Discovery of hydroxamate bioisosteres as KAT II inhibitors with improved oral bioavailability and pharmacokinetics†‡
Jaclyn L.
Henderson
,
Aarti
Sawant-Basak
,
Jamison B.
Tuttle
,
Amy B.
Dounay
,
Laura A.
McAllister
,
Jayvardhan
Pandit
,
Suobao
Rong
,
Xinjun
Hou
,
Bruce M.
Bechle
,
Ji-Young
Kim
,
Vinod
Parikh
,
Somraj
Ghosh
,
Edelweiss
Evrard
,
Laura E.
Zawadzke
,
Michelle A.
Salafia
,
Brian
Rago
,
Ronald S.
Obach
,
Alan
Clark
,
Kari R.
Fonseca
,
Cheng
Chang
and
Patrick R.
Verhoest
*
Neuroscience Medicinal Chemistry, 700 Main Street, Cambridge, MA 12139, USA. E-mail: patrick.r.verhoest@pfizer.com
First published on 13th August 2012
Abstract
A series of kynurenine aminotransferase II (KAT II) inhibitors has been developed replacing the hydroxamate motif with a bioisostere. Triazolinones or triazoles have proven to be effective replacements with significantly improved pharmacokinetics including reduced clearance and increased bioavailability. An X-ray crystal structure of an inhibitor bound in KAT II confirms that the irreversible binding to the co-factor is maintained and that the heterocycles make productive hydrogen bonds to the arginine-399.
Introduction
Treatment of the cognitive disruption in schizophrenia is a high unmet medical need with no therapies approved for these deficits. Evidence in the literature has pointed to a disruption in the tryptophan metabolism pathway with schizophrenic patients showing an elevation in L-kynurenine and kynurenic acid (KYNA) in cerebral spinal fluid (CSF) and post mortem prefrontal cortex.1,2 Kynurenic acid is one of the terminal products in the pathway and is produced from L-kynurenine by the kynurenine aminotransferase (KAT) family of enzymes. The predominant isoform in human brain tissue is the KAT II enzyme.3 Targeting inhibitors of this enzyme is a strategy to reduce central KYNA burden in a disease state (Fig. 1).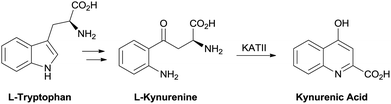 | ||
| Fig. 1 Kynurenic acid biosynthesis. | ||
Kynurenic acid has a diverse and interesting pharmacology. KYNA was first reported as a native antagonist at the glycine site of the N-methyl-D-aspartate (NMDA) receptor and later reported to be an inhibitor of the alpha7 nicotinic ion channel.4,5 Inhibition of both of these pathways in schizophrenics has been hypothesized to contribute to the disease. More recently KYNA has been shown to be a potent agonist at the arylhydrocarbon (AhR) receptor. Endogenous brain levels of KYNA are consistent with its AhR affinity making the AhR receptor and its pharmacology of significant interest in understanding the biological relevance of decreasing KYNA levels in disease.6
Recent literature reports have utilized selective tool compounds (S)-ESBA (1) and BFF-122 (2) via central administration to explore the pharmacology of KAT II inhibition in vivo.7–9 We have reported the first brain penetrant KAT II inhibitor PF-04859989 (3), which can lower KYNA levels by 50% in rat prefrontal cortex following subcutaneous administration.10 This potent series of compounds inhibits KAT II irreversibly via formation of an imine adduct with the aldehyde group of the enzyme co-factor pyridoxal phosphate (PLP), followed by tautomerization to an enamine. This newly formed adduct with PLP cannot exit the active site, resulting in irreversible inhibition of the enzyme without covalent linkage to the protein (Fig. 2).
