Truncated militarinone fragments identified by total chemical synthesis induce neurite outgrowth†
Fabian
Schmid
,
Henning J.
Jessen‡
,
Patrick
Burch
and
Karl
Gademann
*
University of Basel, Department of Chemistry, St. Johanns-Ring 19, 4056 Basel, Switzerland. E-mail: karl.gademann@unibas.ch; Tel: +41 61 267 11 44
First published on 13th August 2012
Abstract
A group of natural products produced by entomopathogenic fungi has recently been shown to induce neurite outgrowth in the absence of nerve growth factor (NGF) in pheochromocytoma PC-12 cell culture. After completion of a total synthesis program, we continued our efforts in reducing complexity by synthetically preparing truncated analogs. Using this approach we were able to not only significantly simplify the structures but also decrease the minimum concentration needed for neuritogenic activity by a factor of 20. This activity was suppressed in the presence of an ERK1/2 inhibitor suggesting activation of the mitogen activated protein kinase (MAPK) pathway as a potential mode of action of these pyridone alkaloids.
Introduction
The capacity of natural products as powerful modulators of biological systems has been recognized for centuries, and they remain a prime source of drug discovery.1 This potential can be leveraged by organic synthesis and, in particular, total synthesis of natural products offers the opportunities to obtain powerful derivatives that are not accessible from the natural product itself.2 These approaches (sometimes referred to as ‘chemical editing’ or ‘diverted total synthesis’)3 complement the long-standing goal of mapping out the pharmacophores of natural products and lead to successful drugs in the clinic (octreotide from somatostatin and eribulin from halichondrin).4 Through such diverted total synthesis efforts, we have shown that the antitumor polyketide anguinomycin (1) can be truncated and that fragment 2 retains most of the biological activity with regard to the inhibition of nuclear export (Fig. 1).5 In this study, we report on the truncation of pyridone natural products such as 3 or 4, leading to an optimized fragment of type 5 that induces neurite outgrowth at lower concentrations.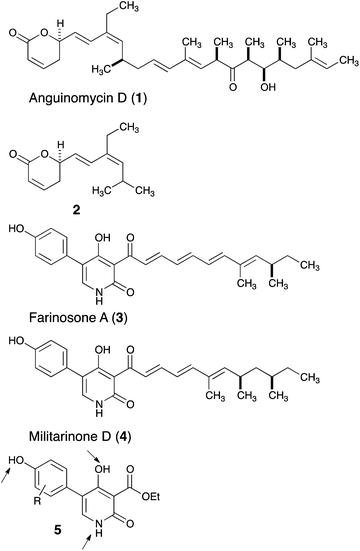 | ||
| Fig. 1 Natural products 1, 3 and 4 and their truncated analogs 2 and 5. | ||
Various pyridone alkaloids have been isolated from entomopathogenic fungi.6 Representative members of this class such as farinosone A (3) and militarinone D (4) have been shown to significantly induce neurite outgrowth in the absence of nerve growth factor (NGF).7 This phenotypic observation could be of interest to target neuritic atrophy, which can be considered a hallmark of neurodegenerative diseases, by reconstruction of neuronal networks using small molecules.8
In the context of our program directed towards the synthesis of neuritogenic natural products,9 we have shown that different naturally occurring pyridone alkaloids can be assembled in a modular way from a bifunctional pyridone core intermediate.9c Starting from this advanced intermediate, we succeeded in the stereoselective preparation of the natural products farinosone A (3), pre-tenellin B, militarinone D, torrubiellone C, putative natural products and their enantiomers in synthetic form.9c–e Interestingly, all of these compounds induced neurite outgrowth in the PC-12 cell line at concentrations of 20 μM. In addition, the activity was found to be generally independent of the length of the side chain as well as the absolute configuration of the stereogenic centers.9c–e Based on these structural insights, we wanted to (1) evaluate whether the side chain could be completely truncated while retaining activity and (2) perform structure activity relationship studies on the biaryl pyridone system to improve potency.
