Synthesis and evaluation of stable substrate analogs as potential modulators of cyclodiphosphate synthase IspF†
J.
Kipchirchir Bitok
and
Caren Freel
Meyers
*
Department of Pharmacology and Molecular Sciences, The Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA. E-mail: cmeyers@jhmi.edu
First published on 20th August 2012
Abstract
Stable IspF substrate analogs were synthesized. In the presence of substrate analogs, the E. coli IspF–MEP complex shows activities distinct from IspF, and bisphosphonates (BP) behave differently than their diphosphate (DP) counterparts. Bisphosphonate analogs activate and/or stabilize IspF, and only the closest structural substrate analog weakly inhibits the IspF–MEP complex.
In Nature, the distinct mevalonate (MVA)1,2 and 2C-methyl-D-erythritol phosphate (MEP) pathways3–5 produce dimethylallyl pyrophosphate (DMAPP) and isopentenyl pyrophosphate (IPP) for biosynthesis of the essential isoprenoid class of natural products (Fig. S1†). While the MVA pathway is operational in plants, archaea, fungi and animals, the MEP pathway is utilized by higher plants, algae, bacteria and parasites.3–5 The MEP pathway is widespread in pathogenic organisms, including M. tuberculosis and P. falciparum, and is therefore being pursued as a potential target for development of anti-infective agents.6–8 Cyclodiphosphate synthase IspF catalyzes the fifth step in the MEP pathway, converting 4-diphosphocytidyl-2C-methyl-D-erythritol 2-phosphate (CDPME2P) to the cyclic diphosphate 2C-methyl-D-erythritol 2,4-cyclopyrophosphate (MEcPP) (Fig. 1).9,10In vitro, IspF also catalyzes formation of 2C-methyl-D-erythritol 3,4-cyclomonophosphate (MEcP) from the IspD product, 4-diphosphocytidyl-2C-methyl-D-erythritol (CDPME, Fig. S2†).10
Importantly, IspF is essential in pathogenic organisms;11–13 however, efforts to target this interesting enzyme have produced modest inhibitors,14–16 with the most potent thiazolopyrimidine class exhibiting inhibitory activities in the low micromolar range.15,16 These efforts by Diederich and colleagues to identify inhibitors underscore the challenges in targeting the highly polar active site of IspF.
Our interest in targeting isoprenoid biosynthesis in human pathogens has motivated studies to understand pathway regulation and to identify new IspF inhibitors. A recent investigation in our laboratory toward understanding pathway regulation shows that the upstream IspC product, 2C-methyl-D-erythritol 4-phosphate (MEP), enhances and sustains activity of IspF in a possible feed-forward regulatory mechanism. The turnover efficiency of IspF (KCDPME2Pm = 339 ± 32 μM; kcat = 61 ± 3 min−1) increases 4.8-fold in the presence of MEP (in the presence of 500 μM MEP, KCDPME2Pm = 93.8 ± 11; kcat = 80.7 ± 5 min−1 and ACMEP50 = 133 ± 33 μM), and the IspF–MEP complex displays altered sensitivity to inhibitors.17
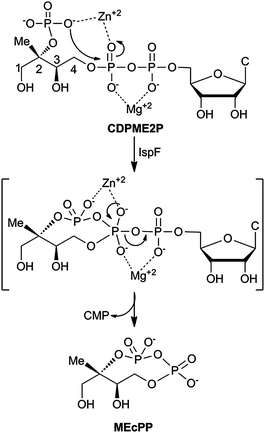 | ||
| Fig. 1 IspF-catalyzed conversion of CDPME2P to MEcPP. C = cytosine. | ||
Here we report the design, synthesis and evaluation of stable substrate analogs as potential active site probes and modulators of IspF activity. In light of our recent findings, we have also evaluated these compounds as modulators of the more active IspF–MEP complex, which may be the more physiologically relevant enzyme form.17 Our results provide further evidence that IspF and the IspF–MEP complex display distinct characteristics in the presence of potential modulators. Contrary to our expectations, stable bisphosphonate substrate analogs are not potent inhibitors. Bisphosphonate analogs bearing the methylerythritol motif enhance and/or sustain activity of IspF, while the closest structural homolog bearing a 2-phosphoryl group exhibits weak inhibitory activity and only against the IspF–MEP complex. Further, this study reveals striking differences in the behaviors of bisphosphonate-containing substrate analogs compared to their diphosphate counterparts and suggests that the cyclodiphosphate synthase modulating activities of bisphosphonates may be driven by the distinct steric and/or electronic properties of the bisphosphonate scaffold (Fig. 2).
