Self-assembly of graphene oxide on the surface of aluminum foil
Qingye
Liu
a,
Meng
He
a,
Xiaojuan
Xu
*a,
Lina
Zhang
a and
Junping
Yu
b
aDepartment of Chemistry, Wuhan University, Wuhan 430072, P. R. China. E-mail: xuxj626@263.net; Fax: +86 27 68754067; Tel: +86 27 87216311
bWuhan Institute of Virology, Chinese Academy of Science, Wuhan 430071, P. R. China
First published on 18th September 2012
Abstract
We have demonstrated a special phenomenon, that the self-assembly of graphene oxide (GO) occurred on the matte surface of Al foil. Paper-like GO and GO composite films, including GO–polysaccharide and GO–poly(vinyl alcohol), were formed through water/air and water/Al interfacial interactions and the hydrophobicity of GO which is partially reduced in the presence of the Al foil. GO was first confirmed to be partially reduced on the surface of Al foil by wide angle X-ray diffraction, X-ray photoelectron spectroscopy, and Raman spectroscopy. Additionally, a three-dimensional (3D) macrostructural GO hydrogel composed of GO sheets in a random order was observed at the interface between the GO aqueous solution and Al foil. The mechanism through which the hydrogel formed was attributed to the initial absorption of the GO sheets on the surface of the Al foil through the liquid/solid interfacial and hydrogen-bonding interactions at the liquid/solid interface, and then the self-assembly though π–π stacking, Al3+ cross-linking, and the hydrophobicity of the partially reduced GO. We have described a very simple and mild method to prepare GO-based thin films and 3D-macroscopic hydrogels with partially reduced GO.
1. Introduction
It is well known that graphene possesses excellent physical and chemical properties, such as high electronic1 and thermal conductivity,2 extraordinary mechanical strength,3 a large specific surface area4 and so forth, leading to its many applications, including biosensors,5 electrochemical energy storage,6 solar cells,7 and field emission devices.8 As an important precursor of graphene,9,10 graphene oxide (GO) has attracted increasing attention in recent years11–15 due to its hydrophobic basal plane and a great deal of hydrophilic oxygen-containing groups, including epoxy, hydroxyl, carboxyl and carbonyl groups, which lead to its amphiphilic properties and thus its chemical modification and strong ability of self-assembly.14,16,17 Great efforts to obtain graphene-based composites in solution and the incorporation into hosts have also yielded promising results by utilizing its special properties. For example, the well-controlled structure of materials from GO aqueous solution, such as layered free-standing paper-like materials and macroscopic three-dimensional (3D) hydrogels, have been well developed. These materials, with well-defined structures, are of great importance for achieving high-performance applications in various technological fields such as optoelectronics, energy storage and catalysis, as indicated by Xu and Shi.18Many techniques have been developed to produce macroscopic GO-based functional materials with such structures, including the Langmuir–Blodgett technique to prepare monolayer films,14 layer-by-layer deposition,19–21 and vacuum-assisted evaporation-induced self-assembly,22–26 to prepare paper-like films, and also template-guided assembly and hydrothermal processes to prepare the 3D structure of GO hydrogels27 Recently, another method to prepare the paper-like materials has been successfully developed through solvent evaporation at the liquid/air interface.28 The self-assembly of GO was carried out mostly in solution or at the interfaces. In particular, self-assembly at interfaces to prepare GO-based materials from GO solutions has attracted the attention of many researchers, and consequently, most of the investigations have been focused on the liquid/air or liquid/liquid interfacial interactions. By contrast, the GO thin film or macroscopic hydrogel formation at the liquid/solid interface were seldom reported, apart from one very recent publication which demonstrated that a hydrogel-like GO-based macrostructure formed on the surface of hydrophilic porous anodic aluminium oxide (AAO).29 Moreover, the popularly accepted method of vacuum-assisted evaporation-induced self-assembly to fabricate the film by vacuum filtration usually takes 12 hours or more. Therefore, in this paper, we aim to propose an easy, less time and energy-consuming self-assembly method to prepare paper-like GO-based thin films and macroscopic hydrogels at the liquid/solid interface with Al foil as the solid surface under mild conditions. More interestingly, partial reduction of GO was synchronously observed at the liquid/solid interface in our method. Namely, we can endow the material with new functions, such as conductivity. Additionally, we mixed the GO solution with the synthetic polymer poly(vinyl alcohol) (PVA) and the natural polymer of the beta-glucan lentinan (LNT), which was extracted from mushrooms named Lentinus edodes, respectively, to successfully prepare GO–PVA and GO–LNT films via the same method. To the best of our knowledge, no such results have yet been reported on this interesting phenomenon of the self-assembly of GO and GO–polymer composites on Al foil, which may provide an ideal platform for the controlled formation of GO-based functional materials and enlarge the applications in the electronic field.