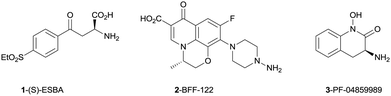 | ||
| Fig. 2 Known inhibitors of KAT II. | ||
Strategy
Although PF-04859989 showed excellent brain penetration and activity in vivo, it suffered from poor pharmacokinetic properties due to rapid O-glucuronidation of the hydroxamate. As shown in the Table 2, PF-04859989 was metabolized to its glucuronide as the major metabolic pathway in human hepatocyctes and human liver microsomes supplemented with UDPGA. Following IV administration to rats, the clearance is significantly greater than blood flow (174 mL min−1 kg−1) with a low oral bioavailability 2–18%. The short systemic half-life of the molecule could be a benefit of an irreversible inhibitor since the pharmacodynamic effect is driven by the protein resynthesis/clearance rate. However, improvement in bioavailability and reduction in glucuronidation would result in less pharmacokinetic interpatient variability and a reduced dose.Our strategy to reduce the clearance and improve bioavailability centered around identifying hydroxamate bioisosteres. We utilized the knowledge from the X-ray crystal structure of 3 in the KAT II active site (Fig. 3). The key interactions that needed to be maintained were the primary amine–PLP adduct as well as identification of a heterocycle that could hydrogen bond with Arg-399 and Asp-202, as the hydroxamate efficiently does. Our modelling suggested that a triazolinone or a triazole replacement could make these desired interactions and maintain selectivity and potency since the KAT II active site is slightly larger than the other family members.11 While 3 had less desirable pharmacokinetics due to glucuronidation, it is an exquisitely efficient inhibitor which allowed for the addition of some size and lipophilicity to replace the optimal hydroxamate to gain potency through other interactions. We had previously identified that incorporation of an O-aryl group para to the hydroxamate had improved our potency and incorporated this knowledge in the design of our bio-isostere replacements.12
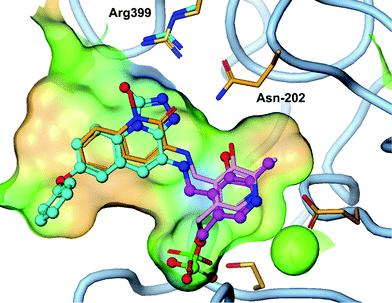 | ||
| Fig. 3 Triazolinone compound 4 (cyan) overlayed with 3 (orange) shows similar hydrogen bond to Arg-399 and Asn-202 as the hydroxamate (PDB 4GDY). | ||
Synthesis
As shown in Scheme 1, the triazolinone 4 could be accessed via a short sequence from ortho-nitroaryl bromides. After installation of the aryl ether moiety and reduction of the nitro group, a Negishi reaction and concomitant cyclisation provided the key lactam intermediate.13 This is in contrast to the synthesis of hydroxamate derivatives in which the amino acid side chain was installed prior to careful reduction of the nitro group.14 Conversion of the lactam to the thio lactam and subsequent methylation allowed for condensation with ethyl hydrazine carboxylate. Unexpectedly, racemization of the amino-bearing stereocenter occurred during this step. The synthesis was completed by cyclisation and deprotection.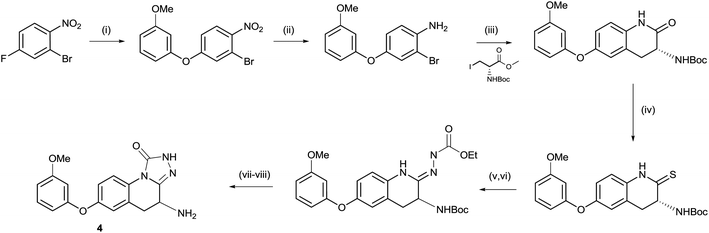 | ||
Scheme 1 Synthesis of compound 5. (i) 3-Methoxyphenol, Cs2CO3, MeCN, 25 °C, 12 h, 84%; (ii) Fe, NH4Cl, THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH:H2O 2:1:1, 70 °C, 3 h, 90%; (iii) Zn, I2, DMF then Pd(OAc)2, XPhos, 60 °C, 12 h, 55%; (iv) P2S5, Na2CO3, THF, reflux, 12 h, 75%; (v) MeI, K2CO3, THF, 15 °C, 20 h, 31%; (vi) ethyl hydrazinecarboxylate, EtOH, 70 °C, 4 h, 44%; (vii) DMF, 150 °C, 2 h, 53%; and (viii) HCl, Et2O, 0 °C, 0.5 h, 69%. :MeOH:H2O 2:1:1, 70 °C, 3 h, 90%; (iii) Zn, I2, DMF then Pd(OAc)2, XPhos, 60 °C, 12 h, 55%; (iv) P2S5, Na2CO3, THF, reflux, 12 h, 75%; (v) MeI, K2CO3, THF, 15 °C, 20 h, 31%; (vi) ethyl hydrazinecarboxylate, EtOH, 70 °C, 4 h, 44%; (vii) DMF, 150 °C, 2 h, 53%; and (viii) HCl, Et2O, 0 °C, 0.5 h, 69%. | ||