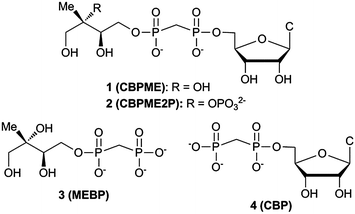 | ||
| Fig. 2 Stable substrate analogs as potential modulators of IspF activity. CBP (4) serves as a bisphosphonate control and potential mimic of CDP. | ||
Results
Design of substrate analogs 1 and 2
Structural and biochemical studies support the proposed mechanisms of cyclization to give MEcPP or MEcP from CDPME2P and CDPME, respectively;10,18,19 however, little is known about the transition state structures or conformational dynamics along these reaction coordinates. Bisphosphonate 2 (CBPME2P) is the closest structural homolog of the natural IspF substrate, incorporating a stable P–C–P linkage to mimic the diphosphate group in CDPME2P. Similarly, bisphosphonate 1 (CBPME) is a close structural homolog to IspD product CDPME, known to undergo IspF-catalyzed conversion to MEcP.10 We reasoned that 1 and 2 would mimic the binding properties of CDPME or CDPME2P, respectively, and/or intermediates along the corresponding reaction coordinates, but would be unable to complete cyclization to release CMP, and thereby act as inhibitors. Further, studies using 1 and 2 as active site probes could reveal distinctive features of IspF at the transition states toward formation of MEcP or MEcPP and provide a basis for the development of transition state analogs as selective inhibitors.Design of substrate analog 3
Rationally designed IspF inhibitors incorporate the CDP moiety14 or a cytidyl-like component15 to bind in the rigid, well-conserved “pocket III” of the IspF active site, and a non-polar component to bind in the more flexible hydrophobic “pocket II”.14 We reasoned that the unique methylerythritol scaffold could also drive recognition of selective IspF inhibitors. Thus, methylerythritol bisphosphonate 3 (MEBP) lacks a cytidyl component but retains the methyerythritol component that is also recognized by IspF.10,19 The stable P–C–P linkage was incorporated as a diphosphate mimic, with the idea that a bisphosphonate prodrug strategy could eventually be pursued for intracellular delivery of MEBP should it display potent inhibition of cyclodiphosphate synthase activity.20Synthesis
Substrate analogs 1 and 2 were synthesized starting from the commercially available tetramethyl bisphosphonate 5 ester (Scheme 1). Removal of a single methyl group by treatment with PhSH and diisopropylethylamine afforded trimethyl ester 6.21,22 Chlorination of crude 6 using Ghosez's reagent23,24 and subsequent reaction of monochloridate 7 with benzylidine protected methylerythritol 825 afforded compound 9.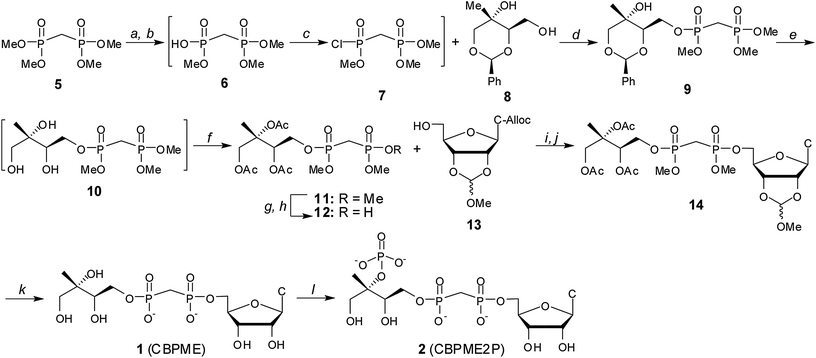 | ||
