 4–6 was examined. As already mentioned common polyimides are not soluble in ethanol. In contrast to that the precursors 4–6 showed good solubility in ethanol. Thus a 20 w% solution of each compound in ethanol was used for all coating experiments. The bis(trialkoxysilylalkyl)pyromellitic diimides form films simply by heating up above 200 °C due to self-condensation. However, films which were produced this way had no smooth surface and showed many cracks. To optimize this process the bisimides were prehydrolyzed with hydrochloric acid. For compound 3 0.1 N HCl was used and the A/W was one. At least 120 °C were required to obtain solid films. A maximum film thickness of 0.9 mm was obtained. The obtained films were transparent, yellow or brown and flexible as shown in Fig. 8(a) and (b). | ||
| Fig. 8 (a) Films obtained from precursors 4–6, (b) the flexibility of the resulting films obtained from precursor 4, (c) the flexibility of the resulting films obtained from precursor 6. | ||
By using the same conditions as for precursor 4, 5 delivered also films (Fig. 8a). But in contrast to compound 4 the quality of the resulting film was low, i.e. the obtained films contained blisters, which caused swelling of the film and a decreased flexibility was observed. To solve this problem water and HCl content was varied. Decreasing water and/or HCl content did not yield any films. Only raising the HCl concentration from 0.1 to a 1 N HCl gave better results.
In contrast to compound 4 and 5 bisimid 6 formed no solid film by using 0.1 N HCl and an A/W ratio of one at 140 °C. A temperature of at least 200 °C was required to get solid layers with a non-sticky surface. At temperatures below 200 °C the system remained liquid. At room temperature the hybrid polymer formed from precursor 6 remained liquid but the viscosity raised. Solid films were obtained at a temperature of 140 °C using 1 N HCl and an A/W of one. These films showed no defects. They are transparent and flexible as shown in Fig. 8(a) and (c). In contrast to the obtained films from precursor 4 the maximum thickness was 0.2 mm. Attempts to obtain films with higher thicknesses were not successful. During the curing process these layers started to swell.
Further results of the ability of precursors 4–6 as coating material especially for the electrical insulation of copper wire will be published elsewhere.
Conclusion
Bis(trialkoxysilylalkyl)pyromellitic diimides 3–6 were synthesized according to the first step of the Gabriel-synthesis. Different silanes including γ-iodopropyltrimethoxysilane, γ-chloropropyltriethoxysilane, β-chloroethyltriethoxysilane and α-chloromethyltriethoxysilane and pyromellitic diimide were used as starting materials. The imide 1 was obtained from a reaction of pyromellitic dianhydride and hexamethyldisilazane (HMDS) followed by hydrolysis of the trimethylsilyl groups and thermal imidization. Since pyromellitic diimide is relatively unreactive towards trialkoxyhalogenoalkylsilanes the imide was converted to the respective dipotassium salt 2. The salt 2 and the different tri(alkoxy)halogenoalkylsilanes reacted in DMF to give the respective bis(trialkoxysilylalkyl)pyromellitic diimides 3–6 in very good yields. The desired imide formed most rapidly using γ-iodopropyltrimethoxysilane. The reactions with the chlorinated compounds proceeded significantly slower.All obtained substances were characterized using FT-IR spectroscopy, 1H, 13C and 29Si NMR spectroscopy as well as elemental analysis. The crystal structures of 3 and 6 were determined by single crystal X-ray diffraction. Both compounds exhibit disordered alkoxy substituents, nevertheless indicating the expected molecular structures with typical bond lengths and angles.
Finally, the hydrolysis and condensation behavior of the imides were studied using 29Si and 1H NMR spectroscopy. The 29Si NMR spectra indicate hydrolysis signals shifted downfield compared to the respective imide, while signals due to condensation products are shifted upfield. In all investigated cases within four hours only singly condensated products could be observed. These results provide a basis for the use of the title precursor molecules for the preparation of novel hybrid materials. They were applied for the formation of transparent and flexible hybrid films with a thickness up to 900 μm. Furthermore, we obtained electrically insulating coatings on copper wires. Details on these results will be published elsewhere.