In order to access the triazole motif, a modified route was employed, shown in Scheme 2. For the pyridyl containing core it was necessary to perform the Negishi reaction prior to reduction of the nitro group, due to the low yields otherwise afforded in this step.15 The nitro group could be rapidly reduced to the amine using zinc, which cyclised spontaneously to the lactam in situ, with none of the corresponding hydroxamate being observed. As in the case of the triazolone, conversion to the thio-lactam was necessary for efficient, acid catalyzed condensation with formic hydrazide, with cyclisation to the triazole occurring in the same step. Again, racemisation of the amine-bearing center was observed.
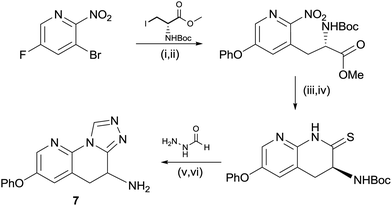 | ||
| Scheme 2 Synthesis of compound 7. (i) Phenol, Cs2CO3, MeCN, 60 °C, 2 h, 99%; (ii) Zn, I2, DMF then Pd(OAc)2, XPhos, 60 °C, 12 h, 28%; (iii) Zn, NH4Cl, THF:MeOH 1:2, 60 °C, 48 h, 73%; (iv) P2S5, Na2CO3, THF, reflux, 1 h, 76%; (v) acetic acid, cyclohexanol, 160 °C, 4 h, 66%; and (vi) HCl, 2-propanol, 25 °C, 2 h, 68%. | ||
Biological results and discussion
The strategy to maintain inhibition of the KAT II enzyme with triazolinone and triazole hydroxamate replacements proved fruitful. The IC50 for the for the triazolone analogues 4 and 5 showed activity less than 100 nM while the triazoles were slightly less effective with an IC50 of 100–200 nM. The triazoles had the advantage of reducing a hydrogen bond donor compared to the hydroxamate and triazolinone which is a desired improvement in a physicochemical property to increase brain penetration.16 To thoroughly understand the SAR of the irreversible inhibition, second order rate constants kinact/Ki were determined to remove the time dependency portion of the potency measure.17 The bioisosteres maintained good potency with inhibition rate constants between 2000 and 5000 M−1 s−1 (Table 1).| Entry | Structure | IC50 (nM) | k inact (min−1) | K i (μM) | k inact/Ki (M−1 s−1) |
|---|---|---|---|---|---|
| a n.d. – not defined. | |||||
| 3 |

|
23 | 0.0156 | 0.0142 | 18500 |
| 4 |

|
84 | n.d. | n.d. | n.d. |
| 5 |

|
65 | 0.0155 | 0.0051 | 5060 |
| 6 |

|
201 | 0.0141 | 0.0815 | 2915 |
| 7 |

|
122 | 0.0203 | 0.0167 | 2160 |
An X-ray crystal structure of 4 in the KAT II enzyme confirms the hypothesis that a triazolinone enhances interaction with Arg-399 and Asn-202 and explains the observed potency. As designed, the triazolinone makes the desired hydrogen bond interactions with Arg-399 and Asn-202, accepting three hydrogen-bonds from Arg-399, and Asn-202. The structure shows a slight backwards shift in the Arg-399 position versus the crystal structure of 3 to accommodate the large heterocyclic ring. We envision that the triazole isostere such as 7 also makes similar hydrogen bonds to the key residues as a hydroxamate replacement. This structure clearly shows that the amine moiety forms a covalent bond with PLP in the enzyme without forming a covalent bond to the protein. This is a desirable interaction since a significant potency gain is achieved without the potential for hapten mediated immune toxicity.18
To confirm the metabolic attributes of the heterocycles, 5 was incubated in human hepatocyctes (HHep) and human liver microsomes (HLMs); metabolic profile was identified using LC-MS/MS as shown in Fig. 4.
