Macrocellular Pd@ionic liquid@organo-Si(HIPE) heterogeneous catalysts and their use for Heck coupling reactions†
Nicolas Brunab, Peter Hesemannc, Guillaume Laurentd, Clément Sanchezd, Marc Birotb, Hervé Deleuze*b and Rénal Backov*a
aUniversité de Bordeaux, Centre de Recherche Paul Pascal, UPR 8641 CNRS, F-33600 Pessac, France. E-mail: backov@crpp-bordeaux.cnrs.fr
bUniversité de Bordeaux, Institut des Sciences Moléculaires, UMR 5255 CNRS - Univ. Bordeaux, F-33400 Talence, France. E-mail: h.deleuz@ism.u-bordeaux1.fr
cInstitut Charles Gerhardt de Montpellier, UMR 5253 CNRS - ENSCM - Univ. Montpellier 1-2, F-34000 Montpellier, France
dLaboratoire de Chimie de la Matière Condensée de Paris, UMR 7574 CNRS - ENSCP - Collège de France, F-75231 Paris, France
First published on 7th September 2012
Abstract
Supported ionic liquid phases (SILPs) within macrocellular silica-based foams are prepared by a simple impregnation in organic solvents. Thin ionic liquid layers with thicknesses of 6 to 12 nm were obtained. The SILs mobility has been evidenced through NMR solid state spectroscopy. Subsequently, palladium salts are efficiently trapped within the SILPs, while an in situ palladium hydrogenation allowed an efficient reduction and formation of 10 nm diameter palladium metal nanoparticles. These hybrid foams are used as heterogeneous macrocellular catalysts for the Heck coupling reaction of iodobenzene and cyclohexyl acrylate, where palladium leaching appears to be very low. Despite recyclability minimized through the entrapment of detrimental ammonium salts within the macroporous network during the reaction, competitive TONs and TOFs were reached, while separation of the products can be reached at ease, due to the fact that both palladium species and by-products are trapped within the monolithic foams.
Introduction
Due to environmental and ecological issues, optimization of chemical synthetic processes in laboratories and industry is essential.1 Beyond yield and selectivity, environmental considerations, such as catalyst recovery or volatile organic compounds (VOCs) substitution, have to be considered in detail. Thus, both design of original catalytic supports and eco-friendly alternatives to VOCs constitute attractive answers. In this context, ionic liquids (ILs) appear as a rising option to substitute VOCs and preserve catalytic entities from leaching.2 Ionic liquids, which are composed entirely of ions, can be distinguished from molten salts by a liquidus state below 100 °C combined with a relatively low viscosity. Particularly, quaternary ammonium and phosphonium salts exhibit, for instance, a stable wide electrochemical window, high ionic mobility, negligible vapour pressure, and excellent chemical and thermal stabilities,3 offering a broad range of applications, from electrochemistry to catalysis.4 However, economical aspects still restrict industrial uses.Therefore, supported ionic liquid phases (SILPs)5 onto porous hosts have recently received much attention as media for reuse of semi-heterogeneous catalysts, such as palladium compounds for carbon–carbon coupling reactions.6 In these systems, metallic complexes or nanoparticles can be easily stabilized within adsorbed ionic liquid phases strongly reducing catalyst leaching. Moreover, SILPs enable us to work with smaller amounts of costly ionic liquid than conventional homogeneous or multiphasic catalytic systems,4f potentially leading to large-scale reactors conception. In most of the works reported so far, SILPs are limited to thin films of less than 1 nm either grafted or simply adsorbed on the host surface.7 This typical thickness corresponds more closely to a monomolecular layer than to a real liquid supported phase. Furthermore, supports are most often mesoporous silica powders6a–c,8 or monolithic ionogels,6d,9 within which ionic liquids are encapsulated, limiting thereby the accessibility of catalysts. Consequently, the design of supported ionic liquid phases onto macrocellular monoliths would appear as a promising alternative to conventional heterogeneous catalysts in order to optimize accessibility and mass transport. To address this issue, our group has recently developed the design of macrocellular silica-based hybrid foams made from concentrated direct emulsion.10 These macrocellular materials, named Si(HIPE) (HIPE is an acronym for High Internal Phase Emulsion), can be easily functionalized to stabilize for instance palladium particles,11a,b europium complexes11c or crude enzymes.11d,e Nevertheless, in spite of efficient stabilisation, catalyst leaching cannot be totally avoided, leading to an inevitable loss of activity.11a
With the aim to circumvent this feature, we describe in the present contribution a facile route to obtain SILPs onto Si(HIPE) monoliths by impregnation under soft vacuum. This approach has allowed the facile immobilization of either Palladium salts or metallic nanoparticles, depending on the post-treatment applied, in the supported ionic liquid layer. Catalytic performances toward the Heck coupling reaction between iodobenzene and cyclohexylacrylate, according to the supported ionic liquid, and the encapsulated catalyst, are also discussed.
Experimental
Materials
Tetradecyltrimethylammonium bromide 98% (TTAB) and sodium hydride (NaH) were purchased from Alfa Aesar. Tetraethylorthosilicate 98% (TEOS), imidazole, (3-chloropropyl)triethoxysilane and palladium acetate (Pd(OAc)2) were purchased from Aldrich. Acetone, toluene, isopropanol, ether, and dodecane 99% were purchased from Rectapur. Tetrahydrofuran (THF) and hydrochloric acid 37% (HCl) were purchased from AnalaR Normapur. Ionic liquids 1-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide, hexafluorophosphate and tetrafluoroborate (labelled later in the text, respectively, as [Bmim]NTf2, [Bmim]PF6 and [Bmim]BF4) were all purchased from Solvionic SA. 3,4-Epoxycyclohexylmethyl-3,4-epoxycyclohexyl carboxylate (ERL 4221), dipropyleneglycoldiglycidylether (DER 736), nonenyl succinic anhydride (NSA) and dimethylaminoethanol (DMAE) were purchased from Sigma. Except opposite indication, reactants were used as received without further purification.Syntheses
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) to extract the oily phase. Drying of the materials for a week at room temperature was followed by a thermal treatment at 650 °C (heating rate of 2 °C min−1) for 6 h, with a 2 h-plateau at 200 °C to remove all organic compounds.