Experimental part
Reagents and solvents
All reagents were used as received. All solvents were dried using standard methods. Pyromellitic dianhydride (PMDA) was purchased from Alfa Aesar. Hexamethyldisilazane (HMDS) and all silanes were obtained from ABCR. Acetone, dimethylsulfoxide (DMSO), dimethylformamide (DMF), hexane and deuterated DMSO (d6-DMSO) were purchased from Aldrich Chemical Co.Syntheses and spectroscopic investigations
Syntheses were carried out under an inert atmosphere of dry argon using standard Schlenk techniques. 1H, 13C and 29Si NMR spectra (solution) were recorded on a Bruker DPX 400 spectrometer. For kinetic investigations a scan number of 40 was chosen. FT-IR spectra were recorded on a Nicolet 380 using 32 scans. Elemental analyses were performed with an Elementar C, H, N analyser.Single-crystal X-ray structure determination
Data collection of 3 and 6 was performed on a STOE IPDS-2 diffractometer (image plate) equipped with a low temperature device (T = 200(2) K), with graphite-monochromatized MoKα radiation (λ = 0.71073 A°) using ω and φ scans. Reflections were corrected for background, Lorentz and polarization effects. Preliminary structure models were derived by application of direct methods57 and the structures were refined by full-matrix least-squares calculation based on F2 for all reflections using SHELXL.57 All hydrogen atoms were included in the models in calculated positions and were refined as constrained to the bonding atoms. Treatment of disorders for 3: the Si(OMe)3 group is disordered in two positions (89:11) including SADI restraints on the geometry, EXYZ and EADP restraints. For 6: three ethoxy groups are disordered in two positions (92:8; 71:29; 27:73) including SADI restraints on the geometry, EXYZ and EADP restraints. The highest left electron density peak close to Si1 cannot be assigned to a specific disorder.
Syntheses
1H NMR (d6-DMSO): δ (ppm): 11.82 (s, 2H); 8.09 (s, 2H).
13C NMR (d6-DMSO): δ (ppm): 167.46, 137.78, 117.09.
IR: ν (cm−1): 3197, 1771, 1712, 1694, 1306.
Anal. Calcd. for C10H4N2O4 C: 55.57, H: 1.87, N: 12.96%; found C: 55.64, H: 1.89, N: 12.92%.
1H NMR (d6-DMSO): δ (ppm): 7.16.
13C NMR (d6-DMSO): δ (ppm): 183.93, 142.10, 110.20.
IR: ν (cm−1): 1699, 1619, 1592, 1288.
Anal. Calcd. for K2C10H2N2O4 C: 41.08, H: 0.69, N: 9.59%; found C: 40.62, H: 0.74, N: 9.31%.
1H NMR (d6-DMSO): δ (ppm): 8.15 (s, 2H); 3.60 (t, 4H); 3.45 (s, 18H); 1.69 (m, 4H); 0.62 (m, 4H).
13C NMR (d6-DMSO): δ (ppm): 166.35, 137.07, 117.03, 50.03, 40.41, 21.15, 6.01.
29Si NMR (d6-DMSO): δ (ppm): −42.25.
IR: ν (cm−1): 1768, 1704, 1071.
Anal. Calcd. for C22H32N2O10Si2 C: 48.87, H: 5.97, N: 5.18%; found C: 48.92, H: 5.95, N: 5.23%.
1H NMR (d6-DMSO): δ (ppm): 8.15 (s, 2H); 3.75 (q, 12H); 3.61 (t, 4H); 1.70 (m, 4H); 1.14 (t, 18H); 0.62 (m, 4H).
13C NMR (d6-DMSO): δ (ppm): 166.36, 136.94, 177.07, 57.74, 40.45, 21.42, 18.15, 7.23.