Scheme 1 Synthesis of stable substrate analogs 1 and 2. Conditions: (a) PhSH, iPr2Net, 12–14 h; (b) DOWEX 50WX8 H+ form; (c) (CH3)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(Cl)N(CH3)2, CH2Cl2, 40 °C; (d) iPr2Net, DMAP, 51–65% over 3 steps; (e) Pd–C, H2 (g), MeOH: (f) Ac2O, iPr2NEt, DMAP, 72–82% over 2 steps; (g) PhSH, Et3N; (h) DOWEX 50WX8 H+ form, 76%; (i) Ph3P, DIAD, THF, 62–78%; (j) PD(Ph3P)4, pTSO2Na, THF/ddH2O, 88–94%; (k) (i) PhSH, Et3N, DMF, 70 °C, 9d, (ii) MeOH/H2O, pH 2.0, 24 h, (iii) 2 C(Cl)N(CH3)2, CH2Cl2, 40 °C; (d) iPr2Net, DMAP, 51–65% over 3 steps; (e) Pd–C, H2 (g), MeOH: (f) Ac2O, iPr2NEt, DMAP, 72–82% over 2 steps; (g) PhSH, Et3N; (h) DOWEX 50WX8 H+ form, 76%; (i) Ph3P, DIAD, THF, 62–78%; (j) PD(Ph3P)4, pTSO2Na, THF/ddH2O, 88–94%; (k) (i) PhSH, Et3N, DMF, 70 °C, 9d, (ii) MeOH/H2O, pH 2.0, 24 h, (iii) 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.5 MeOH:H2O:Et3N, 18 h, 39%; (l) IspE, ATP, PEP, PK, 1 h, 43%. :1:0.5 MeOH:H2O:Et3N, 18 h, 39%; (l) IspE, ATP, PEP, PK, 1 h, 43%. | ||
Benzylidine-protected methylerythritol-based precursors have been successfully converted to MEP and MEcPP.25 However, removal of the benzylidine protecting group from late stage intermediates en route to substrate analog 1 proved difficult in this system, consistent with the findings reported by Narayanasamy, et al.26 Consequently, intermediate 9 was subjected to hydrogenolysis, and the resulting methylerythritol bisphosphonate tetraester 10 was acetylated to give intermediate 11. Treatment of 11 with PhSH/Et3N followed by cation exchange afforded bisphosphonic acid 12, which was coupled to the protected nucleoside 1327–30 using Mitsunobu conditions. Palladium-catalyzed removal of the allyloxycarbonyl (alloc) protecting group from the 4-amino group of the cytosine base was accomplished as described27,31 to afford compound 14. Compound 14 was converted to the IspE substrate analogue CBPME (1) in three tandem steps, and CBPME (1) was converted to CBPME2P (2) by the action of kinase IspE (Fig. S3†).32
Substrate analog 3 (MEBP) was prepared using a similar approach starting from tetrabenzyl bisphosphonate 15 (Scheme 2).33,34 Removal of a single benzyl group in the presence of quinuclidine,35 followed by conversion to the corresponding bisphosphonic acid and finally chlorination23,24 afforded intermediate 16. Bisphosphonic monochloridate 16 was coupled to benzylidene protected methylerythritol 825 to give compound 17. Palladium-catalyzed hydrogenolysis of 17 afforded MEBP (3).