2. Experimental section
2.1. Samples
GO was prepared through a modified Hummers method.30,31 In brief, the graphite powder was first oxidized into graphite oxide using KMnO4/H2SO4, and then the graphite oxide was exfoliated into GO sheets by ultrasonication in water. The pH of the as-obtained yellow-brown aqueous of GO (5.0 mg mL−1) was evaluated to be 2.1 by a pH-meter (BEBCH/PHS-25, ±0.01).LNT, a natural neutral beta-glucan, was isolated from the fruiting bodies of Lentinus edodes cultivated in the Fujian province of China according to the previously published work.32 PVA was a commercial product with the repeating unit number of 1750 ± 50.
The GO/Al film was prepared by coating the uniform GO solution (5.0 mg mL−1) on the matte surface of Al foil, and then subjected to 50–60 °C in an oven for drying. The GO–LNT and GO–PVA films were obtained by placing the uniform mixtures on the surface of the Al foil for drying in oven. All the test film samples were obtained by direct scraping from the surface of the Al foil without further treatment. However, in order to avoid the effect of trace Al, the films used in the performance of electrical conductivity were treated in a diluted HCl solution. The Al foil will be dissolved, and the films can be obtained by rinsing them in water several times to remove the Al ions, followed by lyophilization. The GO/Al hydrogel was formed by immersing the Al foil in the above-prepared GO solution for 12 h, and the hydrogel, without mobility, was then observed at the GO/Al interface. The dry GO/Al hydrogel was obtained through lyophilization.
2.2. Characterization
Atomic force microscopy (AFM) tests were carried out using a PicoScan 2500 PicoSPM II Controller (PicoPlus, Molecular Imaging, Tempe, AZ) with a silicon probe of k = 40 N m−1 and a 300 kHz resonant frequency. The GO was exfoliated into monolayered sheets in water by ultrasonication (200 W) at room temperature for 2 h, and a 10 μL drop with concentration of 0.02 mg mL−1 was deposited onto the freshly cleaved mica and allowed to dry in air at room temperature in a small covered Petri dish prior to imaging with AFM. The instrument was operated in a tapping mode, and the measurements were performed in air at ambient pressure and humidity.Transmission electron microscopy (TEM) images were acquired using a JEM-2100 transmission electron microscope (JEOL Ltd., Japan) operated at 200 kV. The specimens for TEM were prepared by placing the GO solution (∼0.02 mg mL−1) on the carbon-coated copper grids, with the coating repeated 3 times, and then allowed to dry in air for two days at room temperature.
Scanning electron microscopy (SEM) measurements of the GO/Al films, GO–LNT/Al, GO–PVA/Al films, GO/Al hygrogel and Al foil were performed on a Sirion 200 field emission scanning electron microscope (FEI Co., Eindhoven, The Netherlands) at an accelerating voltage of 12 kV.
Wide angle X-ray diffraction (WAXD) measurements of graphite, GO, GO/Al, GO–PVA and GO–LNT films were carried out on a WAXD diffractometer (D8-Advance, Bruker, USA). The patterns with Cu Kα radiation (λ = 0.15406 nm) at 40 kV and 30 mA were recorded in the region of 2θ from 5° to 45°.