:1 v/v) to extract the oily phase. Drying of the materials for a week at room temperature was followed by a thermal treatment at 650 °C (heating rate of 2 °C min−1) for 6 h, with a 2 h-plateau at 200 °C to remove all organic compounds.Catalytic Heck coupling reactions
Catalytic tests were realized in 20 mL glass tubes with a side-armed frit. Typically, a piece of hybrid foam (100 mg) was firstly introduced in the reactor, closed with a septum. Then, triethylamine (11 mmol), diluted in octane (1.0 mL), was injected via a syringe. The reaction media was gently purged with argon for 10 min, and then immersed into a preheated oil bath at 136 °C. Then a mixture of iodobenzene (10 mmol), cyclohexyl acrylate (12 mmol) and dodecane (as a GC internal standard; 5 mmol), diluted in octane (3.0 mL), was quickly added. Stirring was carried out by octane boiling. Aliquots were periodically withdrawn from the reaction mixture and diluted with ice-cooled THF (1.0 mL) before GC analysis. Between each catalytic cycle, the pieces of monolith were profusely rinsed out with octane, then immersed for a few minutes in an aqueous NaOH solution (0.05 M, 10 mL), to remove any insoluble side-product (mainly triethylammonium iodide). Monolithic catalysts were finally dried at 80 °C in an oven.Instrumentation
Scanning electron microscopy (SEM) observations were performed with a Hitachi TM-1000 apparatus at 15 kV. The specimens were gold–palladium-coated in a vacuum evaporator prior to examination. Transmission electron microscopy (TEM) experiments were performed with a Jeol 2000 FX microscope (accelerating voltage of 200 kV). High Resolution TEM (HR-TEM) micrographs were obtained with a Jeol 2200 FS microscope. Ultramicrotomy were performed, using a Reichert-Jung Ultracut E. Typically, a piece of hybrid foam (20 mg) was previously immersed in 5.0 mL of ERL 4221 for 3 h. Then, a mixture of ERL 4221 (18 mL), DER 736 (14 mL), NSA (48 mL) and DMAE (0.6 mL) was prepared. Afterwards, 1.0 mL of this mixture was added to the impregnation solution. Then, polymerisation was performed at 70 °C for 10 h in an oven, and the impregnated sample was finally cut to get 70 nm thin slices.Intrusion/extrusion mercury measurements were performed using a Micromeritics Autopore IV 9500 porosimeter, this to reach the scaffolds macrocellular cells characteristics. Surface areas and pore characteristics on a mesoscale were obtained with a Micromeritics ASAP 2010 apparatus. Small angle X-ray scattering (SAXS) experiments were carried out with a 18 kW rotating anode X-ray source (Rigaku-200) with use of a Ge (111) crystal as a monochromator. The scattered radiation was collected on a two-dimensional detector (Imaging Plate system from Mar Research, Hamburg). The sample to detector distance was 500 mm.
FTIR spectra were obtained with a Nicolet 750 FTIR spectrometer. Solid-state NMR was carried out on a Bruker Avance III 300 operating at 300 MHz for 1H, 75 MHz for 13C and 60 MHz for 29Si. Samples were packed into a 7 mm zirconia rotor, put into a standard Bruker double-resonance probe and spun at 5 kHz. 13C Power Gated (PG) spectra were obtained using a 90° pulse of 5 μs, a relaxation delay (RD) of 10 s, 256 scans, and a low power decoupling of ν1H = 2.5 kHz during relaxation. 13C High Power DECoupling (HPDEC) spectra were acquired under the same conditions without decoupling during the relaxation. 13C cross polarization (CP) spectra used ν1H = ν1C = 50 kHz, RD = 1 s and 26624 scans. In all 13C experiments a decoupling of ν1H = 50 kHz was applied during the acquisition. The spectra were simulated with the DMFIT program.12 X-ray photoelectron spectroscopy (XPS) experiments were performed using an Escalab VG 220i XL apparatus. X-ray diffraction (XRD) measurements were carried out using X'pert MPD Philips using Cu Kα radiation with an average current of 50 mA. Chromatography experiments were performed using a Varian 3800 and the Star chromatography software. The capillary column was a DB5 (5% phenyl groups) column. Iodobenzene (IDB) conversion was calculated from the peak area ratio of IDB relative to dodecane (D) as obtained from GC analysis. Elemental analyses of carbon, nitrogen and palladium were determined by laser ablation inductively coupled plasma mass spectrometry (ICP-MS) and carried out by the SCA of the Centre National de la Recherche Scientifique (Solaize, France). Thermogravimetric analyses (TGA) were carried out under an oxygen flux (5 mL min−1) using a heating rate of 5 °C min−1. The apparatus was a Setaram TAG-1750 thermogravimetric analyser.
Results and discussion
Syntheses and characterization
Macrocellular monoliths were obtained using a concentrated direct emulsion, i.e. dispersion of oil in water, where the hydrophilic continuous phase contains hydrolyzed silane precursors. For synthetic details see the Experimental section.![Pore size distribution of connecting windows obtained by mercury porosimetry. (a) g-imidazole-Si(HIPE) and (b) Pd(OAc)2@[Bmim]NTf2@g-imidazole-Si(HIPE). The inset corresponds to a SEM micrograph of the Pd(OAc)2@[Bmim]NTf2@g-imidazole-Si(HIPE) macrocellular foam.](/image/article/2013/NJ/c2nj40527j/c2nj40527j-f1.gif) | ||
| Fig. 1 Pore size distribution of connecting windows obtained by mercury porosimetry. (a) g-imidazole-Si(HIPE) and (b) Pd(OAc)2@[Bmim]NTf2@g-imidazole-Si(HIPE). The inset corresponds to a SEM micrograph of the Pd(OAc)2@[Bmim]NTf2@g-imidazole-Si(HIPE) macrocellular foam. | ||
| Si(HIPE) | g-imidazole-Si(HIPE) | Pd(OAc)2@[Bmim]NTf2@g-imidazole-Si(HIPE) | |
|---|---|---|---|
| Porosity (%) | 92 | 91 | 89 |
| Intrusion volume (cm3 g−1) | 10.9 | 10.4 | 5.6 |
| Bulk density (g cm−3) | 0.08 | 0.09 | 0.16 |
| Skeletal density (g cm−3) | 0.85 | 0.99 | 1.50 |
Nevertheless, looking more precisely at the mercury porosimetry data collected in Table 1, it is obvious that both bulk and skeletal densities increase after functionalization and more importantly after impregnation steps. Moreover, the intrusion volume is strongly affected by the IL-impregnation step, since a large decrease can be pointed out in the material Pd(OAc)2@[Bmim]NTf2@g-imidazole-Si(HIPE). These characteristics suggest an effective encapsulation and/or coating of ionic liquid phases within the silica-based porous framework. To confirm the presence of the ionic liquid phase within the macroporous framework, hybrid foams were thoroughly characterized at meso- and microscopic scales.