Results and discussion
Synthesis of the pyridone derivatives
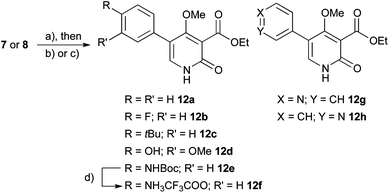
The first goal consisted of the evaluation of the three different H-bond donors (denoted by arrows for lead structure 5 in Fig. 1) on activity. This was achieved by synthesizing pyridones methylated in the H-bond donor positions following a cross-coupling/methylation/demethylation strategy from the advanced intermediates 7 and 8. This gave access to different permutations of methylation patterns (Scheme 1), but also allowed easy introduction of new biaryl scaffolds via a cross-coupling approach (vide supra).![Synthesis of pyridones with different methylation patterns. Reagents and conditions: (a) (4-methoxyphenyl)boronic acid, K2CO3, 5–10% Pd(PPh3)4, glyme–H2O–DMF 9 : 1 : 0.5, 60 °C, 16 h; (b) TBAF, THF, 60 °C, 9 h; (c) {4-[(4-methoxybenzyl)oxy]phenyl}boronic acid, K2CO3, 5–10% Pd(PPh3)4, glyme–H2O–DMF 9 : 1 : 0.5, 60 °C, 16 h; (d) LiI, THF, reflux; (e) MeI, K2CO3, CH3CN; (f) TFA (5% in DCM), rt, 30 min.](/image/article/2013/MD/c2md20181j/c2md20181j-s1.gif) | ||
Scheme 1 Synthesis of pyridones with different methylation patterns. Reagents and conditions: (a) (4-methoxyphenyl)boronic acid, K2CO3, 5–10% Pd(PPh3)4, glyme–H2O–DMF 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.5, 60 °C, 16 h; (b) TBAF, THF, 60 °C, 9 h; (c) {4-[(4-methoxybenzyl)oxy]phenyl}boronic acid, K2CO3, 5–10% Pd(PPh3)4, glyme–H2O–DMF 9:1:0.5, 60 °C, 16 h; (d) LiI, THF, reflux; (e) MeI, K2CO3, CH3CN; (f) TFA (5% in DCM), rt, 30 min. :1:0.5, 60 °C, 16 h; (b) TBAF, THF, 60 °C, 9 h; (c) {4-[(4-methoxybenzyl)oxy]phenyl}boronic acid, K2CO3, 5–10% Pd(PPh3)4, glyme–H2O–DMF 9:1:0.5, 60 °C, 16 h; (d) LiI, THF, reflux; (e) MeI, K2CO3, CH3CN; (f) TFA (5% in DCM), rt, 30 min. | ||
The synthesis of pyridones with different methyl substitutions began with the preparation of the pyridone core starting from ethyl cyanoacetate (6) following literature procedures (Scheme 1).9c,10 As previously shown,9c the pyridone could not be directly functionalized using the Suzuki–Miyaura cross-coupling,11 possibly due to inactivation of the catalyst by the bidentate pyridone serving as ligand. Therefore, the SEM protecting group was introduced, yielding both the N- and O-protected derivatives 7 and 8 quantitatively as a 3:2 mixture.12 Both of these compounds were useful in the course of the project, amenable to cross-coupling reactions, and therefore no attempts were made to obtain a single entity. Furthermore, they could be readily prepared and separated on a gram scale. The N-protected pyridone 8 was functionalized with either (4-methoxyphenyl)boronic acid (conditions a) or {4-[(4-methoxybenzyl)oxy]phenyl}boronic acid (conditions c) to allow access to different methyl substitution patterns. For these building blocks quantitative yields could be obtained using optimized conditions. The efficiency of the transformations proved to be highly dependent on the composition of the degassed solvent mixture with glyme–water–DMF (9:1:0.5) as the optimal system. Catalyst screening revealed bis(tri-tert-butylphosphine)palladium as the most efficient catalyst (complete conversion after 1 h at rt, 2% catalyst loading), whereas the PEPPSI-IPr-catalyst, for example, did not yield the product. As the former catalyst had to be handled in a glove-box for good results, we eventually decided to use Pd(PPh3)4 for our transformations, although at the cost of prolonged reaction times (16 h), higher temperatures (60 °C) and catalyst loadings (5–10%). After removal of the SEM-group with TBAF under reflux, the pyridones 9a and 10 were obtained in quantitative yield. Interestingly, compound 9a was found to crystalize in two polymorphs, with the ethyl-carboxylate moiety rotated about 180° in the two polymorphs (see ESI† for more details). N-Selective methylations were carried out with methyl iodide and potassium carbonate, giving rise to the N-methyl pyridones 9c and 11c. Furthermore, selective cleavage of the C(4)-methoxy ethers with lithium iodide (LiI) allowed for the construction of pyridone 9b and N-methyl pyridone 9d. Also the fully deprotected pyridone 11b could be obtained under these conditions. It is interesting to note that in this series, the rigid biaryl scaffold is decorated with a variety of H-bond donors and acceptors, thus allowing us to evaluate the role of each group. Furthermore, ClogP values shifted from −0.4 for pyridone 11b to 0.5 for methylated pyridone 9c. As will be discussed later in detail, we found that a key structural feature for neuritogenic activity was represented by the 4′-hydroxy-biaryl unit with the 4-hydroxy group of the pyridone core being less important. Therefore, we focused our synthetic efforts on the modification of the peripheral phenyl group and retaining the methyl ether.