X-Ray photoelectron spectroscopic (XPS) measurements of the GO, GO/Al films and hydrogel were performed on an X-ray photoelectron spectroscopy (XSAM800, Kratos, UK) using a monochromated Mg Kα (1253.6 eV) source at 16 mA × 12 kV.
Raman measurements were performed on a Confocal Raman microspectrometer (RM-1000, Renishaw, England) with an Ar+ excitation laser beam with a wavelength of 514.5 nm. The powders of the GO and GO/Al films were placed on a clean SiO2/Si substrate with a scanning range from 500 to 2000 cm−1.
Thermogravimetric analysis (TGA) tests of the GO, GO–LNT/Al, GO–/Al films, LNT and PVA were performed on a TG instrument (Perkin–Elmer Co., USA) in an atmosphere of nitrogen at a heating rate of 10 °C min−1 from 30 to 700 °C.
Dynamic frequency sweep measurements of the storage modulus (G′) and loss modulus (G′′) for GO and the AO/Al hydrogel were conducted at a strain of 10% for sample solutions. The GO/Al hydrogel was formed by immersing the Al foil in the prepared GO aqueous for a storage time of 5 days. The rheometer used was an ARES-RFS III strain-controlled type (TA Instruments, USA), equipped with two force transducers, allowing the torque to be in the range of 0.004 to 1000 g cm and a double-concentric cylinder geometry consisting of a rotating outer cylinder (cup) and an inner cylinder (bob). Temperature control was achieved by using a circulating fluid bath (a julabo FS18 cooling/heating bath). The sample solutions were loaded into the outer cup and allowed to equilibrate for 20 min in order to achieve temperature equilibration and stress relaxation.
3. Results and discussion
3.1. Paper-like self-assembly of GO sheets on the matte surface of Al foil
The typical AFM and TEM images (Fig. 1) of GO by drying GO/water solutions on the surface of solid substrates mica and carbon-coated copper grids were obtained to confirm the topography and the size of the monolayered GO sheet. Although some overlapped layers existed, as shown in the AFM image (Fig. 1a), the thickness could still be evaluated to be about 1.0 nm from the height profile, indicative of the formation of a single layered GO sheet in water.11 The TEM image (Fig. 1b) shows the production of the GO film with a large area and some wrinkle regions.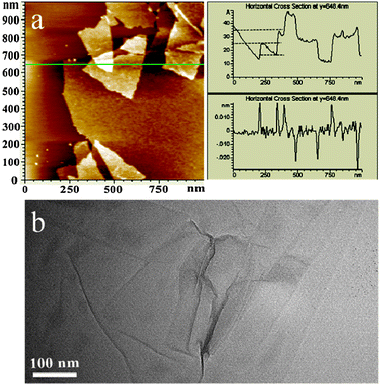 | ||
| Fig. 1 AFM image of GO sheets on a mica surface (left) and the height profile of the AFM image (right) (a). HRTEM image of the GO sheets (b). | ||
The prepared GO water solution (5.0 mg mL−1) was very stable without notable precipitation after a long storage time. By casting the GO solution on the matte surface of the Al foil, and then subjecting it to a mild temperature (40–60 °C) at ambient pressure for drying, a black thin film GO/Al (Fig. 2a,b) was thus formed. The SEM images of the cross-section of the GO/Al film (Fig. 2c,d) suggested that the GO sheets re-assembled into a layer-by-layer macroscopic structure on the Al foil. The effective-assembly could even be performed in concentrated solution, with a concentration of 10.0 mg mL−1, which is much higher than the reported concentration of 3.0 mg mL−1.28 However, the GO/Al film prepared at the lower concentration of 5.0 mg mL−1 looked in better order than the one prepared at the higher concentration. Therefore, we can control the ordered structure of the GO films by adjusting the concentration of GO. Moreover, the thickness and the area of the film could also be controlled by the volume of the used GO/water solution and its concentration.