![(a) TEM micrograph of a SILPs onto Si(HIPE) macrocellular hybrid material. (b) SAXS spectrum obtained for ■ a native silica monolith, ○ an hybrid foam g-imidazole-Si(HIPE), and ● a SILP onto Si(HIPE) monoliths, Pd(OAc)2@[Bmim]NTf2@g-imidazole-Si(HIPE).](/image/article/2013/NJ/c2nj40527j/c2nj40527j-f2.gif) | ||
| Fig. 2 (a) TEM micrograph of a SILPs onto Si(HIPE) macrocellular hybrid material. (b) SAXS spectrum obtained for ■ a native silica monolith, ○ an hybrid foam g-imidazole-Si(HIPE), and ● a SILP onto Si(HIPE) monoliths, Pd(OAc)2@[Bmim]NTf2@g-imidazole-Si(HIPE). | ||
TEM observations highlight a pore diameter close to 2 nm ordered in a vermicular mesostructure. SAXS data confirm the presence of a vermicular structure, exhibiting a unit cell parameter of 3.3 nm (corresponding to the constant peak centred at a wave vector q = 0.19 Å−1), including both pore diameter and hybrid silica-based wall in between. Despite the proof of a constant mesostructure, these techniques do not allow quantifying the accessibility of the mesoporosity in this series of hybrid foams. With this purpose, we performed nitrogen sorption measurements before and after impregnation of the ionic liquid phase. Thereby, it clearly appears that the functionalization step induces a decrease of the meso- and micropores surface area (Table 2, entries 1 and 2), while the ionic liquid impregnation generates a large drop of the micro- and mesoporous contributions (Table 2, entries 3–6), which is consistent with the increase of skeletal densities depicted by mercury porosimetry experiments (Table 1).
| Entry | Hybrid foams Si(HIPE) | Nitrogen sorption data | ||
|---|---|---|---|---|
| BET surface areaa (m2 g−1) | Mesoporous surface areab (m2 g−1) | Total pore volumec (cm3 g−1) | ||
| a Brunauer, Emmett and Teller method.b Mesoporous surface area extracted from the BJH method (Barret, Joyner and Halenda).c Single point adsorption total pore volume. | ||||
| 1 | Si(HIPE) | 620 | 209 | 0.31 |
| 2 | g-imidazole-Si(HIPE) | 462 | 139 | 0.39 |
| 3 | Pd(OAc)2@[Bmim]NTf2@g-imidazole-Si(HIPE) | 133 | 79 | 0.13 |
| 4 | Pd(OAc)2@[Bmim]NTf2@Si(HIPE) | 110 | 63 | 0.10 |
| 5 | Pd(OAc)2@[Bmim]BF4@g-imidazole-Si(HIPE) | 117 | 77 | 0.11 |
| 6 | Pd(OAc)2@[Bmim]BF4@Si(HIPE) | 187 | 119 | 0.19 |
Hence, the encapsulation and/or coating of SILPs seems to be efficient within the macrocellular silica-based monoliths. To check the integrity of both the organosilane molecules grafted and the ionic liquid coated, a qualitative study by Fourier-transformed infrared spectroscopy (FTIR), together with quantitative studies by elemental analysis and solid-state NMR, was performed.
First, the integrity of imidazole groups within the silica-based foams was checked by FTIR spectroscopy (Fig. 3). It is necessary to differentiate absorption bands related to the silica framework from those associated with functional organic groups. Concerning silica (Fig. 3c), we can distinguish three absorption bands centered at 1080, 800 and 460 cm−1, corresponding, respectively, to Si–O–Si antisymmetric and symmetric stretching modes and rocking motions, and a broader band between 3200 and 3600 cm−1, associated with υ(OH) and stretching modes of the physisorbed water molecules and silanol groups.
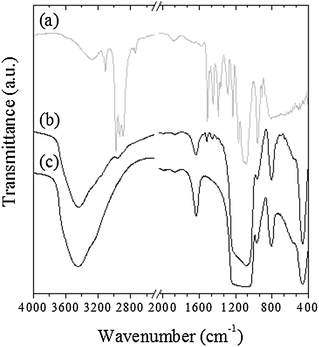 | ||
| Fig. 3 FTIR spectra focused on 4000–400 cm−1 wavenumber region. (a) imidazole triethoxysilane precursor, (b) g-imidazole-Si(HIPE) hybrid foam and (c) native Si(HIPE). | ||
In addition to these major absorption bands of the silica framework itself, signals related to imidazole groups (Fig. 3a) are clearly noticeable in the FTIR spectrum of the g-imidazole-Si(HIPE) hybrid foam (Fig. 3b). Thus, υ(CH2) symmetrical and antisymmetrical stretching modes and δ(CH2) rocking motions, related to the silicopropyl chain (propyl segment between Si atom and imidazole group), can be noticed, respectively, at 2928, 2870 and 1451 cm−1. In addition, vibration modes associated with imidazole heterocycles, υ(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), υ(C–N), υ(C–C) and υ(CC), are noticeable from 1549 to 1390 cm−1 for the spectrum associated with g-imidazole-Si(HIPE) hybrid foam (Fig. 3b). A small shoulder around 3100 cm−1, corresponding probably to υ(C–H) vibration modes, is also visible (Fig. 3b). Moreover, a shoulder centred at 2957 cm−1, assigned to υ(CH3) asymmetrical valence vibration, suggests the presence of ethoxy groups, coming from an incomplete hydrolysis of the organosilane precursors, or the presence of residual isopropanol/toluene molecules of the impregnation solution (see Experimental section). The presence of imidazole groups is also confirmed by 13C solid-state NMR study (Fig. 4).
N), υ(C–N), υ(C–C) and υ(CC), are noticeable from 1549 to 1390 cm−1 for the spectrum associated with g-imidazole-Si(HIPE) hybrid foam (Fig. 3b). A small shoulder around 3100 cm−1, corresponding probably to υ(C–H) vibration modes, is also visible (Fig. 3b). Moreover, a shoulder centred at 2957 cm−1, assigned to υ(CH3) asymmetrical valence vibration, suggests the presence of ethoxy groups, coming from an incomplete hydrolysis of the organosilane precursors, or the presence of residual isopropanol/toluene molecules of the impregnation solution (see Experimental section). The presence of imidazole groups is also confirmed by 13C solid-state NMR study (Fig. 4).
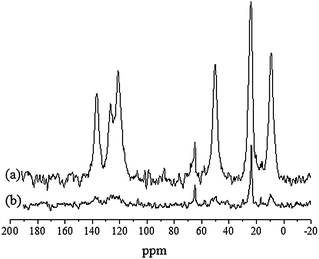 | ||
| Fig. 4 13C solid-state NMR study of the g-imidazole-Si(HIPE). (a) Cross polarization (CP) spectrum. (b) High-power decoupling (HPDEC) spectrum. | ||
Thus, the cross polarization (CP) spectrum shows six major peaks centred at 9.0, 23.8, 50.0, 121.0, 126.6 and 136.5 ppm (Fig. 4a). The first three correspond to the Si–(CH2)3 silicopropyl chain and the others to the imidazole ring. A little narrow peak is visible at 64.9 ppm, more intense in the high-power decoupling (HPDEC) quantitative spectrum in which another intense narrow peak is visible at 23.6 ppm (Fig. 4b). Both correspond to isopropanol, which is confirmed by the relative decrease in the CP spectrum, showing the high mobility of these species. Henceforth, it seems obvious that the methyl groups shown by FTIR comes from isopropanol trapped within the hybrid foam. Subsequently, to check the local chemical environment around silicon atoms, and quantify both grafting ratio and condensation degree of imidazole groups within silica-based foams, 29Si solid-state NMR study was carried out (Fig. 5).