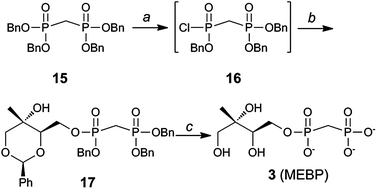 | ||
| Scheme 2 Synthesis of MEBP 3. Conditions: (a) (i) Quinuclidine, toluene, 100 °C, 1 h; (ii) 5% HCl; (iii) (CH3)2CC(Cl)N(CH3)2, CH2Cl2, 40 °C; (b) 8, iPr2NEt, DMAP, 51–65% over 3 steps; (c) H2, Pd/C, MeOH, 70–80 p.s.i., 2.5 h, 66%. | ||
Biochemical evaluation of IspF modulating activity of stable substrate analogs
Bisphosphonate-containing substrate analogs 1 (CBPME), 2 (CBPME2P), 3 (MEBP) and control compound 4 (CBP) were evaluated as modulators of IspF activity and the more active IspF–MEP complex.CBPME ( 11111111111111111 ) and CBPME2P ( 2222222222222222 ). The stable IspF substrate analogs CBPME and CBPME2P are, in principle, incapable of completing the cyclization reaction to release CMP and were therefore expected to exhibit inhibitory activity against the enzyme. Contrary to our expectations, the closest structural homolog to the natural substrate, CBPME2P, lacks inhibitory activity against IspF (column d, Fig. 3). Rather, CBPME2P prevents loss of enzyme activity to some degree (column l, Fig. 3), as evidenced by a retention of ∼60% activity following a 30 minute pre-incubation (a 7.6-fold enhancement in the rate of CMP formation, compared to control after the same pre-incubation, Table S1†). In contrast, CBPME2P exhibits weak, time-dependent inhibition of the IspF–MEP complex (37% inhibition following 30 min pre-incubation, column p in Fig. 3, Table S1†). These results are consistent with our previous findings that IspF and the IspF–MEP complex display distinct sensitivities to inhibitors,17 and suggest CBPME2P exhibits a surprisingly weak affinity for the enzyme.
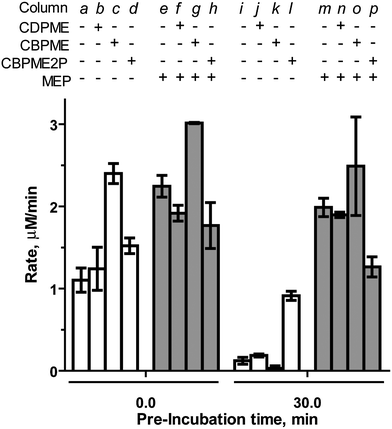 | ||
| Fig. 3 Effects of CDPME, CBPME or CBPME2P on the rate of CMP formation catalyzed by IspF or the IspF–MEP complex, in the presence of 100 μM CDPME2P. Substrate analogs and MEP are added to a final concentration of 500 μM. | ||
CBPME (1) is a close structural analog of the IspE substrate CDPME, also known to undergo IspF-catalyzed cyclization to give MEcP (Fig. S2†).10 In contrast to its phosphorylated counterpart (CBPME2P), CBPME enhances IspF activity 2.2-fold (column c in Fig. 3, Table S1†), but fails to stabilize the enzyme through a 30 minute pre-incubation (column k). This is also in contrast to its diphosphate counterpart, CDPME, which neither enhances activity nor stabilizes IspF (columns b and k in Fig. 3).17 CBPME appears to subtly enhance activity of the IspF–MEP complex, producing a modest 1.3-fold increase in the rate of CMP formation compared to the IspF–MEP complex alone (column g in Fig. 3, Table S1†), and this enhanced activity is not significantly affected following pre-incubation of CBPME with the IspF–MEP complex (column o, Fig. 3). This is again in contrast to its diphosphate counterpart, CDPME, which has no apparent effect on the IspF–MEP complex (Fig. 3, column n).