As we had chosen an approach employing cross-coupling reactions, we were now able to rapidly introduce different aryl groups derived from commercially available boronic acids and boronic esters. For this purpose, O-SEM protected pyridine 7 or N-SEM protected pyridone 8 were subjected to the previously optimized reaction conditions9c with different building blocks, resulting in yields of the target compounds ranging from 20% for electron deficient systems to 95% for electron rich systems (Scheme 2). Afterwards, the SEM protecting group was removed by either addition of TFA in CH2Cl2 or TBAF in THF, thus yielding the desired pyridones 12a–h including amine 12f after deprotection of 12e with TFA as the TFA salt. The pyridines 12g/h were obtained in low yields owing to their hydrophilicity (ClogP = −1.0), which complicated purifications but provided sufficient material for the bioassays. As initial studies had shown no decrease in activity for compounds where the polyene sidechain was replaced by an ethyl carboxylate moiety, no further modifications at the 3-position were performed for this study. It is also interesting to note that N-methylated 4-hydroxy-pyridones are intrinsically fluorescent compounds emitting blue light.
 | ||
| Scheme 2 Preparation of differently aryl-substituted pyridones. Reagents and conditions: (a) corresponding boronic acid, K2CO3, 5–10% Pd(PPh3)4, glyme–H2O–DMF 9:1:0.5, 60 °C, 16 h; (b) TBAF, THF, 60 °C, 9 h; (c) 2 equiv. TFA, DCM, 5 min; (d) 50% TFA in DCM, 10 min. | ||
Investigation of the neuritogenic properties in the PC-12 assay
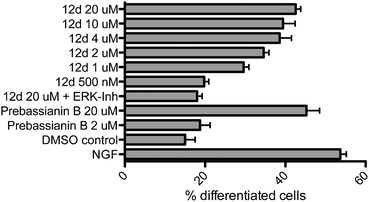
All compounds were examined concerning their ability to induce neurite outgrowth in the PC-12 assay7d,9e,13 (rat pheochromocytoma). Briefly, PC-12 cells were grown in DMEM (Dulbecco's modified Eagle medium, high glucose + L-glutamine) containing antibiotics (penicillin, streptomycin) under sterile conditions. Cells were seeded into collagene coated 24 well plates (105 cells per well) after passage through a 21-gauge needle and after two days fresh medium containing the different compounds was added. The cells were incubated for three more days in a humidified atmosphere at 37 °C (5% CO2) with the compounds, then fixed with formaldehyde, stained with Giemsa stain and analyzed under a microscope. Pictures from three to five randomly chosen areas were taken (see Fig. 2), the fraction of differentiated cells was calculated with more than 500 cells in total and the results were analyzed by ANOVA. Errors are indicated as the standard error of the mean. As negative and positive controls, cells were incubated with DMSO (vector = 0.1%) or NGF 7S (nerve growth factor from the murine submaxillary gland 10 ng mL−1), respectively. For the active compounds, dilution series ranging from 500 nM to 20 μM were evaluated, which constituted the benchmark concentration obtained from earlier analyses of the natural products such as farinosone A (3). To get further insight into the mode of action of these compounds, the most efficient one was additionally incubated with PC-12 cells in the presence of 5 μM ERK1/2 inhibitor (extracellular regulated kinase). The results are summarized in Fig. 3 and 4. | ||
| Fig. 2 Micrographs of the incubated PC-12 cells: 12d 20 μM (left), NGF positive control (middle) and DMSO blank (right). | ||
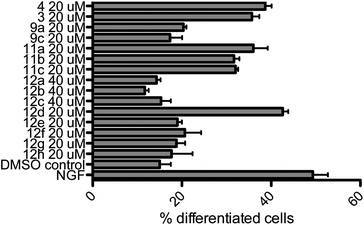 | ||
| Fig. 3 Neuritogenic activity of the pyridones in the PC-12 assay. Nerve growth factor (NGF) control: 10 ng mL−1. DMSO control: 0.1%. Incubation period: 3 days. Number of counted cells: >500. Error bars denote SEM. | ||
 | ||
| Fig. 4 Neuritogenic activity of 12d in the PC-12 assay. Nerve growth factor (NGF) control: 10 ng mL−1. DMSO control: 0.1%. Incubation period: 3 days. Number of counted cells: >500. Error bars denote SEM. | ||
A hypothesis for the pharmacophore can be delineated from these experimental observations. Compounds 9a and 9c did not display any neuritogenic activity at 20 μM. In contrast, compounds 11a–c were all active at this concentration. This underlined the importance of the unprotected para-hydroxy group on the phenyl moiety. Further evidence was provided by the inactivity of phenyl- or tert-butyl modified pyridone 12a/c even at concentrations of 40 μM. In contrast to all these inactive compounds, the initial hypothesis of whether the complex polyene side chains of the natural products can be truncated was corroborated by the activity of the truncated analogs 11a–c, leading to compounds that were as active as the natural products. Our next aim was to improve this activity.