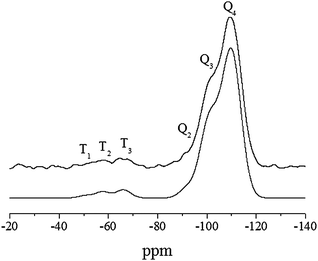 | ||
| Fig. 5 29Si HPDEC solid-state NMR study of the g-imidazole-Si(HIPE) hybrid foam (deconvoluted bottom). | ||
At this stage, we have to specify that 29Si sites are labelled with the conventional Tn and Qn notation. Thus, T refers to functional RSiO3-units resulting from hydrolysis and condensation of organosilane precursors, while Q species are associated with SiO4-units from the silica framework. In this notation, n corresponds to the number of bridging oxygen atoms surrounding the silicon and R to the organic chain (i.e., imidazole groups). Therefore, in this study, Tn signals clearly confirm the presence of imidazole-based organosilane derivatives. Then, after a Lorentzian/Gaussian deconvolution of these spectra,13 a molar ratio of around 5% in Tn species was determined. This feature was also supported by elemental analysis of nitrogen (Table 3). Moreover, due to the presence of both T2 and T1 species, we can consider that the organosilane precursors used in this study were subjected to an incomplete condensation. This feature can be mainly explained from geometrical constraints on the surface of the silica framework. Thus, considering the lack of catalyst in the impregnation solution, partial hydrolysis of ethoxy groups and condensation is largely promoted by superficial silanol groups and residual physisorbed water. Henceforth, steric obstructions, involving a limited access to superficial groups, are likely to prevent organosilanes from complete condensation. Nevertheless, organosilane derivatives are mainly grafted onto the inorganic structure by one or two oxygen atoms, suggesting a rigid anchoring.
Non-covalent anchoring and integrity of ionic liquid phases onto macrocellular foams surface was firstly highlighted by FTIR spectroscopy (Fig. 6).
![FTIR spectra focused on 4000–900 cm−1 wavenumber region. (a) [Bmim]NTf2 ionic liquid, (b) hybrid foam Pd(OAc)2@[Bmim]NTf2@g-imidazole-Si(HIPE) and (c) g-imidazole-Si(HIPE).](/image/article/2013/NJ/c2nj40527j/c2nj40527j-f6.gif) | ||
| Fig. 6 FTIR spectra focused on 4000–900 cm−1 wavenumber region. (a) [Bmim]NTf2 ionic liquid, (b) hybrid foam Pd(OAc)2@[Bmim]NTf2@g-imidazole-Si(HIPE) and (c) g-imidazole-Si(HIPE). | ||
Thus, for each sample, the 1-butyl-3-methylimidazolium cation [Bmim]+ IRTF typical signature (Fig. 6a) was clearly displayed. Antisymmetrical and symmetrical υ(CH2) stretching modes, together with δ(CH2) rocking motions, related to carbons of the silicopropyl chain, are, respectively, noticeable at 2941, 2854 and 1465 cm−1 (Fig. 6b). Concerning the sample previously functionalized with imidazole groups (Fig. 6b), these signals, coming from both chemically grafted imidazoles and physisorbed [Bmim]+ cations non-covalently anchored, are largely enhanced, compared with the initial g-imidazole-Si(HIPE) hybrid foam (Fig. 6c). Moreover, concerning the SILPs within macrocellular silica-based foams, two intense signals centred at 2968 and 2882 cm−1, related to antisymmetrical and symmetrical υ(CH3) stretching modes from short alkyl chain segments of the [Bmim]+ cation, are clearly visible (Fig. 6b). In addition, υ(CN), υ(C–N), υ(C–C) and υ(CC) vibration modes associated with the imidazole ring are discernible between 1575 and 1390 cm−1. Two intense peaks at 3162 and 3128 cm−1, probably corresponding to υ(C–H) stretching modes, are also revealed. Concerning the anionic part of ILs, a few signals are also noticeable for the SILPs. In the case of bis(trifluoromethanesulfonyl)imide anion NTf2−, two peaks between 1354 and 1138 cm−1 suggest the presence of both sulfonamide (–SO2–N) and trifluoromethyl (–CF3) groups. The integrity of the ionic liquid has also been checked by 1H and 13C solid-state NMR (not shown herein). Due to the high ionic mobility of this phase, very narrow lines are observed and the spinning side bands have almost disappeared, even on the 1H spectra at a spinning speed of 5 kHz. All the characteristic resonances are observed, including the CF3 quadruplet of the counter-ion that is not visible under CP.
Secondly, stoichiometries of this SILPs onto Si(HIPE) series were determined combining elemental and thermogravimetric analysis (Table 4).
| Entry | Hybrid foams Pd(OAc)2@Organo-Si(HIPE) | Wt% | |||
|---|---|---|---|---|---|
| Pda | Ca | Na | H2Ob | ||
| a Palladium, carbon and nitrogen contents determined by elemental analysis.b Water physisorbed content determined by thermogravimetric analysis. | |||||
| 1 | Pd(OAc)2@[Bmim]NTf2@g-imidazole-Si(HIPE) | 0.72 | 18.75 | 5.92 | 1.20 |
| 2 | Pd(OAc)2@[Bmim]NTf2@Si(HIPE) | 0.68 | 15.92 | 4.85 | 1.40 |
| 3 | Pd(OAc)2@[Bmim]BF4@g-imidazole-Si(HIPE) | 0.91 | 17.64 | 4.84 | 1.80 |
| 4 | Pd(OAc)2@[Bmim]BF4@Si(HIPE) | 0.99 | 20.28 | 5.18 | 2.00 |
| 5 | Pd(OAc)2@[Bmim]PF6@Si(HIPE) | 0.71 | 17.91 | 4.57 | 1.50 |
The molar ratio Pd/ILs, experimentally fixed at 1/20, was maintained for each sample, whatever the synthetic route in use. Thus, it appears, to a first extent, that previous covalent grafting of imidazole groups onto the inorganic support did not influence the encapsulation of the palladium salt. However, it seems that a previous functionalization enhances subsequent [Bmim]NTf2 physisorption, while generates a decrease in the amount of [Bmim]BF4 onto the silica-based support (Table 5). This feature suggests that the anchoring of hydrophobic ionic liquids ([Bmim]NTf2) is probably enhanced via a potential π–π stacking with imidazole groups, while the coating of hydrophilic ILs ([Bmim]BF4) is promoted via hydrogen bonds between superficial oxygen of the silica framework and the imidazolium cation.