29Si NMR (d6-DMSO): δ (ppm): −44.11.
IR: ν (cm−1): 1768, 1704, 1097, 1071.
Anal. Calcd. for C28H44N2O10Si2 C: 53.82, H: 7.10, N: 4.48%; found C: 53.62, H: 6.83, N: 4.40%.
1H NMR (d6-DMSO): δ (ppm): 8.13 (s, 2H); 3.77 (q, 12H); 3.69 (m, 4H); 1.15 (t, 18H); 1.08 (m, 4H).
13C NMR (d6-DMSO): δ (ppm): 165.78, 136.93, 116.86, 52.80, 33.25, 18.08, 10.28.
29Si NMR (d6-DMSO): δ (ppm): −47.94.
IR: ν (cm−1): 1768, 1698, 1100, 1071.
Anal. Calcd. for C26H40N2O10Si2 C: 52.33, H: 6.76, N: 4.69%; found C: 51.72, H: 6.50, N: 5.02%.
1H NMR (d6-DMSO): δ (ppm): 8.22 (s, 2H); 3.81 (q, 12H); 3.21 (s, 4H); 1.13 (t, 18H).
13C NMR (d6-DMSO): δ (ppm): 165.79, 136.95, 116.94, 58.30, 23.35, 17.94.
29Si NMR (d6-DMSO): δ (ppm): −53.82.
IR: ν (cm−1): 1765, 1712, 1065.
Anal. Calcd. for C28H44N2O10Si2 C: 41.08, H: 0.69, N: 9.59%; found C: 40.62, H: 0.74, N: 9.31%.
Film preparation
Acknowledgements
We would like to thank Dipl.-Chem. Benjamin Gutschank (University of Essen-Duisburg) for his help in measuring NMR spectra and Torsten Pieper (Westphalian University of Applied Sciences) for his administrative help. This project is supported by a grant (FKZ1752X08) from the FH3 BMBF program in the context of cooperative project “Entwicklung einer nanoskaligen Kupferlackdrahtbeschichtung für den Einsatz in der Automobil- und Medizintechnik (NanoCuL)”.References
- C. E. Sroog, J. Polym. Sci., Part D: Macromol. Rev., 1976, 11, 161–208 Search PubMed.
- A. V. Reddy, J. Appl. Polym. Sci., 2000, 75, 1721–1727 Search PubMed.
- J. Preston and Y. Tropsha, Polym. Eng. Sci., 1994, 34, 305–307 Search PubMed.
- A. B. Frazier, Trans. Ind. Electron., 1995, 42, 442–448 Search PubMed.
- G. Ragosta, M. Abbate, P. Musto and G. Scarinzi, J. Mater. Sci., 2012, 47, 2637–2647 Search PubMed.
- S. Mehdipour-Ataei and N. Bahri-Laleh, Iran. Polym. J., 2008, 17, 95–124 Search PubMed.
- D. Scola and J. H. Vontell, Polym. Compos., 1988, 9, 443–452 Search PubMed.
- N. Miyadera, T. Kuroda, T. Takahashi, R. Yamamoto, M. Yamaguchi, S. Yagi and S. Koibuchi, Mol. Cryst. Liq. Cryst., 2003, 406, 39–49 Search PubMed.
- C. Calebrese, L. Hui, L. S. Schadler and J. K. Nelson, IEEE Trans. Dielectr. Electr. Insul., 2011, 18, 938–945 Search PubMed.
- M. Mesaki, Y. Tatematsu and H. Goda, Furukawa Rev., 2002, 22, 1–4 Search PubMed.
- P. M. Budd and N. B. McKeown, Polym. Chem., 2010, 1, 63–68 RSC.
- S. V. Vinogradovja, S. Vygodskijv, V. Korsak and T. N. Spirina, Acta Polym., 1979, 30, 3–14 Search PubMed.