MEBP ( 333333333333333 ) and CBP ( 444444444444444444 ). MEBP (3) lacks the cytidyl component recognized by “pocket III” in the IspF active site, but retains the unique methylerythritol group recognized by “pocket II” and was therefore expected to display inhibitory activity. CBP (4) was used as a bisphosphonate-containing control and stable counterpart for the known inhibitor CDP. Interestingly, MEBP enhances and sustains activity of IspF. At a final concentration of 500 μM, MEBP modestly increases the rate of CMP formation by 1.6-fold (Table S2†). However, in the presence of 1000 μM MEBP, IspF activity is further enhanced to 1.8-fold above control (column b, Fig. 4), comparable to enhancement observed by the structurally related activator MEP.17 At this higher concentration, MEBP also exhibits profound stabilizing effects, retaining 54% activity (column j, Fig. 4) and representing an 8.7-fold increase in the rate of CMP formation compared to the control experiment after the same pre-incubation. Notably, MEBP does not modulate activity of the IspF–MEP complex (Fig. 4 and Table S2†).
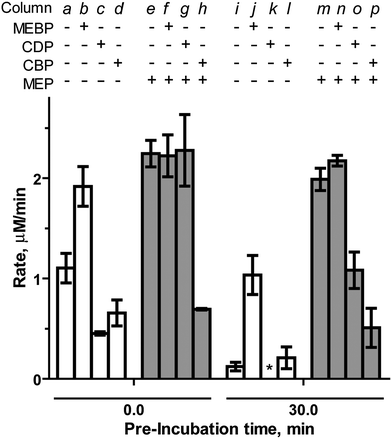 | ||
| Fig. 4 Effects of MEBP (1000 μM) and CBP (700 μM) and CDP (700 μM) on the rate of CMP formation catalyzed by IspF or the IspF–MEP complex in the presence of 100 μM CDPME2P. *No activity was observed in the presence of CDP following a 30 minute pre-incubation. | ||
As expected, CBP inhibits IspF (IC50 = 797.3 ± 57 μM, Fig. S4†) with an activity comparable to CDP (IC50 = 768 μM).17 However, while the introduction of MEP ablates inhibitory activity of CDP (column g in Fig. 4),17 CBP exhibits enhanced inhibitory activity against the IspF–MEP complex (IC50 = 343.4 ± 24 μM, Fig. S4†). Further, CDP displays apparent time-dependent inhibition of the IspF–MEP complex (46% inhibition following pre-incubation, column o) whereas the more pronounced inhibitory activity of CBP against the IspF–MEP complex does not appear to be time-dependent (69% inhibition at 0 minutes versus 74% inhibition following pre-incubation with IspF–MEP).
Discussion
The MEP pathway is indispensable in many pathogenic organisms;11,12 thus the enzymes of this metabolic pathway present new targets for the development of anti-infective agents. The fifth enzyme, cyclodiphosphate synthase IspF, has been the focus of efforts to develop inhibitors of isoprenoid biosynthesis.11–16 Rationally designed IspF inhibitors incorporate recognition elements for the cytidine binding pocket whereas compounds identified from a high throughput screen inhibit via an unknown mechanism.16 The present study attempted new approaches for the design of IspF inhibitors, and resulted in unexpected yet enlightening outcomes. Despite a lack of potent inhibition by any of the IspF substrate analogs presented here, this study emphasizes several important points that should be considered in the design and evaluation of future IspF inhibitors. First, and consistent with our previous findings,17 we provide new examples which highlight the striking differences in the behaviors of IspF and the IspF–MEP complex in the presence of modulators. It is clear that a better understanding of the mechanism and physiological relevance of IspF activation by MEP is required to fully appreciate IspF as a potential drug target, and attention should be given to the conditions under which IspF inhibitors are screened.Second, although we postulated that incorporation of the methylerythritol element in potential IspF modulators could direct binding to “pocket II” of the IspF active site and cause inhibition, it appears the methylerythritol component instead serves as a recognition element to enhance and/or sustain IspF activity. In the present study, this is demonstrated best by MEBP, which appears to behave as a lower affinity MEP analog to enhance and sustain IspF activity.