In order to investigate the role of the phenol moiety, we investigated the replacement of the OH group by F, NHR or NH2 groups as found in the pyridones 12b, 12e and f, but none of these modifications provided active compounds, even at concentrations of 40 μM. Another group of compounds investigated were the pyridines 12g/h; again, these compounds were found to be inactive, further underlining the importance of the phenolic OH-group for activity. An interesting result was achieved with the catechol derived structure 12d. Already in our first assays at 20 μM concentration, we observed a convincing induction of neurite outgrowth as can be seen from the micrographs shown in Fig. 2.
Catechol derived pyridone 12d induced neurite outgrowth with a comparable efficiency to the natural products farinosone A (3) and prebassianin B9c at 20 μM. Furthermore, this neuritogenic effect could be observed down to concentrations of 2 μM of 12d. At a concentration of 1 μM an effect could still be observed, whereas at 500 nM the neuritogenic effect was not observable anymore. In contrast, prebassianin B treated cultures were devoid of significant neurite outgrowth below 2 μM concentrations.
As a control compound, phenyl pyridine 13 (Fig. 5) was prepared and tested, but this compound did not significantly induce neuritogenesis at the 20 μM concentration.
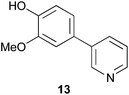 | ||
| Fig. 5 Phenyl pyridine 13. | ||
Neurite outgrowth promoted via NGF activation of the MAP kinase can be suppressed by small organic molecules.14 In particular, it has been shown that the commercially available inhibitor PD98059 selectively inhibits activation of MEK1 and MEK2 with IC50 values of 4 μM and 50 μM, respectively.15 Thus, we decided to investigate the influence of inhibition of the MAP kinase pathway on neurite outgrowth induced by compound 12d.
The neuritogenic effect of pyridone 12d (20 μM) was completely blocked by co-incubation with the commercially available ERK1/2 inhibitor PD98059 (5 μM), thus suggesting the activation of the MAP-kinase pathway by these pyridone alkaloid derived structures as one pathway of biological activation. Indeed, the same effect in the presence of the ERK1/2 inhibitor was observed for the natural product militarinone D (4), which is also supported by earlier studies on the activation of MEK/ERK pathways by this metabolite.16 It should be noted that the natural product biaryl phenol 4-O-methylhonokiol, was found to induce neurite outgrowth at similar concentrations via ERK pathway activation in rat embryonic neuronal cells.17
Conclusion
In summary, this study discloses the structural simplification of complex polyene pyridone alkaloid natural products by replacing the lipophilic and configurationally unstable polyene side chain containing stereogenic centers and modifications of the phenyl group. It was found that methylation at the 1- or 4-position did not significantly influence activity. A 4′-hydroxy substituent was found to be necessary for activity, as none of the derivatives bearing a functional group other than 4′-OH were found to be active. These optimization efforts resulted in the catecholate pyridone derivative 12d, which induces neurite outgrowth at markedly lower concentrations compared to its natural product progenitors, while cutting the number of carbon atoms from 26 to 16 and reducing the overall number of synthetic transformations by 12. As already demonstrated in the anguinomycin case,5 truncated natural products can offer advantages of concomitantly increasing biological activity while reducing structural complexity.Acknowledgements
K. G. is a European Young Investigator (EURYI). We gratefully acknowledge financial support by the DFG (Project JE 572/1-1) and the SNF (PE002-117136/1) and thank Dr Neuburger for X-ray analyses.Notes and references
- For an overview, see: D. Newman and G. Cragg, J. Nat. Prod., 2007, 70, 461–477 CrossRef CAS.