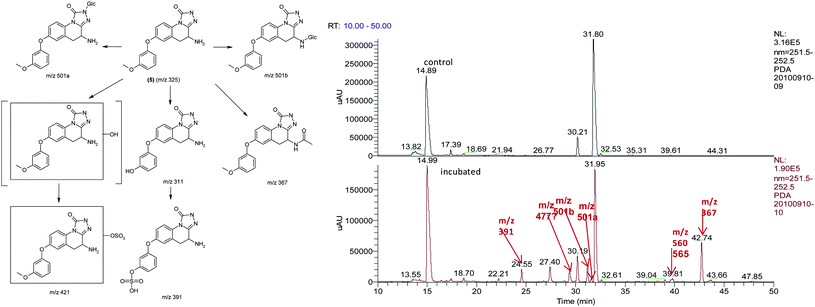 | ||
| Fig. 4 Metabolic identification of 5. | ||
As shown in the LC-MS/MS total ion chromatogram (bottom panel), the peak eluting at 31.80 min corresponding to 5 was the major drug related moiety, suggesting that this compound has low clearance, in in vitro human systems. It was also observed that the major pathways of metabolism were mediated by phase I oxidative mechanisms with direct phase II mediated conjugation i.e. N-glucuronidation at the triazolinone core being a very small component of the total metabolic clearance. Due to the increased confidence from a PK perspective, 5 was profiled in rat and dog PK studies. The intrinsic clearance of 5 was also studied in human, rat and dog liver microsomes.
With confirmation that phase II clearance was reduced from the metabolism identification work with 5, incubation of 7 in HLMs and HHeps demonstrated a prolonged in vitro t1/2 of >120 min. Using the well-stirred model, and incorporation of appropriate binding factors, HLMs predicted a scaled blood clearance of 1.6 mL min−1 kg−1, suggestive of very low turnover in humans. In a similar manner, compound 5 was incubated in HHeps and also demonstrated a prolonged in vitro t1/2 of 114 min, which predicts a low scaled blood clearance of 3.6 mL min−1 kg−1, further confirming low clearance in humans. This was strongly aligned with the metabolic profile of 5. In addition, rat showed a strong in vitro to in vivo correlation in respect to clearance and %F as shown in Table 2. All of the in vitro and in vivo data gave high confidence to the prediction human PK attributes of 5 and 7 using HLMs. Thus, overall oral %F was predicted to be 80–90% in humans contrasting to the low predicted bioavailability of 3.19
| Example 3 (PF-04859989) | Example 5 | Example 7 | |
|---|---|---|---|
| a n.d. not determined. | |||
| t ½ in HHep (min) | n.d. | 114 | 138 |
| t ½ in HLM (min) | n.d. | >104 | >120 |
| Clint in HLM supplemented with UDPGA | 104 | n.d. | n.d. |
| Predicted Clb from HLM | n.d. | 3.6 | |
| Predicted Clb from HLM | n.d. | 1.9 | 1.6 |
| Vdss | 1.05 | 1.4 | 1.4 |
| Predicted plasma elimination t½ (h) | <1 | 10 | 12 |
| Rat IV Clb (mL min−1 kg−1) | n.d. | 78.4 | 20.6 |
| Dog IV Clb (mL min−1 kg−1) | n.d. | 4.5 | n.d. |
| Rat %F | 10.8 | 40 | 95 |
| Dog %F | 18.5 | 100 | n.d. |
| Predicted %F | 10.8 | 92 | 91 |
To further characterize the clinical potential of this series of KAT II inhibitors, it was paramount that brain penetration was maintained. The physicochemical properties of 5 and 7 predicted that they would have good central penetration with each having a CNS MPO scores of 5.17. While these compounds are polar with a ClogP of 1.8(5) and 0.66(7) and a topological polar surface area of 95(5) and 78(7), these are parameters that are in line with marketed CNS drugs and do not impact the potential to generate P-gp substrates. The molecular weight is low for both compounds (324 and 