 | ||
| Fig. 2 Photographs of the GO/Al film deposited on the matte surface of the Al foil after drying at 50–60 °C for 3 h (a), and the GO thin film peeled off from the Al foil (b). SEM images of the cross-sections of the GO/Al films with the GO concentration of 5.0 (c) and 10 mg mL−1 (d). | ||
GO-based polymer composite films have been prepared using layer-by-layer (LBL)19–21 and vacuum-assisted evaporation-induced self-assembly techniques.26 The LBL technique has been mostly applied for polyelectrolytes because the GO sheet has negative surface charges and they can thus interact with the cationic polymers through electrostatic interactions. While the vacuum-assisted filtration technique usually takes 12 h or more to fabricate the films. Based on the above, we aimed to verify the possibility of the formation of a GO-based composite with a layered structure on the surface of Al foil using a time and energy-saving method. We mixed GO aqueous solution with two neutral polymers LNT and PVA, respectively, and cast the uniform mixture on the matte surface of the Al foil. After drying for 3 h at 40–50 °C, the GO-based composites GO–LNT and GO–PVA were thus obtained. The surface of the resulting film with homogeneous color was smooth. SEM images of the cross-section of the films (Fig. 3a,b) revealed that the formed GO–polymer films also exhibited layer-by-layer macroscopic structure. Comparison of the cross-section of the pure GO/Al film (Fig. 2c) with the same GO concentration, the layered structures of the GO–LNT and GO–PVA films were not clearly demarcated, especially for GO–PVA, which showed a relatively smooth cross-section and indistinct dividing lines between the layers. This is due to stronger intermolecular interactions between GO and PVA than those between GO and LNT. This result also indicated that LNT and PVA were retained and intercalated in the intersheet gallery. Further evidence obtained from the XRD patterns also confirmed that LNT and PVA were really incorporated into the intersheet spacing of the GO. The diffraction peaks of GO in the composite films moved to a smaller value of 2θ compared with the pure GO film (Fig. 4a,b), indicating that the polymers were intercalated between adjacent graphene oxide layers resulting in larger intersheet spacings.26 The TGA results (Fig. 3c,d) illustrated that LNT and PVA obviously enhanced the thermal stability in contrast to the pure GO sheets, confirming the co-existence of the polymers in the composite papers.
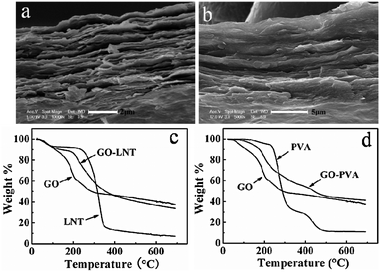 | ||
| Fig. 3 Cross-section SEM images of the (a) GO–LNT and (b) GO–PVA films formed on the matte surface of the Al foil; TGA curves of GO, LNT, PVA, GO–LNT and GO–PVA composites (c,d). The concentration of GO, LNT and PVA were 5.0, 6.6, and 5.0 mg mL−1, respectively. | ||
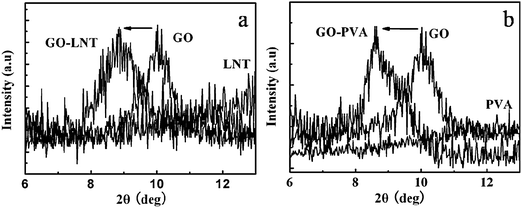 | ||
| Fig. 4 XRD patterns of GO, LNT, PVA, GO–LNT and GO–PVA composites films. | ||
3.2 Partially reduced GO on the surface of Al foil