Finally, incorporation of a stable bisphosphonate scaffold, a technique commonly used to study phosphoryl transfer enzymes, offers little to impart potent inhibitory activity through stabilizing a transition state conformation around the penta-coordinate intermediate in IspF catalysis. Direct comparisons of bisphosphonate to diphosphate counterparts (CDPME vs. CBPME and CDP vs. CBP) suggest that the bisphosphonate linkage is not a suitable isostere for diphosphate in this system. The pKa values for the first and second ionizations are well below physiologic pH for diphosphate and bisphosphonate scaffolds; thus, we presume CDPME and CBPME are both dianionic under the assay conditions. The different activities of CDPME and CBPME might therefore arise from differences in steric properties of diphosphate and bisphosphonate linkages;36 this phenomenon might also explain the unexpected weak inhibition by CBPME2P against the IspF–MEP complex. In contrast, it is possible that the observed differences in inhibitory activities of CDP and CBP may arise from differences in ionization characteristics, as the pKa for the third ionization is higher for methylenebisphosphonate compared to diphosphate.
Notes and references
- J. C. Sacchettini and C. D. Poulter, Science, 1997, 277, 1788 CrossRef.
- I. Buhaescu and H. Izzedine, Clin. Biochem., 2007, 40, 575 CrossRef.
- M. Rohmer, Pure Appl. Chem., 2003, 75, 375 CrossRef CAS.
- V. S. Dubey, R. Bhalla and R. Luthra, J. Biosci., 2003, 28, 637 Search PubMed.
- W. Hunter, C. Bond, M. Gabrielsen and L. Kemp, Biochem. Soc. Trans., 2003, 31, 537 CAS.
- V. S. Dubey, Curr. Sci., 2002, 83, 685 CAS.
- S. N. J. Moreno and Z. Li, Expert Opin. Ther. Targets, 2008, 12, 253 CrossRef CAS.
- M. Rodriguez-Concepcion, Curr. Pharm. Des., 2004, 10, 2391 CrossRef CAS.
- M. Takagi, T. Kuzuyama, K. Kaneda, H. Watanabe, T. Dairi and H. Seto, Tetrahedron Lett., 2000, 41, 3395 CrossRef CAS.
- S. Herz, J. Wungsintaweekul, C. A. Schuhr, S. Hecht, H. Lüttgen, S. Sagner, M. Fellermeier, W. Eisenreich, M. H. Zenk, A. Bacher and F. Rohdich, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 2486 CrossRef CAS.
- L. Buetow, A. Brown, T. Parish and W. Hunter, BMC Struct. Biol., 2007, 7, 68 CrossRef.
- S. Steinbacher, J. Kaiser, J. Wungsintaweekul, S. Hecht, W. Eisenreich, S. Gerhardt, A. Bacher and F. Rohdich, J. Mol. Biol., 2002, 316, 79 CrossRef CAS.
- T. L. Campbell and E. D. Brown, J. Bacteriol., 2002, 184, 5609 CrossRef CAS.
- C. M. Crane, J. Kaiser, N. L. Ramsden, S. Lauw, F. Rohdich, W. Eisenreich, W. N. Hunter, A. Bacher and F. Diederich, Angew. Chem., Int. Ed., 2006, 45, 1069 CrossRef CAS.
- C. Baumgartner, C. Eberle, F. Diederich, S. Lauw, F. Rohdich, W. Eisenreich and A. Bacher, Helv. Chim. Acta, 2007, 90, 1043 CrossRef CAS.