| Entry | Hybrid foams Pd(OAc)2@organo-Si(HIPE) | Stoichiometries |
|---|---|---|
| 1 | Pd(OAc)2@[Bmim]NTf2@g-imidazole-Si(HIPE) | Pd0.009LI0.19 SiO1.96(C6N2H9)0.04·0.09H2O |
| 2 | Pd(OAc)2@[Bmim]NTf2@Si(HIPE) | Pd0.008LI0.16 SiO2·0.09H2O |
| 3 | Pd(OAc)2@[Bmim]BF4@g-imidazole-Si(HIPE) | Pd0.008LI0.14 SiO1.96(C6N2H9)0.04·0.09H2O |
| 4 | Pd(OAc)2@[Bmim]BF4@Si(HIPE) | Pd0.01LI0.23 SiO2·0.12H2O |
| 5 | Pd(OAc)2@[Bmim]PF6@Si(HIPE) | Pd0.008LI0.21 SiO2·0.10H2O |
Finally, average SILPs thickness onto the surface of the macropores of the hybrid foams were determined (Table S1, ESI†), combining nitrogen sorption and mercury porosimetry data together with calculated stoichiometries. Depending on the starting porous support used, that is, the unmodified Si(HIPE) or the hybrid g-imidazole-Si(HIPE), different adsorption behaviours are assumed. In the first case, a complete filling of the mesoporous volume by the ionic liquid phase is supposed, while we consider that the remaining ionic liquid forms a homogeneous thin film on the surface of macropores (surface of 65 m2 g−1, determined by mercury porosimetry). In contrast, in the case of SILP@g-imidazole-Si(HIPE), considering the size of the grafted molecules, making the smallest pores less accessible, and the viscosity of the ionic liquid impregnated, the ILs cannot fill the mesoporous volume. However, we still assume that the nanometric ILs film onto the macropores is homogeneous. Therefore, depending on the samples, the thickness of the SILPs is estimated between 6 and 12 nm. In summary, the presence of macropores in the support allows us to reach thickness of ILs coating largely higher than the ones reported so far in the literature, particularly for mesoporous silica-based powders.
Reduction of the palladium(II) acetate species encapsulated within the supported ionic liquid phase onto palladium(0) nanoparticles have been performed, with a view to compare the catalytic performances of both species toward the Heck coupling reaction. The starting material was Pd(OAc)2@[Bmim]NTf2@g-imidazole-Si(HIPE)s.
![XPS and pictures carried out on Pd(OAc)2@[Bmim]NTf2@g-imidazole-Si(HIPE) (a–b) before and (c–d) after treatment under 10 bar of hydrogen at 80 °C during 1 h. (e–f) X-ray diffraction spectrum obtained for the Pd0@[Bmim]NTf2@g-imidazole-Si(HIPE).](/image/article/2013/NJ/c2nj40527j/c2nj40527j-f7.gif) | ||
| Fig. 7 XPS and pictures carried out on Pd(OAc)2@[Bmim]NTf2@g-imidazole-Si(HIPE) (a–b) before and (c–d) after treatment under 10 bar of hydrogen at 80 °C during 1 h. (e–f) X-ray diffraction spectrum obtained for the Pd0@[Bmim]NTf2@g-imidazole-Si(HIPE). | ||
Weak contributions at lower binding energies, corresponding to palladium oxide PdO (341.8 and 336.6 eV) and zerovalent palladium (metallic) (340.6 and 335.2 eV), can also be noticed. Different methods, such as self-induced reduction in alcoholic solvent or hydrogenation under high pressure, are mentioned in the literature to generate palladium metallic nanoparticules encapsulated within the SILPs. However, the first method induced a fast reduction followed by a precipitation of the metallic particles during the impregnation step of the silica-based porous support. Therefore, hydrogenation of Pd(OAc)2@[Bmim]NTf2@Si(HIPE) hybrid foams was preferred. After this treatment, Pd(OAc)2@[Bmim]NTf2@Si(HIPE) monoliths turned black, highlighting an efficient reduction of the palladium salt (Fig. 7d). This feature is clearly supported from the XPS spectrum (Fig. 7c), which displays a shift of the two main peaks related to Pd 3d3/2 and Pd 3d5/2, initially centred at 343.5 and 338.3 eV, toward lower binding energies (respectively 340.6 and 335.2 eV). After deconvolution of the raw spectrum, a weak superficial oxidation of the Pd0 nanoparticles can be noticed, via the presence of two contributions related to PdO (341.8 and 336.6 eV). These characteristics have been supported by XRD (Fig. 7e–f). Indeed, beyond an intense and broad peak associated with amorphous silica, centered at 2θ = 21° (Fig. 7e), clear signals related to palladium metallic Pd0 and palladium oxide PdIIO are also noticed. Particularly, XRD peaks centered at 39.8°, 46.3° and 68° (Fig. 7f) correspond, respectively, to (111), (200) and (220) planes of the typical face centered cubic (fcc) structure of palladium metal. Therefore, the in situ reduction of palladium(II) yields metallic palladium nanoparticles. This feature can be induced by an enhancement of the mass transfer at the gas-SILP interfaces, related to the interconnected macroporous framework together with the nanometric thickness of the ionic liquid layer. Moreover, the presence of palladium metallic nanoparticles was highlighted by TEM (Fig. 8a), while the planes of the fcc structure were noticed via HR-TEM (Fig. 8b). The particle size distribution was determined for the Pd0@[Bmim]NTf2@g-imidazole-Si(HIPE) hybrid foam, by TEM observations carried out on 180 particles (Fig. 8c). A broad distribution, probably related to the absence of the stabilizing agent, underlines an average diameter of (11 ± 6) nm. Nevertheless, due to the experimental procedure to prepare TEM grids (see Experimental section), this technique did not offer the opportunity to observe the supported ionic liquid phase immobilized within the silica-based foams.
![(a) TEM, (b) HR-TEM, and (c) particle size distribution of the Pd0@[Bmim]NTf2@g-imidazole-Si(HIPE) hybrid foam. The particle size distribution was determined on 180 nanoparticles.](/image/article/2013/NJ/c2nj40527j/c2nj40527j-f8.gif) | ||
| Fig. 8 (a) TEM, (b) HR-TEM, and (c) particle size distribution of the Pd0@[Bmim]NTf2@g-imidazole-Si(HIPE) hybrid foam. The particle size distribution was determined on 180 nanoparticles. | ||
In order to circumvent this drawback, a microtome section has been carried out on Pd0@[Bmim]NTf2@g-imidazole-Si(HIPE) samples (Fig. 9). TEM performed on the 70 nm thin sections thus prepared, beyond the observation of the macroscopic structure made of aggregated hollow spheres (Fig. 9a), underlined the presence of palladium metal nanoparticles and clusters on the macrocellular scaffold (Fig. 9b). A more careful observation of the micrograph (Fig. 9b) allows us to notice that the nanoscopic clusters are embedded within a viscous and heterogeneous thin layer, probably the SILP. Nevertheless, the observation of the SILP is still delicate, since the 1,3-dialkylimidazolium salts are soluble in the mixture used to impregnate the hybrid foam before the microtome cutting process. To get more information concerning the supported ionic liquid phase and the interaction with the palladium nanoparticles, a mobility study had to be carried out by solid-state NMR.