Since the Al foil is composed of a reducing Al element, will it react with GO to produce a reduced GO? To confirm this, we first checked the electrical conductivity of the GO film. Very interestingly, different sides of the GO film showed different results; the reverse side (contacting with the Al foil) showed an electrical conductivity of 18.75 S m−1, evaluated by a 4-point probe method, while the up-side, far from the surface of the Al foil, showed large electrical resistance, which could not even be detected by the instrument. We thus deduced that the reverse side was partially reduced. As depicted in Fig. 5a, graphite, GO, and GO film show different XRD patterns. The peak of graphite at 2θ = 26.6° corresponds to a layer-to-layer distance (d-spacing) of about 0.335 nm. However, the sharp peak of GO at around 10.2° (d-spacing 0.865 nm) indicated damage of the regular crystalline structure of graphite due to oxidation, which is typical for a layered structure of GO nanosheets.31 For the GO/Al film, the peak at 2θ = 10.2° became weak, and a new broad and weak diffraction peak centered at around 23.4° (d-spacing 0.380 nm) appeared, which was closer to the typical diffraction peak of graphite (002).31 This newly occurring peak suggested that the GO was partially reduced by the Al foil. The d-spacing of the GO/Al film was slightly greater than the pristine natural graphite, indicating the presence of a small amount of residual oxygen-containing functional groups or other structural defects. The partial reduction of GO by Al foil was further confirmed by XPS. Fig. 5b, c shows the C 1s XPS spectra of GO and GO/Al film. Four different peaks of GO centered at 284.1, 286.2, 287.6 and 289.6 eV, corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C–C in aromatic rings, C–O (epoxy and alkoxy), CO, and COOH groups, respectively.31 After partial reduction, the intensities of the peaks (peak 2, 3, and 4), which were assigned to oxygen functional groups, were much lower than those of GO, indicating that most of the oxygen functional groups were removed.33
C/C–C in aromatic rings, C–O (epoxy and alkoxy), CO, and COOH groups, respectively.31 After partial reduction, the intensities of the peaks (peak 2, 3, and 4), which were assigned to oxygen functional groups, were much lower than those of GO, indicating that most of the oxygen functional groups were removed.33
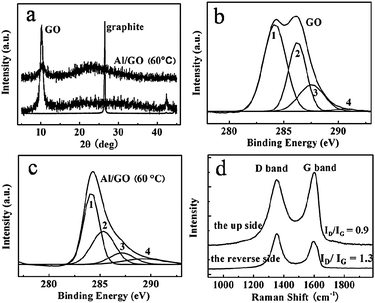 | ||
| Fig. 5 XRD patterns of graphite, GO, and the GO/Al film (a). XPS spectra of C 1s of GO (b), and the GO/Al film (c). The peaks 1, 2, 3, and 4 correspond to CC/C–C in aromatic rings, C–O (epoxy and alkoxy), CO, and COOH groups, respectively. (d) Raman spectra of the up and reverse side of the GO/Al film. The reverse side is the one directly contacting with the Al foil. | ||
Raman spectra was also taken to confirm the partial reduction of GO by the Al foil. As shown in Fig. 5d, the up-side of GO exhibited two intense peaks at 1356 and 1608 cm−1, corresponding to the D and G bands, respectively. By contrast, the D and G bands of the reverse side were red-shifted to 1351 and 1597 cm−1, and the ratio of ID/IG (1.3) was higher than that of the up side (ID/IG = 0.9), implying that the reduction of the reverse side of the GO film occurred due to the presence of Al. The variation of the peak position and relative intensities of the G and D bands revealed the changes of the electronic conjugation state.34,35 However, the exact mechanism of the reduction by the Al foil was not clearly understood in the present work. One possible reason might be related to the electron transfer from the Al foil to GO, because the standard reduction potential value of Al/Al3+ (−1.68 V, Al = Al3+ + 3e−)36,37 was higher than hydrazine (−1.16 V, N2H4 + 4OH− = N2 + 4H2O + 4e−).38 The partial destruction of the Al foil as shown in Fig. 6 confirmed the occurrence of the reduction.