- Reviews: (a) R. M. Wilson and S. J. Danishefsky, J. Org. Chem., 2006, 71, 8329–8351 CrossRef CAS; (b) A. M. Szpilman and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 9592–9628 CrossRef CAS; (c) K. Gademann and S. Sieber, Chimia, 2011, 65, 835–838 CrossRef CAS.
- See for examples: (a) A. Fürstner, D. Kirk, M. D. B. Fenster, C. Aissa, D. De Souza and O. Müller, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8103–8108 CrossRef; (b) T. Oskarsson, P. Nagorny, I. J. Krauss, L. Perez, M. Mandal, G. Yang, O. Ouerfelli, D. Xiao, M. A. S. Moore, J. Massagué and S. J. Danishefsky, J. Am. Chem. Soc., 2010, 132, 3224–3228 CrossRef CAS; (c) A. Fürstner, E. Kattnig, G. Kelter and H. H. Fiebig, Chem.–Eur. J., 2009, 15, 4030–4043 CrossRef; (d) J. Gagnepain, E. Moulin, C. Nevado, M. Waser, A. Maier, G. Kelter, H. H. Fiebig and A. Fürstner, Chem.–Eur. J., 2011, 17, 6973–6984 CrossRef CAS.
- Review: J.-Y. Wach and K. Gademann, Synlett, 2012, 163–170 CAS.
- (a) S. Bonazzi, O. Eidam, S. Güttinger, J.-Y. Wach, I. Zemp, U. Kutay and K. Gademann, J. Am. Chem. Soc., 2010, 132, 1432–1442 CrossRef CAS; (b) S. Bonazzi, S. Güttinger, I. Zemp, U. Kutay and K. Gademann, Angew. Chem., Int. Ed., 2007, 46, 8707–8710 CrossRef CAS.
- Review: H. J. Jessen and K. Gademann, Nat. Prod. Rep., 2010, 27, 1168–1185 RSC.
- (a) K. Schmidt, W. Gunther, S. Stoyanova, B. Schubert, Z. Li and M. Hamburger, Org. Lett., 2002, 4, 197–199 CrossRef CAS; (b) K. Schmidt, U. Riese, Z. Li and M. Hamburger, J. Nat. Prod., 2003, 66, 378–383 CrossRef CAS; (c) Y. Cheng, B. Schneider, U. Riese, B. Schubert, Z. Li and M. Hamburger, J. Nat. Prod., 2004, 67, 1854–1858 CrossRef CAS; (d) U. Riese, E. Ziegler and M. Hamburger, FEBS Lett., 2004, 577, 455–459 CrossRef CAS; (e) Y. Cheng, B. Schneider, U. Riese, B. Schubert, Z. Li and M. Hamburger, J. Nat. Prod., 2006, 69, 436–438 CrossRef CAS.
- Reviews: (a) C. Tohda, T. Kuboyama and K. Komatsu, Neurosignals, 2005, 14, 34–45 CrossRef CAS; (b) R. M. Wilson and S. J. Danishefsky, Acc. Chem. Res., 2006, 39, 539–549 CrossRef CAS; (c) P. M. Joyner and R. H. Cichewicz, Nat. Prod. Rep., 2011, 28, 26–47 RSC; (d) J. Qi, Y. Luo and L. Gao, Mini-Rev. Med. Chem., 2011, 11, 658–677 CrossRef CAS . Selected recent examples: ; (e) X. Cheng, N. Harzdorf, Z. Khaing, D. Kang, A. M. Camelio, T. Shaw, C. E. Schmidt and D. Siegel, Org. Biomol. Chem., 2012, 10, 383–393 RSC; (f) J. Xu, L. Trzoss, W. K. Chang and E. A. Theodorakis, Angew. Chem., 2011, 123, 3756–3760 CrossRef; (g) M. Li, K.-S. Tsang, S.-T. Choi, K. Li, P.-C. Shaw and K.-F. Lau, ChemBioChem, 2011, 12, 449–456 CrossRef CAS; (h) P. Wang, X. Ran, H. Luo, J. Hu, R. Chen, Q. Ma, H. Dai, Y. Liu, M. Xie and J. Zhou, Org. Lett., 2011, 13, 3036–3039 CrossRef CAS; (i) H. Yasui, N. Ito, T. Yamamori, H. Nakamura, J. Okano, T. Asanuma, T. Nakajima, M. Kuwabara and O. Inanami, Free Radical Res., 2010, 44, 645–654 CrossRef CAS; (j) H. Imagawa, H. Saijo, T. Kurisaki, H. Yamamoto, M. Kubo, Y. Fukuyama and M. Nishizawa, Org. Lett., 2009, 11, 1253–1255 CrossRef CAS.