- J. G. Geist, S. Lauw, V. Illarionova, B. Illarionov, M. Fischer, T. Gräwert, F. Rohdich, W. Eisenreich, J. Kaiser, M. Groll, C. Scheurer, S. Wittlin, J. L. Alonso-Gómez, W. B. Schweizer, A. Bacher and F. Diederich, ChemMedChem, 2010, 5, 1092 CrossRef CAS.
- J. K. Bitok and C. F. Meyers, ACS Chem. Biol., 2012 DOI:10.1021/cb300243w.
- T. Sgraja, L. E. Kemp, N. Ramsden and W. N. Hunter, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2005, 61, 625 Search PubMed.
- S. B. Richard, J. Ferrer, M. E. Bowman, A. M. Lillo, C. N. Tetzlaff, D. E. Cane and J. P. Noel, J. Biol. Chem., 2002, 277, 8667 CrossRef CAS.
- M. R. Webster, M. Zhao, M. A. Rudek, C. L. Hann and C. L. J. Freel Meyers, Med. Chem., 2011, 54, 6647 CrossRef CAS.
- B. Müller, T. J. Martin, C. Schaub and R. R. Schmidt, Tetrahedron Lett., 1998, 39, 509 CrossRef CAS.
- H. Streicher and H. Busse, Bioorg. Med. Chem., 2006, 14, 1047 CrossRef CAS.
- R. Norlin, L. Juhlin, P. Lind and L. Trogen, Synthesis, 2005, 1765 CrossRef CAS.
- J. G. Bendall, A. N. Payne, T. E. O. Screen and A. B. Holmes, Chem. Commun., 1997, 1067 RSC.
- M. Urbansky, C. E. Davis, J. D. Surjan and R. M. Coates, Org. Lett., 2004, 6, 135 CrossRef CAS.
- P. Narayanasamy, H. Eoh and D. C. Crick, Tetrahedron Lett., 2008, 49, 4461 CrossRef CAS.
- S. C. Tobias and R. F. Borch, Mol. Pharmaceutics, 2004, 1, 112 CAS.
- V. Nahum, G. Zündorf, S. A. Lévesque, A. R. Beaudoin, G. Reiser and B. Fischer, J. Med. Chem., 2002, 45, 5384 CrossRef CAS.
- B. E. Griffin, M. Jarman, C. B. Reese and J. E. Sulston, Tetrahedron, 1967, 23, 2301 CrossRef CAS.
- S. S. Jones, B. Rayner, C. B. Reese, A. Ubasawa and M. Ubasawa, Tetrahedron, 1980, 36, 3075 CrossRef CAS.
- W. Wu, J. Sigmond, G. J. Peters and R. F. Borch, J. Med. Chem., 2007, 50, 3743 CrossRef CAS.
- V. Illarionova, J. Kaiser, E. Ostrozhenkova, A. Bacher, M. Fischer, W. Eisenreich and F. Rohdich, J. Org. Chem., 2006, 71, 8824 CrossRef CAS.
- P. C. B. Page, M. J. McKenzie and J. A. Gallagher, Synth. Commun., 2002, 32, 211 CrossRef CAS.
- P. C. B. Page, M. J. McKenzie and J. A. Gallagher, J. Org. Chem., 2001, 66, 3704 CrossRef.
- M. Saady, L. Lebeau and C. Mioskowski, J. Org. Chem., 1995, 60, 2946 CrossRef CAS.
- M. J. Rogers, R. G. G. Russell, G. M. Blackburn, M. P. Williamson and D. J. Watts, Biochem. Biophys. Res. Commun., 1992, 189, 414 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The methylerythritol phosphate (MEP) pathway; IspF reaction conditions; the synthesis of CBPME, CBPME2P and MEBP; tabulated and normalized rates of CMP formation; CBP inhibition of IspF or IspF–MEP complex. See DOI: 10.1039/c2md20175e |
| This journal is © The Royal Society of Chemistry 2013 |