- D. J. Liaw and B. Y. Liaw, Chem. Mater., 1998, 10, 734–739 Search PubMed.
- K. Choi, J. Junga, H. Kim, B. Sohn, W. Zin and M. Ree, Polymer, 2004, 45, 1517–1524 Search PubMed.
- L. Mascia and A. Kioul, Polymer, 1995, 36, 3649–3659 CrossRef CAS.
- G. Ragosta and P. Musto, Polym. Lett., 2009, 3, 413–428 Search PubMed.
- S. M. Alam, T. Kawauchi and T. Takeichi, High Perform. Polym., 2010, 22, 742–760 Search PubMed.
- M. A. Wahab, II Kim, W.-J. Cho and C.-S. Ha, Mol. Cryst. Liq. Cryst., 2004, 417, 127–134 Search PubMed.
- S. Pandey and S. B. Mishra, J. Sol–Gel. Sci. Technol., 2011, 59, 73–94 Search PubMed.
- J. H. Kang, K. Cho and C. E. Park, Polymer, 2001, 42, 2513–2520 Search PubMed.
- G. Kickelbeck, Hybrid Materials: Synthesis, Characterization, and Applications, Wiley-VCH, 2007 Search PubMed.
- J. H. Wengrovius, V. M. Powell and J. L. Webb, J. Org. Chem., 1994, 59, 2813–2817 Search PubMed.
- O. Dautel, G. Wantz, R. Almairac, D. Flot, L. Hirsch, J.-P. Lere-Porte, J.-P. Parneix, F. Serein-Spirau, L. Vignau and J. J. E. Moreau, J. Am. Chem. Soc., 2006, 128, 4892–4901 CrossRef CAS.
- S. T. Hobson and K. J. Shea, Chem. Mater., 1997, 9, 616–623 CrossRef CAS.
- L. Bes, A. Rousseau, B. Boutevin, R. Mercier and B. Sillion, Macromol. Chem. Phys., 2001, 202, 933–942 Search PubMed.
- C. L. Homrighausen, B. J. Kennedy and E. J. Schutte, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4922–4932 Search PubMed.
- L. Bes, A. Rousseau, B. Boutevin and R. Mercier, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2602–2619 Search PubMed.
- M. S. Gibson and R. W. Bradshawi, Angew. Chem., 1968, 1, 986–996 Search PubMed.
- K. Soai, A. Ookawa and K. Kato, Bull. Chem. Soc. Jpn., 1982, 55, 1671–1672 CAS.
- H. Meyer and K. Steiner, Monatsh. Chem., 1913, 35, 391–405 Search PubMed.
- C. Chiriac, M. Nechifor and F. Tanasa, Rev. Roum. Chim., 2007, 52, 883–886 Search PubMed.
- T. Seckin, I. Özdemir and S. Köytepe, J. Inorg. Organomet.Polym. Mater., 2009, 19, 143–151 CrossRef CAS.
- I. V. Bulgarovskaya, L. A. Novakovskaya, Yu. G. Fedorov and Z. V. Zvonkova, Kristallografiya, 1976, 21, 515–519 Search PubMed.
- K. Kacprzak, Synth. Commun., 2003, 33, 1499–1507 Search PubMed.
- Y. Hase, Spectrosc. Lett., 1979, 12, 75–81 Search PubMed.
- I. Sideridou-Karayannidou and G. P. Karayannidis, J. Macromol. Sci., Chem., 1987, 24, 689–700 Search PubMed.