 | ||
| Fig. 6 SEM images of the matte surface of the Al foil before (a), and after (b) being subjected to the GO membrane formation process at 50–60 °C for 3 h. | ||
Taken together, a simple, gentle, time and energy-saving means to prepare GO-based paper-like films with synchronous reduction on the surface of Al foil was developed, which shows an advantage of avoiding annealing treatment at high temperature in inert gas as reported in the literature.39 The ability for layered material formation on the surface of the Al foil is prominent. By casting the GO solution on the Al foil, the liquid/air interface and liquid/solid interface formed. It has been confirmed that GO is amphiphilic and surface active, the GO sheets were inclined to re-arrange at the water/air and water/Al interfaces, and aggregated into a continuous film with a layer-by-layer structure during heating in the present experiments.16,28,40 Therefore, the formation of a thin film on the Al foil during evaporation could be easily understood. Additionally, at the water/Al interface, the reducing Al partially reduced the oxygen-containing groups, leading to an increase in the hydrophobicity of GO. At the same time, Al was also transferred into Al3+ in the acidic medium of pH = 2.1 to produce H2 gas. Both the upward induction of gas flow and the hydrophobicity of partially reduced GO accelerated the migration of GO sheets to the water/air interface. In a word, reduction, the strong water/air and water/Al interfacial interactions led to the paper-like GO-based thin film formation on the surface of the Al foil.
3.3. Gel formation of GO in the presence of Al foil
A macroscopic GO hydrogel on the surface of the Al foil, through self-assembly of GO sheets, was also observed (Fig. 7b). By immersing the Al foil in a GO aqueous solution, with a concentration of 5.0 mg mL−1 (Fig. 7a), a GO hydrogel formed on the Al foil surface. Furthermore, there were more gels on the matte side than on the shiny one. The formed GO hydrogel in the vial lost mobility, and could stand in an upturned container test after 12 h, as depicted in Fig. 7b. A SEM image of the GO/Al hydrogel showed a 3D macrostructure network of overlapped GO sheets in a random manner (Fig. 7d). The dynamic rheological data in Fig. 7e confirmed that both the GO aqueous solution and GO/Al mixture exhibited gel characteristics with a weak dependence on the angular frequency of G′ and G′ > G′′ in the whole frequency range from 0.1–100 rad s−1. However, the G′ values of the GO/Al hydrogel were largely enhanced by roughly four times over those of the original GO solution. All the data demonstrated that the macroscopic GO hydrogel formed on the surface of the Al foil. Moreover, the GO hydrogel was also confirmed to be partially reduced by XPS (Fig. 8), similar to the results of the GO thin film shown in Fig. 5c.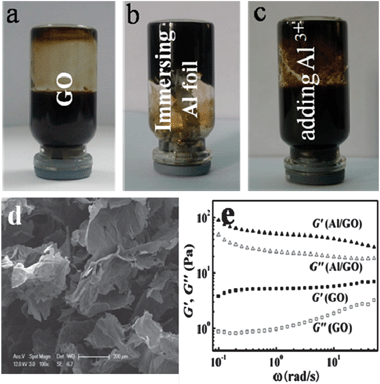 | ||
| Fig. 7 Digital photos of the GO aqueous solution (a), hydrogel formation by immersing the Al foil in GO aqueous solution for 12 h at 25 °C (b), and GO aqueous solution containing 50 mM AlCl3 standing for 12 h at 25 °C (c). The concentration of the GO solution mentioned above was 5.0 mg mL−1. (d) SEM image of GO/Al hydrogel. (e) Dynamic rheological behavior of GO solution (5.0 mg mL−1) and the GO/Al hydrogel. | ||
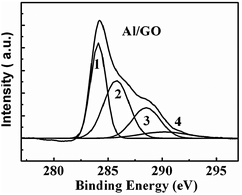 | ||
| Fig. 8 XPS spectrum of C 1s of the GO/Al hydrogel. The peaks 1, 2, 3, and 4 correspond to CC/C–C in aromatic rings, C–O (epoxy and alkoxy), CO, and COOH groups, respectively. | ||