![(a–b) TEM of 70 nm-thickness thin sections of the Pd0@[Bmim]NTf2@g-imidazole-Si(HIPE).](/image/article/2013/NJ/c2nj40527j/c2nj40527j-f9.gif) | ||
| Fig. 9 (a–b) TEM of 70 nm-thickness thin sections of the Pd0@[Bmim]NTf2@g-imidazole-Si(HIPE). | ||
![Solid-state NMR 13C, (A) HPDEC, (B) PG of (a) [Bmim]NTf2@Si(HIPE), (b) Pd0@[Bmim]NTf2@Si(HIPE) and (c) Pd0@[Bmim]NTf2@g-imidazole-Si(HIPE).](/image/article/2013/NJ/c2nj40527j/c2nj40527j-f10.gif) | ||
| Fig. 10 Solid-state NMR 13C, (A) HPDEC, (B) PG of (a) [Bmim]NTf2@Si(HIPE), (b) Pd0@[Bmim]NTf2@Si(HIPE) and (c) Pd0@[Bmim]NTf2@g-imidazole-Si(HIPE). | ||
These results can also be confirmed using 13C CP experiments (Fig. 11). Despite the high number of scans, a very low signal to noise ratio is obtained, because of the mobility averaging of the 1H–13C dipolar interaction. By addition and reduction of the palladium (Fig. 11b), we can notice a widening of the resonances and an increase of the signal to noise ratio, which both confirm the mobility decrease. This feature is much more pronounced for the Pd0@[Bmim]NTf2@g-imidazole-Si(HIPE) sample obtained after grafting of imidazole species and reduction of palladium (Fig. 11c), confirming the influence of the grafting on the SILP mobility.
![Solid-state NMR 13C CP of (a) [Bmim]NTf2@Si(HIPE), (b) Pd0@[Bmim]NTf2@Si(HIPE) and (c) Pd0@[Bmim]NTf2@g-imidazole-Si(HIPE).](/image/article/2013/NJ/c2nj40527j/c2nj40527j-f11.gif) | ||
| Fig. 11 Solid-state NMR 13C CP of (a) [Bmim]NTf2@Si(HIPE), (b) Pd0@[Bmim]NTf2@Si(HIPE) and (c) Pd0@[Bmim]NTf2@g-imidazole-Si(HIPE). | ||
Overall, stabilisation of palladium-based catalysts within supported ionic liquid phases within macrocellular silica-based monoliths is effective and has been proved by different techniques.
Catalytic properties toward Heck coupling reaction
The use of ionic liquids to replace organic solvents in metal catalysed reactions has recently gained much attention and great progress has been accomplished in this area in the last few years. Also, we have to underline that to really assess heterogeneous catalyst efficiency; it is important working in worst conditions that is to say in a non-reductive atmosphere. As a direct consequence olefin reductions occurring with hydrogen have already been tested within Pd@PolyHIPE materials15a and show high efficiency, efficiency extrinsically enhanced by hydrogen that helps maintaining the Palladium nanoparticles at the zero-valent state. Therefore, we turned our choice toward chemical reactions occurring in a non-reductive atmosphere (Myzoroki–Heck11a,b or Suzuki–Myaura C–C coupling reactions15b) where Palladium nanoparticles will have to endure both oxidation and leaching processes. In this context, it is significant that the first well documented palladium-catalysed reaction performed in ILs was the Heck reaction.16 To increase catalyst performance and develop more sustainable and environmental benign reaction conditions, immobilisation of Pd(OAc)2 in silica gel pores, with the aid of [Bmim]PF6 and the application of this catalytic system in recyclable Heck reaction has been reported.6a,b The use of a hydrocarbon solvent was necessary to prevent removal of the IL layer from the silica. Catalytic performances of different heterogeneous catalysts disclosed in this work have been tested in the Heck coupling reaction (Scheme 1) using the same reagents and similar experimental conditions (excepted that dodecane was replaced by octane) than in the work cited above in order of comparison (Fig. 12). | ||
| Scheme 1 Heck coupling reaction addressed in this study: (A) Iodobenzene, (B) Acrylate cyclohexyl, (C) Benzyl cyclohexyl acrylate, (D) Triethylammonium iodate. | ||
![Catalytic properties of the SILPs within silica-based hybrid foams obtained for the first cycle toward Heck coupling reaction of iodobenzene and cyclohexyl acrylate. (a) Pd(OAc)2@[Bmim]NTf2-Si(HIPE), (b) Pd(OAc)2@[Bmim]NTf2@g-imidazole-Si(HIPE), (c) Pd(OAc)2@[Bmim]PF6-Si(HIPE), (d) Pd0@[Bmim]NTf2@g-imidazole-Si(HIPE), and (e) Pd(OAc)2@[Bmim]BF4-Si(HIPE).](/image/article/2013/NJ/c2nj40527j/c2nj40527j-f12.gif) | ||
| Fig. 12 Catalytic properties of the SILPs within silica-based hybrid foams obtained for the first cycle toward Heck coupling reaction of iodobenzene and cyclohexyl acrylate. (a) Pd(OAc)2@[Bmim]NTf2-Si(HIPE), (b) Pd(OAc)2@[Bmim]NTf2@g-imidazole-Si(HIPE), (c) Pd(OAc)2@[Bmim]PF6-Si(HIPE), (d) Pd0@[Bmim]NTf2@g-imidazole-Si(HIPE), and (e) Pd(OAc)2@[Bmim]BF4-Si(HIPE). | ||
Divergences in catalytic performances appear according to the IL's counterion used (BF4, PF6 or NTf2) when using palladium acetate as catalytic species : the conversion level of iodobenzene after 24 hours is almost two times lower when performed in an hydrophilic supported ionic liquid such as [Bmim]BF4, than in [Bmim]PF6 and [Bmim]NTf2 that are more hydrophobic. Constraints associated with wettability differences on the surface of the SILPs with the apolar co-solvent used, e.g. octane, can explain this behaviour. Indeed, a hydrophilic layer induces most probably a more difficult diffusion of the reagents from octane toward SIL than a hydrophobic layer. As a direct consequence, the accessibility to the palladium-based catalysts is largely restricted.