279) and is the dominant physicochemical property predictor for central exposure.20 Compound 5 has an MDR efflux ratio of 1.51 and 7 showed an MDR efflux ratio of 1.24 in a P-gp over expressing cell line suggesting that these compounds would have no brain impairment in vivo.21 As was predicted based on the physicochemical properties and in vitro MDR efflux ratios, both compounds 5 and 7 displayed excellent brain penetration with all three compartments, free plasma, free brain and cerebral spinal fluid essentially in equilibrium (Fig. 5).22
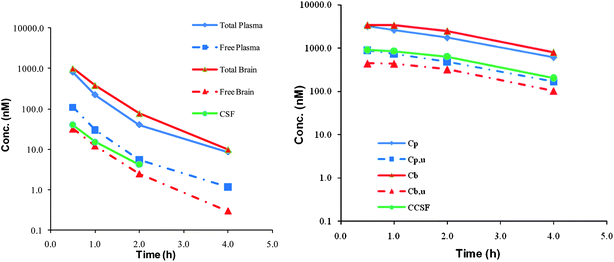 | ||
| Fig. 5 Brain penetration data for compound 5 (left) and 7 (right). Cp = total plasma, Cpu = free plasma, Cb = total brain, Cbu = free brain and CCSF = concentration cerebral spinal fluid. | ||
Ultimately, clinical candidate selection relies on predicted efficacious dose, which represents the culmination of all pharmacokinetic and pharmacodynamic properties. Based on aforementioned favorable pharmacokinetic properties (Table 2), in vitro potency and pharmacodynamic understanding of the KAT II system (to be published), 0.6 mg QD of 5 and 0.2 mg QD dosing of 7 was predicted to be able to lower KYNA level by 50% at steady state. This represented a significant improvement over compound 3 (corresponding dose: 40 mg), which can be attributed to a projected clearance reduction and subsequently improved oral bioavailability.
Conclusion
In conclusion, the design to replace the hydroxamate motif within the previously reported centrally penetrant KAT II inhibitors proved successful. Both the triazolinone and triazole proved to be competent bioisosteres for the hydroxamate. X-ray crystal structure of 4 bound in KAT II confirms that these compounds form a covalent adduct with PLP and are irreversible inhibitors. Additionally the triazolinone and triazole make productive hydrogen bonds to Arg-399 and Asp-202. Metabolite identification studies confirm that the use of the heterocycles blocks the primary glucoronidation route of metabolism of 3. This structural change results in excellent brain penetration and significantly improved projected human pharmacokinetic properties. Ultimately this produces a significantly lower dose projection for 5 and 7 even though they have intrinsically less potency at the KAT II enzyme than 3.References
- K. R. Linderholm, E. Skogh, S. K. Olsson, M. Dahl, M. Holtze, G. Engberg, M. Samuelsson and S. Erhardt, Schizophr. Bull., 2010, 38, 426–432 CrossRef.
- R. Schwarcz, A. Rassoulpour, H. Q. Wu, D. Medoff, C. A. Tamminga and R. C. Roberts, Biol. Psychiatry, 2001, 50, 521–530 CrossRef CAS.
- Q. Han, T. Cai, D. A. Tagle and J. Li, Cell. Mol. Life Sci., 2010, 67, 353–368 CrossRef CAS.
- M. Kessler, T. Terramani, G. Lynch and M. A. Baudry, J. Neurochem., 1989, 52, 1319–1328 CrossRef CAS.
- C. Hilmas, E. F. Pereira, M. Alkondon, A. Rassoulpour, R. Schwarcz and E. X. Albuquerque, J. Neurosci., 2001, 21, 7463–7473 CAS.