As mentioned in the introduction, it has been found that there are strong liquid/solid interfacial interactions between the porous AAO and the GO aqueous solution, promoting a fast sedimentation of GO on the alumina surface and final GO hydrogel formation.29 In the present case, a liquid/solid (water/Al) interface also formed. Did the macroscopic hydrogel also form through liquid/solid interfacial interactions? What is the possible mechanism through which the GO hydrogel formed? Since the pH of GO in water was determined to be 2.1, some Al could thus be dissolved in the GO aqueous solution as Al3+. Additionally, GO could be partially reduced on the surface of the Al foil as evidenced in Fig. 8, the elementary substance of Al was also transferred into Al3+ through the electron transfer. Therefore, Al3+ could certainly exist in the solution. It has been supposed that the Al3+ can act as a cross-linker due to the interaction between the negative charges of GO and Al3+.37 To clarify the role of Al3+ in the GO gel formation, such a control experiment was also performed, where Al3+ (AlCl3) was directly added into GO aqueous solution. However, no remarkable hydrogel formed after standing for 12 h (Fig. 7c), suggesting that the gel formation of GO on the surface of the Al foil was not mainly determined by the cross-linker of Al3+. Yet, it is clearly seen that more GO hung on the wall of the vial than the pure GO aqueous solution (Fig. 7a,c), implying that Al3+ cross-linking also occurred. Besides the Al3+ cross-linking, what else contributed to the GO gel formation? As we know, GO sheets act like a surfactant, which can be adsorbed on the interfaces and lower the surface or interfacial tension, leading to its self-assembly on the Al surface.16 Moreover, some hydroxyl groups always exist on the surface of the Al foil (little oxidization on the Al surface), leading to the hydrogen-bonding interaction between hydroxyl on the Al foil and the oxygen-containing groups of the GO sheets.29 Then, the GO sheets self-assembled together through π–π stacking41,42 and Al3+ cross-linking.37 Meanwhile, some gel was also partially reduced by the Al foil (Fig. 8), resulting in an increase in the hydrophobicity of GO and thus promotion of the self-assembly of GO. Taken together, multiple interactions finally resulted in the gel formation of GO on the surface of the Al foil.
4. Conclusions
The self-assembly of GO could be easily carried out through the liquid/air and liquid/solid interfacial interactions in the presence of Al foil. The paper-like structure of GO materials could be readily prepared by casting the GO aqueous solution on the surface of the Al foil. Furthermore, the layered paper-like GO-based polymer composites were also obtained through flexible deposition. The mechanism through which the paper-like thin film formed was believed to be due to multiple factors, including the evaporation, upward gas (H2) flow, and the increasing hydrophobicity, resulting from GO which is partially reduced by the Al foil. Additionally, by immersing the Al foil in the GO aqueous solution, a GO gel formed with a random sheet distribution. Moreover, the GO gels were also partially reduced by the Al foil. The gel formation mechanism was also ascribed to multiple contributions, including liquid/solid interfacial, hydrogen-bonding, Al3+ cross-linking, and hydrophobicity interactions.Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation (grant 20874078), the Youth Technology Chenguang Project of Wuhan (200950431193), and the National Basic Research Program of China (973 Program, 2010CB732203).References
- X. Du, I. Skachko, A. Barker and E. Y. Andrei, Nat. Nanotechnol., 2008, 3, 491–495 CrossRef CAS.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS.
- H. C. Schniepp, J. L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud′homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS.
- Y. Wang, Y. Shao, D. W. Matson, J. Li and Y. Lin, ACS Nano, 2010, 4, 1790–1798 CrossRef CAS.
- W. Lv, D. M. Tang, Y. B. He, C. H. You, Z. Q. Shi, X. C. Chen, C. M. Chen, P. X. Hou, C. Liu and Q. H. Yang, ACS Nano, 2009, 3, 3730–3736 CrossRef CAS.
- N. Yang, J. Zhai, D. Wang, Y. Chen and L. Jiang, ACS Nano, 2010, 4, 887–894 CrossRef CAS.
- F. Xia, D. B. Farmer, Y. Lin and P. Avouris, Nano Lett., 2010, 10, 715–718 CrossRef CAS.
- K. P. Loh, Q. Bao, G. Eda and M. Chhowalla, Nat. Chem., 2010, 2, 1015–1024 CrossRef CAS.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS.