Catalytic performances of the different (Si)HIPEs SILs on the Heck coupling between iodobenzene and cyclohexyl acrylate are reported in Table 6 and compared with results of similar literature experiments.6a,b Best performances are obtained with Pd(OAc)2 entrapped in the hydrophobic [Bmim]NTf2 (Table 6, entry 1), grafting of imidazole moieties onto the (Si) HIPE surface seems to have no apparent effect (Table 6, entry 2).
| Entry | Hybrid foams | Molar ratio Pd/iodobenzene | Conversion (%) | Time (h) | TON | TOF (h−1) |
|---|---|---|---|---|---|---|
| a From data reported in ref. 6a and b. | ||||||
| 1 | Pd(OAc)2@[Bmim]NTf2@Si(HIPE) | 0.00064 | 65 | 24 | 1015 | 42 |
| 2 | Pd(OAc)2@[Bmim]NTf2@g-imidazole-Si(HIPE) | 0.00068 | 62 | 24 | 910 | 38 |
| 3 | Pd0@[Bmim]NTf2@g-imidazole-Si(HIPE) | 0.00064 | 43 | 24 | 670 | 28 |
| 4 | Pd(OAc)2@[Bmim]PF6@Si(HIPE) | 0.00067 | 56 | 24 | 835 | 35 |
| 5 | Pd(OAc)2@[Bmim]BF4@Si(HIPE) | 0.00093 | 36 | 24 | 390 | 16 |
| 6 | Pd(OAc)2@[Bmim]PF6@silica gela | 0.00130a | 94a | 8.5a | 720a | 85a |
As mentioned above, the use of an hydrophilic IL such as [Bmim]BF4 leads to reduced performances. Pd0 appears to be less efficient than Pd(OAc)2 (Table 6, entries 2 and 3), an inactivation induced by the formation of “black” palladium during the reduction step may be the main cause of this behavior. Finally, the best results obtained in this work appears somewhat lower than data reported by H. Hagiwara et al.6a,b for a similar catalytic system using SILPs within mesoporous silica-based hybrid under similar experimental conditions (entries 1, 4 and 6).6a,b It has to be enlightened that in this latter case, the support used was high specific surface area finely powdered silica which could enhance the diffusion of substrates in solution toward the catalytic species in comparison with millimetre-size monoliths used in the present work. A better exploitation of the monoliths nature of (Si)HIPE-based catalyst would be continuous flow reactors.
Catalysts exhibiting higher performances in the first cycle were reused up to four times as shown in Fig. 13.
![Catalytic properties of the macrocellular hybrid foams obtained on a few cycles toward Heck coupling reaction of iodobenzene and cyclohexyl acrylate. (a) ■ Pd(OAc)2@[Bmim]PF6-Si(HIPE). (b) ▲ Pd(OAc)2@[Bmim]NTf2@Si(HIPE), and Δ Pd(OAc)2@[Bmim]NTf2@g-imidazole-Si(HIPE). The numbers on the right of the curves represent the cycle numbers.](/image/article/2013/NJ/c2nj40527j/c2nj40527j-f13.gif) | ||
| Fig. 13 Catalytic properties of the macrocellular hybrid foams obtained on a few cycles toward Heck coupling reaction of iodobenzene and cyclohexyl acrylate. (a) ■ Pd(OAc)2@[Bmim]PF6-Si(HIPE). (b) ▲ Pd(OAc)2@[Bmim]NTf2@Si(HIPE), and Δ Pd(OAc)2@[Bmim]NTf2@g-imidazole-Si(HIPE). The numbers on the right of the curves represent the cycle numbers. | ||
A significant drop of the conversion yield, from 25 up to 50%, is observed in each experiment between the first and the second use even if catalysts pieces have been washed between each cycle with a 0.05 M NaOH aqueous solution, in order to prevent the detrimental effect associated with the formation of the triethylammonium iodide by-product. Moreover, a random evolution of the conversion yield from use to use can be noticed for the Pd(OAc)2@[Bmim]PF6-Si(HIPE) sample, suggesting a large dependence of the washing step conditions on the catalytic properties. Indeed, an increase of the immersion duration in the basic solution after use 3, leads to a large rise of the conversion in the fourth use, more than 100%. Consequently, the by-product formation seems to be the main detrimental factor of the catalytic system used in this study. This feature is supported by SEM observations performed on hybrid foams after the first catalytic cycle and the first washing step (Fig. 14).
![SEM obtained after the first catalytic cycle toward Heck coupling reaction of iodobenzene and cyclohexyl acrylate for the Pd(OAc)2@[BMIM]NTf2@g-imidazole-Si(HIPE) sample.](/image/article/2013/NJ/c2nj40527j/c2nj40527j-f14.gif) | ||
| Fig. 14 SEM obtained after the first catalytic cycle toward Heck coupling reaction of iodobenzene and cyclohexyl acrylate for the Pd(OAc)2@[BMIM]NTf2@g-imidazole-Si(HIPE) sample. | ||
Beyond the fact that the typical macromorphology of aggregated hollow spheres is preserved despite the use of a basic solution the washing step (Fig. 14a and b), the presence of triethylammonium iodide crystals is clearly highlighted within the macrocellular framework (Fig. 14c). Thus, the hybrid monoliths kept the by-products entrapped in the cells, which could represent a great advantage in view of products separation. This feature has already been reported by S. Volland et al.6d using Pd(OAc)2@SILPs confined in silica gels. A very interesting characteristic of these devices concerns the low leaching of palladium species. Inductively coupled plasma atomic emission spectroscopy (ICP-AES) performed on the catalytic media after the first catalyst use revealed a palladium release between 0.2 and 0.6 molar% of the palladium initially trapped within the SILPs. This result is in good agreement with the values reported by H. Hagiwara et al.6a,b
Conclusion
Overall we have combined emulsion-based porous materials, syntheses of Ils, Palladium nanoparticles heterogeneous nucleation to promote efficient heterogeneous catalysts when test for Heck coupling reactions. With this endeavor this work falls into the Integrative Chemistry synthetic path.17 Particularly, supported ionic liquid phases within emulsion-templated macrocellular silica-based monoliths were prepared by a simple impregnation in organic solvents. Thin ionic liquid layers, from 6 up to 12 nm, were obtained. Moreover, palladium salts were efficiently trapped within the SILPs, while an in situ palladium hydrogenation allowed an efficient reduction and formation of 10 nm diameter palladium metal nanoparticles. These hybrid foams were used as heterogeneous macrocellular catalysts toward Heck coupling reaction of iodobenzene and cyclohexyl acrylate. Despite a minimized recyclability related to the entrapment of detrimental ammonium salts within the macroporous network, catalytic performances appear, competitive with best literature results.Acknowledgements
Acknowledgments are made to Christine Labrugère (CeCaMA – Institute for Solid State Chemistry Bordeaux, ICMCB, Pessac) for XPS experiments, Eric Lebraud (ICMCB) for XRD measurements, Marie-France Achard (CRPP, Pessac) for SAXS, Elisabeth Sellier (CREMEM, University Bordeaux 1) for HR-TEM observations, Isabelle Ly (CRPP) for microtomy experiments and Laetitia Etienne (ICMCB) for ICP-AES assessments. The authors would like to gratefully acknowledge Dr Simona Ungureanu and Annick Babeau-Garcia for fruitful discussions.References
- R. A. Sheldon, J. Chem. Technol. Biotechnol., 1997, 68, 381 CrossRef CAS.