- (a) E. Elamparuthi, C. Fellay, M. Neuburger and K. Gademann, Angew. Chem., Int. Ed., 2012, 51, 4071–4073 CrossRef CAS; (b) C. K. Jana, J. Hoecker, T. M. Woods, H. J. Jessen, M. Neuburger and K. Gademann, Angew. Chem., Int. Ed., 2011, 50, 8407–8411 CrossRef CAS; (c) H. J. Jessen, A. Schumacher, T. Shaw, A. Pfaltz and K. Gademann, Angew. Chem., Int. Ed., 2011, 50, 4222–4226 CrossRef CAS; (d) H. J. Jessen, A. Schumacher, F. Schmid, A. Pfaltz and K. Gademann, Org. Lett., 2011, 13, 4368–4370 CrossRef CAS; (e) H. J. Jessen, D. Barbaras, M. Hamburger and K. Gademann, Org. Lett., 2009, 11, 3446–3449 CrossRef CAS.
- B. Kasum and R. H. Prager, Aust. J. Chem., 1983, 36, 1455–1467 CrossRef CAS . See also: R. C. F. Jones, A. K. Choudhury, J. N. Iley, M. E. Light, G. Loizou and T. A. Pillainayagam, Beilstein J. Org. Chem., 2012, 8, 308–312 CrossRef; R. C. F. Jones, A. K. Choudhury, J. N. Iley, G. Loizou, C. Lumley and V. McKee, Synlett, 2010, 654–658 CrossRef; B. H. Patel, A. M. Mason and A. G. M. Barrett, Org. Lett., 2011, 13, 5156–5159 CrossRef.
- N. Miyaura and A. Suzuki, J. Chem. Soc., Chem. Commun., 1979, 866–867 RSC.
- For a detailed study on 2-pyridone reactivity, see: M. Breugst and H. Mayr, J. Am. Chem. Soc., 2010, 132, 15380–15389 CrossRef CAS.
- (a) L. A. Greene and A. S. Tischler, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 2424–2428 CrossRef CAS; (b) I. Dikic, J. Schlesinger and I. Lax, Curr. Biol., 1994, 4, 702–708 CrossRef CAS; (c) Y. Obara, T. Akoi, M. Kusano and Y. Ohizumi, J. Pharmacol. Exp. Ther., 2002, 301, 803–811 CrossRef CAS.
- L. Pang, T. Sawada, S. T. Decker and A. R. Saltiel, J. Biol. Chem., 1995, 270, 13585–13588 CrossRef CAS.
- D. R. Alessi, A. Cuenda, P. Cohen, D. T. Dudley and A. R. Saltiel, J. Biol. Chem., 1995, 270, 27489–27494 CrossRef CAS.
- P. Küenzi, S. Kiefer, A. Koryakina and M. Hamburger, Apoptosis, 2008, 13, 364–376 CrossRef.
- Y. K. Lee, I. S. Choi, Y. H. Kim, K. H. Kim, S. Y. Nam, Y. W. Yun, M. S. Lee, K. W. Oh and J. T. Hong, Neurochem. Res., 2009, 34, 2251–2260 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full characterisation data and experimental procedures are available. CCDC 888459–888461. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2md20181j |
| ‡ Present address: University of Zürich, Institute of Organic Chemistry, Winterthurerstrasse 190, 8057 Zürich, Switzerland. |
| This journal is © The Royal Society of Chemistry 2013 |