- K. P. Loh, Q. Bao, P. K. Ang and J. Yang, J. Mater. Chem., 2010, 20, 2277–2289 RSC.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- V. K. Singh, A. Shukla, M. K. Patra, L. Saini, R. K. Jani, S. R. Vadera and N. Kumar, Carbon, 2012, 50, 2202–2208 CrossRef CAS.
- J. Kim, L. J. Cote, F. Kim, W. Yuan, K. R. Shull and J. Huang, J. Am. Chem. Soc., 2010, 132, 8180–8186 CrossRef CAS.
- F. Kim, L. J. Cote and J. Huang, Adv. Mater., 2010, 22, 1954–1958 CrossRef CAS.
- Y. Xu and G. Shi, J. Mater. Chem., 2011, 21, 3311–3323 RSC.
- T. Cassagneau and J. H. Fendler, Adv. Mater., 1998, 10, 877–881 CrossRef CAS.
- T. Szabó, A. Szeri and I. Dékány, Carbon, 2005, 43, 87–94 CrossRef CAS.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. B. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457–460 CrossRef CAS.
- H. Chen, M. B. Müller, K. J. Gilmore, G. G. Wallace and D. Li, Adv. Mater., 2008, 20, 3557–3561 CrossRef CAS.
- X. Yang, L. Qiu, C. Cheng, Y. Wu, Z. F. Ma and D. Li, Angew. Chem., Int. Ed., 2011, 50, 7325–7328 CrossRef CAS.
- Y. Xu, H. Bai, G. Lu, C. Li and G. Shi, J. Am. Chem. Soc., 2008, 130, 5856–5857 CrossRef CAS.
- K. W. Putz, O. C. Compton, M. J. Palmeri, S. B. T. Nguyen and L. C. Brinson, Adv. Funct. Mater., 2010, 20, 3322–3329 CrossRef CAS.
- Y. Xu, Q. Wu, Y. Sun, H. Bai and G. Shi, ACS Nano, 2010, 4, 4324–4330 CrossRef CAS.
- C. Chen, Q. H. Yang, Y. Yang, W. Lv, Y. Wen, P. X. Hou, M. Wang and H. M. Cheng, Adv. Mater., 2009, 21, 3007–3011 CrossRef CAS.
- J. J. Shao, S. D. Wu, S. B. Zhang, W. Lv, F. Y. Su and Q. H. Yang, Chem. Commun., 2011, 47, 5771–5773 RSC.
- W. S. Hummers Jr. and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339–1339 CrossRef CAS.
- J. Zhang, H. Yang, G. Shen, P. Cheng and S. Guo, Chem. Commun., 2010, 46, 1112–1114 RSC.
- L. Zhang, X. Zhang, Q. Zhou, P. Zhang, M. Zhang and X. Li, Polym. J., 2001, 33, 317–321 Search PubMed.
- V. H. Pham, T. V. Cuong, T. D. Nguyen-Phan, H. D. Pham, E. J. Kim, S. H. Hur, E. W. Shin, S. Kim and J. S. Chung, Chem. Commun., 2010, 46, 4375–4377 RSC.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. B. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS.
- J. A. Dean, Lange’s Handbook of Chemistry, McGraw-Hill Book Company, New York, 15th edn, 1999, pp. 741–770 Search PubMed.
- Z. Fan, K. Wang, T. Wei, J. Yan, L. Song and B. Shao, Carbon, 2010, 48, 1686–1689 CrossRef CAS.
- B. L. Cushing, V. L. Kolesnichenko and C. J. O′Connor, Chem. Rev., 2004, 104, 3893–3946 CrossRef CAS.
- C. Vallés, J. David Núñez, A. M. Benito and W. K. Maser, Carbon, 2012, 50, 835–844 Search PubMed.
- Y. Li and Y. Wu, J. Am. Chem. Soc., 2009, 131, 5851–5857 CrossRef CAS.
- Z. Sui, X. Zhang, Y. Lei and Y. Luo, Carbon, 2011, 49, 4314–4321 CrossRef CAS.
- W. Chen and L. Yan, Nanoscale, 2011, 3, 3132–3137 RSC.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2013 |