- Crystal data for 3: C22H32N2O10Si2, M = 540.68, monoclinic, a = 7.7341(6) Å, b = 8.1924(4) Å, c = 20.4587(14) Å, β = 98.448(6)°, V = 1282.22(15) Å3, T = 200(2) K, space group P21/n, Z = 2, μ = 0.196 mm−1, 11991 reflections measured, 2258 independent reflections (Rint = 0.0604), 194 parameters, 23 restraints, R1 (I > 2σ(I)) = 0.0403, wR(F2) (I > 2σ(I)) = 0.1053, R1 (all data) = 0.0468, wR(F2) (all data) = 0.1090, S = 1.085. CCDC 883128. Crystal data for 6: C24H36N2O10Si2, M = 568.73, monoclinic, a = 14.2971(4) Å, b = 7.7793(3) Å, c = 25.6231(7) Å, β = 99.546(2)°, V = 2810.38(15) Å3, T = 200(2) K, space group P21/c, Z = 4, μ = 0.183 mm−1, 36889 reflections measured, 5229 independent reflections (Rint = 0.1208), 374 parameters, 7 restraints, R1 (I > 2σ(I)) = 0.0577, wR(F2) (I > 2σ(I)) = 0.1557, R1 (all data) = 0.0634, wR(F2) (all data) = 0.1604, S = 1.092. CCDC 883129.
- G. De, B. Karmakar and D. Ganguli, J. Mater. Chem., 2000, 10, 2289–2293 RSC.
- B. Orel, R. Jese, A. Vilcnik and U. L. Stanger, J. Sol–Gel Sci. Technol., 2005, 34, 251–265 CrossRef CAS.
- Y. Sugahara, S. Okada, S. Sato, K. Kuroda and C. Kato, J. Non-Cryst. Solids, 1994, 21–28 Search PubMed.
- Y. Sugahara, S. Okada, S. Sato, K. Kuroda and C. Kato, J. Non-Cryst. Solids, 1992, 25–34 Search PubMed.
- R. Pena-Alonso, F. Rubio, J. Rubio and J. L. Oteo, J. Mater. Sci., 2007, 42, 595–603 CrossRef CAS.
- K. J. Shea and D. A. Loy, Acc. Chem. Res., 2001, 34, 707–716 CrossRef CAS.
- H. Saito, Y. Nishio, M. Kobayashi and Y. Sugahara, J. Sol–Gel Sci. Technol., 2011, 57, 51–56 Search PubMed.
- Y. Xu, X. Sun, D. Wu, Y. Sun, Y. Yang, H. Yuan, F. Deng and Z. Wu, J. Solution Chem., 2007, 36, 327–344 Search PubMed.
- M. Asomoza, M. P. Dominguez, S. Solis, V. H. Lara, P. Bosch and T. Lopez, Mater. Lett., 1998, 36, 249–253 Search PubMed.
- I. Artaki, T. W. Zerda and J. Jonas, Mater. Lett., 1985, 3, 493–496 CrossRef CAS.
- I. Artaki, T. W. Zerda and J. Jonas, Mater. Lett., 1986, 81, 381–395 Search PubMed.
- A. H. Boonstra, T. N. M. Bernards and J. J. T. Smits, J. Non-Cryst. Solids, 1989, 109, 141–152 Search PubMed.
- A. H. Boonstra and J. M. E. Baken, J. Non-Cryst. Solids, 1990, 122, 171–182 CrossRef CAS.
- M. Mazur, V. Mlynarik, M. Valko and P. Pelikan, Appl. Magn. Reson, 2000, 18, 187–197 Search PubMed.
- D. W. Schaefer, J. Sol–Gel Sci. Technol., 2009, 51, 23–31 Search PubMed.
- Y. Sugahara, T. Inouea and K. Kuroda, J. Mater. Chem., 1997, 7, 53–59 RSC.
- S. A. Myers, R. A. Assink, D. A. Loy and K. J. Shea, J. Chem. Soc., Perkin Trans., 2000, 2, 545–549 Search PubMed.
- Y. Dubitsky, A. Zaopo, G. Zannoni and L. Zetta, Mater. Chem. Phys., 2000, 64, 45–53 Search PubMed.
- B. D. Ratner, Biomaterials Science: An Introduction to Materials in Medicine, Elsevier, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 883128-883129. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2nj40538e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2013 |