- (a) G. W. Parshall, J. Am. Chem. Soc., 1972, 94, 8716 CrossRef CAS; (b) R. Sheldon, Chem. Commun., 2001, 2399 RSC.
- (a) P. Bonhôte, A.-P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168 CrossRef CAS; (b) J. G. Huddleston, A. E. Visser, W. M. Reichert, H. D. Willauer, G. A. Broker and R. D. Rogers, Green Chem., 2001, 3, 156 RSC.
- (a) P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772 CrossRef; (b) A. P. Umpierre, G. Machado, G. H. Fecher, J. Morais and J. Dupont, Adv. Synth. Catal., 2005, 347, 1404 CrossRef CAS; (c) G. Machado, J. D. Scholten, T. de Vargas, S. R. Teixeira, L. H. Ronchi and J. Dupont, Int. J. Nanotechnol., 2007, 4, 541 Search PubMed; (d) J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667–3692 CrossRef CAS; (e) V. I. Pârvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615 CrossRef CAS; (f) A. J. Carmichael, M. J. Earle, J. D. Holbrey, P. B. Mc-Cormac and K. R. Seddon, Org. Lett., 1999, 1, 997 CrossRef CAS.
- (a) C. P. Mehner, R. A. Cook, N. C. Dispenziere and M. Afeworki, J. Am. Chem. Soc., 2002, 124, 12932–12933 CrossRef CAS; (b) M. H. Valkenberg, C. deCastro and W. F. Hölderich, Green Chem., 2002, 4, 88 RSC.
- (a) H. Hagiwara, Y. Sugawara, K. Isobe and T. Hoshi, Org. Lett., 2004, 6, 2325 CrossRef CAS; (b) H. Hagiwara, Y. Sugawara, K. Isobe, T. Hoshi and T. Suzuki, Org. Lett., 2006, 8, 1749 Search PubMed; (c) P. Han, H. Zhang, X. Qiu, X. Ji and L. Gao, J. Mol. Catal. A: Chem., 2008, 295, 57 CrossRef CAS; (d) S. Volland, M. Gruit, T. Régnier, L. Viau, O. Lavastre and A. Vioux, New J. Chem., 2009, 33, 2015 RSC.
- (a) J. Huang, T. Jiang, H. Gao, B. Han, Z. Liu, W. Wu, Y. Chang and G. Zhao, Angew. Chem., Int. Ed., 2004, 43, 1397–1399 CrossRef CAS; (b) C. P. Mehnert, E. J. Mozeleski and R. A. Cook, Chem. Commun., 2002, 3010 RSC.
- M. S. Sarkar, H. Qiu and M.-J. Jin, J. Nanosci. Nanotechnol., 2007, 7, 3880 Search PubMed.
- (a) F. Shi, Q. Zhang, D. Li and Y. Deng, Chem.–Eur. J., 2005, 11, 5279 CrossRef CAS; (b) A. Vioux, L. Viau, S. Volland and J. Le Bideau, C. R. Chim., 2010, 13, 242 CrossRef CAS.
- F. Carn, A. Colin, M.-F. Achard, M. Birot, H. Deleuze and R. Backov, J. Mater. Chem., 2004, 14, 1370 RSC.
- (a) S. Ungureanu, M. Birot, L. Guillaumme, H. Deleuze, O. Babot, B. Julian, M.-F. Achard, M. I. Popa, C. Sanchez and R. Backov, Chem. Mater., 2007, 19, 5786 CrossRef CAS; (b) S. Ungureanu, H. Deleuze, M. I. Popa, C. Sanchez and R. Backov, Chem. Mater., 2008, 20, 6494 CrossRef CAS; (c) N. Brun, B. Julian-Lopez, P. Hesemann, L. Guillaume, M.-F. Achard, H. Deleuze, C. Sanchez and R. Backov, Chem. Mater., 2008, 20, 7117 CrossRef CAS; (d) N. Brun, A. Babeau-Garcia, H. Deleuze, F. Duran, C. Sanchez, V. Ostreicher and R. Backov, Chem. Mater., 2010, 22, 4555 CrossRef CAS; (e) N. Brun, A. Babeau Garcia, M.-F. Achard, C. Sanchez, F. Durand, G. Laurent, M. Birot, H. Deleuze and R. Backov, Energy Environ. Sci., 2011, 4, 2840 RSC.
- (a) R. K. Harris and M. L. Robbins, Polymer, 1978, 19, 1123 CAS; (b) F. Babonneau, New J. Chem., 1994, 18, 1065 Search PubMed; (c) Y. Chevalier, A.-C. Grillet, M.-I. Rahmi, C. Lière, M. Masure, P. Hémery and F. Babonneau, Mater. Sci. Eng., C, 2002, 21, 143 CrossRef.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J. O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- T. Azais, G. Hartmeyer, S. Quignard, G. Laurent and F. Babonneau, J. Phys. Chem. C, 2010, 114, 8884 CrossRef CAS.
- (a) A. Desforges, H. Deleuze, O. Mondain-Monval and R. Backov, Ind. Eng. Chem. Res., 2005, 44, 8521 CrossRef CAS; (b) A. Desforges, R. Backov, H. Deleuze and O. Mondain-Monval, Adv. Funct. Mater., 2005, 15, 1689 CrossRef CAS.
- D. Kaufmann, M. Nouroozian and H. Henze, Synlett, 1996, 1091 CrossRef CAS.
- (a) R. Backov, Soft Matter, 2006, 2, 452 RSC; (b) N. Brun, S. Ungureanu, H. Deleuze and R. Backov, Chem. Soc. Rev., 2011, 40, 771 RSC; E. Prouzet, Z. Khani, M. Bertrand, M. Tokumoto, V. Gyuot-Ferreol and J.-F. Tranchant, Microporous Mesoporous Mater., 2006, 96, 369 CrossRef CAS; (c) C. Sanchez, C. Boissière, D. Grosso, C. Laberty and L. Nicole, Chem. Mater., 2008, 20, 682 CrossRef CAS; (d) E. Prouzet, S. Ravaine, C. Sanchez and R. Backov, New J. Chem., 2008, 32, 1284 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2nj40527j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2013 |