- B. C. Natale, I. A. Murray, J. C. Schroeder, C. A. Flaveny, T. S. Lahoti, E. M. Laurenzana, C. J. Omiecinski and G. H. Perdew, Toxicol. Sci., 2010, 115, 89–97 CrossRef.
- R. Pellicciari, R. C. Rizzo, G. Costantino, M. Marinozzi, L. Amori, P. Guidetti, H. Q. Wu and R. Schwarcz, ChemMedChem, 2006, 1, 528–531 CrossRef CAS.
- R. Schwarcz, Y. Kajii and S. Ono, 2009, WO2009064836.
- F. Rossi, C. Valentian, S. Garavaglia, K. Sathyasaikumar, R. Schwarcz, K. Okuwaki, S. Ono, Y. Kajii and M. Rizzi, J. Med. Chem., 2010, 52, 5684–5689 CrossRef.
- A. B. Dounay, M. Anderson, B. M. Bechle, B. M. Campbell, M. M. Claffey, A. Evdokimov, E. Evrard, K. R. Fonseca, X. Gan, S. Ghosh, M. M. Hayward, W. Horner, J.-Y. Kim, L. A. McAllister, J. Pandit, V. Paradis, V. D. Parikh, M. R. Reese, S. Rong, M. A. Salafia, K. Schuyten, C. A. Strick, J. B. Tuttle, J. Valentine, H. Wang, L. E. Zawadzke and P. R. Verhoest, ACS Med. Chem. Lett., 2012, 3, 187–192 CrossRef CAS.
- F. Rossi, S. Garavaglla, V. Montalbano, M. Walsh and M. Rizzi, J. Biol. Chem., 2008, 238, 3559–3566 Search PubMed.
- M. M. Claffey, A. B. Dounay, X. Gan, M. M. Hayward, S. Rong, J. B. Tuttle and P. R. Verhoest, 2010, WO2010146488.
- A. J. Ross, H. L. Lang and R. F. W. Jackson, J. Org. Chem., 2010, 75, 245–248 CrossRef CAS.
- L. A. McAllister, B. M. Bechle, A. D. Dounay, E. Evrard, X. Gan, S. Ghosh, J.-Y. Kim, V. D. Parikh, J. B. Tuttle and P. R. Verhoest, J. Org. Chem., 2011, 76, 3484–3497 CrossRef CAS.
- J. B. Tuttle, J. M. Azzarelli, B. M. Bechle, A. B. Dounay, E. Evrard, X. Gan, S. Ghosh, J. Henderson, J.-Y. Kim, V. D. Parikh and P. R. Verhoest, Tetrahedron Lett., 2011, 52, 5211–5213 CrossRef CAS.
- T. T. Wager, X. Hou, P. R. Verhoest and A. Villalobos, ACS Chem. Neurosci., 2010, 1, 435–449 CrossRef CAS.
- R. A. Copeland, Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, Wiley, Hoboken, NJ, 2005, pp. 1–265 Search PubMed.
- A detailed view of the N-terminal loop is in the ESI.†.
- S. R. Obach, J. G. Baxter, T. E. Liston, B. M. Silber, B. C. Jones, F. MacIntyre, D. J. Rance and P. Wastall, J. Pharmacol. Exp. Ther., 1997, 283, 46–58 Search PubMed.
- T. T. Wager, R. Y. Chandrasekaran, X. Hou, M. D. Troutman, P. R. Verhoest, A. Villalobos and Y. Will, ACS Chem. Neurosci., 2010, 1, 420–434 CrossRef CAS.
- B. Feng, J. B. Mills, R. E. Davidson, R. J. Mireles, J. S. Janiszewski, M. D. Troutman and S. M. de Morais, Drug Metab. Dispos., 2008, 36, 268–275 CrossRef CAS.
- J. C. Kalvass, E. R. Olson, M. P. Cassidy, D. E. Selley and G. M. Pollack, J. Pharmacol. Exp. Ther., 2007, 323, 346–355 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2md20166f |
| ‡ This article is part of a MedChemComm ‘New Talents’ issue highlighting the work of outstanding rising scientists in medicinal chemistry research. |
| This journal is © The Royal Society of Chemistry 2013 |